

Abstract
Kinetochore localized Mad1 is essential for generating a “wait anaphase” signal during mitosis, hereby ensuring accurate chromosome segregation. Inconsistent models for the function and quantitative contribution of the two mammalian Mad1 kinetochore receptors: Bub1 and the Rod‐Zw10‐Zwilch (RZZ) complex exist. By combining genome editing and RNAi, we achieve penetrant removal of Bub1 and Rod in human cells, which reveals that efficient checkpoint signaling depends on the integrated activities of these proteins. Rod removal reduces the proximity of Bub1 and Mad1, and we can bypass the requirement for Rod by tethering Mad1 to kinetochores or increasing the strength of the Bub1‐Mad1 interaction. We find that Bub1 has checkpoint functions independent of Mad1 localization that are supported by low levels of Bub1 suggesting a catalytic function. In conclusion, our results support an integrated model for the Mad1 receptors in which the primary role of RZZ is to localize Mad1 at kinetochores to generate the Mad1‐Bub1 complex.
Keywords: Bub1, CRISPR, kinetochore, Mad1, mitosis
Subject Categories: Cell Cycle
Introduction
Accurate segregation of chromosomes during cell division depends on a functional spindle assembly checkpoint (SAC) that in response to improperly attached kinetochores generates a “wait anaphase” signal (Musacchio, 2011; Lischetti & Nilsson, 2015). The generation of this signal depends on the recruitment of checkpoint proteins to the outer kinetochore, which facilitates the formation of the mitotic checkpoint complex (MCC) composed of the checkpoint proteins Mad2 and BubR1/Bub3 bound to Cdc20 (Sudakin et al, 2001; Chao et al, 2012). The MCC is a potent inhibitor of the anaphase‐promoting complex/cyclosome (APC/C) in complex with its mitotic co‐activator Cdc20, and the presence of the MCC therefore delays anaphase onset (Primorac & Musacchio, 2013; Izawa & Pines, 2015; Alfieri et al, 2016; Yamaguchi et al, 2016).
How the kinetochore catalyzes MCC production is a major unresolved question in the field. Answering this requires dissection of the molecular mechanisms of checkpoint protein recruitment to the kinetochore and of the interactions between checkpoint proteins. It is clear that phosphorylation of Met‐Glu‐Leu‐Thr (MELT) repeats in the outer kinetochore protein KNL1 by the checkpoint kinase Mps1 generates binding sites for the checkpoint complexes Bub1/Bub3 and BubR1/Bub3 (London et al, 2012; Shepperd et al, 2012; Yamagishi et al, 2012; Primorac et al, 2013; Vleugel et al, 2013, 2015b; Zhang et al, 2014, 2016). Subsequent phosphorylation of Bub1 by Mps1 then facilitates an interaction between Mad1 and Bub1 a mechanism conserved from yeast to man (London & Biggins, 2014; Mora‐Santos et al, 2016; Faesen et al, 2017; Ji et al, 2017; Qian et al, 2017; Zhang et al, 2017). Mad1 is in a stable complex with Mad2 and the recruitment of Mad1/Mad2 to kinetochores is essential because this complex catalyzes the first step in MCC formation by loading Mad2 onto Cdc20 (De Antoni et al, 2005; Faesen et al, 2017; Ji et al, 2017). Indeed, the kinetochore levels of the Mad1/Mad2 complex have been shown to correlate with the strength of the checkpoint signal (Collin et al, 2013; Dick & Gerlich, 2013). Bub1 is the only Mad1 kinetochore receptor in yeast, but the situation is more complex in higher eukaryotes as a three‐subunit complex composed of Rod, ZW10, and Zwilch (RZZ complex) contributes to Mad1 localization (Basto et al, 2000; Buffin et al, 2005; Kops et al, 2005; Gama et al, 2017; Mosalaganti et al, 2017). The RZZ complex is able to polymerize, hereby generating the outer corona of the kinetochore which recruits checkpoint proteins and proteins regulating microtubule attachment (Pereira et al, 2018; Rodriguez‐Rodriguez et al, 2018; Sacristan et al, 2018). Genetic approaches in Drosophila melanogaster and HAP1 cells as well as antibody injection in human cells have revealed that the RZZ complex is required for checkpoint signaling (Basto et al, 2000; Chan et al, 2000; Raaijmakers et al, 2018). However, an accurate quantitative characterization is still missing and the limited effect of Rod removal in non‐transformed cells suggests a minor role of the complex in checkpoint signaling (Silió et al, 2015). Furthermore, the RZZ complex could have additional functions in the checkpoint beyond Mad1 recruitment (Grohme et al, 2018). Bub1 was identified as an essential component of the checkpoint in budding yeast and was subsequently shown to be important in multiple model organisms (Hoyt et al, 1991; Roberts et al, 1994; Basto et al, 2000; Vanoosthuyse et al, 2004; Meraldi & Sorger, 2005; Perera et al, 2007; Klebig et al, 2009). However, two recent studies using CRISPR/Cas9‐mediated Bub1 gene deletion in HAP1 and RPE1 cells revealed a minor role of Bub1 in checkpoint signaling (Currie et al, 2018; Raaijmakers et al, 2018). These recent studies question a conserved function for Bub1 in the SAC.
One reason for the reported discrepancies on the contribution of the RZZ complex and Bub1 to Mad1 localization and checkpoint signaling could be differences between the large number of cell lines and model systems used to study these proteins. Alternatively, since very efficient depletion is needed to uncover the actual contribution of certain checkpoint proteins (Meraldi & Sorger, 2005), a different explanation could be that small variations in checkpoint protein depletion efficiency strongly influence observed phenotypes.
The aim of this study was to quantitatively compare penetrant Bub1 and Rod depletions and dissect their functions on the molecular level, hereby clarifying their respective contributions and roles in the checkpoint.
Results
RZZ‐mediated localization of Mad1 is required for an efficient response to unattached kinetochores
To investigate the precise role of the RZZ complex in SAC signaling, we first identified a Rod RNAi oligo and established a depletion protocol that efficiently depleted the RZZ component ZW10 to below 5% on kinetochores in HeLa cells and to ≈1% as determined by quantitative Western blot (Figs 1A, D and E, and 2A). We analyzed the strength of the checkpoint by time‐lapse microscopy in the presence of nocodazole—a microtubule poison that generates unattached kinetochores. Measuring the time from nuclear envelope breakdown (NEBD) to mitotic exit provides a quantitative readout of checkpoint activity and depletion of Rod clearly decreased the activity of the SAC (control‐depleted cells mean arrest time = 790 min, Rod‐depleted cells = 220 min; Fig 1B and C). Importantly, co‐transfection with an RNAi‐resistant Venus‐Rod construct fully restored the checkpoint response in Rod‐depleted cells confirming that the observed Rod RNAi phenotype was due to depletion of Rod (Fig 1C). Analysis of Venus‐Rod localization revealed that it localized to kinetochores immediately after NEBD consistent with a role in the checkpoint (Fig 1B). A Rod construct lacking the N‐terminal β‐propeller domain required for polymerization of RZZ was less efficient in supporting checkpoint signaling (Gama et al, 2017; Fig 1C). Depletion of Rod in RPE1 and U2OS cells also decreased the duration of the arrest in nocodazole (Fig 1F).
Figure 1. Rod is required for Mad1 localization and checkpoint response to unattached kinetochores.
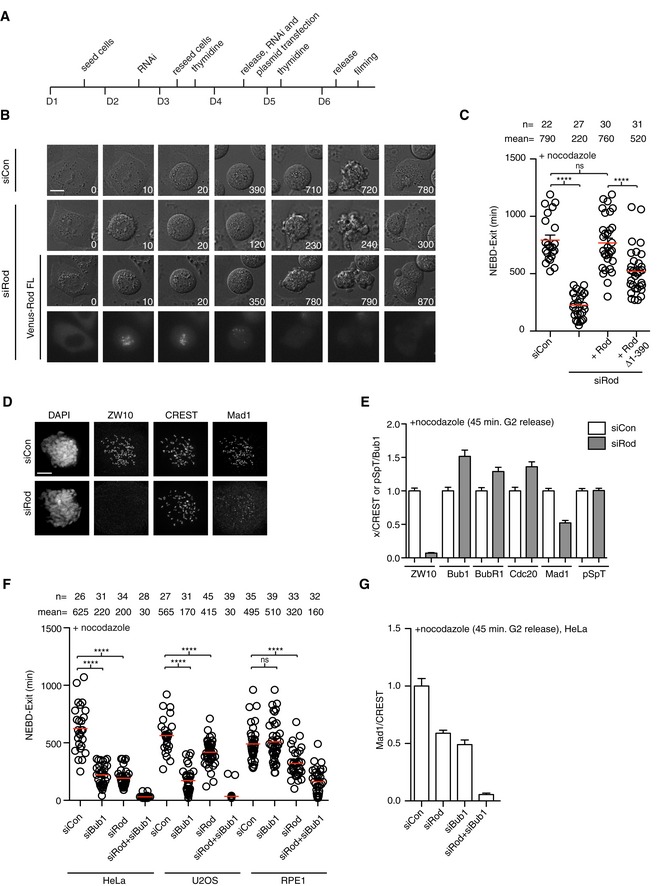
- Schematic of Rod RNAi depletion and synchronization protocol.
- Representative still images of control, Rod‐depleted cells, and Rod‐depleted cells supplemented with RNAi‐resistant Venus‐Rod treated with nocodazole and time in minutes indicated. Scale bar, 5 μm.
- Time from NEBD to mitotic exit for the indicated conditions with each circle representing a single cell analyzed and mean time indicated by red line and standard error of mean shown. The number of cells counted and mean time indicated above. A Mann–Whitney U‐test was used for statistical analysis (ns: non‐significant, ****P ≤ 0.0001). A representative result from at least three independent experiments is shown.
- Immunofluorescence analysis of ZW10 and Mad1 upon depletion of Rod. Scale bar, 5 μm.
- Quantification of kinetochore intensities of the indicated proteins in control‐ or Rod‐depleted cells with the signal normalized to CREST levels and Bub1 pSpT normalized to Bub1. Bar indicates mean and standard error of mean is shown by line. At least 200 kinetochores from 10 cells were analyzed and representative result from at least two independent experiments is shown.
- Time from NEBD to mitotic exit in control‐depleted, Rod‐depleted, Bub1‐depleted, and Rod‐ and Bub1‐co‐depleted HeLa, U2OS, and RPE1 cells. Cells were treated with nocodazole. (Mann–Whitney U‐test, ns: non‐significant, ****P ≤ 0.0001). A representative result from at least three independent experiments is shown.
- Mad1 kinetochore levels in the indicated conditions normalized to level of CREST in HeLa cells. Cells were fixed at 45 min after releasing from RO3306 arrest into nocodazole. Bar indicates mean and standard error of mean is shown by line. At least 200 kinetochores from 10 cells were analyzed and representative result from at least two independent experiments is shown.
Figure 2. Penetrant Rod removal by combing CRISPR/Cas9 and RNAi.
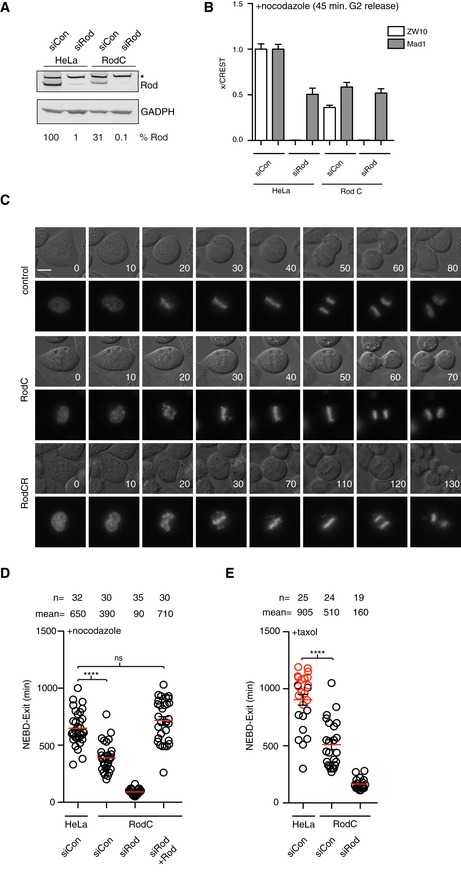
-
AWestern blot analysis of whole‐cell extract for the indicated conditions. Quantitative analysis of Rod levels using LI‐COR technology is indicated below. The asterisk indicates an unspecific band.
-
BAnalysis of ZW10 and Mad1 kinetochore levels in the indicated cells and conditions. Kinetochore signal is normalized to CREST signal. Bar indicates mean and standard error of mean is shown by line. At least 200 kinetochores from 10 cells were analyzed and representative result from at least two independent experiments shown.
-
CRepresentative still images of unperturbed mitosis for parental cells, Rod C cells, and Rod CR cells with time in minutes indicated. CFP‐Histone 3 was used here as the chromosome marker. Scale bar, 5 μm.
-
D, ETime from NEBD to exit of indicated conditions with cells treated with nocodazole or taxol. Red circles represent cells still arrested in mitosis when the filming stopped (Mann–Whitney U‐test, ns: non‐significant, ****P ≤ 0.0001). Mean (red line) and standard error of mean (black bar) indicated. A representative result from at least three independent experiments is shown.
Consistent with the reported role of the RZZ complex in recruitment of Mad1 to kinetochores, analysis by immunofluorescence revealed that Mad1 levels were reduced by 50% in Rod‐depleted cells (45 min after releasing from RO3306 into nocodazole) while all other checkpoint proteins analyzed were not decreased (Figs 1D and E, and EV1A). Importantly, the phosphorylation sites in Bub1 required for Bub1‐Mad1 complex formation (Ji et al, 2017; Qian et al, 2017; Zhang et al, 2017) were not affected by Rod depletion arguing that the reduction in Mad1 levels upon Rod depletion was not an indirect effect on Bub1 phosphorylation (Figs 1D and E, and EV1A). Depletion of both Rod and Bub1 efficiently removed Mad1 from kinetochores in HeLa cells and resulted in a strong SAC defect in HeLa, U2OS, and RPE1 cells (Fig 1F and G).
Figure EV1. Rod and Bub1 depletions.
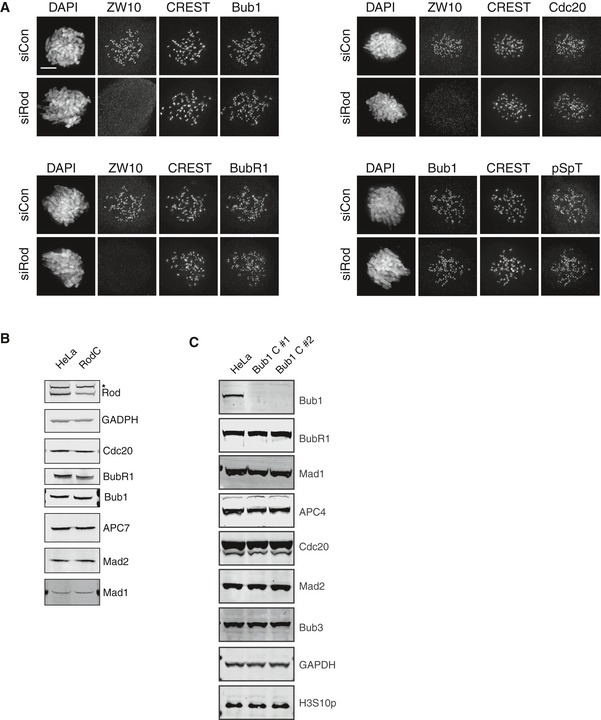
-
AImmunofluorescence images of control‐depleted cells and Rod‐depleted cells with ZW10, Bub1, Bub1 pSpT, BubR1, and Cdc20 kinetochore staining. The quantification is in Fig 1E. Scale bar, 5 μm.
-
B, CAnalysis of protein levels by Western blot analysis of whole‐cell extract from HeLa, Rod C, and Bub1 C cells. * in (B) indicates unspecific band recognized by Rod antibody.
Given that low levels of Rod might be sufficient to support SAC signaling, we wanted to address SAC strength upon penetrant removal of Rod. To do this, we used CRISPR/Cas9 technology to target Rod exon 2 in HeLa cells. We never obtained HeLa Rod null clones suggesting that the RZZ complex is essential for viability of HeLa cells. However, multiple clones had reduced levels of Rod while all other SAC proteins or APC/C components analyzed were not affected (Figs 2A and EV1B). We will refer to these cell lines as Rod C throughout. During unperturbed mitosis, alignment of chromosomes in the Rod C cell line was delayed shortly compared to the parental HeLa cell line (Fig 2C and Table 1). However, despite the delay in alignment, the cells waited with initiating anaphase until all chromosomes had congressed. When we depleted Rod by RNAi in the Rod C cell line (referred to as Rod CR), we observed more penetrant removal of Rod compared to parental HeLa cells treated with Rod RNAi, while the effect on Mad1 kinetochore levels was subtle (Fig 2A and B). Despite this small difference in Rod levels, the defect in SAC signaling was more penetrant in Rod CR cells compared to Rod RNAi cells (mean time in nocodazole t = 220 min in Rod RNAi, t = 90 min in Rod CR). Importantly, the defect in checkpoint strength could be restored by exogenous expression of Venus‐Rod attesting to a specific removal of Rod (Fig 2D). Similarly, in taxol‐arrested cells we observed a clear requirement for Rod in checkpoint signaling and Rod C cells clearly arrested less efficiently in taxol consistent with this drug more readily unmasks perturbations to SAC signaling (Collin et al, 2013; Fig 2E). In Rod CR cells, we observed severe defects in chromosome alignment and cells exiting with unaligned chromosomes consistent with a weakened checkpoint response and the reported role of the RZZ complex in chromosome alignment (Starr et al, 1998; Savoian et al, 2000; Fig 2C and Table 1).
Table 1.
Analysis of mitotic timing and segregation errors during an unperturbed mitosis
| Condition | NEBD‐exit (min) | Alignment delay (%) | Missegregation at exit (%) | n |
|---|---|---|---|---|
| HeLa | 40 | 5 | 5 | 42 |
| Rod C | 50 | 8 | 5 | 52 |
| Rod CR | 105 | 90 | 55a | 42 |
| Bub1 C | 90 | 56 | 5 | 41 |
| Bub1 CR | 85 | 100 | 100b | 31 |
| Rod CR + Bub1 RNAi | 35 | 100 | 100 | 14 |
| Bub1 CR + Rod RNAi | 30 | 100 | 100 | 17 |
Few unaligned chromosomes at exit.
Large number of unaligned chromosomes at exit.
In conclusion, penetrant Rod removal reveals that checkpoint signaling strongly depends on the RZZ complex.
Bub1 is required for the checkpoint response to unattached kinetochores in different cell lines
Based on our results on Rod, we reasoned that the reported differences on the role of Bub1 in the checkpoint might be caused by small variations in protein levels remaining. We therefore decided to investigate this using CRISPR/Cas9 to lower Bub1 levels and hereby sensitizing cells to Bub1 RNAi depletion as we had done for Rod. We targeted exon 2 and obtained multiple Bub1 cell lines (Bub1 C) that based on Western blot analysis appeared to be Bub1 knockout cell lines (Fig EV1C). Surprisingly, these cell lines had a normal checkpoint response in nocodazole but a slightly weakened response to taxol (Fig 3A and B). However, a number of observations argued that the Bub1 C cell lines still expressed residual levels of Bub1 sufficient to support checkpoint signaling. Firstly, when Bub1 C was exposed to several independent Bub1 RNAi oligos (referred to Bub1 CR), we observed a clear impairment of the SAC response in nocodazole that could be rescued by exogenous Bub1 but not Bub1 lacking its Mad1 binding domain (Bub1ΔCD1; Figs 3A and EV2A). Secondly, by immunofluorescence analysis of mitotic Bub1 C cells we observed very weak kinetochore staining with a phospho‐specific Bub1 antibody and this staining disappeared upon subsequent Bub1 RNAi depletion (Fig EV2B). Thirdly, mass spectrometry analysis of Bub3 or BubR1 mitotic purifications, which are stable binding partners of Bub1, revealed multiple Bub1 peptides present in Bub1 C, although with reduced intensity compared to parental cells (Figs 3C and D, and EV2C, Table EV1). Importantly, in Bub1 CR we only detected few low intensity peptides revealing further reduction in Bub1 levels (Fig 3C and D, Table EV1). Contamination between samples was excluded by mass spectrometry analysis of purifications run in parallel but only treated with buffer (Table EV1). We estimate that 4% Bub1 are left in the Bub1 C cell lines based on Bub1 peptide intensities in BubR1 purifications. As expected, the level of MCC components in Bub3 and BubR1 purifications was not affected in agreement with an almost normal checkpoint in Bub1 C cells (Figs 3C and EV2C). MCC levels are also normal in Bub1 CR cells as we collected mitotic arrested cells shortly after entry. Collectively, these results argue that very low levels of Bub1 are sufficient for generating a functional checkpoint signal. Given that CRISPR/Cas9‐mediated HAP1 and RPE1 Bub1 KO cells have been generated and reported to have an almost fully functional checkpoint response (Currie et al, 2018; Raaijmakers et al, 2018), we investigated if these cells might also have residual Bub1 left accounting for the functional checkpoint. We therefore obtained these cell lines and depleted Bub1 by 4 different RNAi oligos and monitored checkpoint strength in nocodazole (Fig EV2A). Similar to our HeLa Bub1 C cell lines, we observed that Bub1 depletion impaired the SAC response in HAP1 and RPE1 Bub1 KO cells suggesting that residual Bub1 are left in these cell lines and this is why they have a normal checkpoint response. This was further confirmed by mass spectrometry analysis of BubR1 purifications from these cell lines that revealed multiple Bub1 peptides (Fig 3C and D, and Table EV1). Interestingly, in both our HeLa Bub1 C and in the RPE1 and HAP1 Bub1 KO cells we did not detect any peptides in the very N‐terminal part of Bub1, which is where the gRNAs are targeting (Fig EV3 and Table EV1). This might suggest that this part of Bub1 is missing in the cell lines.
Figure 3. Bub1 is required for SAC signaling from unattached kinetochores.
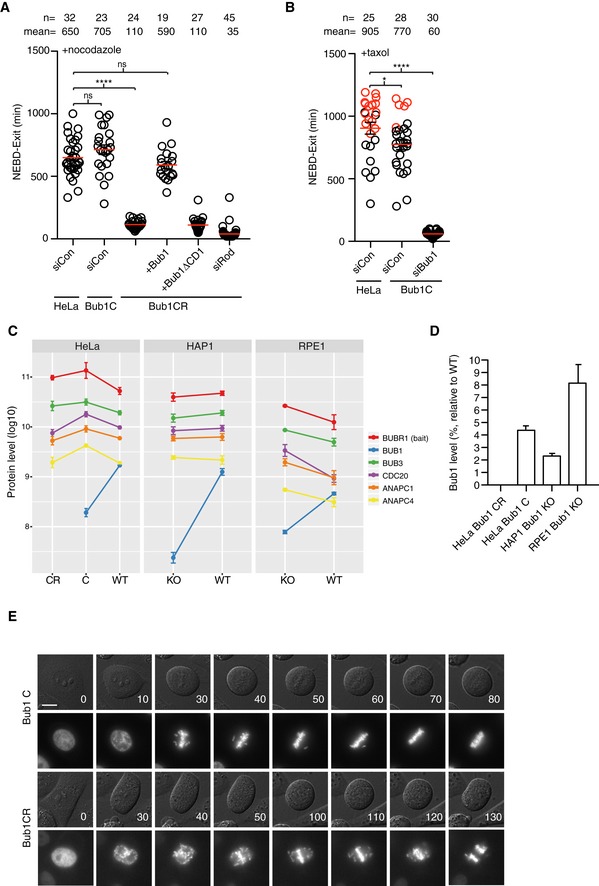
-
A, BTime from NEBD to exit in indicated conditions with cells treated with nocodazole or taxol. Red circles represent cells still arrested in mitosis when the filming stopped (Mann–Whitney U‐test, ns: non‐significant, *P ≤ 0.05, ****P ≤ 0.0001). Mean (red line) and standard error of mean (black bar) indicated. A representative result from at least three independent experiments is shown.
-
CMass spectrometry analysis of BubR1 IPs from the indicated cell lines. Relative protein quantification values (MaxLFQ, log10) are plotted across conditions. Data from analysis of three technical repeats of BubR1 purifications with standard deviation indicated.
-
DEstimated protein levels of Bub1 in the indicated conditions relative to parental cell lines. In Bub1 CR, we only detected few peptides from three purifications. Standard deviation indicated.
-
ERepresentative still images of unperturbed mitosis for parental cells, Bub1 C cells, and Bub1 CR cells with time in minutes indicated. CFP‐Histone 3 was used as the chromosome marker. Scale bar, 5 μm.
Figure EV2. Live cell and mass spectrometry analysis of Bub1 knockout cell lines.
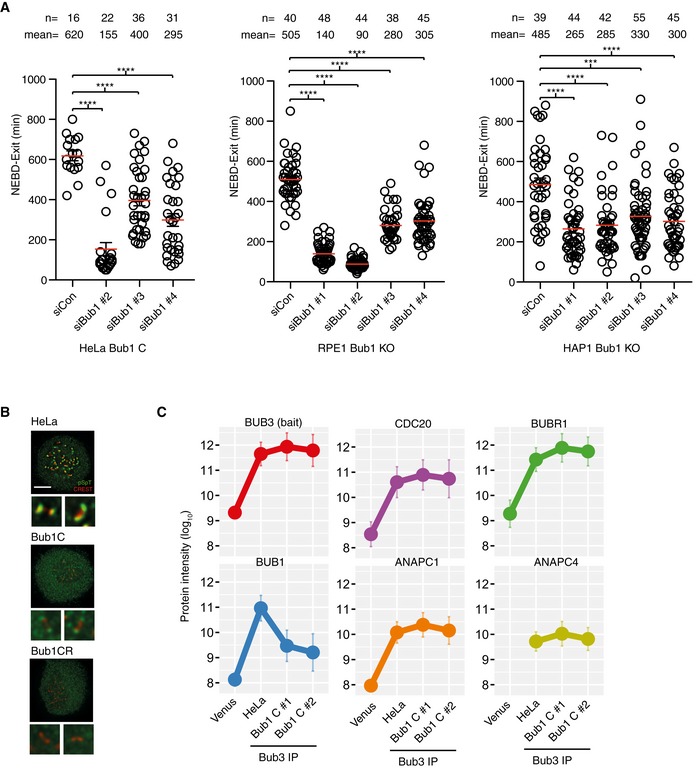
- HeLa Bub1C, HAP1 Bub1 KO, and RPE1 Bub1 KO cells were treated with 3 or 4 Bub1 RNAi oligos, and the time from NEBD to exit was measured in the presence of nocodazole. Mean (red line) and standard error of mean (black bar) indicated (Mann–Whitney U‐test, ns: non‐significant, ***P ≤ 0.0001, ****P ≤ 0.00001). Representative result from two independent experiments is shown.
- Staining of indicated cell lines with Bub1 pSpT antibody. Scale bar, 5 μm.
- Relative protein quantification values (MaxLFQ, log10) are plotted across conditions for Venus‐Bub3 purifications from indicated cell lines with standard deviation shown. Mass spectrometry analysis was performed on three technical replicates of Venus purification.
Figure EV3. Bub1 peptides detected by mass spectrometry.

The primary sequence of Bub1 is shown and peptides identified in HeLa and HeLa Bub1 C cells shown from Venus Bub3 purifications. The site targeted by the gRNA and RNAi oligos is indicated.
Depletion of Bub1 by RNAi in Bub1 C cells allowed us to determine the contribution of Bub1 to the SAC and its role in chromosome segregation. In both nocodazole‐ and taxol‐arrested cells, the checkpoint was strongly impaired although not fully abrogated which is due to Rod‐mediated recruitment of Mad1 (mean time in nocodazole t = 705 min in Bub1 C, t = 110 min in Bub1 CR; mean time in taxol t = 770 min in Bub1 C, t = 60 min in Bub1 CR; Fig 3A and B). In Bub1 C cells, chromosome alignment was delayed, but cells rarely entered anaphase with unaligned chromosomes due to a functional checkpoint (Fig 3E and Table 1). However, the complete removal of Bub1 resulted in massive alignment problems and cells exiting mitosis with many unaligned chromosomes suggesting a near complete loss of checkpoint activity (Fig 3E and Table 1). Although Bub1 is known to be important for chromosome alignment (Meraldi & Sorger, 2005), we are not aware of previous studies reporting such a strong defect upon Bub1 removal.
In summary, we conclude that Bub1 is required for the efficient response to unattached kinetochores in human cells and that 2–4% of Bub1 are sufficient for almost normal checkpoint activity. Furthermore, our quantitative comparison of Rod and Bub1 CR phenotypes suggests that Rod and Bub1 activities are integrated, as their combined individual contributions to SAC strength are considerably lower than that of the control situation.
Tethering of Mad1 to kinetochores bypasses the requirement for Rod
As both Rod and Bub1 are responsible for Mad1 kinetochore localization and checkpoint activation, we wanted to address whether Mad1 kinetochore localization is their only function or whether they have additional roles in the checkpoint. To address this, we analyzed checkpoint signaling in cells where Mad1 was artificially tethered to kinetochores through fusion to KNL1 or Ndc80 and analyzed checkpoint strength in Bub1 CR, Rod CR or by RNAi depletion (Fig 4A and B). While the Ndc80‐Mad1 or Mad1‐KNL1 fusion completely bypassed the requirement for Rod in SAC signaling, Bub1 was still required. This result suggests that RZZ is mainly required for Mad1 localization while Bub1 has additional checkpoint functions. In agreement with the Mad1‐tethering experiments, we observed cells exiting from nocodazole arrest with reduced but relatively stable Venus‐tagged Mad1 localized to kinetochores in cells lacking Bub1 (Fig 4C). This was in contrast to the Mad1 localization in cells lacking Rod where we observed continuously reduced levels of Mad1 at kinetochores with the kinetochore signal disappearing after approximately 1 h.
Figure 4. Mad1 localization and kinetochore dynamics.
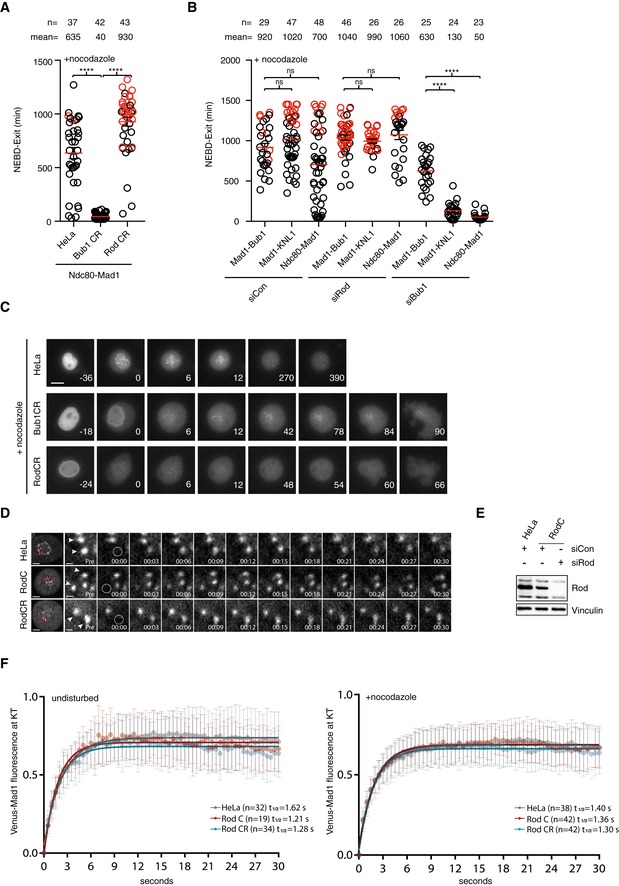
-
A, BTethering of Mad1 (Mad1 485–718) to Ndc80, KNL1 (Zhang et al, 2017), or Bub1 1–553ΔCD1 and the time from NEBD to exit was measured in each condition. Red circles represent cells still arrested in mitosis when the filming stopped. Mean (red line) and standard error of mean (black bar) indicated (Mann–Whitney U‐test, ns: non‐significant, ****P ≤ 0.0001). A representative result from at least three independent experiments is shown.
-
CLocalization of Venus‐tagged Mad1 in parental HeLa cells, Bub1 CR cells, and Rod CR cells in the presence of nocodazole. Scale bar, 5 μm.
-
DStill images from FRAP experiments using Venus‐tagged Mad1 in the indicated conditions. Image before bleach is shown and then time following bleach. Arrowheads indicate kinetochore pairs. Scale bar, 5 μm for whole‐cell image and 1 μm for the zoom in on kinetochores.
-
EAnalysis of Rod levels in cells used for FRAP experiment.
-
FVenus‐tagged Mad1 kinetochore intensity versus time from bleaching shortly after NEBD in unperturbed cells (left) or in cells arrested with nocodazole (right). Data combined from three independent experiments and standard deviation indicated.
The inability of Mad1 to remain at kinetochores in cells lacking Rod suggested that the RZZ complex might affect the turnover of Mad1 at kinetochores. To test this, we analyzed the dynamics of Venus‐tagged Mad1 in unperturbed and nocodazole‐treated cells using fluorescence recovery after photobleaching (FRAP). We only analyzed kinetochore pairs where we could continuously detect the unbleached kinetochore of the pair to ensure that the pair of kinetochores remained in focus (Figs 4D, and EV4A and B). In both unperturbed cells and in nocodazole‐treated cells, we could not detect any effect on Mad1 dynamics upon removal of Rod arguing that the RZZ complex mainly acts to localize Mad1 to kinetochores without affecting its turnover (Fig 4E and F). Similarly, Mad1 dynamics was not affected by the removal of Bub1 arguing that Mad1 dynamically associates with both kinetochore receptors (Fig EV4C).
Figure EV4. Mad1 dynamics and Mad1 localization in Bub1 mutants.
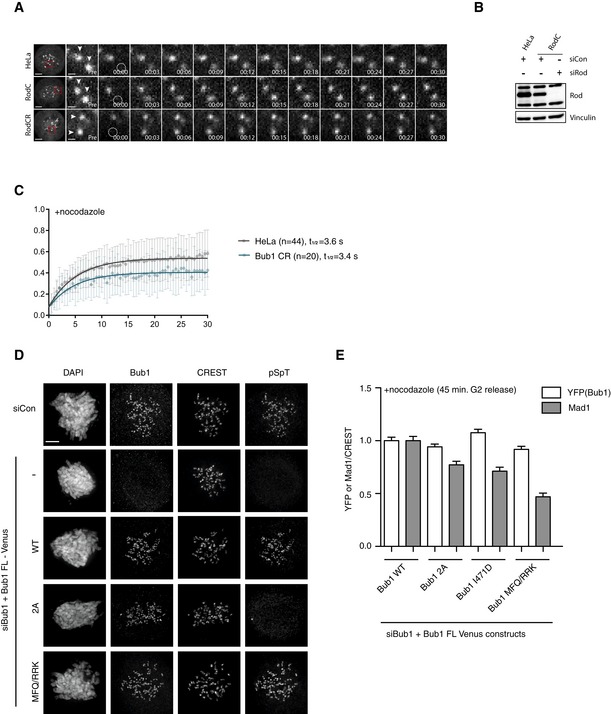
- Still images from FRAP experiments for unperturbed mitosis using Venus‐tagged Mad1 in the indicated conditions. Arrowheads indicate kinetochore pairs. Image before bleach is shown and then time following bleach. The quantification is shown in Fig 4F (left). Scale bar, 5 μm for whole‐cell image and 1 μm for kinetochore zoom in.
- The analysis of Rod levels in cells used for FRAP experiment is shown on the right.
- Quantification of Venus‐tagged Mad1 recovery following bleaching in HeLa or Bub1 CR cells with standard deviation indicated. Data combined from three independent experiments.
- Immunofluorescence analysis of Bub1 phosphorylation in the indicated Bub1 constructs. Cells were depleted of Bub1 and complemented with the indicated Bub1 mutants and analyzed by immunofluorescence 45 min after release from a Cdk1 block by RO3306 into nocodazole. Kinetochore Bub1 and phosphorylated Bub1 were stained by corresponding antibodies. Scale bar, 5 μm.
- Kinetochore levels of Mad1 and YFP (Bub1 fusion protein) for the indicated Bub1 constructs normalized to CREST. Bar indicates mean and standard error of mean is shown by line. At least 200 kinetochores from 10 cells were analyzed and representative result from at least two independent experiments shown.
In summary, the experiments suggest that the main function of the RZZ complex in the checkpoint is to localize Mad1 to kinetochores.
Increasing the interaction between Bub1 and Mad1 bypasses the requirement for Rod
Collectively, our data reveal a critical importance of the Mad1‐Bub1 interaction as well as RZZ‐mediated Mad1 kinetochore localization for SAC signaling. Given that our quantitative analysis of Bub1 CR and Rod CR cells indicated an integrated function of the two checkpoint components, we hypothesized that RZZ localizes Mad1 on kinetochores to facilitate the interaction between Bub1 and Mad1. To further investigate this, we first determined whether the RZZ complex is required for the interaction between Mad1 and Bub1 using our recently established proximity‐dependent ligation assay (Zhang et al, 2017). In this assay, Mad1 is fused to the BirA* biotin ligase resulting in biotinylation of proteins in proximity of Mad1 upon addition of biotin to the cell culture. A point mutant unable to bind Bub1 in vitro fails to biotinylate Bub1 in this assay showing that the assay reports on the Mad1‐Bub1 interaction in cells. If Rod and Bub1 operate in separate pathways and localize and interact with Mad1 independently of each other, then the prediction would be that depletion of Rod should not affect the proximity of Mad1 and Bub1. Strikingly, the removal of Rod almost completely abolished biotinylation of Bub1 in nocodazole‐arrested cells supporting a model in which Rod positively influences Mad1‐Bub1 interaction (Fig 5A). If the hypothesis that Rod stimulates Mad1‐Bub1 interaction is correct, then we would predict that increasing the strength of the Mad1‐Bub1 interaction might bypass the requirement for Rod in generating a checkpoint signal. To test this directly, we sought of ways to artificially stimulate the Mad1‐Bub1 interaction. Interestingly, plants and algae lack the RZZ complex and one of their three Bub1 like proteins contains multiple repeats of the CD1 domain likely to increase the strength of the Mad1‐Bub1 interaction (Di Fiore et al, 2016; van Hooff et al, 2017). To mimic this in human cells, we generated a Bub1 construct containing four repeats of the CD1 domain (Bub1 4XCD1). As a control, a similar construct but with the four CD1 domains lacking the phosphorylation sites (S459A, T461A) mediating Mad1 interaction was used (4XCD1 2A). Bub1‐4XCD1 very efficiently recruited Mad1 to kinetochores and supported efficient checkpoint signaling in Rod CR cells while Bub1 4XCD1 2A or Bub1 1–529 with only one CD1 did not (Fig 5B, D and E). In a complementary approach, Mad1 was fused directly to Bub1 and this also bypassed the requirement for Rod (Figs 4B and 5C). Bub1 CR complemented with Bub1 4xCD1 mounted a SAC response similar to Bub1 1–529 despite recruiting four times more Mad1 to kinetochores suggesting that additional factors are affecting SAC strength (Fig 6A and B). To determine whether Bub1 4xCD1 had a dominant effect on mitotic progression, we analyzed mitotic timing and chromosome segregation in HeLa cells expressing this construct but observed no effect (Fig 6C and D).
Figure 5. Increased Mad1‐Bub1 interaction bypasses the requirement for Rod.
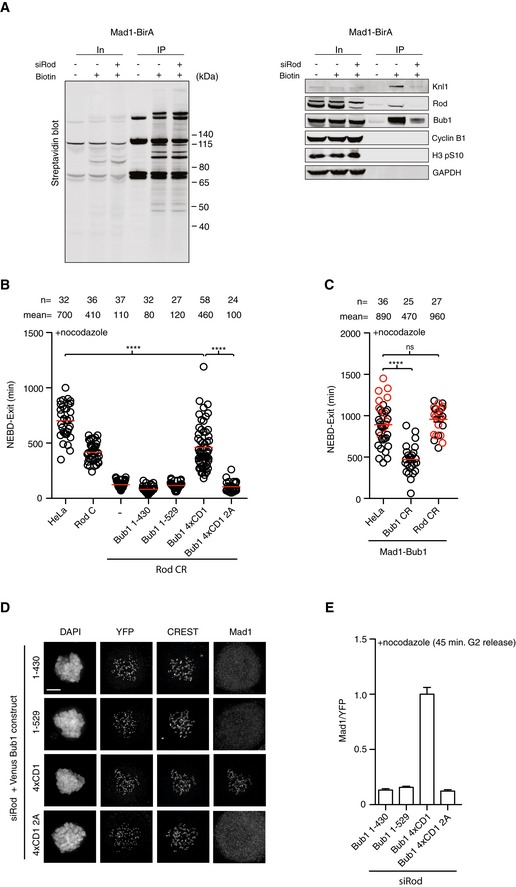
-
AHeLa cell line stably expressing Mad1‐BirA was depleted of Rod and synchronized in mitosis using thymidine and nocodazole. Biotin was added to media where indicated and biotinylated proteins were purified and analyzed by Western blot with the indicated antibodies. Representative of two independent experiments.
-
BThe duration from NEBD to exit in each indicated conditions was recorded by time‐lapse microscopy. Mean (red line) and standard error of mean (black bar) indicated (Mann–Whitney U‐test, ****P ≤ 0.0001). A representative result from at least three independent experiments is shown.
-
CMitotic duration in the indicated conditions expressing Mad1‐Bub1 (Mad1 485–718 fused to Bub1 1–553ΔCD1) fusion protein. The time from NEBD to exit is indicated. Red circles represent cells still arrested in mitosis when the filming stopped. Mean (red line) and standard error of mean (black bar) indicated. (Mann–Whitney U‐test, ns: non‐significant, ****P ≤ 0.0001). A representative result from at least three independent experiments is shown.
-
D, ECells depleted of Rod using RNAi and transfected with the indicated Bub1 constructs. (D) Representative immunofluorescence images of cells stained for YFP and Mad1 in each condition. Scale bar, 5 μm. (E) Mad1 kinetochore levels were measured and normalized to Bub1 (YFP) level. Bar indicates mean and standard error of mean is shown by line. At least 200 kinetochores from 10 cells were analyzed and representative result from at least two independent experiments is shown.
Figure 6. Analysis of Bub1 4xCD1 in Bub1 CR and parental cells.
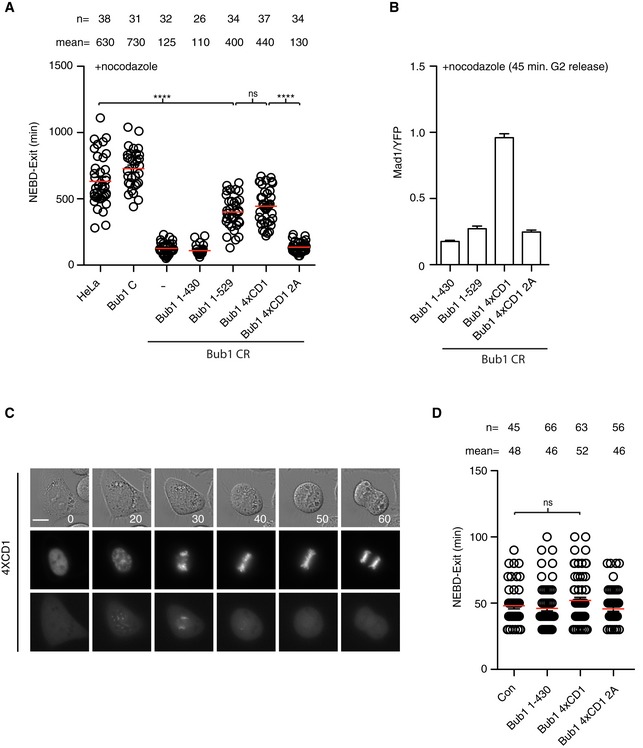
-
ABub1 CR cells complemented with the indicated Bub1 constructs and time from NEBD to exit in nocodazole measured by time‐lapse microscopy. Mean (red line) and standard error of mean (black bar) indicated (Mann–Whitney U‐test, ns: non‐significant, ****P ≤ 0.0001). A representative result from at least three independent experiments is shown.
-
BKinetochore levels of Mad1 in Bub1 CR cells complemented with the indicated YFP tagged Bub1 constructs. Mad1 levels are normalized to YFP signal. Bar indicates mean and standard error of mean is shown by line. At least 200 kinetochores from 10 cells were analyzed and representative result from at least two independent experiments shown.
-
C, DHeLa cells transfected with the indicated Bub1 constructs and time from NEBD to exit determined during an unperturbed mitosis. (C) Still images of unperturbed mitosis of HeLa cells transfected with Venus‐Bub1 4xCD1. CFP‐Histone 3 was used as the chromosome marker (the middle panel), and Venus‐Bub1 4xCD1 was shown in the bottom panel. Scale bar, 5 μm. (D) The time from NEBD to exit was analyzed in each condition. Mean (red line) and standard error of mean (black bar) indicated (Mann–Whitney U‐test, ns: non‐significant). A representative result from at least three independent experiments is shown.
Altogether our results support a model where the role of the RZZ complex in the SAC is to localize Mad1 at kinetochores allowing for a functional checkpoint generating Mad1‐Bub1 interaction.
A distinct region of Bub1 stimulates RZZ localization to kinetochores
To further understand the interplay between the RZZ complex and Bub1 in the SAC, we explored the reported role of Bub1 in localizing the RZZ complex to kinetochores (Caldas et al, 2015; Zhang et al, 2015), which we reconfirmed in live cell assays (Fig 7A). From previous work, it was unclear if the ability of Bub1 to stimulate RZZ localization was simply due to Bub1‐mediated recruitment of Mad1 or a separate function of Bub1. To test this, we determined whether the Mad1 binding and RZZ recruitment activities of Bub1 can be uncoupled. We first analyzed RZZ localization in two Bub1 mutants, Bub1 S459A/T461A (Bub1 2A) and I471D, unable to bind Mad1 (Zhang et al, 2017). In both of the Bub1 mutants, RZZ localization, as determined by kinetochore levels of ZW10, was normal while Mad1 levels were reduced by 20–30% (Figs 7B–D, and EV4D and E). This argues that Mad1 binding to Bub1 is unlikely to account for the function of Bub1 in stimulating recruitment of RZZ. Interestingly, the Bub1 MFQ/RKK mutant, also unable to bind Mad1, strongly reduced Bub1‐dependent recruitment of RZZ suggesting that a distinct region of CD1 contributes to RZZ localization (Fig 7B–D). Furthermore, the region downstream of CD1, amino acids 485–521, was required for RZZ localization which shows that these residues together with CD1 stimulate RZZ localization (Fig 7C and D). Indeed, fusing the kinetochore targeting domain of Bub1 to a region encompassing CD1 and the downstream region was sufficient for RZZ recruitment (Fig 7E and F).
Figure 7. Bub1 recruitment of RZZ to kinetochores.
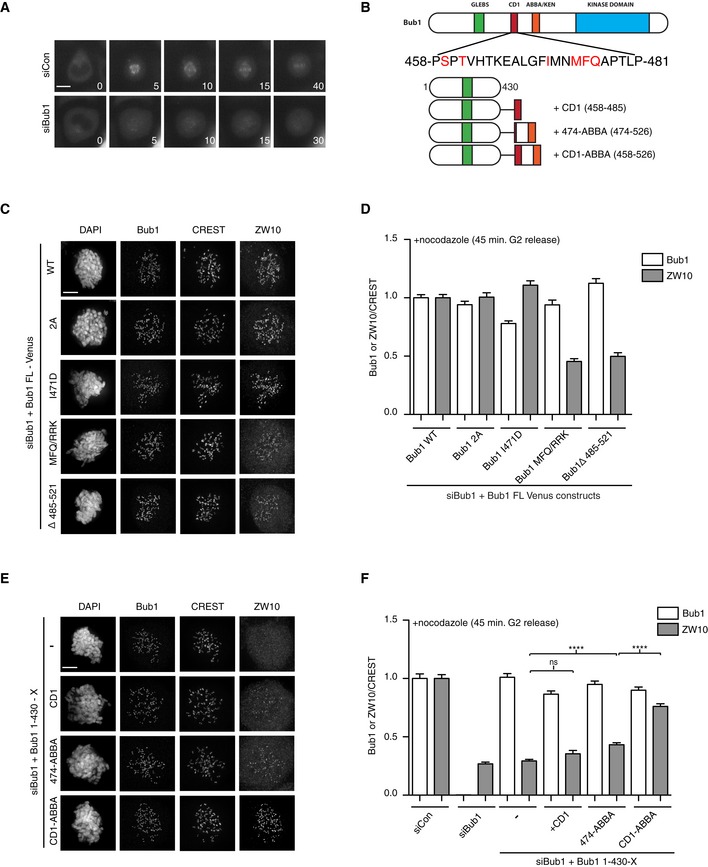
-
ALocalization of Venus‐Rod during unperturbed mitosis in control‐depleted or Bub1‐depleted cells.
-
BPrimary structure of Bub1 with sequence of CD1 shown and residues mutated in red. Schematic of fusion constructs used in panels (E and F).
-
CHeLa cells were depleted of Bub1 using RNAi and complemented with indicated Venus‐Bub1 constructs. Immunofluorescence images were shown for Bub1 and ZW10 localization.
-
DQuantification of Bub1 and ZW10 kinetochore levels normalized to CREST in the indicated conditions.
-
E, FAs in (C and D) but with the indicated Bub1 fusion proteins (Student's t‐test used for statistical comparison, ns: non‐significant, ****P ≤ 0.0001).
In conclusion, Bub1 stimulation of RZZ localization is independent of the Bub1‐Mad1 interaction, but the region in Bub1 responsible for RZZ localization is partly overlapping with the Mad1 interaction domain.
Discussion
By integrating CRISPR/Cas9 and RNAi depletion, we obtain penetrant depletion of two essential checkpoint proteins to reveal their contribution to checkpoint signaling. This approach avoids issues such as adaption during selection or that residual protein sufficient to support function remains. This can be a general approach to obtain penetrant depletion of essential genes if lower protein levels are sufficient for viability. We provide evidence that reported RPE1 and HAP1 Bub1 knockout cells express residual levels of Bub1, explaining why the response to unattached kinetochores is not strongly affected in these cells (Currie et al, 2018; Raaijmakers et al, 2018). Our results show that both Bub1 and Rod are required for efficient checkpoint signaling in human cells and we do not favor that there is a fundamental difference between transformed or untransformed cells in checkpoint architecture. These observations are consistent with recent work from the Jallepalli laboratory (Rodriguez‐Rodriguez et al, 2018).
It is evident from our work that very penetrant depletion of Bub1 and Rod is needed to reveal their contribution to checkpoint signaling. The Mad1‐tethering experiments support that the main function of the RZZ complex is to localize Mad1 to kinetochores. Although the RZZ complex is less efficient in inducing a SAC arrest in the absence of Bub1, it is capable of delaying mitosis approximately threefold likely through RZZ‐mediated Mad1 localization. This is consistent with our mass spectrometry analysis of BubR1 purifications from mitotic Bub1 CR cells that reveal similar MCC levels as controls. At present, we cannot rule out that undetectable amounts of Bub1 remaining in Bub1 CR are responsible for the delay seen in these cells. However, we do not favor this because all the mitotic timings are tightly clustered around 110 min in nocodazole which is very similar to that obtained when dominant negative versions of Bub1 are expressed (Zhang et al, 2017). Why RZZ in the absence of Bub1 cannot maintain a prolonged SAC arrest despite MCC generation is unclear to us but could possible reflect a change in SAC silencing activities with time.
Our analysis of Bub1 C cells shows that in HeLa cells Bub1 levels have to be reduced below 4% to have a penetrant effect on the SAC suggesting that Bub1 has a catalytic role. What could this catalytic function be? The ability of Bub1 to bind Cdc20 has been suggested to be an important determinant for MCC production (Di Fiore et al, 2015; Faesen et al, 2017; Ji et al, 2017). However, our previous work and analysis in Bub1 CR (Bub1 1–529, Fig 6A) shows that Cdc20 binding is insufficient to explain the function of Bub1 beyond Mad1 kinetochore recruitment (Zhang et al, 2015, 2017). Another contributing factor could be that binding of Bub1 to Mad1 increases the catalytic efficiency of the Mad1/Mad2 complex consistent with the fact that the Bub1 CD1 domain is still required when Mad1 is tethered to kinetochores in fission yeast (Heinrich et al, 2014). Bub1 could also act to position Mad1 close to KNL1 allowing for efficient MCC generation, and this proximity to KNL1 is not achieved in Mad1‐tethering experiments after Bub1 depletion. Recent established in vitro reconstitution systems and our Bub1 C cell lines will be important tools to further explore the function of Bub1 (Faesen et al, 2017; Ji et al, 2017).
We propose that the function of Bub1 in Mad1 recruitment is twofold and highly integrated with the RZZ complex. The central Mad1 binding region of Bub1 stimulates RZZ localization, hereby ensuring efficient Mad1 kinetochore localization. It is important to point out that Bub1 only stimulates RZZ recruitment, and if sufficient time is provided, then in the absence of Bub1 the RZZ complex will localize and generate a fibrous corona that can recruit Mad1 (Caldas et al, 2015; Vleugel et al, 2015a). The stimulation of RZZ localization by Bub1 might act to facilitate its own interaction with Mad1 as suggested by our BioID experiment and the observation that increased Bub1‐Mad1 interaction bypasses the requirement for Rod. Since Bub1 and Mad1 are present at nanomolar concentrations at kinetochores, while measured affinities are in the micromolar range, it is possible that RZZ, by increasing the local concentration of Mad1, allows for more efficient Bub1‐Mad1 complex formation (Simonetta et al, 2009; Ji et al, 2017; Zhang et al, 2017).
We favor that the amount of Mad1‐Bub1 complex at kinetochores is an important determinant of SAC strength, but our experiments with Bub1 4xCD1 also suggest that additional parameters control the overall strength of the checkpoint. The amount of Mad1‐Bub1 complex likely changes over time as the amount of MCC that needs to be produced for establishing and maintaining the SAC have different thresholds during mitosis. An initial high level of Bub1 phosphorylation might ensure that a large fraction of Mad1 is bound to Bub1 at early stages of mitosis to ensure a high level of MCC production to establish the checkpoint (Qian et al, 2017). During checkpoint maintenance, less Mad1‐Bub1 is needed to maintain MCC levels and the bulk of Mad1 is bound to RZZ, but Bub1 is still needed for efficient SAC signaling as shown by our Mad1‐tethering results.
In conclusion, our work reveals that the overall conceptual architecture of the checkpoint is conserved from yeast to man and likely the unique requirement for RZZ in mammalian cells reflects the weak affinity of Mad1 for Bub1.
Materials and Methods
Cell culture and RNAi
HeLa, U2OS, RPE1, and HAP1 cells were cultivated respectively in DMEM or DMEM F12 HAM or IMDM medium (Invitrogen) supplemented with 10% fetal bovine serum and antibiotics. For Bub1 or Rod RNAi and rescue experiments, two times of RNAi were performed using Lipofectamine 2000 or RNAi Max (Invitrogen) according to manufactures instructions. The first Bub1 RNAi was performed 48 h before filming or fixation with the second at 24 h before in the presence of thymidine (2.5 mM). The first Rod RNAi was performed 96 h before filming or fixation with the second at 48 h before. Thymidine was used afterward to synchronize cells. Plasmid co‐transfection was done in the first transfection for Bub1 and in the second transfection for Rod. For Bub1 and Rod co‐depletion, the first Bub1 RNAi was performed at the same time as the second Rod RNAi at 48 h before filming or fixation. RNAi oligos targeting Bub1 (5′ GAGUGAUCACGAUUUCUAA 3′) or luciferase (5′ CGUACGCGGAAUACUUCGA 3′; Sigma) or Rod (5′ GGAAUGAUAUUGAGCUGCUAACAAA 3′; Thermofisher) were used for RNAi depletions. Ambion silencer select RNAi oligos against Bub1: Bub1 #2 (5′ GAGUCAAAUAUGGAACGAAtt 3′), Bub1 #3 (5′ GGCCCUACGUAAUAGGCUAtt 3′), Bub1 #4 (5′ GGAUUACCACAGCCUAAAAtt 3′).
Immunofluorescence and quantification
This was done essentially as described in Zhang and Nilsson (2018). Cells growing on coverslips were synchronized with a double thymidine block followed by RO3306 (10 nM) block. After washing with PBS, the cells were treated with nocodazole (200 ng/ml) for 45 min and fixed with 4% paraformaldehyde in PHEM buffer (60 mM PIPES, 25 mM HEPES, pH 6.9, 10 mM EGTA, 4 mM MgSO4) for 20 min at room temperature. Fixed cells were extracted with 0.5% Triton X‐100 in PHEM buffer for 10 min. The antibodies used for cell staining include Bub1 (Abcam, ab54893, 1:400), Bub1 pSpT (home made, 1:200), Rod (gift from Gordon Chan), CREST (Antibodies Incorporated, 15‐234, 1:400), BubR1 (homemade), Cdc20 (Millipore, MAB3775, 1:200), ZW10 (Abcam, ab21582, 1:200), Mad1 (Santa Cruz, sc65494, 1:200). All the fluorescent secondary antibodies are Alexa Fluor Dyes (Invitrogen, 1:1,000). Z‐stacks 200 nm apart were recorded on a Deltavision Elite microscope (GE Healthcare) using a 100× oil objective followed by deconvolution using Softworx prior to quantification. Protein intensity on kinetochores was quantified by drawing a circle closely along the rod‐like CREST staining covering the interested outer kinetochore protein staining on both ends. The intensity values from the peak three continuous stacks were subtracted of the background from neighboring areas and averaged. The combined intensity was normalized against the combined CREST fluorescent intensity. At least 200 kinetochores from 10 cells were measured per condition. At least two independent experiments were performed for each immunofluorescence assay, and representative experiment is shown in figures.
CRISPR/Cas9‐mediated gene knockout
GeneArt Platinum Cas9 Nuclease (Thermofisher) with synthesized gRNA was used for gene editing. Pre‐designed DNA primers for gRNA template assembly were purchased (Thermofisher) with forward primer IVT‐TAATACGACTCACTATAGTACAAGGGCAATGACC and reverse primer IVT‐TTCTAGCTCTAAAACAGAGGGTCATTGCCCTTGT for Bub1; with forward primer IVT‐TAATACGACTCACTATAGCTTTTCTTGAACCGAC and reverse primer IVT‐TTCTAGCTCTAAAACGAGTGTCGGTTCAAGAAAA for Rod. Guide RNA was synthesized according to the instruction from GeneArt Precision gRNA Synthesis Kit (Thermofisher). 625 ng of gRNA and 2,500 ng of Cas9 nuclease (GeneArt Platinum Cas9 Nuclease, Thermofisher) were transfected into 4.5 × 105 HeLa cells by Lipofectamine CRISPRMAX transfection reagent. Twenty‐four hours later, the cells were diluted and re‐cultivated for single colony isolation. 20–100 single colonies were isolated and amplified followed by examination of the kinetochore signals of interested protein by immunofluorescence. The ones with reduced signals compared to the parental HeLa cells were maintained for further characterization.
Cloning
Rod cDNA was purchased from Kazusa DNA Res Inst (ORK05777). The internal BamHI site was eliminated by Gibson Assembly, and the cDNA was cloned into pcDNA5/FRT/TO N‐Venus vector by BamHI and NotI sites. Venus‐Bub1 1–430 and its variants were cloned into the same vector by KpnI and NotI sites. 4xCD1 WT or 2A DNA was synthesized by GeneArt (Thermofisher) and cloned into Venus‐Bub1 1–430 construct. Details will be provided upon request.
Live cell imaging
Live cell imaging was performed on a Deltavision Elite system using a 40× oil objective (GE Healthcare). Cells were transfected in 6‐well plate and re‐seeded in 8‐well Ibidi dishes (Ibidi) 1 day before the filming. Growth media was changed to Leibovitz's L‐15 (Life Technologies) before filming started. Appropriate channels were recorded for 18–24 h, and data were analyzed using Softworx (GE Healthcare). Statistical analysis was done using Prism software. At least three repeats were performed for each live cell imaging experiment, and a representative experiment is shown. For nocodazole treatment, 30 ng/ml of nocodazole was applied to cells just before filming. For taxol treatment, 100 nM of taxol was applied to cells before filming.
Western blot analysis of cell lines
Mitotic cells induced by nocodazole (200 ng/ml) were collected and lysed in lysis buffer containing 10 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA, and 0.5% NP‐40. After centrifugation at 18,000 g for 10 min, the supernatant was applied to SDS–PAGE followed by Western blot with interested antibodies. The antibodies used in this study include APC4 (homemade; Sedgwick et al, 2013), APC7 (Bethyl, A302‐551A, 1:2,000), Bub1 (abcam, ab54893, 1:1,000, and sheep anti‐Bub1 from Stephen Taylor 1:1,000; Taylor et al, 2001), Bub3 (BD Transduction Lab), BubR1 (homemade; Zhang et al, 2016), Cdc20 (Santa Cruz, sc13162, 1:1,000; Hein & Nilsson, 2014), GAPDH (Santa Cruz, sc25778, 1:2,000), H3S10p (Milipore 06‐570), Mad1 (Sigma, M8069, 1:1,000; Fava et al, 2011), Mad2 (Bethyl, A300‐301A, 1:1,000), and Rod (gift from Reto Gassmann; Gama et al, 2017). Fluorescent labeled secondary antibodies (1:5,000, LI‐COR Biosciences) were used for quantitative analysis by LI‐COR Odyssey imaging system (LI‐COR Biosciences).
Purification of biotinylated protein complexes
This was done essentially as in Zhang et al (2017). Stable HeLa cell lines expressing the Mad1 BirA fusion protein were exposed to 0.1 ng/ml doxycycline for 18 h to obtain near endogenous Mad1 expression levels. Cells were arrested in mitosis by a double thymidine block and subsequent nocodazole (150 ng/ml) treatment for 12 h. Biotinylation of proximity interactors was induced by the addition of a final concentration of 25 μM of biotin simultaneously with the addition of nocodazole. Rod siRNA knockdown was performed as described above. Mitotic cells were collected and washed three times in PBS before lysed in RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P‐40, 0.25% Na‐deoxycholate, 0.1% SDS) containing protease inhibitors (Roche). Cell lysate was clarified by centrifugation and incubated overnight at 4°C with High Capacity Streptavidin Resin (Thermo Scientific). Streptavidin beads were washed once with RIPA buffer followed by two washes with water containing 2% SDS and a final wash with RIPA buffer. Biotinylated proteins were eluted from the streptavidin beads with 2× Laemmli LDS sample buffer containing 1 mM of biotin before separated on 4–12% Bis‐Tris NuPage gels (Life Technologies). After separation, proteins were examined by Western blot using following antibodies: Cyclin B1 (554177, 1:1,000, BD Pharmingen), H3 pS10 (06‐570, 1:1,000, Millipore), GAPDH (sc‐25778, 1:500, Santa Cruz Biotech.), Bub1 (ab54893, 1:1,000, abcam), Knl1 (produced in house, 1:1,000; Zhang et al, 2016), Rod (1:2,000, gift from Reto Gassmann).
FRAP analysis of Mad1
For Fluorescence Recovery After Photobleaching (FRAP) measurements, HeLa, RodC, or Bub1C cells were cultured in DMEM medium supplemented with 10% fetal bovine serum, double transfected with control, Rod RNAi, or Bub1 RNAi oligos respectively as described above, and co‐transfected with Venus‐Mad1 or Mad1‐Venus. The day before filming, cells were seeded in 35 mm glass‐bottomed dishes (14 mm, No. 1.5, MatTek Corporation) in the presence of thymidine. Cells were then released in the absence or presence of 3.3 μM Nocodazole for 7–11 h before starting the live cell imaging. Time‐lapse imaging was performed at 37°C using a Plan‐Apochromat 63×/1.4NA with differential interference contrast oil objective mounted on an inverted Zeiss Axio Observer Z1 microscope (Marianas Imaging Workstation from Intelligent Imaging and Innovations Inc., Denver, CO, USA), equipped with a CSU‐X1 spinning‐disk confocal head (Yokogawa Corporation of America). Images were acquired using an iXon Ultra 888 EM‐CCD camera (Andor Technology). After identification of kinetochore pairs in prometaphase cells, one of the two kinetochores was photobleached with 4 laser pulses using 100% power of the argon laser, and the fluorescence recovery was recorded for 30 s at 500 ms intervals, after 2–6 prebleached images were acquired. RodC and RodCR cells were imaged using 100 ms exposure time of 40% laser power, whereas Bub1CR cells were imaged with 200 ms exposure time. For FRAP calculations, the fluorescence intensity of the bleached area was normalized using the intensity of the whole cell, after background correction. Then, a full‐scale normalization was done and the mean values were plotted and used for exponential curve fitting using GraphPad Prism 7.01 to determine the half‐life and the efficiency of the recovery.
Purification of Bub3 and BubR1 complexes
Parental HeLa cells, Bub1 C clone 1 and clone 2, were transfected with Venus‐Bub3 plasmid, and additional parental cells were transfected with a plasmid expressing Venus alone as a control. Following 24 h of thymidine (2.5 mM) arrest, the cells were released into nocodazole (200 ng/ml) and collected by mitotic shake‐off after 15 h.
The cells were lysed in GFP‐trap lysis buffer (10 mM Tris, 150 mM NaCl, 0.5% NP‐40, 1 mM DTT) supplemented with protease and phosphatase inhibitors (Roche). Cell lysates were cleared by spinning for 15 min at 19,500 g. 10 mg of supernatant was applied to 10 μl GFP‐trap (Chromotek) beads and was incubated for 90 min at 4°C with constant rotation. After three times of washes with 500 μl lysis buffer and one time with 1 ml PBS, the beads were frozen at −20°C and analyzed by mass spectrometry afterward.
Bub1 C clone 2, HAP1, and RPE1 BUB1 KO cells were arrested with thymidine (2.5 mM) for 24 h and then released into nocodazole (200 ng/ml) and mitotic cells collected by shake‐off. For the comparative analysis of Bub1 C and Bub1 CR, we released cells from thymidine into RO3306 to synchronize cells in G2 and then release them into nocodazole for approximately 2 h before harvesting. This was to avoid Bub1 CR cells escaping the nocodazole‐induced arrest. Cells were lysed in lysis buffer (10 mM Tris, 150 mM NaCl, 0.5% NP‐40, 1 mM DTT) supplemented with protease and phosphatase inhibitors and cleared for 15 min at 13,500 rpm. Protein concentrations were measured and immunoprecipitations were set up with 10 mg of lysate for HeLa cells and 8 mg of lysate for HAP1 and RPE1 cells and incubated with 25 μl BubR1 antibody‐Protein G beads. Immunoprecipitations were incubated for 70 min at 4°C with constant rotation. Beads were washed three times with 500 μl lysis buffer and two times with PBS. The washed beads were frozen at −20°C and analyzed by mass spectrometry afterward.
Affinity purification and mass spectrometry (AP‐MS)
Elution from GFP‐trap beads was carried out by a partial on‐bead digest. Briefly, 100 μl of elution buffer (2 M urea, 2 mM DTT, 20 μg/ml trypsin, 50 mM Tris, pH 7.5) was added and incubated at 37°C for 30 min at 210 g. Supernatants were transferred into new tubes, alkylated with 25 mM CAA, and further digested over night at room temperature (1,400 rpm). Digestion was stopped the next day by adding 1% trifluoroacetic acid (TFA). Peptides were desalted and purified with styrenedivinylbenzene‐reversed phase sulfonate (SDB‐RPS) StageTips. Briefly, StageTips with two layers of SDB‐RPS were prepared with 100 μl of wash buffer (0.2% TFA in H2O). Peptides were directly loaded on top and centrifuged for 5 min at 500 g. After one further wash with 150 μl of wash buffer, peptides were eluted with 50 μl of elution buffer (80% ACN and 1% ammonia) and vacuum‐dried. Dried peptides were finally reconstituted in 2% acetonitrile (ACN) and 0.1% TFA in water and kept at −20°C until MS analysis.
Liquid chromatography‐mass spectrometry (LC‐MS) analysis
This was done essentially as in Doll et al (2017). Nanoflow LC–MS/MS analysis of tryptic peptides was conducted on a quadrupole Orbitrap mass spectrometer (Q Exactive HF‐X, Thermo Fisher Scientific, Rockford, IL, USA; Kelstrup et al, 2018) coupled to an EASYnLC 1200 ultra‐high‐pressure system (Thermo Fisher Scientific) via a nano‐electrospray ion source. About 0.5 μg of peptides was loaded on a 50 cm HPLC column (75 μm inner diameter, New Objective, USA; in‐house packed using ReproSil‐Pur C18‐AQ 1.9 μm silica beads; Dr Maisch GmbH, Germany). Peptides were separated using a linear gradient from 2 to 20% B in 55 min and stepped up to 40% in 40 min followed by a 5 min wash at 98% B at 350 nl/min where solvent A was 0.1% formic acid in water and solvent B was 80% acetonitrile and 0.1% formic acid in water. The total duration of the run was 100 min. Column temperature was kept at 60°C by an in‐house‐developed oven. The mass spectrometer was operated in “top‐15” data‐dependent mode, collecting MS spectra in the Orbitrap mass analyzer (60,000 resolution, 300–1,650 m/z range) with an automatic gain control (AGC) target of 3E6 and a maximum ion injection time of 25 ms. The most intense ions from the full scan were isolated with an isolation width of 1.4 m/z. Following higher‐energy collisional dissociation (HCD) with a normalized collision energy (NCE) of 27, MS/MS spectra were collected in the Orbitrap (15,000 resolution) with an AGC target of 1E5 and a maximum ion injection time of 28 ms. Precursor dynamic exclusion was enabled with a duration of 30 s.
Raw file processing and bioinformatic analyses
This was done essentially as in Coscia et al (2018). MS raw files were processed with the MaxQuant software (Cox & Mann, 2008; version 1.5.0.38). The integrated Andromeda search engine (Cox et al, 2011) was used for peptide and protein identification at an FDR of < 1%. The human UniProtKB database (October 2017) was used as forward database and the automatically generated reverse database for the decoy search. “Trypsin” was set as the enzyme specificity. We required a minimum number of 7 amino acids for the peptide identification process. Proteins that could not be discriminated by unique peptides were assigned to the same protein group (Cox & Mann, 2008). Label‐free protein quantification was performed using the MaxLFQ (Cox et al, 2014) algorithm. Protein ratios were determined based on median peptide ratios, and only common peptides were used for pair‐wise ratio calculations. The “match‐between‐runs” feature of MaxQuant was enabled to transfer peptide identifications across runs based on high mass accuracy and normalized retention times. Prior to data analysis, proteins, which were found as reverse hits or only identified by site‐modification, were filtered out.
All statistical and bioinformatic analyses were performed using Perseus (Tyanova et al, 2016) or the R framework (https://www.r-project.org/). For peptide alignment onto the primary BUB1 protein sequence, we use the Open‐pFIND software for raw file analysis and data visualization (Chi et al, 2018).
Author contributions
GZ performed all experiments except the FRAP experiments performed by CGB and MB, preparation of samples for mass spectrometry performed by DHG, analysis of samples performed by mass spectrometry FC and MM, and Mad1 BioID experiment performed by TK. JN supervised the work and all authors contributed to discussions and data interpretation and writing the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
We thank Gordon Chan, Reto Gassman, Stephen Taylor, Rene Medema, and Jonathan Millar for providing reagents and Norman Davey, Prasad Jallepalli, Jonathan Millar, Rene Medema, and Geert Kops for fruitful discussions and for communication unpublished data. The Novo Nordisk Foundation Center for Protein Research, University of Copenhagen, is supported financially by the Novo Nordisk Foundation (grant agreement NNF14CC0001). In addition, this work was supported by grants from the Danish Cancer Society (R72‐A4351‐13‐S2 and R124‐A7827‐15‐S2), a grant from the Danish Council for Independent Research (DFF‐4183‐00388), and a grant from the Novo Nordisk Foundation (NNF16OC0022394) to J.N. and (NNF15CC0001) to M.M. A grant from Danish Cancer Society (R146‐A9322) and the Lundbeck Foundation (R215‐2015‐4081) supported work in the laboratory of MB. G.Z. is supported by Tai Shan Scholar program from Shandong, China.
The EMBO Journal (2019) 38: e100977
See also: P Meraldi (April 2019)
Contributor Information
Gang Zhang, Email: gang.zhang@cpr.ku.dk.
Jakob Nilsson, Email: jakob.nilsson@cpr.ku.dk.
Data Availability
The mass spectrometry data from this publication have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset) via the PRIDE partner repository with the dataset identifier PXD012235.
References
- Alfieri C, Chang L, Zhang Z, Yang J, Maslen S, Skehel M, Barford D (2016) Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 536: 431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Gomes R, Karess RE (2000) Rough deal and Zw10 are required for the metaphase checkpoint in Drosophila . Nat Cell Biol 2: 939–943 [DOI] [PubMed] [Google Scholar]
- Buffin E, Lefebvre C, Huang J, Gagou ME, Karess RE (2005) Recruitment of Mad2 to the kinetochore requires the Rod/Zw10 complex. Curr Biol 15: 856–861 [DOI] [PubMed] [Google Scholar]
- Caldas GV, Lynch TR, Anderson R, Afreen S, Varma D, Deluca JG (2015) The RZZ complex requires the N‐terminus of KNL1 to mediate optimal Mad1 kinetochore localization in human cells. Open Biol 5: 150160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan GK, Jablonski SA, Starr DA, Goldberg ML, Yen TJ (2000) Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat Cell Biol 2: 944–947 [DOI] [PubMed] [Google Scholar]
- Chao WCH, Kulkarni K, Zhang Z, Kong EH, Barford D (2012) Structure of the mitotic checkpoint complex. Nature 484: 208–213 [DOI] [PubMed] [Google Scholar]
- Chi H, Liu C, Yang H, Zeng W‐F, Wu L, Zhou W‐J, Wang R‐M, Niu X‐N, Ding Y‐H, Zhang Y, Wang Z‐W, Chen Z‐L, Sun R‐X, Liu T, Tan G‐M, Dong M‐Q, Xu P, Zhang P‐H, He S‐M (2018) Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat Biotechnol 36: 1059 [DOI] [PubMed] [Google Scholar]
- Collin P, Nashchekina O, Walker R, Pines J (2013) The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat Cell Biol 15: 1378–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscia F, Lengyel E, Duraiswamy J, Ashcroft B, Bassani‐Sternberg M, Wierer M, Johnson A, Wroblewski K, Montag A, Yamada SD, López‐Méndez B, Nilsson J, Mund A, Mann M, Curtis M (2018) Multi‐level proteomics identifies CT45 as a chemosensitivity mediator and immunotherapy target in ovarian cancer. Cell 175: 159–170 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie CE, Mora‐Santos M, Smith CA, McAinsh AD, Millar JBA (2018) Bub1 is not essential for the checkpoint response to unattached kinetochores in diploid human cells. Curr Biol 28: R929–R930 [DOI] [PubMed] [Google Scholar]
- De Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L, Mapelli M, Sironi L, Faretta M, Salmon ED, Musacchio A (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol 15: 214–225 [DOI] [PubMed] [Google Scholar]
- Di Fiore B, Davey NE, Hagting A, Izawa D, Mansfeld J, Gibson TJ, Pines J (2015) The ABBA motif binds APC/C activators and is shared by APC/C substrates and regulators. Dev Cell 32: 358–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore B, Wurzenberger C, Davey NE, Pines J (2016) The mitotic checkpoint complex requires an evolutionary conserved cassette to bind and inhibit active APC/C. Mol Cell 64: 1144–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick AE, Gerlich DW (2013) Kinetic framework of spindle assembly checkpoint signalling. Nat Cell Biol 15: 1370–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Dreßen M, Geyer PE, Itzhak DN, Braun C, Doppler SA, Meier F, Deutsch M‐A, Lahm H, Lange R, Krane M, Mann M (2017) Region and cell‐type resolved quantitative proteomic map of the human heart. Nat Commun 8: 1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faesen AC, Thanasoula M, Maffini S, Breit C, Müller F, van Gerwen S, Bange T, Musacchio A (2017) Basis of catalytic assembly of the mitotic checkpoint complex. Nature 542: 498–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava LL, Kaulich M, Nigg EA, Santamaria A (2011) Probing the in vivo function of Mad1:C‐Mad2 in the spindle assembly checkpoint. EMBO J 30: 3322–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama JB, Pereira C, Simões PA, Celestino R, Reis RM, Barbosa DJ, Pires HR, Carvalho C, Amorim J, Carvalho AX, Cheerambathur DK, Gassmann R (2017) Molecular mechanism of dynein recruitment to kinetochores by the Rod‐Zw10‐Zwilch complex and Spindly. J Cell Biol 216: 943–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohme MA, Schloissnig S, Rozanski A, Pippel M, Young GR, Winkler S, Brandl H, Henry I, Dahl A, Powell S, Hiller M, Myers E, Rink JC (2018) The genome of Schmidtea mediterranea and the evolution of core cellular mechanisms. Nature 554: 56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein JB, Nilsson J (2014) Stable MCC binding to the APC/C is required for a functional spindle assembly checkpoint. EMBO Rep 15: 264–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich S, Sewart K, Windecker H, Langegger M, Schmidt N, Hustedt N, Hauf S (2014) Mad1 contribution to spindle assembly checkpoint signalling goes beyond presenting Mad2 at kinetochores. EMBO Rep 15: 291–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hooff JJ, Tromer E, van Wijk LM, Snel B, Kops GJ (2017) Evolutionary dynamics of the kinetochore network in eukaryotes as revealed by comparative genomics. EMBO Rep 18: 1559–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, Totis L, Roberts BT (1991) S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66: 507–517 [DOI] [PubMed] [Google Scholar]
- Izawa D, Pines J (2015) The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature 517: 631–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Gao H, Jia L, Li B, Yu H (2017) A sequential multi‐target Mps1 phosphorylation cascade promotes spindle checkpoint signaling. Elife 6: e22513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelstrup CD, Bekker‐Jensen DB, Arrey TN, Hogrebe A, Harder A, Olsen JV (2018) Performance evaluation of the Q exactive HF‐X for shotgun proteomics. J Proteome Res 17: 727–738 [DOI] [PubMed] [Google Scholar]
- Klebig C, Korinth D, Meraldi P (2009) Bub1 regulates chromosome segregation in a kinetochore‐independent manner. J Cell Biol 185: 841–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJPL, Kim Y, Weaver BAA, Mao Y, McLeod I, Yates JR, Tagaya M, Cleveland DW (2005) ZW10 links mitotic checkpoint signaling to the structural kinetochore. J Cell Biol 169: 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lischetti T, Nilsson J (2015) Regulation of mitotic progression by the spindle assembly checkpoint. Mol Cell Oncol 2: e970484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, Ceto S, Ranish JA, Biggins S (2012) Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr Biol 22: 900–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, Biggins S (2014) Mad1 kinetochore recruitment by Mps1‐mediated phosphorylation of Bub1 signals the spindle checkpoint. Genes Dev 28: 140–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Sorger PK (2005) A dual role for Bub1 in the spindle checkpoint and chromosome congression. EMBO J 24: 1621–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora‐Santos MDM, Hervas‐Aguilar A, Sewart K, Lancaster TC, Meadows JC, Millar JBA (2016) Bub3‐Bub1 binding to Spc7/KNL1 toggles the spindle checkpoint switch by licensing the interaction of Bub1 with Mad1‐Mad2. Curr Biol 26: 2642–2650 [DOI] [PubMed] [Google Scholar]
- Mosalaganti S, Keller J, Altenfeld A, Winzker M, Rombaut P, Saur M, Petrovic A, Wehenkel A, Wohlgemuth S, Müller F, Maffini S, Bange T, Herzog F, Waldmann H, Raunser S, Musacchio A (2017) Structure of the RZZ complex and molecular basis of its interaction with Spindly. J Cell Biol 216: 961–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A (2011) Spindle assembly checkpoint: the third decade. Philos Trans R Soc Lond B Biol Sci 366: 3595–3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira C, Reis RM, Gama JB, Celestino R, Cheerambathur DK, Carvalho AX, Gassmann R (2018) Self‐assembly of the RZZ complex into filaments drives kinetochore expansion in the absence of microtubule attachment. Curr Biol 28: 3408–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera D, Tilston V, Hopwood JA, Barchi M, Boot‐Handford RP, Taylor SS (2007) Bub1 maintains centromeric cohesion by activation of the spindle checkpoint. Dev Cell 13: 566–579 [DOI] [PubMed] [Google Scholar]
- Primorac I, Musacchio A (2013) Panta rhei: the APC/C at steady state. J Cell Biol 201: 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primorac I, Weir JR, Chiroli E, Gross F, Hoffmann I, van Gerwen S, Ciliberto A, Musacchio A (2013) Bub3 reads phosphorylated MELT repeats to promote spindle assembly checkpoint signaling. Elife 2: e01030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, García‐Gimeno MA, Beullens M, Manzione MG, Van der Hoeven G, Igual JC, Heredia M, Sanz P, Gelens L, Bollen M (2017) An attachment‐independent biochemical timer of the spindle assembly checkpoint. Mol Cell 68: 715–730 e5 [DOI] [PubMed] [Google Scholar]
- Raaijmakers JA, van Heesbeen RGHP, Blomen VA, Janssen LME, van Diemen F, Brummelkamp TR, Medema RH (2018) BUB1 is essential for the viability of human cells in which the spindle assembly checkpoint is compromised. Cell Rep 22: 1424–1438 [DOI] [PubMed] [Google Scholar]
- Roberts BT, Farr KA, Hoyt MA (1994) The Saccharomyces cerevisiae checkpoint gene BUB1 encodes a novel protein kinase. Mol Cell Biol 14: 8282–8291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Rodriguez J‐A, Lewis C, McKinley KL, Sikirzhytski V, Corona J, Maciejowski J, Khodjakov A, Cheeseman IM, Jallepalli PV (2018) Distinct roles of RZZ and Bub1‐KNL1 in mitotic checkpoint signaling and kinetochore expansion. Curr Biol 28: 3422–3429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacristan C, Ahmad MUD, Keller J, Fermie J, Groenewold V, Tromer E, Fish A, Melero R, Carazo JM, Klumperman J, Musacchio A, Perrakis A, Kops GJ (2018) Dynamic kinetochore size regulation promotes microtubule capture and chromosome biorientation in mitosis. Nat Cell Biol 20: 800–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoian MS, Goldberg ML, Rieder CL (2000) The rate of poleward chromosome motion is attenuated in Drosophila zw10 and rod mutants. Nat Cell Biol 2: 948–952 [DOI] [PubMed] [Google Scholar]
- Sedgwick GG, Hayward DG, Di Fiore B, Pardo M, Yu L, Pines J, Nilsson J (2013) Mechanisms controlling the temporal degradation of Nek2A and Kif18A by the APC/C‐Cdc20 complex. EMBO J 32: 303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepperd LA, Meadows JC, Sochaj AM, Lancaster TC, Zou J, Buttrick GJ, Rappsilber J, Hardwick KG, Millar JBA (2012) Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr Biol 22: 891–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silió V, McAinsh AD, Millar JB (2015) KNL1‐Bubs and RZZ provide two separable pathways for checkpoint activation at human kinetochores. Dev Cell 35: 600–613 [DOI] [PubMed] [Google Scholar]
- Simonetta M, Manzoni R, Mosca R, Mapelli M, Massimiliano L, Vink M, Novak B, Musacchio A, Ciliberto A (2009) The influence of catalysis on mad2 activation dynamics. PLoS Biol 7: e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr DA, Williams BC, Hays TS, Goldberg ML (1998) ZW10 helps recruit dynactin and dynein to the kinetochore. J Cell Biol 142: 763–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Chan GK, Yen TJ (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol 154: 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ (2001) Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J Cell Sci 114: 4385–4395 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Vanoosthuyse V, Valsdottir R, Javerzat J‐P, Hardwick KG (2004) Kinetochore targeting of fission yeast Mad and Bub proteins is essential for spindle checkpoint function but not for all chromosome segregation roles of Bub1p. Mol Cell Biol 24: 9786–9801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vleugel M, Tromer E, Omerzu M, Groenewold V, Nijenhuis W, Snel B, Kops GJPL (2013) Arrayed BUB recruitment modules in the kinetochore scaffold KNL1 promote accurate chromosome segregation. J Cell Biol 203: 943–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vleugel M, Hoek T, Tromer E, Sliedrecht T, Groenewold V, Omerzu M, Kops GJPL (2015a) Dissecting the roles of human BUB1 in the spindle assembly checkpoint. J Cell Sci 128: 2975–2982 [DOI] [PubMed] [Google Scholar]
- Vleugel M, Omerzu M, Groenewold V, Hadders MA, Lens SMA, Kops GJPL (2015b) Sequential multisite phospho‐regulation of KNL1‐BUB3 interfaces at mitotic kinetochores. Mol Cell 57: 824–835 [DOI] [PubMed] [Google Scholar]
- Yamagishi Y, Yang C‐H, Tanno Y, Watanabe Y (2012) MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat Cell Biol 14: 746–752 [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, VanderLinden R, Weissmann F, Qiao R, Dube P, Brown NG, Haselbach D, Zhang W, Sidhu SS, Peters J‐M, Stark H, Schulman BA (2016) Cryo‐EM of mitotic checkpoint complex‐bound APC/C reveals reciprocal and conformational regulation of ubiquitin ligation. Mol Cell 63: 593–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Lischetti T, Nilsson J (2014) A minimal number of MELT repeats supports all functions of KNL1 in chromosome segregation. J Cell Sci 127: 871–884 [DOI] [PubMed] [Google Scholar]
- Zhang G, Lischetti T, Hayward DG, Nilsson J (2015) Distinct domains in Bub1 localize RZZ and BubR1 to kinetochores to regulate the checkpoint. Nat Commun 6: 7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Mendez BL, Sedgwick GG, Nilsson J (2016) Two functionally distinct kinetochore pools of BubR1 ensure accurate chromosome segregation. Nat Commun 7: 12256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Kruse T, López‐Méndez B, Sylvestersen KB, Garvanska DH, Schopper S, Nielsen ML, Nilsson J (2017) Bub1 positions Mad1 close to KNL1 MELT repeats to promote checkpoint signalling. Nat Commun 8: 15822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Nilsson J (2018) The closed form of Mad2 is bound to Mad1 and Cdc20 at unattached kinetochores. Cell Cycle 17: 1087–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Data Availability Statement
The mass spectrometry data from this publication have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset) via the PRIDE partner repository with the dataset identifier PXD012235.