

Abstract
The diagnosis of bacterial infections remains a major challenge in medicine. Although numerous contrast agents have been developed to image bacteria, their clinical impact has been minimal because they are unable to detect small numbers of bacteria in vivo, and cannot distinguish infections from other pathologies such as cancer and inflammation1–7. Here, we present a family of contrast agents, termed maltodextrin-based imaging probes (MDPs), which can detect bacteria in vivo with a sensitivity two orders of magnitude higher than previously reported, and can detect bacteria using a bacteria-specific mechanism that is independent of host response and secondary pathologies. MDPs are composed of a fluorescent dye conjugated to maltohexaose, and are rapidly internalized through the bacteria-specific maltodextrin transport pathway8–11, endowing the MDPs with a unique combination of high sensitivity and specificity for bacteria. Here, we show that MDPs selectively accumulate within bacteria at millimolar concentrations, and are a thousand-fold more specific for bacteria than mammalian cells. Furthermore, we demonstrate that MDPs can image as few as 105 colony-forming units in vivo and can discriminate between active bacteria and inflammation induced by either lipopolysaccharides or metabolically inactive bacteria.
Bacterial infections cause significant mortality and morbidity worldwide despite the availability of antibiotics. For example, in the United States in 2010, bacterial infections caused 40,000 deaths from sepsis alone and were also the leading cause of limb amputations12,13. A major limitation preventing the effective treatment of bacterial infections is an inability to image them in vivo with accuracy and sensitivity. Consequently, bacterial infections can be diagnosed only after they have become systemic or have caused significant anatomical tissue damage, a stage at which they are challenging to treat owing to the high bacterial burden14,15. There is therefore a great need for the development of contrast agents that can image small numbers of bacteria accurately in vivo.
Here we present a family of contrast agents that are robustly internalized through the bacteria-specific maltodextrin transporter and can image bacterial infections in vivo with unprecedented sensitivity and specificity (see Fig. 1). Maltohexaose is a major source of glucose for bacteria16 and MDPs can therefore deliver millimolar concentrations of imaging probes into bacteria, making it possible to image low numbers of bacteria. MDPs also have high specificity for bacteria because mammalian cells do not express the maltodextrin transporter9 and cannot internalize contrast agents conjugated to maltohexaose. MDPs are composed of α (1–4)-linked glucose oligomers, which are hydrophilic and membrane impermeable17; therefore, MDPs are efficiently cleared from uninfected tissues in vivo, leading to a low background. Furthermore, the lumen of intestinal tissues or the outer layers of the skin are not permeable to glucose oligomers18, and MDPs delivered systemically should therefore not be internalized by the resident bacterial microflora present in healthy subjects. These unique properties should allow MDPs to accurately and sensitively image bacteria in vivo.
Figure 1|. In vivo detection of bacteria with MDPs.

a, Chemical design of MDPs. MDPs are a family of contrast agents that target the maltodextrin transport pathway and can image bacteria in vivo. MDPs are composed of maltohexaose conjugated to an imaging probe. MDPs are internalized as a glucose source and are transported by bacteria at a high rate. Maltodextrin transporters are not present in mammalian cells and MDPs therefore also have specificity for bacteria. b, MDPs image bacteria in vivo with high sensitivity and specificity. MDPs are robustly internalized by bacteria but not by mammalian cells, and can therefore detect low numbers of bacteria in vivo and also distinguish between inflammation and bacterial infections.
The bacterial imaging agents MDP-1 and MDP-2 were synthesized to image bacteria in vitro and in vivo, and are composed of maltohexaose conjugated to either perylene or IR786 (see Fig. 2). MDP-1 and MDP-2 were synthesized by clicking alkyne-functionalized fluorescent dyes onto an azide-functionalized maltohexaose19,20, which was synthesized from maltohexaose in four steps, following the scheme shown in the Supplementary Information. This synthetic strategy introduces the imaging probes at the anomeric carbon of maltohexaose and was selected because maltodextrin transporters tolerate structural modifications at the reducing end ofmaltodextrins21,22.
Figure 2|. Synthesis of MDP-1 and MDP-2.

MDP-1 and MDP-2 were synthesized by conjugation of 1 with either 2 or 3 using the copper (I) catalysed click reaction.
A central problem in imaging bacterial infections is to develop targeting strategies that can deliver large quantities of imaging probes to bacteria. This has been challenging because most present imaging probes target the bacterial cell wall and cannot access the bacterial intracellular volume2–5. Maltodextrin transporters, in contrast, internalize their substrates at a robust rate and MDPs should therefore be capable of reaching a high concentration within bacteria. We therefore investigated the uptake of MDP-1 in gram-positive and gram-negative bacteria, under aerobic and anerobic fermentative conditions. Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus were incubated with a 20 μM concentration of MDP-1 for 1h, washed with PBS, lysed, and the MDP-1 in the cellular supernatant was analysed by fluorescence microscopy. Figure 3a demonstrates that MDPs can deliver large quantities of imaging probes to bacteria, under both aerobic and anaerobic fermentative conditions (see Supplementary Fig. S11). For example, E. coli internalized MDP-1 at a rate sufficient to generate millimolar intracellular concentrations, and followed Michaelis–Menten kinetics, with a Vmax of 2.7 nmolmin –1 per 109 cells and a KM of 1.3 μM (shown in Fig. 3b). Furthermore, pathogenic bacteria such as P. aeruginosa, S. aureus and B. subtilis also robustly internalized MDP-1. To our knowledge, this represents the first demonstration of a targeting strategy that can deliver millimolar concentrations of an imaging probe to bacteria.
Figure 3|. MDPs have specificity for planktonic bacteria and bacterial biofilms.
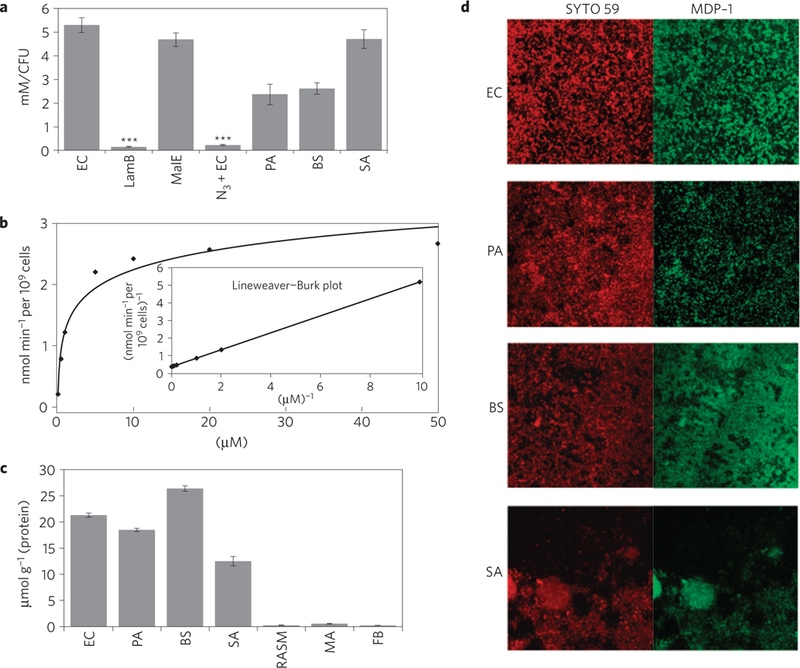
Histogram showing the levels of MDP-1 internalization. Gram-negative and gram-positive bacteria robustly internalize MDP-1. MDP-1 is robustly internalized by E. coli (EC), P. aeruginosa (PA), B. subtilis (BS), S. aureus (SA) and E. coli MalE mutant strains (MalE). The uptake of MDP-1 in E. coli LamB mutant strains (LamB) and metabolically inactive E. coli (EC + N3) is significantly reduced. Results are expressed as mean millimolar concentration per CFU ± standard error of the mean (s.e.m.), for n = 6 per group. The p values between the EC and LamB or EC + N3 were determined by a one-way analysis of variance (ANOVA) using Bonferroni’s post-hoc test, and were found to be statistically significant (p ≤ 0.001). b, Plot showing that the uptake of MDP-1 in E. coli is saturable and follows Michaelis-Menten kinetics, with a Vmax of 2.7 nmol min –1 per109 cells and a KM of 1.3 μM. c, Histogram quantifying the level of MDP-1 transport. MDP-1 has high specificity for bacteria when compared with mammalian cells. Bacteria (E. coli, P. aeruginosa, B. subtilis and S. aureus) transport MDP-1 at a rate three orders of magnitude faster than mammalian cells (rat aortic smooth muscle cells (RASMs), macrophages (MAs) and fibroblasts (FBs)). The results are expressed as mean micromoles per gram of protein ± s.e.m. for n = 6 per group. The p values between each group of bacteria and each group of mammalian cells were determined by a one-way ANOVA using Bonferroni’s post-hoc test, and were found to be statistically significant (p ≤ 0.001). d, Fluorescence micrographs showing that the biofilms (E. coli, P. aeruginosa, B. subtilis and S. aureus) robustly internalize MDP-1.
We performed experiments with LamB mutant E. coli (LamB mutants)23 to determine whether MDP-1 was internalized through the maltodextrin transporter. LamB mutants were incubated with MDP-1 and the internalization of MDP-1 was determined following the procedure described above. Figure 3a demonstrates that LamB mutants do not internalize MDP-1 and that, therefore, MDP-1 enters E. coli through the maltodextrin transport pathway. The uptake of MDP-1 in wild-type E. coli could also be inhibited by an excess of maltose or maltohexaose, further confirming that MDP-1 is internalized by maltodextrin transporters (see Supplementary Fig. S12). Finally, we investigated whether metabolically inactive bacteria (azide-treated) internalized MDP-1. Figure 3a shows that metabolically inactive bacteria do not accumulate MDP-1, demonstrating that MDP-1 is not binding to the bacteria cell surface through non-specific interactions.
A key challenge in imaging bacteria is to develop probes that have high specificity for bacteria. For example, several present bacterial imaging agents detect bacterial infections using mechanisms that have previously been employed to image inflammation or cancer, and thus lack specificity1–3,5. Present imaging strategies therefore have a high rate of false positives and require an invasive biopsy for verification. In contrast, MDPs have the potential to image bacteria with high specificity because mammalian cells do not express maltodextrin transporters9. MDPs should also have low levels of non-specific uptake in mammalian cells because they are hydrophilic and cannot pentrate the membrane17. We therefore investigated the specificity of MDPs towards bacteria. The uptake of MDPs in bacteria and mammalian cells was determined and compared. Bacteria (E. coli, P. aeruginosa, B. subtilis and S. aureus) and mammalian cells (rat aortic smooth muscle cells, macrophages and fibroblasts) were incubated with a 20 μM concentration of MDP-1 for 1 h, washed with PBS,lysed, and the cellular supernatant was analysed for perylene fluorescence signal. Figure 3c shows that MDP-1 has high specificity for bacteria. For example, both gram-positive and gram-negative bacteria internalized MDP-1 at a rate three orders of magnitude faster than mammalian cells. In particular, pathogenic bacteria such as P. aeruginosa and S. aureus internalized 200–300 μmol of MDP-1 per milligram of protein, whereas rat aortic smooth muscle cells and fibroblasts internalized undetectable levels of MDP-1. Furthermore, MDP-2 has a similarly high level of specificity for bacteria when compared with mammalian cells (see Supplementary Fig. S9). We found that MDPs have a thousand times better selectivity for bacteria when compared with mammalian cells and should therefore be able to detect bacteria in vivo with high specificity.
We performed experiments to determine whether MDPs could target bacterial biofilms, a major source of pathology from infectious diseases24–27. Although bacterial biofilms have a significantly altered physiology in comparison with planktonic bacteria, they still consume glucose, and therefore can potentially be imaged by MDPs. We therefore investigated the ability of MDP-1 to image bacterial biofilms. Biofilms were incubated with a 20 μM concentration of MDP-1 for 10 min, and counter-stained with SYTO59, a long-wavelength cell-permeable nucleic acid stain. Figure 3d demonstrates that MDP-1 is actively taken up by a wide variety of bacterial biofilms. In particular, biofilms formed from E. coli (12 ±4 μm thickness), P. aeruginosa (24± 15 μm thickness), B.subtilis (16± 7 μm thickness) and S. aureus (51±30 μm thickness) all avidly internalized MDP-1, demonstrating that maltodextrin transporters are active in bacterial biofilms and can potentially be used in diagnosing diseases associated with bacterial biofilms.
On the basis of these in vitro results we formed a hypothesis that MDPs have the potential to image bacteria in vivo. Accordingly, we investigated the ability of MDP-2 to image bacterial infections in rats. The rats were injected in the left and right thigh muscles, respectively, with E. coli (107 colony-forming units, CFUs) and saline (as a control). After 1 h the rats were injected with MDP-2 (280–350 μl of 1 mM MDP-2 in PBS) through the jugular vein and imaged after 16 h in an IVIS imaging machine. Figure 4a shows that MDP-2 can image bacterial infections in vivo. For example, rat thigh muscles infected with E. coli had a 26-fold increase in fluorescence intensity when compared with uninfected controls, allowing the infected area to be easily visualized in vivo. We further quantified the ability of MDP-2 to target bacteria in vivo by performing a biodistribution study of MDP-2 in rats infected with E. coli (107 CFUs). Figure 4b demonstrates that MDP-2 accumulates in infected muscle tissues and is efficiently cleared from uninfected muscle, having a 42-fold increase in fluorescence intensity between infected and uninfected muscle tissues. MDP-2 did not accumulate in the bacterial microflora of colon tissue, presumably because of the impermeability of the lumen tissue of intestinal tissues to glucose oligomers18. MDP-2 was also efficiently cleared from all the major organs, indicating that it could potentially be used for imaging infections in a wide range of tissues.
Figure 4|. MDP-2 images bacteria in vivo.

a Left: fluorescence image of a rat showing that MDP-2 can image 107 E. coli CFUs in vivo. Middle: histogram showing quantification of fluorescence intensity. E. coli (107 CFUs) infected muscles have a 26-fold increase in fluorescence intensity when compared with uninfected control muscles. Right: micrograph of the histology of E. coli-injected thigh muscles showing that bacteria are present in infected muscles (×20 magnification). b, Histogram showing MDP-2 distribution in rats infected with E. coli. MDP-2 is efficiently cleared from all the major organs and selectively accumulates in infected muscle tissue. Data are plotted as mean fluorescent units (FUs) per gram of tissue ± s.e.m. (n = 6 rats per group). The p values between the infected muscle and the other tissues were determined by a one-way ANOVA using Bonferroni’s post-hoc test, and were found to be statistically significant (p ≤ 0.001). c Left: fluorescence image of a rat showing that MDP-2 can image 105 E. coli CFUs in vivo. Right: histogram showing quantification of fluorescence intensity. E. coli (105 CFUs) infected muscles have a twofold increase in fluorescence intensity when compared with uninfected control muscles. The rat images in a left and c left are representative results of six experiments. Regions of interest (ROI) in a left and c left were identified and integrated using software from the Lumina machine. The results in a middle and c middle are expressed as mean numbers of photons per second per cm2 in the designated ROI ± s.e.m. for n = 6 per group. The statistical significances in a middle and c middle were determined using a two-sample Student f-test (**p ≤ 0.01 and ***p ≤ 0.001).
We also performed experiments to determine the minimum number of bacteria that could be detected by MDP-2 in vivo. E. coli (105 CFUs) were injected into the left rear thigh muscle of rats and imaged with MDP-2 as described above. Figure 4c demonstrates that MDP-2 is capable of detecting as few as 105 bacterial CFUs in vivo. For example, rat thigh muscles infected with 105 bacterial CFUs had a twofold increase in fluorescence intensity when compared with uninfected controls. Present contrast agents for imaging bacteria, such as FIAU (ref. 1), zinc-dipicolylamine probes2 and antimicrobial peptides4, can image only 107–108 bacterial CFUs in vivo; in comparison MDP-2 has a two orders of magnitude higher sensitivity for bacteria in vivo.
Finally, we investigated the specificity of MDP-2 for bacteria in vivo. The development of contrast agents that have high specificity for bacteria has been challenging because most contrast agents also accumulate in inflamed and cancerous tissues1–3,5, owing to their increased metabolic activity and permeability. In contrast, MDP-2 has a thousandfold specificity for bacteria when compared with mammalian cells and clears well from uninfected tissues; therefore, it has the potential to image bacteria with high specificity in vivo. We performed experiments to determine whether MDP-2 could distinguish bacterial infections from both lipopolysaccharide (LPS)-induced inflammation28 and inflammation induced by metabolically inactive bacteria29. We also performed experiments with LamB mutants23 to determine whether MDP-2 internalization in vivo was occurring by transport through the maltodextrin transporter.
We injected rats with 107 CFUs of E. coli in the left thigh muscle and either LPS (1 mg kg–1) or metabolically inactive E. coli in the right thigh muscle, and then imaged them using MDP-2 as described above. Figure 5a shows that MDP-2 can distinguish between bacterial infections and inflammation with high specificity. For example, rat thigh muscles infected with E. coli had a 17-fold increase in fluorescence intensity when compared with LPS-treated tissues. Furthermore, MDP-2 did not accumulate in metabolically inactive E. coli (Fig. 5b), demonstrating that MDP-2 is being actively transported by bacteria in vivo. Finally, we investigated whether MDP-2 was being internalized in vivo through the maltodextrin transporter. LamB mutants (107 CFUs) were injected into rats and the uptake of MDP-2 was compared with that for wild-type E. coli, as described above. Figure 5c demonstrates that LamB mutants did not internalize MDP-2, indicating that MDP-2 is transported in vivo through the maltodextrin transport pathway. The uptake of MDP-2 in E. coli could also be inhibited by an excess of maltose in vivo, further confirming that MDP-2 is being internalized by maltodextrin transporters (see Supplementary Fig. S16).
Figure 5|. MDP-2 images bacteria in vivo using internalization through the maltodextrin transporter.

a Left: image showing that MDP-2 can distinguish between E. coli infection (107 CFUs) and LPS (1 mg kg–1)-induced inflammation. Middle: histogram showing quantification of fluorescence intensity. E. coli-infected tissues had a 17-fold increase in fluorescence intensity when compared with LPS-treated tissues. Right: micrograph showing the histology of E. coli- and LPS-treated muscles demonstrating that both E. coli and LPS induce a large amount of inflammation (×20 magnification). b Left: image showing that MDP-2 is actively transported by bacteria in vivo, and does not accumulate in metabolically inactive bacteria. Middle: histogram showing quantification of fluorescence intensity. E. coli-infected tissues have an 18-fold increase in fluorescence intensity when compared with tissues treated with metabolically inactive bacteria. Right: image showing the histology of thigh muscles injected with either E. coli or metabolically inactive E. coli demonstrating that bacteria are present (×20 magnification). c Left: image showing that MDP-2 is transported by bacteria in vivo, through the maltodextrin transport pathway, and does not accumulate in LamB mutants. Middle: histogram showing quantification of fluorescence intensity. E. coli-infected tissues have a 20-fold increase in fluorescence intensity when compared with tissues treated with LamB-negative E. coli. Right: image showing histology of thigh muscles injected with either E. coli or LamB mutants demonstrating that bacteria are present in infected muscles (×20 magnification). The rat images in a left, b left and c left are representative results of six experiments. Regions of interest in a left, b left and c left were identified and integrated using software from the Lumina machine. The results in a middle, b middle and c middle are expressed as mean numbers of photons per second per cm2 in the designated ROI ± s.e.m. for n = 6 per group. The statistical significances in a middle, b middle and c middle were determined using a two-sample Student f-test (***p ≤ 0.001).
There is a great need to develop contrast agents that can image bacterial infections with high sensitivity and specificity. In this report we demonstrate that MDPs have a unique combination of robust transport and high specificity, and are able to detect as few as 105 CFUs in vivo with high specificity. MDPs have tremendous potential for improving the diagnosis of bacterial infections, given their ability to accurately detect small numbers of bacteria in vivo.
Methods
Synthesis of MDP-1 and MDP-2.
See Fig. 2. MDP-1 and MDP-2 were synthesized by conjugating alkyne-functionalized fluorescent dyes 2 and 3 to azide-functionalized maltohexaose 1, using the click reaction. The synthesis and characterization of the intermediates 1, 2 and 3 are described in the Supplementary Information. The details of the click reaction between 1 and 3 used to generate MDP-2 are described below. The compounds 1 (57.0mg, 0.03mmol) and 3 (39.0mg, 0.06mmol) were dissolved in DMF (5 ml), to which was added CuI (0.6mg, 3.0 μmol) and DIPEA (1.2 mg, 0.01 mmol). The mixture was stirred at room temperature for 24 h under nitrogen and the solvent was removed in vacuo. The residue was redissolved in CH2Cl2 (20 ml) and washed with water (5 ml) and brine (5ml). The organic phase was dried over Na2SO4, filtered and evaporated to dryness in vacuo. The residue was purified by flash column chromatography on silica gel (CH2Cl2/CH3OH, 15/1) to afford the intermediate 15 ina73% yield (55.0 mg, see Supplementary Information for structure and characterization). This intermediate 15 (50.0 mg, 0.02 mmol) was deprotected in a mixture of CH3OH (2 ml) and aqueous LiOH (1.0M, 2 ml) for 24h under nitrogen. The crude MDP-2 was isolated by neutralizing the reaction mixture with Dowex 50W resin, filtering, and concentrating in vacuo. MDP-2 was purified by flash column chromatography on silica gel (CH2Cl2/CH3OH/H2O, 5/5/2) (33.8mg, quantitative). See Supplementary Information for characterization of MDP-2 and details of the synthesis and characterization of MDP-1.
Uptake of MDP-1 and MDP-2 in vitro.
Uptake of MDP-1 and MDP-2 in bacteria (Fig. 3 and SupplementaryFig. S9, respectively).
The uptake of MDP-1 and MDP-2 was investigated in E. coli (ATCC 33456), P. aeruginosa (ATCC 47085), B. subtilis (ATCC 23059), S. aureus (ATCC 6538), metabolically inactive E. coli (sodium azide-treated, see details in the Supplementary Information) and two E. coli mutant strains, which contained either a LamB mutation (JW3992–1) or a MalE mutation (TL212; ref. 30). All bacteria were cultured overnight in Luria–Bertani medium at 37 °C under 5% CO2 in an incubator shaker (Innova 4230, New Brunswick Scientific). Bacteria (100 ¡o! from the overnight culture) were re-suspended in 30 ml fresh Luria–Bertani medium and cultured to an attenuance D600nm = 0.5 in a 250 ml flask in an incubator shaker. Bacteria (3 ml) at steady-state growth were transferred into six-well plates and incubated with 20 μM MDP-1 or MDP-2 in Luria–Bertani medium in an incubator shaker at 37 °C for 1 h. The bacteria were centrifuged at 10,000 r.p.m. for 15 min in 15 ml centrifuge tubes, using a Microfuge 18 centrifuge (Beckman Coulter). The recovered bacterial pellets were washed three times with 10 ml PBS. The bacteria were lysed in 2 ml deionized water by sonication with a Branson Sonifier S-250A (Branson Ultrasonics Corporation), using a constant dutycycle at a 200W output; 10 sonication cycles were performed. The bacterial supernatant (diluted in a 2 ml volume) was isolated by centrifuging at 10,000 r.p.m. for 10 min. The fluorescence intensity of the supernatant was measured in a Shimadzu spectrofluorometer (RF 5301PC) and normalized to either the bacterial protein content or the bacterial cell volume. See Supplementary Information for detailed procedures.
Uptake of MDP-1 in bacterial biofilms.
See Fig. 3d. The uptake of MDP-1 in bacterial biofilms is described in the Supplementary Information.
In vivo imaging of bacterial infections with MDP-2.
In vivo imaging of 105–107 bacterial CFUs (Fig. 4 and SupplementaryFig.S14).
Female Wistar rats (10 weeks, 200–250g, Harlan Laboratories) were anaesthetized with isofluorane and the hair on the thigh and back was removed. A suspension of E. coli (105–107 CFUs) in 250 μL saline was injected into the left rear thigh muscle (injection depth 5 mm), and 250 μL of saline was injected into right rear thigh muscle as a control (injection depth 5 mm). After 1 h the rats were injected with MDP-2 (280–350 μL of 1 mM MDP-2 in PBS) through the jugular vein. Fluorescence images were captured using an IVIS Lumina Imaging System (Caliper Life Sciences) 16h after the MDP-2 injection. The fluorescence intensity from the bacteria or saline injection area (region of interest) was integrated. At the end of the imaging procedure rats were euthanized, by CO2 inhalation, and the bacterial infected and saline-treated muscles were collected and analysed by histology for the presence of bacteria. See Supplementary Information for detailed procedures. Six rats were used for each experimental group.
Animal protocol.
All animal studies were conducted under an animal protocol that was approved by the Animal Use and Care Committee of the Georgia Institute of Technology (IACUC#A10041).
Supplementary Material
Acknowledgements
This project has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN268201000043C, NSF-BES-0546962 Career Award (N.M.) and NIH RO1 HL096796–01 (N.M.).
Footnotes
Additional information
The authors declare no competing financial interests. Supplementary information accompanies this paper on www.nature.com/naturematerials. Reprints and permissions information is available online at http://www.nature.com/reprints. Correspondence and requests for materials should be addressed to N.M.
References
- 1.Bettegowda C et al. Imaging bacterial infections with radiolabeled 1-(2′-deoxy-2′-fluoro-β-D-arabinofuranosyl)-5-iodouracil. Proc. Natl Acad. Sci. USA 102, 1145–1150 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leevy WM et al. Optical imaging of bacterial infection in living mice using a fluorescent near-infrared molecular probe. J. Am. Chem. Soc. 128, 16476–16477 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith BA et al. Optical imaging of mammary and prostate tumors in living animals using a synthetic near infrared zinc(II)-dipicolylamine probe for anionic cell surfaces. J. Am. Chem. Soc. 132, 67–69 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welling MM, Paulusma-Annema A, Balter HS, Pauwels EK & Nibbering PH Technetium-99m labelled antimicrobial peptides discriminate between bacterial infections and sterile inflammations. Eur. J. Nucl. Med. 27, 292–301 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Mahfouz T et al. 18F-fluorodeoxyglucose positron emission tomography contributes to the diagnosis and management of infections in patients with multiple myeloma: A study of 165 infectious episodes. J. Clin. Oncol. 23, 7857–7863 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Leevy WM et al. Noninvasive optical imaging of Staphylococcus aureus bacterial infection in living mice using a Bis-dipicolylamine-Zinc(II) affinity group conjugated to a near-infrared fluorophore. Bioconjug. Chem. 19, 686–692 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rouzet F et al. Technetium 99m-labeled annexin V scintigraphy of platelet activation in vegetations of experimental endocarditis. Circulation 117, 781–789 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Boos W & Shuman H Maltose/maltodextrin system of Escherichia coli: Transport, metabolism, and regulation. Microbiol. Mol. Biol. Rev. 62, 204–229 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gopal S et al. Maltose and maltodextrin utilization by Listeria monocytogenes depend on an inducible ABC transporter which is repressed by glucose.PLoS ONE 5, e10349 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oldham ML, Khare D, Quiocho FA, Davidson AL & Chen J Crystal structure of a catalytic intermediate of the maltose transporter. Nature 450, 515–521 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Brass JM, Bauer K, Ehmann U & Boos W Maltose-binding protein does not modulate the activity of maltoporin as a general porin in Escherichia coli.J. Bacteriol. 161, 720–726 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lipsky BA, Itani K & Norden C Treating foot infections in diabetic patients: A randomized, multicenter, open-label trial of linezolid versus ampicillin-sulbactam/amoxicillin-clavulanate. Clin. Infect. Dis. 38,17–24 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Reiber GE, Pecoraro RE & Koepsell TD Risk factors for amputation in patients with diabetes mellitus. A case-control study. Ann. Intern. Med. 117, 97–105 (1992). [DOI] [PubMed] [Google Scholar]
- 14.Moore EH Atypical mycobacterial infection in the lung: CT appearance. Radiology 187, 777–782 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Erasmus JJ, McAdams HP, Farrell MA & Patz EF Jr Pulmonary nontuberculous mycobacterial infection: Radiologic manifestations. Radiographies 19, 1487–1505 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Dahl MK & Manson MD Interspecific reconstitution of maltose transport and chemotaxis in Eseheriehia eoli with maltose-binding protein from various enteric bacteria. J. Baeteriol 164, 1057–1063 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reuss R et al. Intracellular delivery of carbohydrates into mammalian cells through swelling-activated pathways. J. Membr. Biol. 200, 67–81 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Line BR, Weber PB, Lukasiewicz R & Dansereau RN Reduction of background activity through radiolabeling of antifibrin Fab′ with 99mTc-dextran. J. Nuel. Med. 41, 1264–1270 (2000). [PubMed] [Google Scholar]
- 19.Demko ZP & Sharpless KB A click chemistry approach to tetrazoles by Huisgen 1,3-dipolar cycloaddition: Synthesis of 5-acyltetrazoles from azides and acyl cyanides. Angew. Chem. Int. Ed. Engl. 41, 2113–2116 (2002). [PubMed] [Google Scholar]
- 20.Tornoe CW, Christensen C & Meldal M Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Dippel R & Boos W The maltodextrin system of Eseheriehia eoli: Metabolism and transport. J. Baeteriol. 187, 8322–8331 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freundlieb S, Ehmann U & Boos W Facilitated diffusion of p-nitrophenyl-alpha-D-maltohexaoside through the outer membrane of Escherichia coli. Characterization of LamB as a specific and saturable channel for maltooligosaccharides. J. Biol. Chem. 263, 314–320 (1988). [PubMed] [Google Scholar]
- 23.Baba T et al. Construction of Eseheriehia eoli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reid G Biofilms in infectious disease and on medical devices. Int. J. Antimierob. Agents 11, 223–226 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Author AN Panel discussion on biofilms in urinary tract infection. Int. J. Antimierob. Agents 11, 237–239 (1999). [Google Scholar]
- 26.Hall-Stoodley L, Costerton JW & Stoodley P Bacterial biofilms: From the natural environment to infectious diseases. Nature Rev. Mierobiol. 2, 95–108 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Kolodkin-Gal I et al. D-amino acids trigger biofilm disassembly. Seienee 328, 627–629 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dehoux MJ, van Beneden RP, Fernandez-Celemin L, Lause PL & Thissen JP Induction of MafBx and Murf ubiquitin ligase mRNAs in rat skeletal muscle after LPS injection. FEBSLett. 544, 214–217 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Luo G, Niesel DW, Shaban RA, Grimm EA & Klimpel GR Tumor necrosis factor alpha binding to bacteria: evidence for a high-affinity receptor and alteration ofbacterial virulence properties. Infeet. Immun. 61, 830–835 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larson TJ, Ludtke DN & Bell RM sn-Glycerol-3-phosphate auxotrophy of plsB strains of Escherichia coli: evidence that a second mutation, plsX, is required. J. Baeteriol. 160, 711–717 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.