

Abstract
Efficient prediction and ranking of small molecule binders by their kinetic (kon and koff) and thermodynamic (∆tt) properties can be a valuable metric for drug lead optimization, as these quantities are often indicators of in vivo efficacy. We have previously described a hybrid molecular dynamics, Brownian dynamics, and milestoning model, Simulation Enabled Estimation of Kinetic Rates (SEEKR), that can predict kon’s, koff’s, and ∆tt’s. Here we demonstrate the effectiveness of this approach for ranking a series of seven small molecule compounds for the model system, β-cyclodextrin, based on predicted kon’s and koff’s. We compare our results using SEEKR to experimentally determined rates as well as rates calculated using long-timescale molecular dynamics simulations and show that SEEKR can effectively rank the compounds by koff and ∆tt with reduced computational cost. We also provide a discussion of convergence properties and sensitivities of calculations with SEEKR to establish “best practices” for its future use.
Graphical Abstract

Molecular binding processes are ubiquitous in biology and serve as the fundamental basis for biological complexity. For the drug discovery community, engineering pharmacologically active small molecules is of particular importance. Traditionally, the paradigm for lead optimization is to select for leads with the greatest affinity for a protein, or other, target of interest. However, recent evidence suggests that the kinetics of binding may also be a useful metric for lead selection. It is now thought that both residence times and association rates are key determinants of in vivo efficacy for many drugs.1–5 Similar to computational predictions of binding thermodynamics, molecular simulations can be used to compute binding kinetics.6–8 Methods such as Brownian Dynamics (BD) have been used effectively for estimating molecular association rates.9–12 Molecular dynamics (MD) simulations which explicitly represent all atoms and forces, can also be used to predict binding kinetics. Due to significantly increased model complexity, MD is limited by sampling. Nevertheless, owing to software improvements and the development of commodity hardware such as GPUs and specialty hardware such as Anton,13,14 “brute force” calculation of binding kinetics with MD is now a possibility.15–19 To improve upon “brute force” sampling statistics, many sampling strategies employ force biases or other statistical mechanical techniques to predict both association and dissociation rates of many systems. This includes methods such as: Markov State Models,20–23 metadynamics,24–26 milestoning,27,28,28–32 and other techniques.33–39
Our previous work uses a multiscale MD, BD, and Milestoning approach for the calculation of both association and dissociation rates of receptor-ligand complexes.40,41 Our implementation, “Simulation Enabled Estimation of Kinetic Rates” (SEEKR) is a freely available software package1 that automates the preparation, simulation, and analysis of these multiscale milestoning calculations using existing softwares: NAMD42 for MD simulations and BrownDye12 for BD simulations. Milestoning theory provides the glue for the multiscale scheme by providing a strategy to subdivide, simulate, and subsequently statistically reconnect small regions of simulation space called “milestones”27,40,41,43–52 This approach reduces the compute time required to simulate transition events, is embarrassingly parallel, and is agnostic to the simulation modality used. This allows us to use atomically detailed, yet computationally expensive, fully flexible MD simulations in milestones near the binding site where these interactions are critical for understanding the binding and unbinding, and BD simulations far from the binding site where rigid body dynamics provides a sufficient description at significantly reduced computational cost. For a more thorough description of milestoning theory and the calculation of kinetic quantities, such as kon and koff, we refer the reader to the existing literature.27,40,41,43–52
The effectiveness of the SEEKR scheme for the calculation of kon and koff values has been demonstrated for multiple protein-ligand systems.40,41 However, it has not yet been used for rank ordering sets of compounds by kinetic (kon and koff) and thermodynamic (∆G) values, as would be done in pharmaceutical discovery settings. Here we use SEEKR to estimate kon, koff, and ∆G for a model host-guest system, β-cyclodextrin with seven ligands representing diverse chemical groups (Fig. 1), using two forcefields for β-cyclodextrin GAFF53,54 and Q4MD.55 We compare the SEEKR estimates with previously published “brute force” (long-timescale) MD predictions19 and experimental results.56–61 Using this model system we examine both the accuracy and efficiency of SEEKR compared to long-timescale MD. We further explore the reduction in computational effort required for SEEKR estimates as well as discuss the convergence properties and sensitivity of SEEKR calculations to establish “best practices” for its future use.
Figure 1:
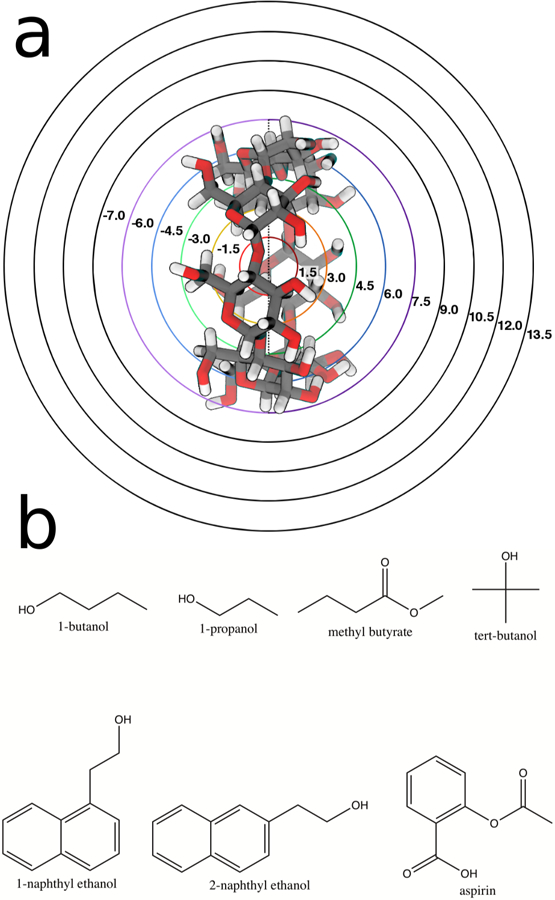
a) β-cyclodextrin with milestones spaced at 1.5 Å increments and b) the seven ligands used in this study. The top four ligands are known to bind more weakly while the bottom three are known to bind more tightly.
SEEKR calculations and the long timescale MD simulations struggle to reproduce both the values and rank ordering of the experimentally determined kon’s (Fig. 2). However, similar qualitative results are seen with the SEEKR calculations and long timescale MD calculations using the same forcefield. On rates calculated using Q4MD are approximately one order of magnitude faster than experimental rates, while the GAFF forcefield produces rates closer to the experimental values, differing by approximately a factor of 3 or less.
Figure 2:

a) Experimental and calculated on rates for SEEKR GAFF and Q4MD forcefields as well as long timescale MD with both forcefields. b) Calculated rank correlation coefficients. Errors are determined with a bootstrapping analysis.
Both methods fail to effectively order the ligands by increasing kon, as demonstrated by low or negative Kendall and Spearman rank correlation coefficients. As the values of all the experimental rates have limited variability (all within half an order of magnitude), the sensitivity of the methods as well as the errors associated with the calculations and experiments makes differentiation and ordering challenging.
Unlike the experimental kon’s, koff’s for the seven guest molecules span multiple orders of magnitude, making them a better target for ranking the compounds with SEEKR. Again, off rates calculated with SEEKR are in good agreement with the long timescale MD simulations using the same forcefield (Fig. 3). Rates calculated using the GAFF forcefield are consistently faster than experiment by approximately one order of magnitude. This trend is seen in both the long timescale MD and SEEKR, but is more pronounced in the SEEKR calculations. The Q4MD forcefield, however, more accurately reproduces the magnitude of the experimental values with both SEEKR and long timescale MD. SEEKR calculations with both Q4MD and GAFF forcefields were effective for ranking the compounds by increasing off rates, as evidenced by high rank correlation values. The smaller magnitudes of the Q4MD values potentially contribute to this forcefield’s difficulty to differentiate between compounds with similar rates, where the larger values associated with the GAFF forcefield allow for more variability in the rate value without changing the overall ordering. Both the GAFF and Q4MD forcefields successfully differentiate the three tighter binding compounds from the four weaker binding, with the tighter binding compounds all having slower off rates and a difference of one order of magnitude between the fastest tightly binding compound and the slowest weakly binding compound. This suggests that SEEKR could be useful for identifying and separating long residence time ligands from shorter residence time ligands and then further discriminating the compounds through ranking by koff.
Figure 3:

a) Experimental and calculated off rates for SEEKR GAFF and Q4MD forcefields as well as long timescale MD with both forcefields. b) Calculated rank correlation coefficients. Errors are determined with a bootstrapping analysis.
An additional benefit of kinetics calculations with SEEKR is that binding free energies can also be obtained from the same simulations (Fig. 4). Binding free energies calculated using the rate constants are most heavily influenced by the koff for these ligands, as this value is more variable, where the kon’s for all ligands are more similar. Therefore, similar trends are observed for the calculated binding free energies as were observed for the off rates. Binding free energies can also be calculated using the stationary probabilities for each milestone, rather than the rate constants, and produce similar results. The GAFF forcefield consistently underestimates the binding free energies in both SEEKR and the long timescale MD, resulting from the consistent underestimation of the magnitudes of the koff’s. The magnitudes of the binding free energies calculated using Q4MD are in much better agreement with the experimental values, differing by 1 kcal or less. SEEKR with both Q4MD and GAFF successfully differentiates the three known tighter binding compounds from the four weaker binding compounds. SEEKR can also further discriminate ligands by its effective ranking by binding free energies, demonstrated by high rank correlation values.
Figure 4:

a) Experimental and calculated binding free energies for SEEKR GAFF and Q4MD forcefields as well as long timescale MD with both forcefields. b) Calculated rank correlation coefficients. Errors are determined with a bootstrapping analysis.
A key aspect of future development of the SEEKR software is the systematic development of methodological best-practices as well as the elucidation of the sensitivity of calculated kinetic parameters to various SEEKR input conditions.
While it is possible to determine the kinetics for small systems like β-cyclodextrin using conventional long timescale MD simulations, increasing system size soon makes this inefficient or even impossible. The convergence of kon and koff were assessed by calculating the rate constants as a function of the reversal trajectory number at increasing intervals of 50 reversal numbers (with 10 trajectories initiated for each reversal number). The reversal number is a direct measure of the equilibrium simulation length, as reversals are initiated from evenly spaced configurations of the equilibrium distribution. In general, both the on and off rates appear converged in less than the maximum number of reversals available. Approximately half the total reversals (4000 of 8000) were sufficient for obtaining reasonably converged rate constants. This suggests that the total simulation cost to obtain a similar result could be as little as 2 µs per ligand, rather than 3.8 µs.
Convergence of the rate constant is a highly complicated quantity dependent on the transition probabilities as well as the incubation times obtained from each milestone. Therefore, a more detailed analysis of the convergence of these quantities on a per milestone level can provide further insight into the overall convergence of a rate calculation within SEEKR. Fig. 5a,b shows the convergence of kon and koff, respectively, as a function of the number of reversals launched for the representative system of Q4MD β-cyclodextrin with aspirin. Reversal number is directly related to the length of equilibrium sampling, the current bottleneck of a SEEKR calculation. While both values appear to converge in fewer than the maximum number of reversals, the dramatic change in koff after reversal number 400 is of note. Further analysis of the per-milestone transition counts (Fig. 5c) and incubation times (Fig. 5d) revealed that this change in koff was due to poor initial sampling of the −1.5 Å milestone, which once sampled decreased the overall koff.
Figure 5.

: Convergence analysis for a representative ligand, aspirin, and β-cyclodextrin with the Q4MD forcefield. Convergence of a) kon, b) koff, c) transition counts, and d) incubation times for each milestone are plotted as a function of the number of reversals launched. Reversal number is directly related to the length of equilibrium sampling, as reversals were launched at 2 ns intervals from the equilibrium trajectory.
This was only observed for the aspirin ligand, one of the bulkiest ligands, where it was extremely unlikely to observe transitions outward from the primary face due to steric effects. Evaluation of these convergence properties on a per milestone basis is a valuable diagnostic tool; identifying which milestones contribute most to the mean first passage time (and therefore kon and koff) and milestones where the ligand spends only a short time, providing detailed molecular insight into the binding and unbinding processes. Furthermore, this analysis is also useful during the simulation process, as the convergence of each milestone can be assessed “on the fly” and individual simulations can be terminated or extended accordingly for each milestone.
We also explore the sensitivity of the calculated rate constants to the milestoning model construction. In particular, the appropriate spacing of milestones is critical for the calculation. Milestones must not be spaced so close such that the velocity of the system cannot decorrelate between transitions.45,46 This assumption is typically valid for molecular dynamics simulations, as velocities typically decorrelate on the subpicosecond timescale.46 However, if milestones are spaced far apart, transitions will require much longer simulations and milestioning sampling efficiency is lost.
For our systems, the incubation times of all milestones are on the order of multiple picoseconds or greater, which is longer than the sub-picosecond timescale typically necessary for decorrelation.46 When the milestone spacing was doubled to 3 Å, simulation efficiency was dramatically reduced, such that few to no transitions between milestones were observed, precluding the calculation of rate constants. These observations suggest that the 1.5 Å spacing used in our simulations was appropriate for the calculation of the desired kinetic parameters.
Our milestoning model differentiates the two faces of the cyclodextrin ring and therefore defines two bound states, corresponding to each face. Investigation into the effect of this on the resulting rate constants revealed that it had only minimal effects. When the two bound states were combined into a single milestone, only small changes to the rate were observed, within the error of both calculations. Furthermore, when the the two faces were not differentiated with unique milestones, minimal change in the calculated rate constants was observed.
It is also important to note that our milestoning model did not explicitly resolve the ligand orientation in any way, and therefore any ligand orientational sampling was achieved entirely through simulation. This resulted in some ligand orientations being unsampled in the deepest milestones where the orientation was sterically restricted to the starting conformation on that milestone. While this is a limitation that will be addressed in future developments of SEEKR, it also highlights that a relatively simplistic model was able effectively calculate kinetic parameters with good agreement to experimental values.
The simplicity of this model has many advantages. The bound state is defined naturally as the innermost milestone and all other milestones can be defined at the same time, including what defines the unbound state. The long timescale MD employed a more empirical definition of the bound state where the ligand was only considered bound when the COM of the ligand was within 7.5 Å of the COM of the β-cyclodextrin for at least 1.0 ns. Similarly the ligand was considered unbound when it left this 7.5 Å bound state for at least 1.0 ns. With the SEEKR approach, minimal prior knowledge of the system is required, as binding and unbinding are determined only from the milestone surfaces. No time cutoff is required, as short excursions that do not result in full binding and unbinding events are captured naturally in the milestoning model. The simplicity of SEEKR milestoning calculation setup, in conjunction with the ability to monitor convergence for each milestone and terminate simulations accordingly, makes this approach well-suited for calculations with multiple ligands as would be necessary in a drug discovery setting.
We present the first successful ranking of a set of seven guest molecules with the β-cyclodextrin host using the SEEKR hybrid MD/BD/Milestoning approach. SEEKR effectively reproduces both the magnitudes and rankings of the experimental off rates56–61 and binding free energies, two quantities of interest in typical drug discovery campaigns.1–4 SEEKR also successfully differentiates the known longer residence time and tighter binding compounds from the weaker binding compounds. Our results are also in good agreement with previously conducted long timescale MD simulations for the same set of ligands.19 In particular SEEKR and long timescale MD simulations using the same forcefield (GAFF or Q4MD) exhibited similar deviations from the experimental values, with GAFF producing consistently faster off rates than experiment and Q4MD producing consistently faster on rates. In general both methods and both forcefields struggled to reproduce the experimental on rate ranking, as all ligands had very similar kon’s. The SEEKR method requires less simulation time (3.8 µs per ligand) than the long timescale MD approach (4.5 – 11 µs per ligand). Furthermore, convergence analysis of the SEEKR calculations suggests that comparable results could be achieved with as little as 2 µs per ligand. In addition, SEEKR’s milestoning approach makes these calculations highly parallel, as the simulations on each milestone are completely independent from all other milestones. We also provide an analysis of the sensitivity of the SEEKR calculations to various factors such as sampling, milestone spacing, and the construction of the milestoning model with the intention of putting forth “best practices” for the use of SEEKR. SEEKR’s effectiveness at ranking compounds for this small model system suggest that it is well suited for ranking compounds of more complex protein-ligand and protein-drug systems, where the efficiency and enhanced sampling advantages of our multiscale MD/BD/milestoning approach will be more apparent.
Computational Methods
GAFF53,54 forcefield parameters for the seven guest molecule along with both GAFF and Q4MD-CD55 parameterizations of β-cyclodextrin were obtained from Tang and Chang.19 For comparison we use identical structures and parameterizations as those used in their study. These initial structures were used by the SEEKR software for preparation of the milestoning simulations. The preparation procedure was the same for each of the seven guest molecules and followed standard SEEKR protocols.41 All systems were solvated with TIP3P waters.62 All BD simulations were performed using the BrownDye software package.12 Electrostatic potentials of the host and guest molecules used as inputs for the BD simulation were calculated with APBS version 1.4.63 A modified version of NAMD 2.12 was used for all MD simulations.42 For all 13 milestones in the MD region, the standard SEEKR procedure for minimization, equilibration and simulation was followed. In total, 2.6 µs of equilibrium sampling (160 ns for 16 milestones) were used and approximately 570 ns of FHPD sampling for a total of 3.2 µs of simulation used in the milestoning model. The total cost per ligand (including simulation discarded for equilibration) was therefore ∼3.8 µs.
Supplementary Material
Acknowledgements
We thank Zhiye Tang and Chia-en Chang for sharing structures and parameters as well as helpful discussions. This work is funded in part by the Director’s New Innovator Award Program NIH DP2-OD007237, the National Biomedical Computation Resource (NBCR) NIH P41-GM103426, and the National Science Foundation through XSEDE supercomputing resources provided via TG-CHE060073 to R.E.A. B.R.J. and C.T.L. also acknowledge support from the NIH Molecular Biophysics Training Program (T32-GM008326).
Footnotes
Supporting Information
The following is made available online: Additional computational details, calculated rate values, convergence plots for all ligands
References
- (1).Schuetz DA; de Witte WEA; Wong YC; Knasmueller B; Richter L; Kokh DB; Sadiq SK; Bosma R; Nederpelt I; Heitman LH et al. Kinetics for Drug Discovery: An Industry-Driven Effort to Target Drug Residence Time. Drug Discov. Today 2017, 22, 896–911. [DOI] [PubMed] [Google Scholar]
- (2).Lu H; Tonge PJ Drug–Target Residence Time: Critical Information for Lead Optimization. Curr. Opin. Chem. Biol 2010, 14, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Copeland RA; Pompliano DL; Meek TD Drug–Target Residence Time and Its Implications for Lead Optimization. Nat. Rev. Drug Discov 2006, 5, 730–739. [DOI] [PubMed] [Google Scholar]
- (4).Copeland RA the Drug-Target Residence Time Model: A 10-Year Retrospective. Nat. Rev. Drug Discov 2016, 15, 87–95. [DOI] [PubMed] [Google Scholar]
- (5).Swinney DC Opinion: Biochemical Mechanisms of Drug Action: What Does It Take for Success? Nat. Rev. Drug Discov 2004, 3, 801–808. [DOI] [PubMed] [Google Scholar]
- (6).De Vivo M; Masetti M; Bottegoni G; Cavalli A Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem 2016, 59, 4035–4061. [DOI] [PubMed] [Google Scholar]
- (7).Amaro RE; Mulholland AJ Bridging Biological and Chemical Complexity in the Search for Cures: Multiscale Methods in Drug Design. Nat Rev Chem 2018, 2, 0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bruce NJ; Ganotra GK; Kokh DB; Sadiq SK; Wade RC New Approaches for Computing Ligand–receptor Binding Kinetics. Curr. Opin. Struct. Biol 2018, 49, 1–10. [DOI] [PubMed] [Google Scholar]
- (9).Northrup SH; Allison SA; McCammon JA Brownian Dynamics Simulation of Diffusion-Influenced Bimolecular Reactions. J. Chem. Phys 1984, 80, 1517–1524. [Google Scholar]
- (10).McCammon JA; Northrup SH; Allison SA Diffusional Dynamics of Ligand-Receptor Association. J. Phys. Chem 1986, 90, 3901–3905. [Google Scholar]
- (11).Zhou H on the Calculation of Diffusive Reaction Rates Using Brownian Dynamics Simulations. J. Chem. Phys 1990, 92, 3092–3095. [Google Scholar]
- (12).Huber GA; McCammon JA Browndye: A Software Package for Brownian Dynamics. Comput. Phys. Commun 2010, 181, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shaw DE; Bowers KJ; Chow E; Eastwood MP; Ierardi DJ; Klepeis JL; Kuskin JS; Larson RH; Lindorff-Larsen K; Maragakis P et al. Millisecond-Scale Molecular Dynamics Simulations on Anton. Proc. Conf. High Perform. Comput Networking, Storage Anal. - SC ‘09. New York, New York, USA, 2009; p 1. [Google Scholar]
- (14).Shaw DE; Grossman JP; Bank JA; Batson B; Butts JA; Chao JC; Deneroff MM; Dror RO; Even A; Fenton CH et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer Int. Conf. High Perform. Comput. Networking, Storage Anal. SC 2014; pp 41–53. [Google Scholar]
- (15).Shan Y; Kim ET; Eastwood MP; Dror RO; Seeliger MA; Shaw DE How Does a Drug Molecule Find Its Target Binding Site? J. Am. Chem. Soc 2011, 133, 9181–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shan Y; Eastwood MP; Zhang X; Kim ET; Arkhipov A; Dror RO; Jumper J; Kuriyan J; Shaw DE Oncogenic Mutations Counteract Intrinsic Disorder in the EGFR Kinase and Promote Receptor Dimerization. Cell 2012, 149, 860–870. [DOI] [PubMed] [Google Scholar]
- (17).Dror RO; Pan AC; Arlow DH; Borhani DW; Maragakis P; Shan Y; Xu H; Shaw DE Pathway and Mechanism of Drug Binding to G-Protein-Coupled Receptors. Proc. Natl. Acad. Sci 2011, 108, 13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pan AC; Borhani DW; Dror RO; Shaw DE Molecular Determinants of Drug-Receptor Binding Kinetics. Drug Discov. Today 2013, 18, 667–673. [DOI] [PubMed] [Google Scholar]
- (19).Tang Z; Chang C.-e. A. Binding Thermodynamics and Kinetics Calculations Using Chemical Host and Guest: A Comprehensive Picture of Molecular Recognition. J. Chem. Theory Comput 2018, 14, 303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Buch I; Giorgino T; De Fabritiis G Complete Reconstruction of an Enzyme-Inhibitor Binding Process by Molecular Dynamics Simulations. Proc. Natl. Acad. Sci 2011, 108, 10184–10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Plattner N; Noé F Protein Conformational Plasticity and Complex Ligand-Binding Kinetics Explored by Atomistic Simulations and Markov Models. Nat. Commun 2015, 6, 7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wu H; Paul F; Wehmeyer C; Noé F Multiensemble Markov Models of Molecular Thermodynamics and Kinetics. Proc. Natl. Acad. Sci 2016, 113, E3221–E3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Doerr S; de Fabritiis G On-The-Fly Learning and Sampling of Ligand Binding by High-Throughput Molecular Simulations. J. Chem. Theory… 2014, 10, 2064–2069. [DOI] [PubMed] [Google Scholar]
- (24).Mollica L; Theret I; Antoine M; Perron-Sierra F; Charton Y; Fourquez J-M; Wierzbicki M; Boutin JA; Ferry G; Decherchi S et al. Molecular Dynamics Simulations and Kinetic Measurements to Estimate and Predict Protein–Ligand Residence Times. J. Med. Chem 2016, 59, 7167–7176. [DOI] [PubMed] [Google Scholar]
- (25).Tiwary P; Limongelli V; Salvalaglio M; Parrinello M Kinetics of Protein–Ligand Unbinding: Predicting Pathways, Rates, and Rate-Limiting Steps. Proc. Natl. Acad. Sci 2015, 112, 201424461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Casasnovas R; Limongelli V; Tiwary P; Carloni P; Parrinello M Unbinding Kinetics of a P38 MAP Kinase Type II Inhibitor from Metadynamics Simulations. J. Am. Chem. Soc 2017, 139, 4780–4788. [DOI] [PubMed] [Google Scholar]
- (27).Elber R. a New Paradigm for Atomically Detailed Simulations of Kinetics in Biophysical Systems. Q. Rev. Biophys 2017, 50, e8. [DOI] [PubMed] [Google Scholar]
- 28).Ma P; Cardenas AE; Chaudhari MI; Elber R; Rempe SB the Impact of Protonation on Early Translocation of Anthrax Lethal Factor: Kinetics from Molecular Dynamics Simulations and Milestoning Theory. J. Am. Chem. Soc 2017, jacs.7b07419. [DOI] [PMC free article] [PubMed]
- (29).Kirmizialtin S; Nguyen V; Johnson KA; Elber R How Conformational Dynamics of DNa Polymerase Select Correct Substrates: Experiments and Simulations. Structure 2012, 20, 618–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yu T-Q; Lapelosa M; Vanden-Eijnden E; Abrams CF Full Kinetics of CO Entry, Internal Diffusion, and Exit in Myoglobin from Transition-Path Theory Simulations. J. Am. Chem. Soc 2015, 137, 3041–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bucci A; Yu T-Q; Vanden-Eijnden E; Abrams CF Kinetics of O 2 Entry and Exit in Monomeric Sarcosine Oxidase Via Markovian Milestoning Molecular Dynamics. J. Chem. Theory Comput 2016, 12, 2964–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ma W; Schulten K Mechanism of Substrate Translocation by a Ring-Shaped ATPase Motor at Millisecond Resolution. J. Am. Chem. Soc 2015, 137, 3031–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Teo I; Mayne CG; Schulten K; Lelièvre T Adaptive Multilevel Splitting Method for Molecular Dynamics Calculation of Benzamidine-Trypsin Dissociation Time. J. Chem. Theory Comput 2016, acs.jctc.6b00277. [DOI] [PMC free article] [PubMed]
- (34).Dickson A; Lotz SD Ligand Release Pathways Obtained with WExplore: Residence Times and Mechanisms. J. Phys. Chem. B 2016, 120, 5377–5385. [DOI] [PubMed] [Google Scholar]
- (35).Dickson A; Lotz SD Multiple Ligand Unbinding Pathways and Ligand-Induced Destabilization Revealed by WExplore. Biophys. J 2017, 112, 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lotz SD; Dickson A Unbiased Molecular Dynamics of 11 Min Timescale Drug Unbinding Reveals Transition State Stabilizing Interactions. J. Am. Chem. Soc 2018, jacs.7b08572. [DOI] [PubMed]
- (37).Chiu SH; Xie L Toward High-Throughput Predictive Modeling of Protein Binding/Unbinding Kinetics. J. Chem. Inf. Model 2016, 56, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wong CF Steered Molecular Dynamics Simulations for Uncovering the Molecular Mechanisms of Drug Dissociation and for Drug Screening: A Test on the Focal Adhesion Kinase. J. Comput. Chem 2018, [DOI] [PubMed]
- (39).Tran DP; Takemura K; Kuwata K; Kitao A Protein-Ligand Dissociation Simulated by Parallel Cascade Selection Molecular Dynamics. J. Chem. Theory Comput 2018, 14, 404–417. [DOI] [PubMed] [Google Scholar]
- (40).Votapka LW; Amaro RE Multiscale Estimation of Binding Kinetics Using Brownian Dynamics, Molecular Dynamics and Milestoning. PLOS Comput. Biol 2015, 11, e1004381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Votapka LW; Jagger BR; Heyneman AL; Amaro RE SEEKR: Simulation Enabled Estimation of Kinetic Rates, a Computational Tool to Estimate Molecular Kinetics and Its Application to Trypsin–Benzamidine Binding. J. Phys. Chem. B 2017, 121, 3597–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Faradjian AK; Elber R Computing Time Scales from Reaction Coordinates by Milestoning. J. Chem. Phys 2004, 120, 10880–10889. [DOI] [PubMed] [Google Scholar]
- (44).Shalloway D; Faradjian AK Efficient Computation of the First Passage Time Distribution of the Generalized Master Equation by Steady-State Relaxation. J. Chem. Phys 2006, 124, 054112. [DOI] [PubMed] [Google Scholar]
- (45).West AMA; Elber R; Shalloway D Extending Molecular Dynamics Time Scales with Milestoning: Example of Complex Kinetics in a Solvated Peptide. J. Chem. Phys 2007, 126, 145104. [DOI] [PubMed] [Google Scholar]
- (46).Vanden-Eijnden E; Venturoli M; Ciccotti G; Elber R on the Assumptions Underlying Milestoning. J. Chem. Phys 2008, 129, 174102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Vanden-Eijnden E; Venturoli M Markovian Milestoning with Voronoi Tessellations. J. Chem. Phys 2009, 130, 194101. [DOI] [PubMed] [Google Scholar]
- (48).Májek P; Elber R Milestoning Without a Reaction Coordinate. J. Chem. Theory Comput 2010, 6, 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kirmizialtin S; Elber R Revisiting and Computing Reaction Coordinates with Directional Milestoning. J. Phys. Chem. A 2011, 115, 6137–6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Cardenas AE; Elber R Computational Study of Peptide Permeation Through Membrane: Searching for Hidden Slow Variables. Mol. Phys 2013, 111, 3565–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Cardenas AE; Shrestha R; Webb LJ; Elber R Membrane Permeation of a Peptide: It Is Better to Be Positive. J. Phys. Chem. B 2015, 119, 6412–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bello-Rivas JM; Elber R Exact Milestoning. J. Chem. Phys 2015, 142, 094102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and Testing of a General Amber Force Field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (54).Wang J; Wang W; Kollman PA; Case DA Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
- (55).Cézard C; Trivelli X; Aubry F; Djedaïni-Pilard F; Dupradeau F-Y Molecular Dynamics Studies of Native and Substituted Cyclodextrins in Different Media: 1. Charge Derivation and Force Field Performances. Phys. Chem. Chem. Phys 2011, 13, 15103. [DOI] [PubMed] [Google Scholar]
- (56).Fukahori T; Nishikawa S; Yamaguchi K Kinetics on Isomeric Alcohols Recognition by α- and β-Cyclodextrins Using Ultrasonic Relaxation Method. Bull. Chem. Soc. Jpn 2004, 77, 2193–2198. [Google Scholar]
- (57).Fukahori T; Kondo M; Nishikawa S Dynamic Study of Interaction Between β-Cyclodextrin and Aspirin by the Ultrasonic Relaxation Method. J. Phys. Chem. B 2006, 110, 4487–4491. [DOI] [PubMed] [Google Scholar]
- (58).Nishikawa S; Fukahori T; Ishikawa K Ultrasonic Relaxations in Aqueous Solutions of Propionic Acid in the Presence and Absence of Beta-Cyclodextrin. J. Phys. Chem. A 2002, 106, 3029–3033. [Google Scholar]
- (59).Nishikawa S; Kondo M Kinetic Study for the Inclusion Complex of Carboxylic Acids with Cyclodextrin by the Ultrasonic Relaxation Method. J. Phys. Chem. B 2006, 110, 26143–26147. [DOI] [PubMed] [Google Scholar]
- (60).Rekharsky MV; Inoue Y Complexation Thermodynamics of Cyclodextrins. Chem. Rev 1998, 98, 1875–1918. [DOI] [PubMed] [Google Scholar]
- (61).Barros TC; Stefaniak K; Holzwarth JF; Bohne C Complexation of Naphthylethanols with β-Cyclodextrin. J. Phys. Chem. A 1998, 102, 5639–5651. [Google Scholar]
- (62).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (63).Baker NA; Sept D; Joseph S; Holst MJ; McCammon JA Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci 2001, 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.