

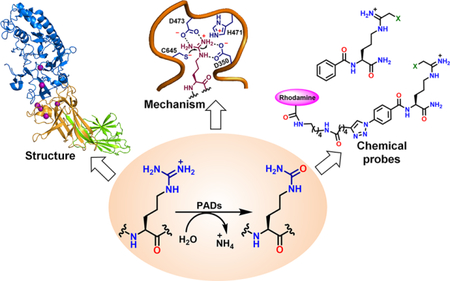
Conspectus
Proteins are well-known to undergo a variety of post-translational modifications (PTMs). One such PTM is citrullination, an arginine modification that is catalyzed by a group of hydrolases called protein arginine deiminases (PADs). Hundreds of proteins are known to be citrullinated and hypercitrullination is associated with autoimmune diseases including rheumatoid arthritis (RA), lupus, ulcerative colitis (UC), Alzheimer’s disease, multiple sclerosis (MS) and certain cancers. In this Accounts, we summarize our efforts to understand the structure and mechanism of the PADs and to develop small molecule chemical probes of protein citrullination. PAD activity is highly regulated by calcium. Structural studies with PAD2 revealed that calcium-binding occurs in a stepwise fashion and induces a series of dramatic conformational changes to form a catalytically competent active site. These studies also identified the presence of a calcium-switch that controls the overall calcium-dependence and a gatekeeper residue that shields the active site in the absence of calcium. Using biochemical and site-directed mutagenesis studies, we identified the key residues (two aspartates, a cysteine, and a histidine) responsible for catalysis and proposed a general mechanism of citrullination. Although all PADs follow this mechanism, substrate-binding to the thiolate or thiol form of the enzyme varies for different isozymes. Substrate specificity studies revealed that PADs 1–4 prefer peptidyl-arginine over free arginine and certain citrullination sites on a peptide substrate.
Using high-throughput screening and activity-based protein profiling (ABPP), we identified several reversible (streptomycin, minocycline and chlorotetracycline) and irreversible (streptonigrin, NSC 95397) PAD-inhibitors. Screening of a DNA-encoded library and lead-optimization led to the development of GSK199 and GSK484 as highly potent PAD4-selective inhibitors. Furthermore, use of an electrophilic, cysteine-targeted haloacetamidine warhead to mimic the guanidinium group in arginine afforded several mechanism-based pan-PAD-inhibitors including Cl-amidine and BB-Cl-amidine. These compounds are highly efficacious in various animal models, including those mimicking RA, UC and lupus. Structure-activity relationships identified numerous covalent PAD-inhibitors with different bioavailability, in vivo stability and isozyme-selectivity (PAD1-selective: D-Cl-amidine; PAD2-selective: compounds 16-20; PAD3-selective: Cl4-amidine; and PAD4-selective: TDFA).
Finally, this Account describes the development of PAD-targeted and citrulline-specific chemical probes. While PAD-targeted probes were utilized for identifying off-targets and developing high-throughput inhibitor screening platforms, citrulline-specific probes enabled the proteomic identification of novel diagnostic biomarkers of hypercitrullination-related autoimmune diseases.
Graphical Abstract
Introduction
The more than 200 posttranslational modifications (PTMs) regulate all aspects of eukaryotic cell signaling. Modifications of arginine are particularly important because arginines play critical roles as substrate specificity determinants and in protein-protein and protein-DNA interactions. Arginine modifications include methylation (forming MMA, SDMA and ADMA), phosphorylation (forming p-Arg), ADP-ribosylation (forming ADP-ribosyl-Arg) and citrullination (forming Cit) (Figure 1A).1,2 During citrullination, the positively-charged guanidinium is hydrolyzed to the neutral urea, which alters the charge and H-bonding potential of this residue, which can impact all of the aforementioned processes. In contrast to most other PTMs, citrullination results in a small mass-change, +0.98 Da, rendering it difficult to disambiguate from the deamidation of neighboring asparagines and glutamines which results in the same mass-change.
Figure 1.
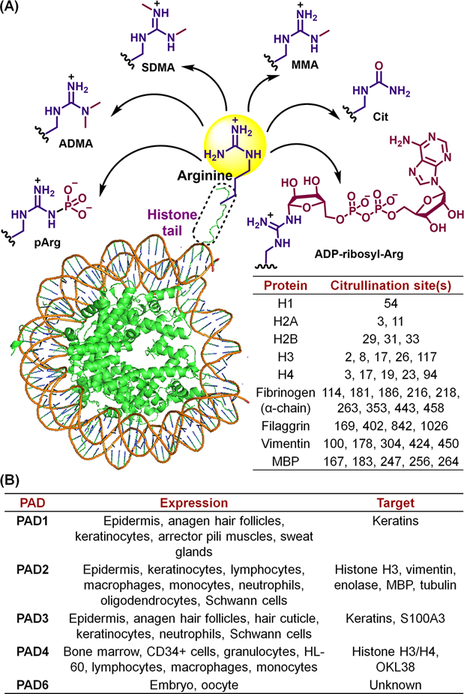
(A) Arginine PTMs. The table highlights representative citrullination sites detected on histones and various other proteins.6 (B) Tissue-specific expression patterns and substrates.
Citrullination is catalyzed by a small family of hydrolases known as the protein arginine deiminases (PADs).1–3 While hundreds of PAD substrates are known, the best characterized are histones. Histones are citrullinated at various sites (Figure 1A), and these PTMs can either activate or repress gene transcription.1 For example, H3R26-citrullination occurs at estrogen receptor α (ERα) target genes and this PTM enhances ERα target gene expression by promoting local chromatin decondensation. By contrast, H3R17 citrullination at the ERα-regulated pS2 promoter leads to transcriptional repression by hindering the gene-transcription-activating effects of R17 methylation. Histone citrullination also plays an important role in DNA damage-induced apoptosis and Neutrophil Extracellular Trap (NET)-formation (or NETosis), a neutrophil-mediated defense mechanism against microbial infection.4,5 Microbial components and/or cytokines promotes the ejection of decondensed chromatin in the form of web-like fibrillar aggregates that can trap pathogens. NETosis can also be induced by several external stimuli including phorbol 12-myristate 13-acetate (PMA) and calcium ionophores.7,8 PAD4 is critical for this process because inhibition or genetic deletion of PAD4 in neutrophils inhibits NETosis.1,2,9,10
The citrullination of fibrinogen, filaggrin, collagen, actin, keratin, β-tubulin and myelin basic protein (MBP) is associated with various physiological processes as well as autoimmune diseases and certain cancers. For example, citrullination is a key driver of rheumatoid arthritis (RA) because PADs are released by neutrophils into joints, where they citrullinate fibrinogen, filaggrin, type II collagen, α-enolase and vimentin. These citrullinated proteins are recognized by anti-citrullinated protein antibodies (ACPA), resulting in the production of pro-inflammatory cytokines and recruitment of additional immune cells that further release PADs into synovial joints, setting up a classic positive-feedback loop.11–14 While ACPA are pathogenic and promote disease progression, they are also important biomarkers to both diagnose and monitor disease progression. Moreover, since ACPA are present 4–5 years before clinical onset they can be used to predict who will develop RA. As such, PADs are novel therapeutic targets to treat RA and other autoimmune disorders associated with aberrant protein citrullination.15,16 Herein, we summarize our efforts to understand the biochemistry of protein citrullination and develop small molecule inhibitors and chemical probes targeting the PADs.
PAD structure and mechanism
Five isozymes – PAD1, PAD2, PAD3, PAD4 and PAD6 – are encoded by five genes arrayed in tandem on chromosome 1.1–3 There is no PAD5 because human PAD5 was originally misannotated and it is now recognized to be the mouse ortholog of PAD4. PADs 1–4 are catalytically active, whereas PAD6 is inactive due to mutations in the active site. Despite their high sequence-similarity, PADs exhibit tissue-specific expression (Figure 1B). While PADs 1, 2 and 4 can enter the nucleus, only PAD4 possesses a canonical nuclear localization signal (NLS; P56PAKKKST63).1,2 Structures of PADs 1, 2 and 4 revealed two N-terminal immunoglobulin domains (IgG1 and IgG2) linked in series to a C-terminal catalytic domain (adopting an α/β propeller fold).17–19 Notably, PAD2 and PAD4 crystallize as head-to-tail homodimers and both active sites are on the same face (Figure 2A).
Figure 2.

(A) Crystal structure of PAD2 (PDB: 4N20). The table highlights aspects of the homodimeric interface. (B-F) Ca2+-binding sites (Ca1–6) in holo-PAD2 (PDB: 4N2C). (G) Overlay of apo- and holo-PAD2 active sites highlighting Ca2+-induced conformational changes. (H) Structure of PAD4C645A in complex with Nα-benzoyl arginine amide (BAA) (PDB: 1WDA).
PAD activity is tightly regulated by calcium, with calcium increasing activity by >10,000-fold.17,19 There are six calcium-binding sites in PAD2 (Ca1–6; Figure 2B–2F). Ca1 and Ca6 are high-affinity sites (KD < 1 µM) as calcium binds these sites even in the absence of added calcium. The Ca6 site (Figure 2F) contains a DxDxDG calcium-binding motif, which is characteristic of EF-hand motifs present in other calcium binding proteins. Notably, this site is conserved amongst all PAD2 orthologues but is absent from PAD1 or PAD4, which bind four or five calcium ions, respectively.17,18 The five other sites are structurally distinct.
Calcium titrations revealed that the Ca3–5 sites bind calcium with moderate affinity (KD~250–260 µM) (Figure 2E) and properly position key catalytic residues (H471, D473 and D351) for catalysis. The exception is C647, which remains 12 Å away from the catalytic center. Ca2 is unoccupied in structures intermediate between apo-PAD2 and holo-PAD2. To obtain the structure of holoPAD2, we mutated F221 and F222 to alanine because these residues bind a hydrophobic pocket on an adjacent monomer in the crystal lattice and we hypothesized that this interaction might prevent calcium binding to Ca2. This was indeed the case and the structure of the F221/222A mutant shows electron density at all six sites and the movement of C647 into a catalytically competent conformation. Moreover, R347 moves out of the active site and W348 flips in to form the substrate-binding cleft (Figure 2G). Therefore, R347 acts as a gatekeeper, or pseudosubstrate, in the absence of calcium-binding at Ca2. Based on these observations, we concluded that Ca3–5 act as a calcium-switch that controls the overall calcium-dependence of the enzyme by governing the formation of the Ca2 site, which in turn controls the formation of a catalytically competent active-site.19
We generated Ca1- (Q350A, E354A, E412A), Ca2- (E352A, D370A, D374A), Ca3–5- (D166A, D177A, D389A) and Ca6- (D123N, D125A) site mutants to validate this hypothesis.19 The most dramatic effect on catalysis was observed with the Ca1 site mutants whose activity (kcat/Km) was decreased by 2,300–3,300-fold with little effect on K0.5 (i.e., the calcium concentration required for half-maximal activity). Ca2 site mutants also exhibited decreased kcat/Km, and minimal effects on K0.5. By contrast, the Ca3–5 mutants showed maximal effects on K0.5 (a 27-fold increase). Mutations in the substrate-binding site remarkably affected catalysis – the W348A mutant was catalytically inactive, whereas the R347A mutant was 50-fold less active than wild-type.19
PADs 1, 3 and 4 also have K0.5 values in the micromolar range (K0.5(PAD1) = 140 µM; K0.5(PAD3) = 550 µM; K0.5(PAD4) = 560 µM) at pH 7.6 when using Nα-benzoyl arginine ethyl ester (BAEE) as the substrate.20,21 Calcium activation is cooperative and the Hill coefficients are >1, consistent with the fact that multiple calcium ions must bind to generate active enzyme. The calcium binding sites are well conserved in PAD1, 2 and 4 (except Ca5, which is absent from PAD1, and Ca6, which is unique to PAD2). Although a PAD3 structure has yet to be published, all PAD isozymes exhibit similar calcium binding kinetics, suggesting that PADs share a common mechanism of calcium activation. Activation is calcium-specific because other metal ions, including barium, magnesium, manganese, strontium, samarium and zinc, do not promote catalysis.20
BAEE, BAA and Nα-benzoyl arginine methyl ester (BAME) (Table 1) are often used model PAD-substrates.1,17 Detailed substrate-specificity studies suggest that BAEE and BAME are preferred for PAD2 and PAD1, respectively, whereas BAA is the best substrate for both PAD1 and PAD4 (Table 1). However, these arginine analogues are poor PAD3 substrates.20–22 The structure of the PAD4C645A-BAA complex highlights how D350 and D473 stabilize the positively charged guanidinium group and place the central guanidinium carbon between C645, positioning it for nucleophilic attack, and H471, which acts as a general acid/base in the first step of the reaction (Figure 2H).17 The carbonyl groups in BAA form a bidentate H-bond with R374, conferring specificity toward peptidyl-arginine substrates over free arginine, which lacks these functionalities.
Table 1.
PAD substrate-specificity.
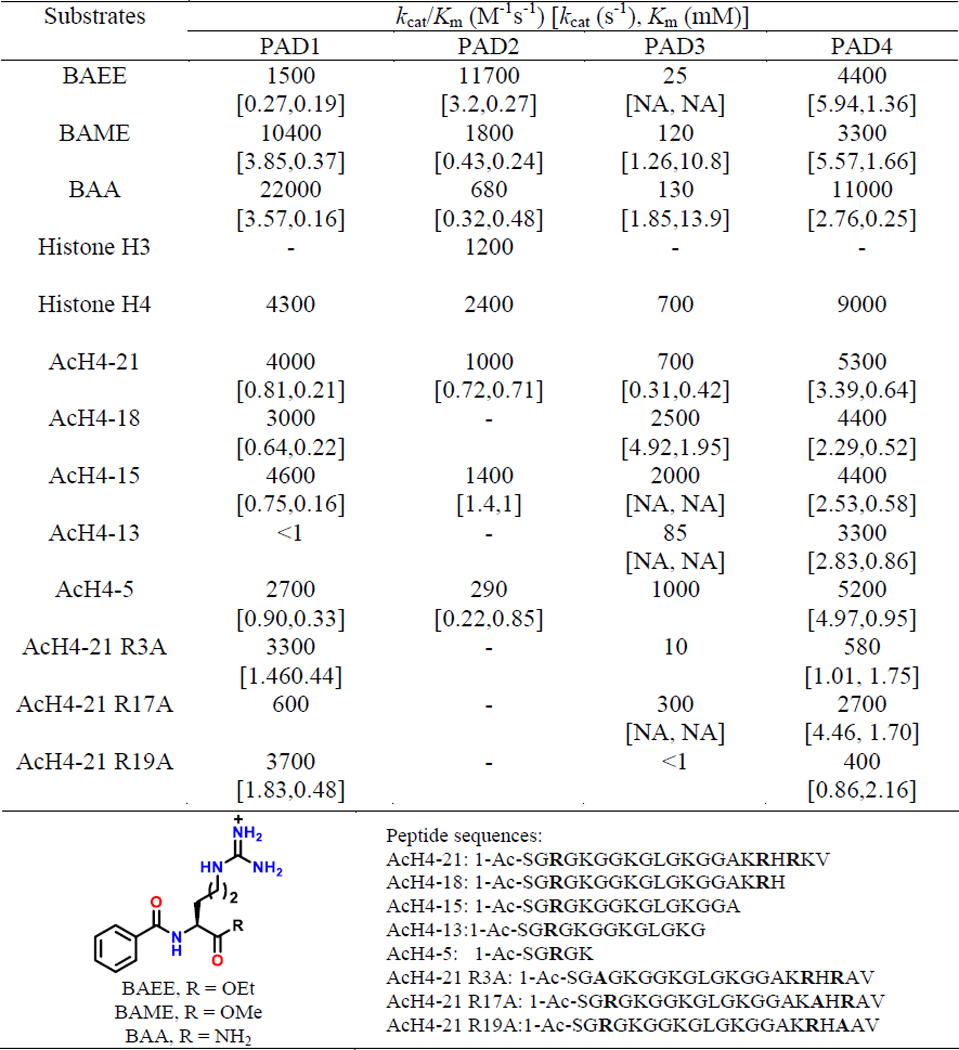 |
In addition to these benzoylated-arginines, we also evaluated the citrullination of histones H3 and H4, two physiological substrates.20–22 The activity of PAD2 towards recombinant histone H3 (kcat/Km = 1200 M-1s-1) is almost 10-fold lower than that observed with BAEE, the best small molecule PAD2 substrate (Table 1).22 Similarly the activity of PAD1 is almost 5-fold lower for histone H4 than BAA.21 Remarkably, histone H4 is a significantly better PAD3-substrate indicating that it has a significantly different substrate scope than the other PADs. Using a library of H4-tail mimics (containing the R3, R17, and R19 citrullination sites), we investigated the role of residues surrounding the citrullination sites in substrate-recognition. The data obtained for the citrullination of the AcH4–21, AcH4–21 R3A, AcH4–21 R17A and AcH4–21 R19A peptides indicate that PAD1 prefers R17, while PAD3 and PAD4 prefer both R3 and R19 (Table 1).21 Furthermore, citrullination of the C-terminal deletion peptides derived from AcH4–21 indicates that long-range interactions and peptide-length contribute minimally to substrate-recognition by PADs 1, 2 and 4. By contrast, the kinetic data for BAA and histone H4 revealed that such interactions are important for substrate-recognition by PAD3.21 Notably these studies did not identify important substrate specificity determinants either upstream or downstream of the site of modification. This finding is consistent with structures of PAD4 in complex with several histone tail-mimics, which show that there are few direct interactions between the residues lining the active site and the side chains of the substrates; most of the key contacts are with the backbone of the substrate.1
Based on detailed biochemical and crystallographic studies, we proposed a general mechanism of catalysis (Figure 3A).17,21,23 The reaction begins with nucleophilic attack of an active-site cysteine (C645 in PAD1 and PAD4; C647 in PAD2; C646 in PAD3) on the central guanidinium carbon (forming INT-1) and the protonation of the departing amine by H471 (generating INT-2). Protonation of the amine may occur either concomitantly or in a stepwise fashion as shown for the conversion of INT-1 into INT-2. Although hydrolysis of INT-3 could afford either ornithine and urea (as in the case of amidinotransferases, a structurally related enzyme family) or citrulline and ammonia, we unambiguously showed that PADs produce citrulline and ammonia in equimolar amounts.20–23 Notably, mutation of any of these four key residues decreases kcat/Km by >5,000-fold, indicating that substrate-binding, nucleophilic attack and general acid-base catalysis are highly cooperative processes.23
Figure 3.

(A) Proposed catalytic mechanism. (B) pH-dependence of kcat/Km with PAD4 (reproduced with permission from ref. 23 Copyright (2007) American Chemical Society).23 Yellow color indicates the fraction of enzyme in the active form. (C) General inactivation mechanisms for 2-iodoacetamide and 2-chloro-acetamidine. (D) Reverse protonation (RP) and substrate-assisted (SA) mechanisms of inactivation.
To gain insights into the protonation states of C645 and H471, we measured PAD4-activity at pH values between 5.7 and 9.0.23 The plot of log kcat/Km versus pH is bell-shaped and pKa values of 7.3 and 8.2 were calculated for the ascending and descending limbs, respectively, (Figure 3B). These two values either represent the deprotonation of C645 and H471, respectively, or in the special case of a reverse-protonation mechanism, the deprotonation of H471 and C645. Detailed inactivation studies with 2-iodoacetamide and 2-chloroacetamidine (Figure 3C) indicated that the descending limb represents the deprotonation of cysteine and, therefore, PAD4 follows a reverse-protonation mechanism, where the inactivator preferentially binds to the thiolate form of the enzyme (Figure 3D). PAD1 and 3 were found to follow a similar mechanism. The observation of a large inverse solvent isotope effect (SIE) on kcat/Km is consistent with a reverse protonation mechanism wherein the active form of the enzyme consists of a thiolate-imidazolium ion pair. Based on the overlap of the titration curves for C645 and H471, only 15% of PAD4 exists in this form at the pH optimum (Figure 3B). By contrast, PAD2 employs a substrate-assisted mechanism wherein the substrate guanidinium generates the reactive thiolate by depressing the pKa of C647 via favorable electrostatic interactions. This mechanism is supported by the observation of both a small inverse SIE and the fact that 2-chloroacetamidine depresses the pKa of C647 by a full log unit relative to 2-iodoacetamide (pKa = 7.2 versus 8.2, respectively) (Figure 3D).22
PAD-inhibitors
Multiple PADs are therapeutic targets. For instance, elevated PAD2 and PAD4 levels, citrullinated proteins, and ACPAs are observed in the blood of RA patients.12,14 Hypercitrullination of MBP by PAD2 and PAD4 promotes its proteolysis, degradation of the myelin sheath, retarded signal transduction, and ultimately multiple sclerosis (MS).16 PAD1 is overexpressed in breast cancer and PAD3 is associated with neurodegeneration.24,25 Protein hypercitrullination is also associated with lupus, Alzheimer’s and Prion’s diseases, and various forms of cancers.16,26,27 Given these strong disease links, numerous reversible and irreversible PAD-inhibitors have been developed.
Reversible PAD-inhibitors
Using an activity-based protein profiling high-throughput screening (ABPP-HTS) platform (described later in the chemical probes section), we identified several potent PAD-inhibitors, including minocycline, chlorotetracycline, ruthenium red, and sanguinarine (Figure 4A).28,29 Among these, ruthenium red was found to be an apo-form selective pan-PAD-inhibitor. GSK121 was identified from a DNA-encoded small molecule library as a PAD4-specific inhibitor. Lead-optimization afforded GSK199 and GSK484 as highly PAD4-selective (>35-fold) reversible inhibitors (Figure 4B).30 Notably, we showed that inhibition is competitive with calcium and the compounds bind preferentially to calcium-free PAD4 (Figure 4C).30 The crystal structure of the PAD4C645A-GSK199 complex confirmed this conclusion and showed that the aminopiperidine interacts with both D473 and H471, and the carbonyl group H-bonds with N588 (Figure 4D).30 PAD4-selectivity is due to the close packing of F634, which is unique to PAD4, against the benzimidazole moiety. GSK106, a control compound methylated on the other benzimidazole nitrogen, lacks potency because this nitrogen is too close (3.6 Å) to the backbone NH of N585 (Figure 4B,D). Notably, an overlay of the PAD4C645A-BAA17 and PAD4C645A-GSK199 complexes highlights how the aminopiperidine moiety in GSK199 and the guanidinium group in BAA bind in the same region (Figure 4E). However, residues 633–645 form a β-hairpin structure in the PAD4C645A-GSK199 complex and pack over the central portion of GSK199. By contrast, they form an α-helical structure in the PAD4C645A-BAA complex. Both GSK199 and GSK484 exhibited remarkable cellular efficacy in preventing citrullination and NET formation in mouse and human neutrophils.30
Figure 4.
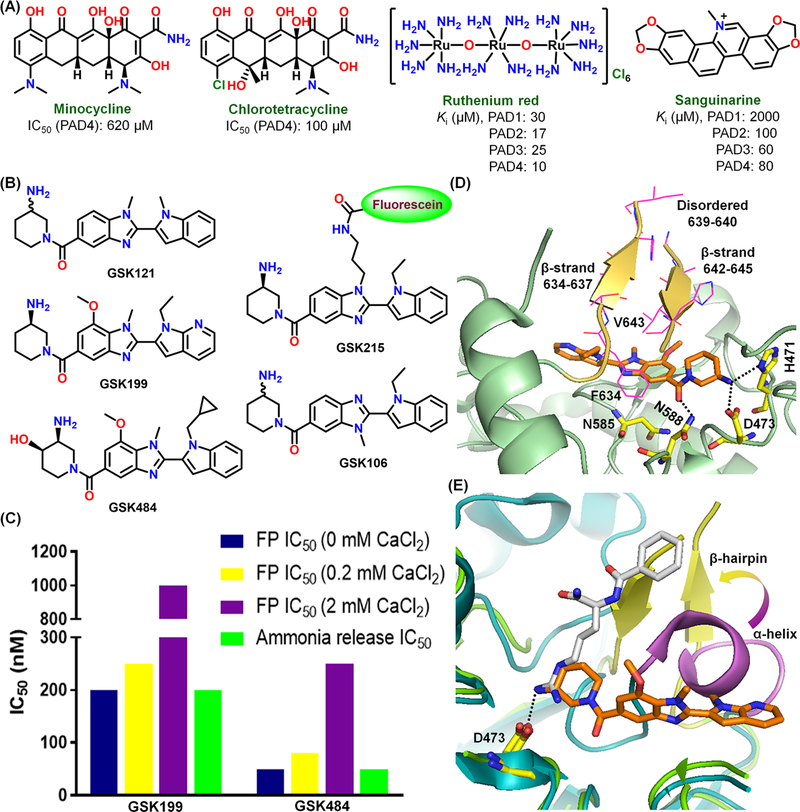
Reversible PAD-inhibitors (A) and allosteric PAD4-selective inhibitors (B). (C) IC50 of GSK199 and GSK484 for PAD4-inhibition assayed with fluorescence polarization (using GSK215) and ammonia release assays at different calcium concentrations. (D) Crystal structure of PAD4C645A-GSK199 complex (PDB: 4X8G). (E) Overlay of PAD4-GSK199 and PAD4-BAA structures (PDB: 1WDA).
Covalent PAD-inhibitors
The ABPP-based HTS (see below) also identified NSC95397 and streptonigrin (Figure 5A) as irreversible PAD-inhibitors.29,31 Interestingly, streptonigrin exhibits excellent potency and PAD4-selectivity, likely due to its substituted pyridyl and phenyl rings.32 Both of these inhibitors contain an α,β-unsaturated carbonyl functionality that covalently modifies the active site cysteine (Figure 5B). Fast et al. showed, and we confirmed, that 2-chloroacetamidine (Figure 3C) irreversibly inhibits PAD4.21–23,33 In parallel, we developed F-amidine and Cl-amidine, which incorporate fluoro- and chloroacetamidine warheads in the BAA scaffold, as pan-PAD inhibitors (Figure 6A).34,35 Detailed kinetic studies revealed that these compounds covalently modify the active-site cysteine in a time- and concentration-dependent manner. Cl-amidine is more potent, consistent with chloride being a better leaving group. Incorporation of a carboxylic acid at the ortho-position of the phenyl group, as in o-F-amidine and o-Cl-amidine, led to superior potency and some isozyme-selectivity (Figure 6A,B).36 Structures of PAD4 bound to F-amidine and o-F-amidine show that these inhibitors form a thioether linkage with C645 while making almost identical contacts within the active site as substrate BAA (Figure 6C, D). The higher potency found for o-F-amidine and o-Cl-amidine is due to a H-bond between the o-carboxylate group in o-F-amidine and the indole NH of W347 (universally conserved amongst all PADs) as well as a water molecule, which H-bonds with Q346.36 Importantly, Cl-amidine and F-amidine inhibited the PAD4-dependent citrullination of p300 in a dose-dependent manner.34,35 o-F-amidine and o-Cl-amidine are 100-fold more potent than Cl-amidine in blocking histone H3 citrullination in HL-60 cells.36
Figure 5.

Chemical structures, potencies (A), and proposed mode of action (B) of NSC 95397 and streptonigrin.
Figure 6.
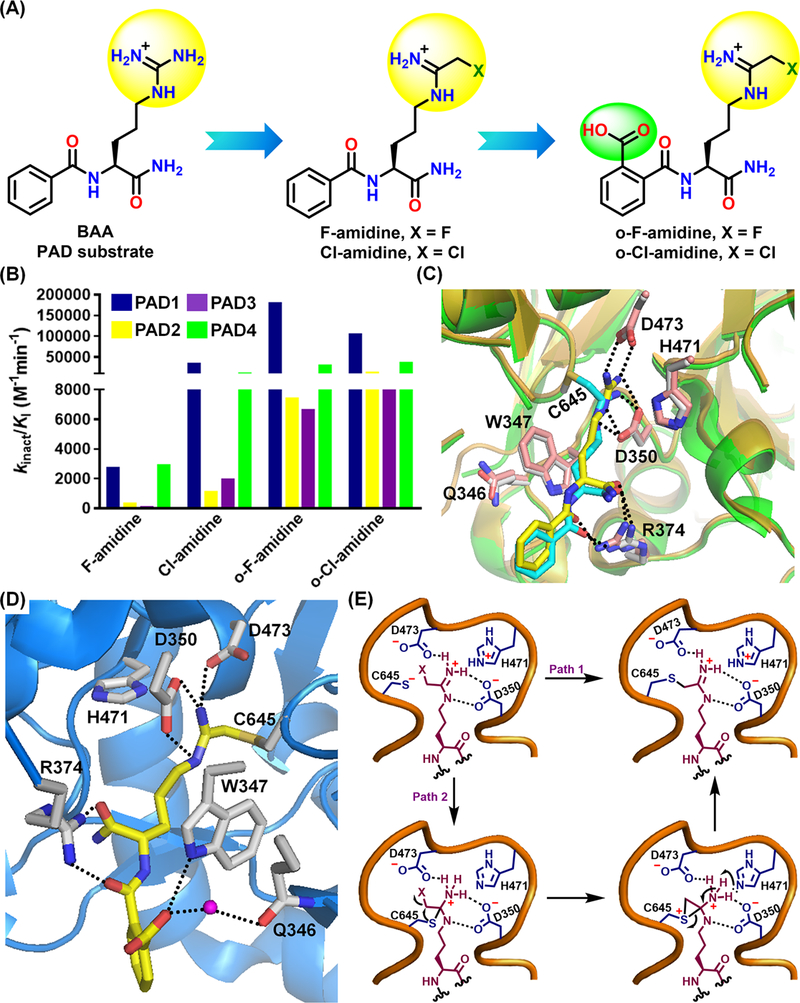
Chemical structures (A) and potencies of F-amidine, Cl-amidine, o-F-amidine and o-Cl-amidine. (B). (C) Overlay of PAD4C645A-BAA (PDB: 1WDA) and PAD4-F-amidine (PDB: 2DW5) structures. (D) Crystal structure of PAD4-o-F-amidine complex (PDB: 3B1U). (E) Mechanism of inactivation for haloacetamidine-based PAD-inhibitors.
Enzyme inactivation follows either an one-step SN2 mechanism (Path 1, Figure 6E) or a multistep pathway involving the formation of a positively-charged sulfonium ring via an intramolecular halide displacement (Path 2, Figure 6E).37 The latter mechanism accounts for the poor leaving group potential of fluoride. Moreover, the pH dependence of enzyme inactivation is bell-shaped, which favors the second mechanism because it accounts for the proton donation step.37 To evaluate the effect of chain-length between the peptide backbone and warhead on potency, we synthesized X2-amidine and X4-amidine (X = Cl, F) with two- and four methylene bridges, respectively, between the chiral α-carbon and warhead (Figure 7A).21,34 While both F2-amidine and Cl2-amidine are poor PAD4-inhibitors, F4-amidine and Cl4-amidine are PAD3-selective inhibitors with decent potency (Figure 7A). Since W347 forms hydrophobic interactions with the alkyl side chain of arginine substrates,17,19 we replaced the alkyl chain with a phenyl group (compounds 1 and 2) hypothesizing that this modification would improve potency by forming π-stacking interactions with W347. However, these compounds are poor PAD4-inhibitors, likely due to an inability to properly position the warhead (Figure 7A).36
Figure 7.

(A) Chemical structures and IC50 of X2-amidine, X4-amidine (X = F, Cl), 1, and 2. (B-C) Chemical structures and potencies of D-F-amidine, D-Cl-amidine, D-o-F-amidine, D-o-Cl-amidine and compounds 3-14.
To improve pharmacokinetic and pharmacodynamic properties, we evaluated the effect of inverting the stereocenter by synthesizing D-F-amidine, D-Cl-amidine, D-o-Cl-amidine and D-o-F-amidine (Figure 7B) as well as a series of tetrazole-containing analogues (compounds 3-14, Figure 7C).38,39 Despite being less potent than the parent compounds, D-Cl-amidine and D-o-F-amidine are PAD1-selective inhibitors, indicating that the stereocenter configuration can alter isozyme-selectivity (Figure 6B, 7B). Relative to Cl-amidine, D-Cl-amidine exhibits better bioavailability, pharmacokinetics, and maximum tolerable dose (i.e., the highest drug dose that does not cause significant toxicity), which is likely due to reduced proteolysis and formation of toxic metabolites.38,39 Notably, the tetrazole-substituted analogues exhibit superior potency with compounds 7-10 having the highest activities. Furthermore, the tert-butyl substitution on the tetrazole increases PAD2-directed potency. For example, 9 (kinact/KI = 120,900 M−1min−1) preferentially inhibits PAD2 (47-fold) relative to its parent compound 7 (kinact/KI = 2,550 M−1min−1) (Figure 7C).39 In contrast to their high in vitro potencies, 7-10 exhibited poor cellular activity which we suspect is due to the negatively-charged carboxylate blocking cellular uptake. However, more hydrophobic compounds, e.g., 13 and 14, are more cytotoxic than Cl-amidine; the EC50 values of 13 and 14 are 45 and 10 µM, respectively, versus 160 µM for Cl-amidine. Furthermore, 14 inhibited NET-formation by mouse neutrophils more efficiently than Cl-amidine and 13 exhibited remarkable microsomal stability relative to Cl-amidine.39
To evaluate other amide bond isosteres, we also synthesized BB-F-amidine and BB-Cl-amidine, which possess an N-terminal biphenyl group to increase hydrophobicity/bioavailability and a C-terminal benzimidazole group that increases proteolytic stability (Figure 8A). These inhibitors exhibited similar in vitro potency as Cl-amidine and F-amidine, however, BB-Cl-amidine is ~20-fold more cytotoxic than Cl-amidine to U2OS cells (EC50 = 8.8 µM for BB-Cl-amidine versus >200 for Cl-amidine) (Figure 8B). Furthermore, BB-Cl-amidine has a significantly longer in vivo half-life than Cl-amidine (Figure 8C), despite similar microsomal stabilities.40 The crystal structure of the PAD4-BB-F-amidine complex revealed that this scaffold forms almost identical contacts with the enzyme active site as Cl-amidine, except that the benzimidazole forms a cation-π interaction with R374 (Figure 8D).41
Figure 8.
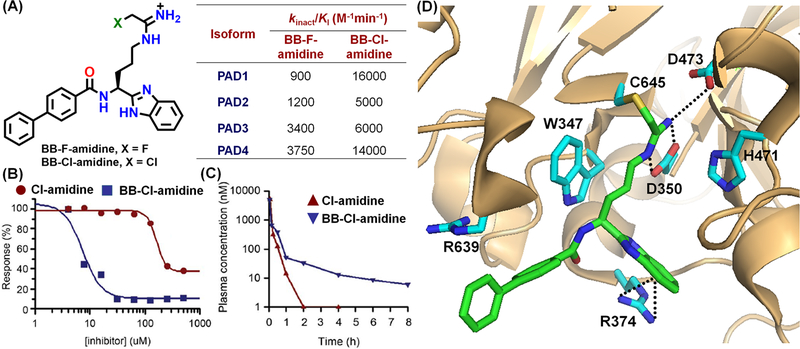
(A) Chemical structures and potencies of BB-F-amidine and BB-Cl-amidine. U2OS cell cytoxicity (B) and in vivo pharmacokinetics (C) of BB-Cl-amidine and Cl-amidine (reproduced with permission from ref. 40 Copyright (2015) BMJ Journals).40 (D) Crystal structure of the PAD4-BB-F-amidine complex (PDB: 5N0M).
Given the enhanced potency afforded by the o-carboxylate in o-Cl-amidine and the tetrazole analogues, we synthesized 15-20, which contain a lactam ring that mimics the o-carboxylate (Figure 9A).41 While the lactam ring and fluoroacetamidine warhead enhance potency and PAD2-selectivity, the benzimidazole substitutions enhanced both properties. The best compounds, 16 (kinact/KI = 210,300 M-1min-1) and 20 (kinact/KI = 365,400 M-1min-1), exhibit high PAD2-selectivity versus PADs 1, 3 and 4 (i.e., 2-, 47- and 15-fold for 16, and 5-, 85- and 85-fold for 20) (Figure 9B).41 Cocrystal structures of PAD4 with 15, 16 and 20 demonstrated that the lactam ring H-bonds with W347 and forces R639, unique to PAD4, to move away from the active site, likely accounting for the PAD2-selectivity (Figure 9C). Superposition of the PAD4-inhibitor complexes on PAD2 suggests that the N-alkyl and alkoxy substitutions orient the benzimidazole ring within a unique hydrophobic pocket on PAD2. Compounds 16-19 potently inhibited histone H3 citrullination in PAD2-overexpressing HEK293TPAD2 cells with EC50’s between 0.4–2.7 µM (Figure 9D).41
Figure 9.
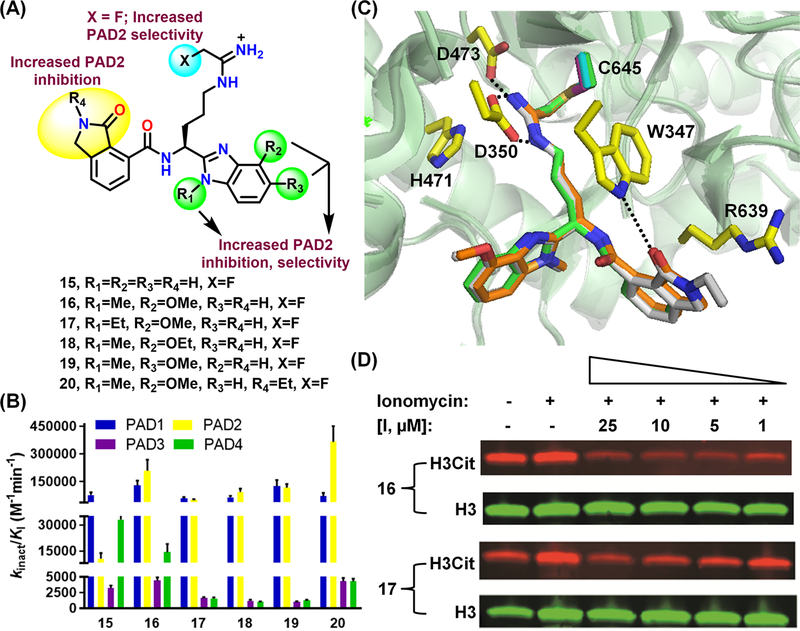
Structures (A) and potencies (B) of compounds 15-20. (C) Structures of PAD4 in complex with 15, 16 and 20 (PDB IDs: 5N1B, 5N0Y, and 5N0Z, respectively). (D) Inhibition of histone H3 citrullination by compounds 16 and 17 in HEK293TPAD2 cells. The ratio of citrullinated H3 (H3Cit) and unmodified H3 was used to determine EC50. Ionomycin is a calcium ionophore. [I, µM] indicates the concentration of inhibitor in micromolar (reproduced with permission from ref. 41 Copyright (2017) American Chemical Society).41
In addition to developing small molecule PAD inhibitors, we identified TDFA from a 264-membered peptide library containing a C-terminal fluoro- or chloroacetamidine-conjugated ornithine.42 TDFA is a PAD4-selective inhibitor (15-, 52- and 65-fold over PADs 1, 2 and 3, respectively) (Figure 10A and B), whereas the chloro-analog, TDCA, was equipotent for PAD1 and PAD4. Interestingly, these peptidic inhibitors contain a carboxylate-containing amino acid next to the fluoroacetamidine-conjugated ornithine similar to o-Cl/F-amidine. The structure of the PAD4-TDFA complex revealed that the aspartate in TDFA forms potential H-bonds with Q346, R639 and R374 as well as W347. By contrast, the carboxylate and lactam ring, respectively, in o-F-amidine and the lactam-substituted compounds (15, 16 and 20) H-bond exclusively with W347, while the C-terminal carboxamide H-bonds with R374 and R372 (Figure 10C). Since R374 is conserved in PAD1 and PAD4, and R639 is unique to PAD4, the PAD4-selectivity over PAD1 likely originates from the interaction with R639. Mutagenesis studies with the R639Q and R374Q mutants validated this conclusion. Notably, TDFA exhibits remarkable potency for the inhibition of histone H3 citrullination in HL-60 cells – 1 nM TDFA was equipotent to 100 µM Cl-amidine and a complete inhibition was observed at 100 nM.42
Figure 10.
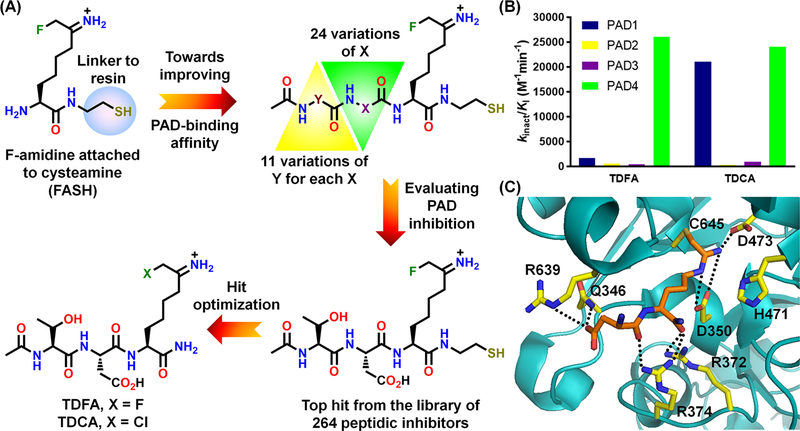
Development (A) and potencies (B) of TDFA and TDCA. (C) Crystal structure of the PAD4–TDFA complex (PDB: 4DKT).
In addition to developing novel PAD-inhibitors with improved potencies and drug-like properties, we exploited the cis/trans isomerism of azobenzenes to modulate inhibitor potency with light (see compounds 21 and 22 in Figure 11A). Irradiation of 21T with 350 nm light, forming 21C, led to an almost 10-fold increase in potency, while irradiation of 22T led to a 45-fold drop in activity (Figure 11A). Detailed kinetic analyses showed that 22T and 22C are irreversible and reversible, respectively, inhibitors of PAD2. Consistent with the in vitro results, 21C inhibits histone H3 citrullination in HEK293TPAD2 cells in a dose-dependent manner, whereas 21T shows no activity even at 100 µM (Figure 11B).43
Figure 11.
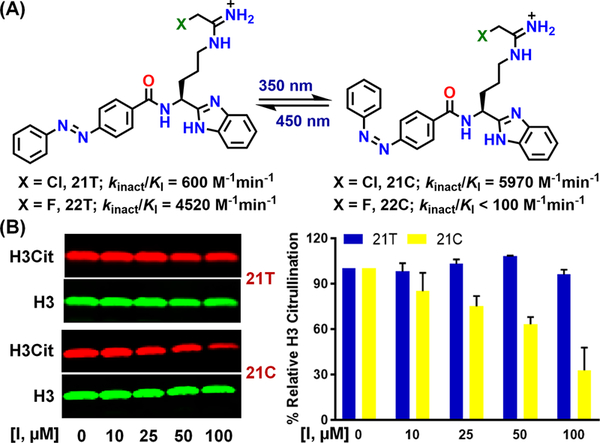
(A) Structures and potencies of photoswitchable inhibitors. (B) Inhibition of histone H3 citrullination by 21T and 21C in HEK293TPAD2 cells (reproduced with permission from ref. 43 Copyright (2018) American Chemical Society).43
Although we have developed numerous PAD-inhibitors with different potencies, isozyme-selectivities, pharmacokinetics, and pharmacodynamics, Cl-amidine and BB-Cl-amidine remain the most thoroughly investigated molecules in animal models of disease. For example, Cl-amidine and BB-Cl-amidine dose-dependently reduce disease severity, joint inflammation and joint damage in the murine collagen-induced arthritis (CIA) model of RA.44,45 Notably, GSK199, a PAD4-selective inhibitor, also reduces disease severity but with lower efficacy than Cl-amidine and BB-Cl-amidine, suggesting that the inhibition of multiple PADs is necessary for maximum therapeutic benefit.46 Cl-amidine was also effective in reducing disease severity, increasing colon length, and improving mobility and activity in the dextran sodium sulfate (DSS) model of ulcerative colitis.47 Efficacy was associated with increased inflammatory cell apoptosis and protection of colonic epithelial cells from DNA damage. Cl-amidine and BB-Cl-amidine treatment also prevented NET formation by neutrophils, interferon (IFN) production and reduced the severity of skin, kidney and vascular disease in the MRL/lpr mouse model of lupus.40 PAD-inhibitors are also efficacious in animal models of spinal cord injury, hypoxic ischemic insult, breast cancer and atherosclerosis.27,48–50
Chemical Probes
Given the pathologies associated with dysregulated PAD activity, it is important to have tools to monitor PAD activity in cells and tissues. Exploiting the covalent nature of our inhibitors, we developed probes that can visualize PAD activity by linking them to rhodamine (see RFA, RCA and RIBFA in Figure 12A).51 RFA also formed the basis for two HTS platforms, in which inhibitor potency is monitored by in-gel fluorescence or fluorescence polarization (FP) (Figure 12B).28,29,31 We also developed alkyne- (Yne) and azide-substituted compounds to enable their bioorthogonal conjugation to either fluorescent tags (FITC-azide and FITC-Yne) or biotin (Figure 12C). These compounds enabled the detection of PAD4-interacting proteins, such as p53, HDAC1 and histone H3, in MCF-7 cells.52
Figure 12.
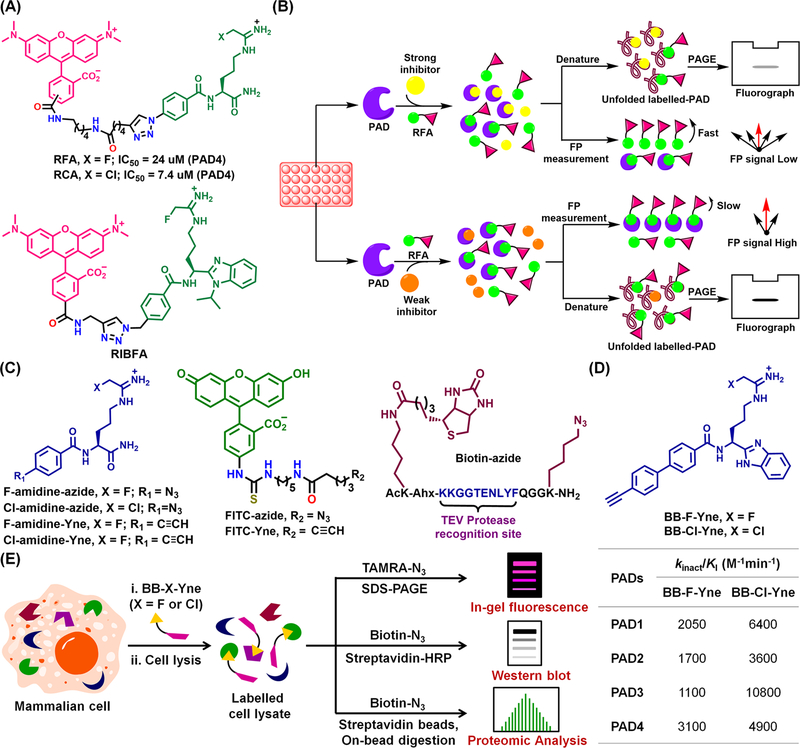
(A) Structures of RFA, RCA and RIBFA. (B) Inhibitor screens using RFA. (C) Structures of Cl/F-amidine-based azide and Yne; fluorescein-based reporter tags (azide and Yne); biotin-azide, in which biotin and the azide functionalities were installed between a TEV protease site. (D) Structures and potencies of BB-F-Yne and BB-Cl-Yne for PAD1–4. (E) Labeling of a PAD by BB-X-Yne (X = F, Cl) in mammalian cells.
Recently, we developed BB-F-Yne and BB-Cl-Yne (Figure 12D) as cell permeable ABPPs.53 These compounds are equipotent to BB-F-amidine and BB-Cl-amidine and can be coupled to TAMRA-N3 or biotin-N3 after PAD inactivation. These probes label recombinant PADs as well as PAD2 in HEK293TPAD2 cells. Using a novel chemoproteomic platform, we identified the off-targets of this scaffold in PAD2 overexpressing HEK293T cells (Figure 12E). Although BB-Cl-Yne has a few off-targets, including heterogeneous nuclear ribonucleoprotein U, clathrin heavy chain 1, bifunctional glutamate/proline-tRNA ligase, tubulin β-chain and actin β-chain, BB-F-Yne is remarkably selective for PAD2; there were essentially no off targets.53 These results highlight the potential of the fluoroacetamidine warhead for developing PAD-selective inhibitors for use in PAD-targeted therapies.
Based on the ability of phenylglyoxal (PG) to selectively condense with the ureido group of citrulline under strongly acidic pH, we developed the citrulline-specific probes rhodamine-PG (Rh-PG) and biotin-PG by attaching rhodamine and biotin, respectively, to PG (Figure 13).54,55 These probes selectively modify citrulline-containing peptides and proteins with remarkable sensitivity, usually in the picomole range. We used Rh-PG to visualize citrullinated proteins present in serum from mice with colitis and biotin-PG to identify >50 proteins involved in mRNA splicing and other cellular process in PAD2 overexpressing HEK293T cells. This platform also identified >150 novel citrullinated proteins, including serine protease inhibitors (SERPINs), serine proteases, transport proteins and complement system components in RA synovial fluid and synovial tissue. We also showed that citrullination can dramatically abolish SERPIN and nicotinamide N-methyltransferase (NNMT) activity.12,56
Figure 13.
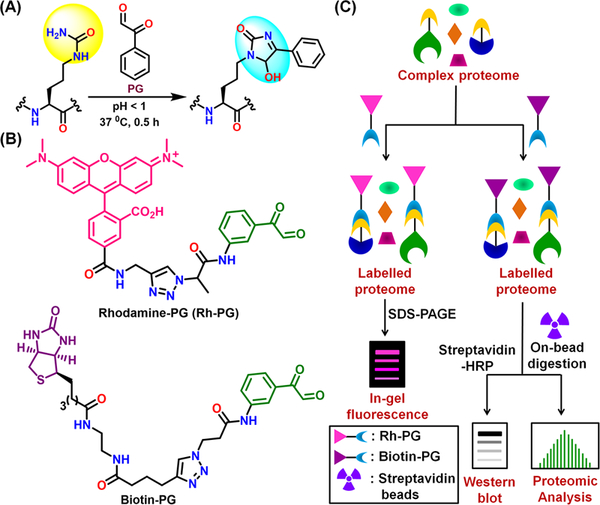
(A) Condensation of phenylglyoxal (PG) with citrulline. (B) Structures of Rh-PG and Biotin-PG. (C) Labeling of citrullinated proteins by Rh-PG, Biotin-PG, and proteomic analysis.
Conclusions
In this account, we summarized our efforts to understand PAD structure, substrate-specificity and catalysis as well as develop inhibitors and chemical probes for these enzymes. Despite our success in these areas, many questions remain unanswered. For example, it is unclear how PADs are activated in cells because near millimolar concentrations of calcium are required for in vitro PAD activity but low micromolar concentrations are found in cells. Moreover, it is unclear whether citrullination is reversible. Although argininosuccinate synthetase and argininosuccinate lyase together convert nonpeptidyl-citrulline to arginine in the urea cycle, the possibility of a similar reaction occurring on peptidyl-citrulline still needs to be explored. Additionally, it will be interesting to investigate the existence of a citrulline-specific ‘reader’ that might interpret the downstream consequences of this modification.
Acknowledgments
Funding Sources
This work was supported in part by NIH grant R35GM118112.
Biographical Information
Santanu Mondal received his Ph.D. from the Indian Institute of Science, Bangalore under the supervision of Prof. G. Mugesh. He is currently a postdoctoral research associate in Prof. Paul R. Thompson’s research group at University of Massachusetts Medical School and is working on the development of PAD-inhibitors.
Paul Thompson is Professor and the Director of Chemical Biology at University of Massachusetts Medical School. His research focuses on the development of novel therapeutics for a range of diseases including cancer, rheumatoid arthritis, and neurodegeneration.
Footnotes
Notes
The authors declare the following competing financial interest(s): P.R.T. founded Padlock Therapeutics and is entitled to payments from Bristol Myers Squibb if certain milestones are met. P.R.T. is a consultant for Celgene and Disarm Therapeutics.
References
- (1).Fuhrmann J; Clancy KW; Thompson PR Chemical biology of protein arginine modifications in epigenetic regulation. Chem. Rev 2015, 115, 5413–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fuhrmann J; Thompson PR Protein Arginine Methylation and Citrullination in Epigenetic Regulation. ACS Chem. Biol 2016, 11, 654–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bicker KL; Thompson PR The protein arginine deiminases: Structure, function, inhibition, and disease. Biopolymers 2013, 99, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brinkmann V; Reichard U; Goosmann C; Fauler B; Uhlemann Y; Weiss DS; Weinrauch Y; Zychlinsky A Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [DOI] [PubMed] [Google Scholar]
- (5).Tanikawa C; Espinosa M; Suzuki A; Masuda K; Yamamoto K; Tsuchiya E; Ueda K; Daigo Y; Nakamura Y; Matsuda K Regulation of histone modification and chromatin structure by the p53-PADI4 pathway. Nat. Commun 2012, 3, 676. [DOI] [PubMed] [Google Scholar]
- (6).Lee CY; Wang D; Wilhelm M; Zolg DP; Schmidt T; Schnatbaum K; Reimer U; Ponten F; Uhlen M; Hahne H; Kuster B Mining the human tissue proteome for protein citrullination. Mol. Cell. Proteomics 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kenny EF; Herzig A; Kruger R; Muth A; Mondal S; Thompson PR; Brinkmann V; Bernuth HV; Zychlinsky A Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Neeli I; Khan SN; Radic M Histone deimination as a response to inflammatory stimuli in neutrophils. J. Immunol 2008, 180, 1895–1902. [DOI] [PubMed] [Google Scholar]
- (9).Hemmers S; Teijaro JR; Arandjelovic S; Mowen KA PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One 2011, 6, e22043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li P; Li M; Lindberg MR; Kennett MJ; Xiong N; Wang Y PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med 2010, 207, 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Burska AN; Hunt L; Boissinot M; Strollo R; Ryan BJ; Vital E; Nissim A; Winyard PG; Emery P; Ponchel F Autoantibodies to posttranslational modifications in rheumatoid arthritis. Mediators Inflamm 2014, 2014, 492873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Tilvawala R; Nguyen SH; Maurais AJ; Nemmara VV; Nagar M; Salinger AJ; Nagpal S; Weerapana E; Thompson PR The Rheumatoid Arthritis-Associated Citrullinome. Cell Chem. Biol 2018, 25, 691–704 e696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Van Steendam K; Tilleman K; Deforce D The relevance of citrullinated vimentin in the production of antibodies against citrullinated proteins and the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2011, 50, 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wright HL; Moots RJ; Edwards SW The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol 2014, 10, 593–601. [DOI] [PubMed] [Google Scholar]
- (15).Jones JE; Causey CP; Knuckley B; Slack-Noyes JL; Thompson PR Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential. Curr. Opin. Drug Discov. Devel 2009, 12, 616–627. [PMC free article] [PubMed] [Google Scholar]
- (16).Witalison EE; Thompson PR; Hofseth LJ Protein Arginine Deiminases and Associated Citrullination: Physiological Functions and Diseases Associated with Dysregulation. Curr. Drug Targets 2015, 16, 700–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Arita K; Hashimoto H; Shimizu T; Nakashima K; Yamada M; Sato M Structural basis for Ca(2+)-induced activation of human PAD4. Nat. Struct. Mol. Biol 2004, 11, 777–783. [DOI] [PubMed] [Google Scholar]
- (18).Saijo S; Nagai A; Kinjo S; Mashimo R; Akimoto M; Kizawa K; Yabe-Wada T; Shimizu N; Takahara H; Unno M Monomeric Form of Peptidylarginine Deiminase Type I Revealed by X-ray Crystallography and Small-Angle X-ray Scattering. J. Mol. Biol 2016, 428, 3058–3073. [DOI] [PubMed] [Google Scholar]
- (19).Slade DJ; Fang P; Dreyton CJ; Zhang Y; Fuhrmann J; Rempel D; Bax BD; Coonrod SA; Lewis HD; Guo M; Gross ML; Thompson PR Protein arginine deiminase 2 binds calcium in an ordered fashion: implications for inhibitor design. ACS Chem. Biol 2015, 10, 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kearney PL; Bhatia M; Jones NG; Yuan L; Glascock MC; Catchings KL; Yamada M; Thompson PR Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 2005, 44, 10570–10582. [DOI] [PubMed] [Google Scholar]
- (21).Knuckley B; Causey CP; Jones JE; Bhatia M; Dreyton CJ; Osborne TC; Takahara H; Thompson PR Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3. Biochemistry 2010, 49, 4852–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dreyton CJ; Knuckley B; Jones JE; Lewallen DM; Thompson PR Mechanistic studies of protein arginine deiminase 2: evidence for a substrate-assisted mechanism. Biochemistry 2014, 53, 4426–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Knuckley B; Bhatia M; Thompson PR Protein arginine deiminase 4: evidence for a reverse protonation mechanism. Biochemistry 2007, 46, 6578–6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Qin H; Liu X; Li F; Miao L; Li T; Xu B; An X; Muth A; Thompson PR; Coonrod SA; Zhang X PAD1 promotes epithelial-mesenchymal transition and metastasis in triple-negative breast cancer cells by regulating MEK1-ERK1/2-MMP2 signaling. Cancer Lett 2017, 409, 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).U KP.; Subramanian V; Nicholas AP; Thompson PR; Ferretti P Modulation of calcium-induced cell death in human neural stem cells by the novel peptidylarginine deiminase-AIF pathway. Biochim. Biophys. Acta 2014, 1843, 1162–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chang X; Han J; Pang L; Zhao Y; Yang Y; Shen Z Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 2009, 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).McElwee JL; Mohanan S; Griffith OL; Breuer HC; Anguish LJ; Cherrington BD; Palmer AM; Howe LR; Subramanian V; Causey CP; Thompson PR; Gray JW; Coonrod SA Identification of PADI2 as a potential breast cancer biomarker and therapeutic target. BMC Cancer 2012, 12, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Knuckley B; Luo Y; Thompson PR Profiling Protein Arginine Deiminase 4 (PAD4): a novel screen to identify PAD4 inhibitors. Bioorg. Med. Chem 2008, 16, 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lewallen DM; Bicker KL; Madoux F; Chase P; Anguish L; Coonrod S; Hodder P; Thompson PR A FluoPol-ABPP PAD2 high-throughput screen identifies the first calcium site inhibitor targeting the PADs. ACS Chem. Biol 2014, 9, 913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lewis HD; Liddle J; Coote JE; Atkinson SJ; Barker MD; Bax BD; Bicker KL; Bingham RP; Campbell M; Chen YH; Chung CW; Craggs PD; Davis RP; Eberhard D; Joberty G; Lind KE; Locke K; Maller C; Martinod K; Patten C; Polyakova O; Rise CE; Rudiger M; Sheppard RJ; Slade DJ; Thomas P; Thorpe J; Yao G; Drewes G; Wagner DD; Thompson PR; Prinjha RK; Wilson DM Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol 2015, 11, 189–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Knuckley B; Jones JE; Bachovchin DA; Slack J; Causey CP; Brown SJ; Rosen H; Cravatt BF; Thompson PR A fluopol-ABPP HTS assay to identify PAD inhibitors. Chem. Commun. (Camb) 2010, 46, 7175–7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dreyton CJ; Anderson ED; Subramanian V; Boger DL; Thompson PR Insights into the mechanism of streptonigrin-induced protein arginine deiminase inactivation. Bioorg. Med. Chem 2014, 22, 1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Stone EM; Schaller TH; Bianchi H; Person MD; Fast W Inactivation of two diverse enzymes in the amidinotransferase superfamily by 2-chloroacetamidine: dimethylargininase and peptidylarginine deiminase. Biochemistry 2005, 44, 13744–13752. [DOI] [PubMed] [Google Scholar]
- (34).Luo Y; Arita K; Bhatia M; Knuckley B; Lee YH; Stallcup MR; Sato M; Thompson PR Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry 2006, 45, 11727–11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Luo Y; Knuckley B; Lee YH; Stallcup MR; Thompson PR A fluoroacetamidine-based inactivator of protein arginine deiminase 4: design, synthesis, and in vitro and in vivo evaluation. J. Am. Chem. Soc 2006, 128, 1092–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Causey CP; Jones JE; Slack JL; Kamei D; Jones LE; Subramanian V; Knuckley B; Ebrahimi P; Chumanevich AA; Luo Y; Hashimoto H; Sato M; Hofseth LJ; Thompson PR The development of N-alpha-(2-carboxyl)benzoyl-N(5)-(2-fluoro-1-iminoethyl)-l-ornithine amide (o-F-amidine) and N-alpha-(2-carboxyl)benzoyl-N(5)-(2-chloro-1-iminoethyl)-l-ornithine amide (o-Cl-amidine) as second generation protein arginine deiminase (PAD) inhibitors. J. Med. Chem 2011, 54, 6919–6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Knuckley B; Causey CP; Pellechia PJ; Cook PF; Thompson PR Haloacetamidine-based inactivators of protein arginine deiminase 4 (PAD4): evidence that general acid catalysis promotes efficient inactivation. Chembiochem 2010, 11, 161–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bicker KL; Anguish L; Chumanevich AA; Cameron MD; Cui X; Witalison E; Subramanian V; Zhang X; Chumanevich AP; Hofseth LJ; Coonrod SA; Thompson PR D-amino acid based protein arginine deiminase inhibitors: Synthesis, pharmacokinetics, and in cellulo efficacy. ACS Med. Chem. Lett 2012, 3, 1081–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Subramanian V; Knight JS; Parelkar S; Anguish L; Coonrod SA; Kaplan MJ; Thompson PR Design, synthesis, and biological evaluation of tetrazole analogs of Cl-amidine as protein arginine deiminase inhibitors. J. Med. Chem 2015, 58, 1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Knight JS; Subramanian V; O’Dell AA; Yalavarthi S; Zhao W; Smith CK; Hodgin JB; Thompson PR; Kaplan MJ Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis 2015, 74, 2199–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Muth A; Subramanian V; Beaumont E; Nagar M; Kerry P; McEwan P; Srinath H; Clancy K; Parelkar S; Thompson PR Development of a Selective Inhibitor of Protein Arginine Deiminase 2. J. Med. Chem 2017, 60, 3198–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jones JE; Slack JL; Fang P; Zhang X; Subramanian V; Causey CP; Coonrod SA; Guo M; Thompson PR Synthesis and screening of a haloacetamidine containing library to identify PAD4 selective inhibitors. ACS Chem. Biol 2012, 7, 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Mondal S; Parelkar SS; Nagar M; Thompson PR Photochemical Control of Protein Arginine Deiminase (PAD) Activity. ACS Chem. Biol 2018, 13, 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kawalkowska J; Quirke AM; Ghari F; Davis S; Subramanian V; Thompson PR; Williams RO; Fischer R; La Thangue NB; Venables PJ Abrogation of collagen-induced arthritis by a peptidyl arginine deiminase inhibitor is associated with modulation of T cell-mediated immune responses. Sci. Rep 2016, 6, 26430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Willis VC; Gizinski AM; Banda NK; Causey CP; Knuckley B; Cordova KN; Luo Y; Levitt B; Glogowska M; Chandra P; Kulik L; Robinson WH; Arend WP; Thompson PR; Holers VM N-alpha-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J. Immunol 2011, 186, 4396–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Willis VC; Banda NK; Cordova KN; Chandra PE; Robinson WH; Cooper DC; Lugo D; Mehta G; Taylor S; Tak PP; Prinjha RK; Lewis HD; Holers VM Protein arginine deiminase 4 inhibition is sufficient for the amelioration of collagen-induced arthritis. Clin. Exp. Immunol 2017, 188, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Chumanevich AA; Causey CP; Knuckley BA; Jones JE; Poudyal D; Chumanevich AP; Davis T; Matesic LE; Thompson PR; Hofseth LJ Suppression of colitis in mice by Cl-amidine: a novel peptidylarginine deiminase inhibitor. Am. J. Physiol. Gastrointest. Liver Physiol 2011, 300, G929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ghari F; Quirke AM; Munro S; Kawalkowska J; Picaud S; McGouran J; Subramanian V; Muth A; Williams R; Kessler B; Thompson PR; Fillipakopoulos P; Knapp S; Venables PJ; La Thangue NB Citrullination-acetylation interplay guides E2F-1 activity during the inflammatory response. Sci. Adv 2016, 2, e1501257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Knight JS; Luo W; O’Dell AA; Yalavarthi S; Zhao W; Subramanian V; Guo C; Grenn RC; Thompson PR; Eitzman DT; Kaplan MJ Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res 2014, 114, 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lange S; Rocha-Ferreira E; Thei L; Mawjee P; Bennett K; Thompson PR; Subramanian V; Nicholas AP; Peebles D; Hristova M; Raivich G Peptidylarginine deiminases: novel drug targets for prevention of neuronal damage following hypoxic ischemic insult (HI) in neonates. J. Neurochem 2014, 130, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Luo Y; Knuckley B; Bhatia M; Pellechia PJ; Thompson PR Activity-based protein profiling reagents for protein arginine deiminase 4 (PAD4): synthesis and in vitro evaluation of a fluorescently labeled probe. J. Am. Chem. Soc 2006, 128, 14468–14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Slack JL; Causey CP; Luo Y; Thompson PR Development and use of clickable activity based protein profiling agents for protein arginine deiminase 4. ACS Chem. Biol 2011, 6, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Nemmara VV; Subramanian V; Muth A; Mondal S; Salinger AJ; Maurais AJ; Tilvawala R; Weerapana E; Thompson PR The Development of Benzimidazole-Based Clickable Probes for the Efficient Labeling of Cellular Protein Arginine Deiminases (PADs). ACS Chem. Biol 2018, 13, 712–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bicker KL; Subramanian V; Chumanevich AA; Hofseth LJ; Thompson PR Seeing citrulline: development of a phenylglyoxal-based probe to visualize protein citrullination. J. Am. Chem. Soc 2012, 134, 17015–17018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lewallen DM; Bicker KL; Subramanian V; Clancy KW; Slade DJ; Martell J; Dreyton CJ; Sokolove J; Weerapana E; Thompson PR Chemical Proteomic Platform To Identify Citrullinated Proteins. ACS Chem. Biol 2015, 10, 2520–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Nemmara VV; Tilvawala R; Salinger AJ; Miller L; Nguyen SH; Weerapana E; Thompson PR Citrullination Inactivates Nicotinamide- N-methyltransferase. ACS Chem. Biol 2018, 13, 2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]