

Abstract
Here we critically evaluate the role of elevated hematocrit as the principal determinant of thrombotic risk in polycythemia and erythrocytosis, defined by an expansion of red cell mass. Since red cell volume determination is no longer readily available, in clinical practice, polycythemia and erythrocytosis are defined by elevated hemoglobin and hematocrit. Thrombosis is common in Chuvash erythrocytosis and polycythemia vera. Although the increased thrombotic risk is assumed to be due to the elevated hematocrit and an associated increase in blood viscosity, thrombosis does not accompany most types of erythrocytosis. We review studies indicating that the occurrence of thrombosis in Chuvash erythrocytosis is independent of hematocrit, that the thrombotic risk is paradoxically increased by phlebotomy in Chuvash erythrocytosis, and that, when compared to chemotherapy, phlebotomy is associated with increased thrombotic risk in polycythemia vera. Inherited and environmental causes that lead to polycythemia and erythrocytosis are accompanied by diverse cellular changes that could directly affect thrombotic risk, irrespective of the elevated hematocrit. The pressing issue in these disorders is to define factors other than elevated hematocrit that determine thrombotic risk. Defining these predisposing factors in polycythemia and erythrocytosis should then lead to rational therapies and facilitate development of targeted interventions.
Introduction
Polycythemia and erythrocytosis
There are several different parameters for diagnosis of polycythemia and erythrocytosis based on a blood count: the number of red blood cells, the hematocrit, and the hemoglobin concentration. Elevations in these measures can occur on a primary or secondary basis (Table 1).1 Primary polycythemia results from functional abnormalities intrinsic to erythroid progenitors, causing them to be hypersensitive to or independent of erythropoietin. This category includes polycythemia vera (PV), which is associated with acquired somatic mutations in the Janus kinase 2 gene (JAK2), dominantly inherited primary familial and congenital polycythemia or erythrocytosis, caused by germline gain-of-function erythropoietin receptor (EPOR) mutations,2 and erythrocytosis due to SH2B3 mutations.3,4 Primary familial and congenital polycythemia or erythrocytosis predisposes patients to cardiovascular disorders, perhaps due to chronic augmented erythropoietin signaling in all tissues bearing EPOR.2 In contrast, in secondary erythrocytosis, functionally normal erythroid progenitors are exposed to increased levels of circulating erythropoiesis-stimulating factors. In most instances, the erythropoiesis-stimulating factor is erythropoietin, but cobalt, insulin growth factor 1, increased angiotensin signaling and manganese may also stimulate erythropoiesis.1,5,6
Table 1.
Classification of polycythemia and erythrocytosis.
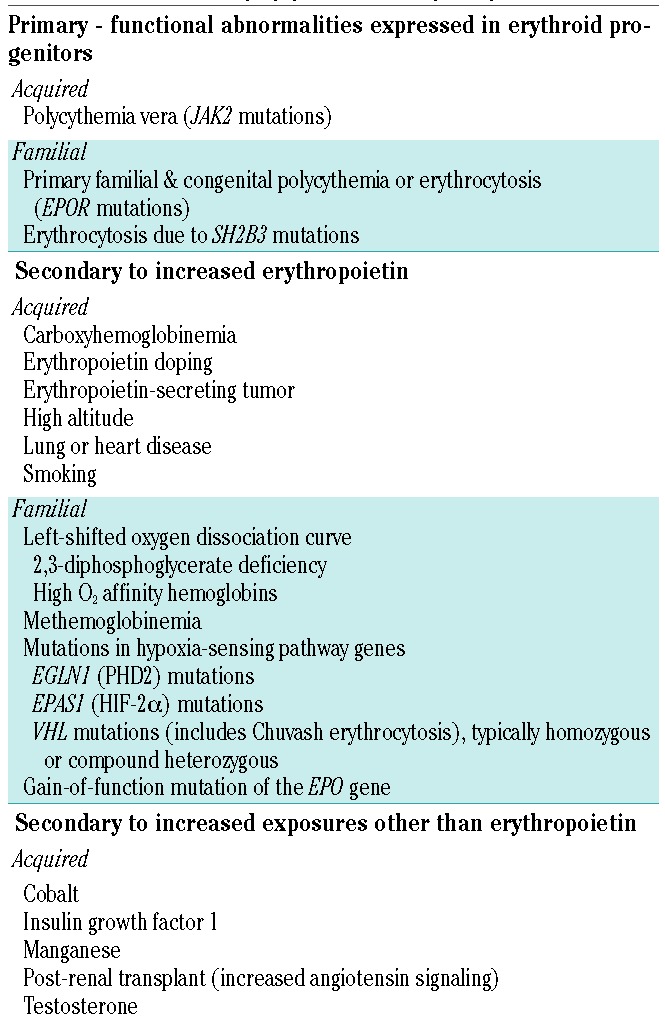
Acquired causes of secondary erythrocytosis include erythrocytosis of pulmonary disease, high altitude erythrocytosis, Eisenmenger syndrome, smoking, carboxyhemoglobinemia, erythropoietin-producing tumors, doping with erythropoietin, post- renal transplant erythrocytosis, exogenous testosterone use, and cobalt and manganese toxicities.1,5,6 Congenital secondary erythrocytosis can be caused by high oxygen affinity hemoglobin variants, inherited low 2,3-diphosphoglycerate leading to high hemoglobin oxygen affinity, congenital methemoglobinemia, and a recently described gain-of-function mutation of the gene encoding erythropoietin (EPO).7 Other congenital conditions include rare germline mutations in hypoxia sensing pathway genes, including loss of function mutations of VHL encoding von Hippel Lindau (VHL) protein and EGLN1 encoding prolyl hydroxylase 2 (PHD2), and gain-of-function mutations of EPAS1 encoding hypoxia inducible factor (HIF)-2α.1
Chuvash erythrocytosis (CE) is an autosomal recessive condition, endemic to Chuvashia in Russia and Ischia in Italy, which results from homozygosity for a C→T missense mutation of VHL (VHL c.598C>T or VHLR200W).8–10 The mutated protein impairs interactions of VHL with the HIF-α subunits, thereby reducing the rate of ubiquitin-mediated HIF-α degradation by the proteasome. As a result, the levels of HIF-1 and HIF-2 heterodimers increase, leading to increased expression of their target genes, including EPO, vascular endothelial growth factor (VEGF), glucose transporter 1 (GLUT1), tissue factor (F3) and a plethora of other genes.9,11,12 In endothelial cells, more than 3% of genes are upregulated by HIF-1.13 CE erythroid progenitors are hypersensitive to erythropoietin, a feature of primary polycythemia, but affected subjects also have increased erythropoietin levels mediated by increased HIF-2, a feature of secondary erythrocytosis.9,14 Similar combined features of both primary and secondary elevations in hematocrit are seen in certain other germline mutations of VHL (loss-of-function mutations) and EPAS1 (gain-of-function mutations).1
Viscosity, hematocrit and blood volume
Both PV and erythrocytosis secondary to hypoxia or upregulated hypoxia sensing are characterized by an increased red cell mass and total blood volume, but the two conditions may at times be divergent with regard to plasma volume. The plasma volume is increased in PV, potentially causing the hematocrit to underestimate the degree of erythrocytosis, whereas the plasma volume may not be increased in all types of erythrocytosis secondary to hypoxia or to upregulated hypoxia sensing.15,16 Some clinical manifestations of erythrocytosis, such as headaches and tinnitus, appear to be related to increased viscosity of blood resulting from the expanded red cell mass and elevated hematocrit. An increase in blood viscosity at higher hematocrits with blood volume in the normal range impairs blood flow and reduces the transport of oxygen.17 In vitro, the viscosity of blood increases exponentially with an increase in hematocrit. However, mitigating factors in patients with erythrocytosis serve to improve oxygen transport, a process that is dependent on both cardiac output and hemoglobin concentration.18 Most importantly, the increase in blood volume accompanying erythrocytosis enlarges the vascular bed, decreases peripheral resistance and increases cardiac output. In addition, the blood flow is axial, with a central core of circulating red cells sliding over a peripheral layer of lubricating plasma. Therefore, optimum oxygen transport with increased blood volume occurs at a higher hematocrit value than with normal blood volume,18,19 and a moderate increase in hematocrit may be beneficial despite the increased viscosity. This may not hold true when there is a more pronounced increase in hematocrit, a circumstance in which high viscosity causes reduced blood flow19,20 that may be responsible for cerebral and cardiovascular impairment in some high-altitude dwellers21 or in patients with severely elevated hematocrit.22,23 In those instances, hematocrit has been reported to reach extreme values, sometimes exceeding 90%.24 In normovolemic individuals, cerebral blood flow decreases at a certain point of hematocrit elevation.25 However, blood flow is also influenced by the oxygen demand of tissues through incompletely understood mechanisms26 and cerebral blood flow remains high at high hematocrits when oxygen delivery is impaired. This was elegantly illustrated in six patients with high hemoglobin oxygen-affinity variants whose cerebral blood flow was 81% higher than that of 11 subjects of comparable age, matched for hematocrit and viscosity, but without the hemoglobin variant.27 Furthermore, cerebral blood flow decreases at much higher levels of hematocrit with any accompanying increased percentage of fetal hemoglobin,28 which also has high oxygen-affinity.29
Elevated hematocrit and thrombosis
Thrombotic events are well documented in patients with PV and CE, apparently less so in those with primary familial and congenital polycythemia or erythrocytosis and HIF-2α gain-of-function mutations, but not in patients with secondary erythrocytosis such as Eisenmenger syndrome,30,31 other cyanotic heart disorders,32,33 high altitude dwellers, and subjects with high oxygen-affinity hemoglobins. Several lines of evidence suggest that an isolated elevation in hematocrit does not, per se, lead to thrombosis. For example, cerebral infarction in young children with cyanotic heart disease is attributed to iron deficiency and relative anemia rather than to erythrocytosis.34,35 In the Framingham study hematocrit was associated with risk of stroke but this association disappeared in multivariate analysis when smoking, a well-established risk factor for stroke,36 was removed.37 In a UK study of 7,346 men, an increased risk of stroke was not seen at higher hematocrit levels (≥51%) in normotensive men but was apparent in hypertensive individuals.38 Coronary blood flow is decreased in secondary erythrocytosis,22 but there is equivocal evidence as to whether the risk of coronary thrombosis is increased in patients with a high hematocrit.23,39,40 Secondary erythrocytosis reportedly does not pose a thrombotic risk in surgical patients.41
Studies of the influence of elevated hematocrit on the risk of thrombosis in animal models of PV and erythrocytosis secondary to elevated erythropoietin have failed to find a consistent positive relationship.42–45 A study of a murine model in which erythrocytosis was induced by transfusing packed red blood cells, with evaluation of thrombotic risk 24 hours later, found that an elevated hematocrit promoted arterial thrombus formation.46 However, acute erythrocytosis induced by transfusion may not reflect the physiology of the chronic elevation of hematocrit seen in PV and secondary erythrocytosis.47 Furthermore, it is not certain how well the ferric chloride-induced thrombosis model in mice reflects thrombosis formation in humans. Thus, in this review, we focus on thrombosis in human conditions of chronic elevation in hematocrit.
Chuvash erythrocytosis and polycythemia vera share thrombosis as the principal cause of morbidity and mortality
Chuvash erythrocytosis
The propensity to thrombosis is even higher in CE than in PV.48 Although endemic in Chuvashia and Ischia, CE is distributed worldwide.8,9,49 This form of erythrocytosis is characterized by a high risk of both arterial and venous thrombosis in subjects living near sea level. It protects from anemia in heterozygotes50 but causes augmented hypoxia sensing with elevated hematocrit in homozygotes.12,51 The VHLR200W variant is not associated with tumors characteristic of the VHL tumor predisposition syndrome. Thrombosis largely accounts for the morbidity and mortality of CE although affected individuals have lower body mass index, systolic blood pressure, glucose and HbA1c levels, and white blood cell and platelet counts compared to controls.48,52,53 The high rate of thrombosis in CE begins in childhood51 and increases with age.48 However, higher hematocrit is not an independent predictor of thrombotic risk in either children or adults.48,51 Furthermore, a history of therapeutic phlebotomy in CE is associated with an increased risk of thrombosis.48 Thus, the thrombotic risk in CE appears to be independent of viscosity, but rather to be related to changes in the upregulated hypoxic responses associated with the homozygous VHL598C>T mutation. We found many HIF-regulated transcripts to be differentially upregulated in CE peripheral blood mononuclear cells, including IL1B, encoding interleukin 1β (2.1-fold), TSP1, encoding thrombospondin-1 (1.5-fold), NLRP3, encoding NLR family pyrin domain containing 3 (1.4-fold), SER-PINE1, encoding plasminogen activator inhibitor-1 (PAI-1) (1.2-fold), and F3 encoding tissue factor (1.1-fold).11 We also found differential gene expression in granulocytes and reticulocytes, and increased TSP-1 concentrations in plasma.48 Thus, increased HIF may cause a pro-thrombotic milieu in CE.54–56 The positive association of phlebotomy with thrombosis in CE parallels observations in the Polycythemia Vera Study Group (PVSG) 01 and 05 studies.57 We postulate that the heightened thrombotic risk is likely due to upregulation of HIF-controlled prothrombotic genes such as tissue factor54–56,58 and thrombospondin.48 It is likely that other HIF-regulated plasma or vascular factors also play contributory roles.59 In aggregate, these data demonstrate that the thrombotic risk in CE is independent of hematocrit.
Polycythemia vera
Thrombosis is the most common complication of PV.60–62 One-half to three-quarters of these events are arterial.63 Ischemic strokes and transient ischemic attacks account for the majority of thrombotic complications, followed in frequency by myocardial infarction, deep vein thrombosis, and pulmonary embolism. Cerebral venous thrombosis and splanchnic thrombosis, including Budd-Chiari syndrome, occur with increased frequency in PV. While it is not unusual for Budd-Chiari syndrome to present as the first indicator of PV, we have been unable to find the exact prevalence of this complication in any large published study of PV. Endogenous erythroid colony formation and the JAK2V617F mutation may be found in patients with splanchnic thrombosis years before an increase in hematocrit.64,65 In fact, the majority of “idiopathic” Budd-Chiari syndrome patients have the JAK2V617F mutation despite a normal hematocrit.64 The association of Budd-Chiari syndrome and PV is so strong that many experts advocate screening for PV with JAK2V617F mutation analysis in all patients who present with hepatic vein or portal/mesenteric thrombosis, regardless of hematocrit.66,67 It should be noted that in PV the hematocrit may be normal despite a marked elevation in red cell mass and total blood volume and that the hematocrit in the splanchnic veins may not be the same as that in the peripheral veins from where the blood sample is drawn. Furthermore, the peripheral hematocrit may be deceptively normal due to an increase in plasma volume in the presence of splenomegaly.15,68
The rationale for phlebotomy in PV was provided by a retrospective analysis of 69 patients in whom elevated hematocrit was controlled by phlebotomy and thrombocytosis by busulfan or other forms of chemotherapy.69 Over 15 years of observation, the incidence of thrombosis was proportional to the elevation in hematocrit,69 but it is not clear how much of the control of the hematocrit was related to phlebotomy versus chemotherapy-related suppression of hematopoiesis.
The prospective, randomized PVSG 01 and 05 studies demonstrated that phlebotomy to control hematocrit was associated with a higher thrombotic risk compared to chemotherapy.57 The PVSG 01 study was the first randomized trial of PV patients.57 Enrollment in the study occurred between 1967 and 1974. All patients (n = 431) were initially treated with phlebotomy to reduce the hematocrit to <45% and then randomized to treatment with phlebotomy alone (n = 134), chlorambucil (n = 141) or 32P (n = 156) to maintain the hematocrit <45%. Phlebotomy was administered in the chlorambucil and 32P arms if the hematocrit was >45% despite the chemotherapy regimen. In 1987, with a maximal follow-up of 19 years, 37.8% of the patients had experienced thrombosis as a major study outcome and 14.8% had died from thrombosis. Overall, therapeutic phlebotomy was independently and significantly associated with an increased risk of thrombosis compared to chemotherapy, but hematocrit level was not independently associated with thrombotic risk. The increased risk of thrombosis in patients undergoing phlebotomy compared to that in patients treated with myelosuppressive therapy seemed to be limited to the first 3 years of therapy.57 The increased thrombotic risk did not seem to be related to poorer disease control as reflected by hematocrit and platelet count: in a retrospective analysis that paired patients with thrombosis to those without thrombosis within the same treatment group, neither hematocrit nor platelet count was associated with thrombosis.70 As of 1987, 10.2% of the patients in the PVSG 01 study had developed acute leukemia and 11.8% had died from a hematologic malignancy. Acute leukemia was much more common in the 32P arm (9.6%) and the chlorambucil arm (13.5%) than in the phlebotomy alone arm (1.5%), and this contributed to the finding that the overall survival of patients treated with phlebotomy was comparable to that of patients treated with 32P and slightly better than that of patients treated with chlorambucil.70,71
The increased risk of thrombosis with phlebotomy compared to chemotherapy observed in the PVSG 01 study was followed up in the PVSG 05 study. Patients were initially phlebotomized to achieve a hematocrit ≤40% and then randomized to treatment with phlebotomy and the combination of aspirin (300 mg) and dipyridamole (75 mg) three times daily (n = 88) versus 32P (n = 90) to maintain the hematocrit <45%.57 The study was stopped at a median followup of <2 years when seven (8.0%) patients in the phlebotomy, aspirin and dipyridamole group had experienced a major thrombosis versus two (2.2%) in the 32P group, providing further evidence of a higher rate of thrombosis with therapeutic phlebotomy versus chemotherapy for PV.
The European Collaboration on Low-Dose Aspirin in the Polycythemia Vera study (ECLAP), which included 1,638 patients from 12 countries and 94 centers, found no difference in thrombotic complications for patients with hematocrits within the range of 40–55%; however, there were not enough subjects with hematocrits >55% for evaluation.72 Evaluation of a cohort of 1,042 patients with PV in the ECLAP trial demonstrated an advantage of hydroxyurea therapy over phlebotomy with respect to the proportion of fatal/nonfatal cardiovascular events: 13.2% in the phlebotomy group versus 7.9% in the hydroxyurea group (P=0.006).73 An important attempt to clarify this issue was a prospective study by the Cytoreductive Therapy in Polycythemia Vera (CYTO-PV) Collaborative Group of the effect of hematocrit on thrombosis in PV patients. This study showed that patients treated with phlebotomy and hydroxyurea to a hematocrit <45% (n = 182) had a lower rate of thrombosis compared to that of patients treated to a target hematocrit of 45–50% (n = 183).74 Hydroxyurea is the most common myelosuppressive agent used in the treatment of PV;75,76 it is effective at controlling erythrocyte, leukocyte, and platelet counts without inducing acute leukemia, and it decreased the risk of thrombosis during the first few years of therapy compared to that in a historical cohort treated with phlebotomy alone.57 By 6 months into the CYTO-PV study,74 fewer patients in the high-hematocrit group were receiving hydroxyurea (47% versus 59%) and among those receiving hydroxyurea the mean daily dose was 12% lower in the high-hematocrit group. This eventuated in a higher white blood cell count in the high-hematocrit group throughout the study (P<0.001). Although absence of hydroxyurea therapy77 and high leukocyte counts78 are independent correlates of thrombotic risk in PV, the rate of thrombosis was greater in the higher hematocrit group whether or not the patient was treated with chemotherapy and whether or not the white blood cell count was elevated.74 Thus, we cannot rule out the possibility that hematocrit may contribute to increased risk of thrombosis in PV along with other PV-associated prothrombotic factors. However, some of the authors of the CYTO-PV and ECLAP studies reanalyzed the study population73 and concluded that there is a “greater antithrombotic protection of hydroxyurea over phlebotomy against arterial thrombosis while the two treatments produce similar results in the protection from venous thrombosis.”79
In the PVSG 01 study, a history of previous thrombosis and older age were independent risk factors for thrombosis after controlling for therapeutic phlebotomy versus chemotherapy.57,70 Currently, the age of the patient (>60 years) and previous thrombotic events are universally acknowledged risk factors for major vascular complications in PV.60 The proportion of activated neutrophils is increased in PV,80 and it is possible that neutrophils may be an important factor in PV-associated thrombosis.81 In a multivariate analysis of the relationship of peripheral blood cell counts with thrombosis in PV subjects, an increased number of leukocytes was the most significant correlate of increased thrombotic risk.78 A study of 1,545 patients by the International Working Group - Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) found that survival of PV patients correlated negatively with leukocytosis, older age, venous thrombosis, and atypical karyotype.82 It was also reported that PV may be associated with tissue factor expression in polymorphonuclear leukocytes in the absence of any in vitro challenge, and that expression is decreased after treatment with hydroxyurea.83 An additional risk for thrombotic events in PV may be environmental hypoxia. We found that PV patients residing in Salt Lake City at approximately 1,400 meters have a higher rate of arterial and venous thromboses than that of patients residing at sea level in Baltimore,84 even though they are only exposed to modest hypoxia.85 In a multivariate analysis, living in Salt Lake City was an independent thrombotic risk factor in PV.84 This may be explained by the recent observation that hypoxia decreases protein S levels in normal subjects by an HIF-1-mediated mechanism.86
Conclusion
Certain disorders with elevated hematocrit, such as PV, CE, primary familial and congenital polycythemia or erythrocytosis (EPOR mutation), and EPAS1 gain-of-function mutations, are associated with thrombotic complications. These conditions are characterized by diverse cellular and metabolic changes that could be directly associated with thrombotic risk, irrespective of hematocrit level. The challenge in these conditions is to elucidate factors for the thrombotic risk other than the elevated hematocrit, and to define what, if any, role that viscosity plays in thrombotic risk. Defining these thrombosis-predisposing factors would provide the basis for identifying and developing novel targeted therapies for these disorders. The evidence we have presented here points to favoring the use of myelosuppressive therapy for intermediate- and high-risk PV, as this approach has been proven to decrease the risk of thrombosis in PV. Furthermore, we trust that the urge to correct any abnormal laboratory data by a therapeutic intervention should be tempered by consideration of the risk-benefit ratio of any such intervention. The routine practice of phlebotomy for elevated hematocrit, with its inevitable iron deficiency (which leads to inhibition of PHD2, increased HIF, and increased erythropoietin) and potential detrimental thrombotic effects, should be re-evaluated. We hope that this review will encourage more studies to pursue the challenge of defining the specific molecular basis of thrombosis in diverse types of polycythemia and erythrocytosis. Improved knowledge of the pathophysiology of these entities should be extended to the development of targeted approaches for the prevention and therapy of thrombotic complications. A review of potential molecular mechanisms contributing to thrombosis in myeloproliferative neoplasms was published at the time of the submission of this manuscript.87
Supplementary Material
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health, R01HL137991 (to JP), and institutional funds from the University of Illinois at Chicago.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/104/4/653
References
- 1.Prchal JT. Primary and secondary erythrocytosis. In: Lichtman MA, Williams WJ, eds. Williams Hematology 9th Edition. New York: McGraw Hill Medical; 2015. [Google Scholar]
- 2.Prchal JT, Semenza GL, Prchal J, Sokol L. Familial polycythemia. Science. 1995;268(5219):1831–1832. [DOI] [PubMed] [Google Scholar]
- 3.McMullin MF, Wu C, Percy MJ, Tong W. A nonsynonymous LNK polymorphism associated with idiopathic erythrocytosis. Am J Hematol. 2011;86(11):962–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giani FC, Fiorini C, Wakabayashi A, et al. Targeted application of human genetic variation can improve red blood cell production from Stem Cells. Cell Stem Cell. 2016;18 (1):73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuschl K, Mills PB, Parsons H, et al. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia–a new metabolic disorder. J Inherit Metab Dis. 2008;31(2):151–163. [DOI] [PubMed] [Google Scholar]
- 6.Quadri M, Federico A, Zhao T, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90(3):467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zmajkovic J, Lundberg P, Nienhold R, et al. A gain-of-function mutation in EPO in familial erythrocytosis. N Engl J Med. 2018;378(10):924–930. [DOI] [PubMed] [Google Scholar]
- 8.Polyakova LA. [Familial erythrocytosis among inhabitants of the Chuvash ASSR. Problem Gematolog I Pereliv Krovi]. 1974;10:30–36. [PubMed] [Google Scholar]
- 9.Ang SO, Chen H, Hirota K, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32(4):614–621. [DOI] [PubMed] [Google Scholar]
- 10.Perrotta S, Nobili B, Ferraro M, et al. Von Hippel-Lindau-dependent polycythemia is endemic on the island of Ischia: identification of a novel cluster. Blood. 2006;107(2): 514–519. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Zhang W, Ma SF, et al. Iron deficiency modifies gene expression variation induced by augmented hypoxia sensing. Blood Cells Mol Dis. 2014;52(1):35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordeuk VR, Sergueeva AI, Miasnikova GY, et al. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. 2004;103(10):3924–3932. [DOI] [PubMed] [Google Scholar]
- 13.Manalo DJ, Rowan A, Lavoie T, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105(2):659–669. [DOI] [PubMed] [Google Scholar]
- 14.Hickey MM, Lam JC, Bezman NA, Rathmell WK, Simon MC. von Hippel-Lindau mutation in mice recapitulates Chuvash polycythemia via hypoxia-inducible factor-2alpha signaling and splenic erythropoiesis. J Clin Invest. 2007;117(12):3879–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spivak JL. Myeloproliferative neoplasms. N Engl J Med. 2017;376(22):2168–2181. [DOI] [PubMed] [Google Scholar]
- 16.Sergeyeva A, Gordeuk VR, Tokarev YN, Sokol L, Prchal JF, Prchal JT. Congenital polycythemia in Chuvashia. Blood. 1997;89(6): 2148–2154. [PubMed] [Google Scholar]
- 17.Dintenfass L. A preliminary outline of the blood high viscosity syndromes. Arch Intern Med. 1966;118(5):427–435. [PubMed] [Google Scholar]
- 18.Thorling EB, Erslev AJ. The “tissue” tension of oxygen and its relation to hematocrit and erythropoiesis. Blood. 1968;31(3):332–343. [PubMed] [Google Scholar]
- 19.Fan FC, Chen RY, Schuessler GB, Chien S. Effects of hematocrit variations on regional hemodynamics and oxygen transport in the dog. Am J Physiol. 1980;238(4):H545–522. [DOI] [PubMed] [Google Scholar]
- 20.Pearson TC, Humphrey PRD, Thomas DJ, et al. Hematocrit, blood viscosity, cerebral blood flow, and vascular occlusion. In: Lowe GDO, ed. Clinical Aspects of Blood Viscosity and Cell Deformability. New York: Springer-Verlag; 1981. [Google Scholar]
- 21.Monge C. Life in the Andes and chronic mountain sickness. Science. 1942;95(2456): 79–84. [DOI] [PubMed] [Google Scholar]
- 22.Kershenovich S, Modiano M, Ewy GA. Markedly decreased coronary blood flow in secondary polycythemia. Am Heart J. 1992;123(2):521–523. [DOI] [PubMed] [Google Scholar]
- 23.Conley CL, Russell RP, Thomas CB, Tumulty PA. Hematocrit values in coronary artery disease. Arch Intern Med. 1964;113: 170–176. [DOI] [PubMed] [Google Scholar]
- 24.Jefferson JA, Escudero E, Hurtado ME, et al. Excessive erythrocytosis, chronic mountain sickness, and serum cobalt levels. Lancet. 2002;359(9304):407–408. [DOI] [PubMed] [Google Scholar]
- 25.Thomas DJ, du Boulay GH, Marshall J, et al. Cerebral blood-flow in polycythaemia. Lancet. 1977;2(8030):161–163. [DOI] [PubMed] [Google Scholar]
- 26.Borzage MT, Bush AM, Choi S, et al. Predictors of cerebral blood flow in patients with and without anemia. J Appl Physiol (1985). 2016;120(8):976–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wade JP, du Boulay GH, Marshall J, et al. Cerebral blood flow, haematocrit and viscosity in subjects with a high oxygen affinity haemoglobin variant. Acta Neurol Scand. 1980;61(4):210–215. [DOI] [PubMed] [Google Scholar]
- 28.Cui MH, Billett HH, Suzuka S, et al. Fetal hemoglobin improves cerebral blood flow and decreases brain inflammation in transgenic-sickle Mice. Blood. 2016;128(22):3639. [Google Scholar]
- 29.Papassotiriou I, Kister J, Griffon N, et al. Modulating the oxygen affinity of human fetal haemoglobin with synthetic allosteric modulators. Br J Haematol. 1998;102(5): 1165–1171. [DOI] [PubMed] [Google Scholar]
- 30.Vongpatanasin W, Brickner ME, Hillis LD, Lange RA. The Eisenmenger syndrome in adults. Ann Intern Med. 1998;128(9):745–755. [DOI] [PubMed] [Google Scholar]
- 31.Martin-Garcia AC, Arachchillage DR, Kempny A, et al. Platelet count and mean platelet volume predict outcome in adults with Eisenmenger syndrome. Heart. 2018; 104(1):45–50. [DOI] [PubMed] [Google Scholar]
- 32.Thorne SA. Management of polycythaemia in adults with cyanotic congenital heart disease. Heart. 1998;79(4):315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perloff JK, Marelli AJ, Miner PD. Risk of stroke in adults with cyanotic congenital heart disease. Circulation. 1993;87(6):1954–1959. [DOI] [PubMed] [Google Scholar]
- 34.Phornphutkul C, Rosenthal A, Nadas AS, Berenberg W. Cerebrovascular accidents in infants and children with cyanotic congenital heart disease. Am J Cardiol. 1973;32(3): 329–334. [DOI] [PubMed] [Google Scholar]
- 35.Cottrill CM, Kaplan S. Cerebral vascular accidents in cyanotic congenital heart disease. Am J Dis Child. 1973;125(4):484–487. [DOI] [PubMed] [Google Scholar]
- 36.Shinton R, Beevers G. Meta-analysis of relation between cigarette smoking and stroke. BMJ. 1989;298(6676):789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kannel WB, Gordon T, Wolf PA, McNamara P. Hemoglobin and the risk of cerebral infarction: the Framingham study. Stroke. 1972;3(4):409–420. [DOI] [PubMed] [Google Scholar]
- 38.Wannamethee G, Perry IJ, Shaper AG. Haematocrit, hypertension and risk of stroke. J Intern Med. 1994;235(2):163–168. [DOI] [PubMed] [Google Scholar]
- 39.Mayer GA. Hematocrit and coronary heart disease. Can Med Assoc J. 1965;93(22):1151–1153. [PMC free article] [PubMed] [Google Scholar]
- 40.Hershberg PI, Wells RE, McGandy RB. Hematocrit and prognosis in patients with acute myocardial infarction. JAMA. 1972;219(7):855–860. [PubMed] [Google Scholar]
- 41.Lubarsky DA, Gallagher CJ, Berend JL. Secondary polycythemia does not increase the risk of perioperative hemorrhagic or thrombotic complications. J Clin Anesth. 1991;3(2):99–103. [DOI] [PubMed] [Google Scholar]
- 42.Paffett-Lugassy N, Hsia N, Fraenkel PG, et al. Functional conservation of erythropoietin signaling in zebrafish. Blood. 2007;110(7): 2718–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shibata J, Hasegawa J, Siemens HJ, et al. Hemostasis and coagulation at a hematocrit level of 0.85: functional consequences of erythrocytosis. Blood. 2003;101(11):4416–4422. [DOI] [PubMed] [Google Scholar]
- 44.Lamrani L, Lacout C, Ollivier V, et al. Hemostatic disorders in a JAK2V617F-driven mouse model of myeloproliferative neoplasm. Blood. 2014;124(7):1136–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strassel C, Kubovcakova L, Mangin PH, et al. Haemorrhagic and thrombotic diatheses in mouse models with thrombocytosis. Thromb Haemost. 2015;113(2):414–425. [DOI] [PubMed] [Google Scholar]
- 46.Walton BL, Lehmann M, Skorczewski T, et al. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood. 2017;129(18):2537–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prchal JT. Secondary polycythemia erythrocytosis. Chapter 57 In: Kaushansky K, Lichtman MA, Prchal JT, et al., eds. Williams Hematology 9th Edition. New York, NY: McGraw Hill; 2015:871–888. [Google Scholar]
- 48.Sergueeva A, Miasnikova G, Shah BN, et al. Prospective study of thrombosis and thrombospondin-1 expression in Chuvash polycythemia. Haematologica. 2017;102(5): e166–e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu E, Percy MJ, Amos CI, et al. The worldwide distribution of the VHL 598C>T mutation indicates a single founding event. Blood. 2004;103(5):1937–1940. [DOI] [PubMed] [Google Scholar]
- 50.Miasnikova GY, Sergueeva AI, Nouraie M, et al. The heterozygote advantage of the Chuvash polycythemia VHLR200W mutation may be protection against anemia. Haematologica. 2011;96(9):1371–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sergueeva AI, Miasnikova GY, Polyakova LA, Nouraie M, Prchal JT, Gordeuk VR. Complications in children and adolescents with Chuvash polycythemia. Blood. 2015;125(2):414–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McClain DA, Abuelgasim KA, Nouraie M, et al. Decreased serum glucose and glycosylated hemoglobin levels in patients with Chuvash polycythemia: a role for HIF in glucose metabolism. J Mol Med (Berl). 2013;91(1):59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoon D, Okhotin DV, Kim B, et al. Increased size of solid organs in patients with Chuvash polycythemia and in mice with altered expression of HIF-1alpha and HIF-2alpha. J Mol Med. 2010;88(5):523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stavik B, Espada S, Cui XY, et al. EPAS1/HIF-2 alpha-mediated downregulation of tissue factor pathway inhibitor leads to a pro-thrombotic potential in endothelial cells. Biochim Biophys Acta. 2016;1862(4):670–678. [DOI] [PubMed] [Google Scholar]
- 55.Sun L, Liu Y, Lin S, et al. Early growth response gene-1 and hypoxia-inducible factor-1alpha affect tumor metastasis via regulation of tissue factor. Acta Oncol. 2013;52(4):842–851. [DOI] [PubMed] [Google Scholar]
- 56.Narita I, Shimada M, Yamabe H, et al. NF-kappaB-dependent increase in tissue factor expression is responsible for hypoxic podocyte injury. Clin Exp Nephrol. 2016;20(5):679–688. [DOI] [PubMed] [Google Scholar]
- 57.Berk P, Wasserman L, Fruchtman S. Treatment of polycythemia vera. A summary of clinical trials conducted by the Polycythemia Study Group. In: Wasserman L, Berk P, Berlin N, eds. Polycythemia Vera and the Myeloproliferative Disorders. Philadelphia: WB Saunders; 1995. [Google Scholar]
- 58.Reeves BN, Song J, Kim SJ, et al. Upregulation of tissue factor may contribute to thrombosis in PV and ET. American Society of Hematology Annual Meeting San Diego, CA; 2018. [Google Scholar]
- 59.Gordeuk VR, Chung DW, Shah BN, et al. Thrombosis and von Willebrand factor in Chuvash polycythemia. Blood. 2017;130(Suppl 1):2377.29187376 [Google Scholar]
- 60.Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23(10):2224–2232. [DOI] [PubMed] [Google Scholar]
- 61.Falanga A, Marchetti M. Thrombotic disease in the myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2012;2012:571–581. [DOI] [PubMed] [Google Scholar]
- 62.Wehmeier A, Daum I, Jamin H, Schneider W. Incidence and clinical risk factors for bleeding and thrombotic complications in myeloproliferative disorders. A retrospective analysis of 260 patients. Ann Hematol. 1991;63(2):101–106. [DOI] [PubMed] [Google Scholar]
- 63.Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114–124. [DOI] [PubMed] [Google Scholar]
- 64.De Stefano V, Fiorini A, Rossi E, et al. Incidence of the JAK2 V617F mutation among patients with splanchnic or cerebral venous thrombosis and without overt chronic myeloproliferative disorders. J Thromb Haemost. 2007;5(4):708–714. [DOI] [PubMed] [Google Scholar]
- 65.De Stefano V, Teofili L, Leone G, Michiels JJ. Spontaneous erythroid colony formation as the clue to an underlying myeloproliferative disorder in patients with Budd-Chiari syndrome or portal vein thrombosis. Semin Thromb Hemost. 1997;23(5):411–418. [DOI] [PubMed] [Google Scholar]
- 66.Colaizzo D, Amitrano L, Tiscia GL, et al. The JAK2 V617F mutation frequently occurs in patients with portal and mesenteric venous thrombosis. J Thromb Haemost. 2007;5(1):55–61. [DOI] [PubMed] [Google Scholar]
- 67.Reikvam H, Tiu RV. Venous thromboembolism in patients with essential thrombocythemia and polycythemia vera. Leukemia. 2012;26(4):563–571. [DOI] [PubMed] [Google Scholar]
- 68.Spivak JL. Polycythemia vera: myths, mechanisms, and management. Blood. 2002;100(13):4272–4290. [DOI] [PubMed] [Google Scholar]
- 69.Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary proliferative polycythaemia. Lancet. 1978;2(8102):1219–1222. [DOI] [PubMed] [Google Scholar]
- 70.Berk PD, Goldberg JD, Donovan PB, Fruchtman SM, Berlin NI, Wasserman LR. Therapeutic recommendations in polycythemia vera based on Polycythemia Vera Study Group protocols. Semin Hematol. 1986;23(2):132–143. [PubMed] [Google Scholar]
- 71.Berlin NI, Wasserman LR. Polycythemia vera: a retrospective and reprise. J Lab Clin Med. 1997;130(4):365–373. [DOI] [PubMed] [Google Scholar]
- 72.Di Nisio M, et al. Barbui T, Di Gennaro L. The haematocrit and platelet target in polycythemia vera. Br J Haematol. 2007;136(2): 249–259. [DOI] [PubMed] [Google Scholar]
- 73.Barbui T, Vannucchi AM, Finazzi G, et al. A reappraisal of the benefit-risk profile of hydroxyurea in polycythemia vera: a propensity-matched study. Am J Hematol. 2017;92(11):1131–1136. [DOI] [PubMed] [Google Scholar]
- 74.Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22–33. [DOI] [PubMed] [Google Scholar]
- 75.Dingli D, Tefferi A. Hydroxyurea: the drug of choice for polycythemia vera and essential thrombocythemia. Curr Hematol Malig Rep. 2006;1(2):69–74. [DOI] [PubMed] [Google Scholar]
- 76.Barbui T, Finazzi G. Evidence-based management of polycythemia vera. Best Pract Res Clin Haematol. 2006;19(3):483–493. [DOI] [PubMed] [Google Scholar]
- 77.Fruchtman SM, Mack K, Kaplan ME, Peterson P, Berk PD, Wasserman LR. From efficacy to safety: a Polycythemia Vera Study Group report on hydroxyurea in patients with polycythemia vera. Semin Hematol. 1997;34(1):17–23. [PubMed] [Google Scholar]
- 78.Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood. 2007;109(6):2446–2452. [DOI] [PubMed] [Google Scholar]
- 79.Barbui T, De Stefano V, Ghirardi A, Masciulli A, Finazzi G, Vannucchi AM. Different effect of hydroxyurea and phlebotomy on prevention of arterial and venous thrombosis in polycythemia vera. Blood Cancer J. 2018;8(12):124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Falanga A, Marchetti M, Evangelista V, et al. Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood. 2000;96(13):4261–4266. [PubMed] [Google Scholar]
- 81.Vannucchi AM. Insights into the pathogenesis and management of thrombosis in polycythemia vera and essential thrombocythemia. Intern Emerg Med. 2010;5(3):177–184. [DOI] [PubMed] [Google Scholar]
- 82.Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maugeri N, Giordano G, Petrilli MP, et al. Inhibition of tissue factor expression by hydroxyurea in polymorphonuclear leukocytes from patients with myeloproliferative disorders: a new effect for an old drug? J Thromb Haemost. 2006;4(12):2593–2598. [DOI] [PubMed] [Google Scholar]
- 84.Zangari M, Fink L, Tolomelli G, et al. Could hypoxia increase the prevalence of thrombotic complications in polycythemia vera? Blood Coagul Fibrinolysis. 2013;24(3):311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ruiz-Arguelles GJ. Altitude above sea level as a variable for definition of anemia. Blood. 2006;108(6):2131; author reply 2131–2132. [DOI] [PubMed] [Google Scholar]
- 86.Pilli VS, Datta A, Afreen S, Catalano D, Szabo G, Majumder R. Hypoxia downregulates protein S expression. Blood. 2018;132(4):452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bar-Natan M, Hoffman R. New insights into the causes of thrombotic events in patients with myeloproliferative neoplasms raise the possibility of novel therapeutic approaches. Haematologica. 2019;104(1):3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.