

Abstract
DNA end resection initiates DNA double‐strand break repair by homologous recombination. MRE11‐RAD50‐NBS1 and phosphorylated CtIP perform the first resection step via MRE11‐catalyzed endonucleolytic DNA cleavage. Human NBS1, more than its homologue Xrs2 in Saccharomyces cerevisiae, is crucial for this process, highlighting complex mechanisms that regulate the MRE11 nuclease in higher eukaryotes. Using a reconstituted system, we show here that NBS1, through its FHA and BRCT domains, functions as a sensor of CtIP phosphorylation. NBS1 then activates the MRE11‐RAD50 nuclease through direct physical interactions with MRE11. In the absence of NBS1, MRE11‐RAD50 exhibits a weaker nuclease activity, which requires CtIP but not strictly its phosphorylation. This identifies at least two mechanisms by which CtIP augments MRE11: a phosphorylation‐dependent mode through NBS1 and a phosphorylation‐independent mode without NBS1. In support, we show that limited DNA end resection occurs in vivo in the absence of the FHA and BRCT domains of NBS1. Collectively, our data suggest that NBS1 restricts the MRE11‐RAD50 nuclease to S‐G2 phase when CtIP is extensively phosphorylated. This defines mechanisms that regulate the MRE11 nuclease in DNA metabolism.
Keywords: DNA end resection, DNA repair, homologous recombination, nuclease, phosphorylation
Subject Categories: DNA Replication, Repair & Recombination
Introduction
The maintenance of genomic integrity is critically important as cells are constantly exposed to various genotoxic agents. DNA double‐strand breaks (DSBs) are difficult to repair due to the loss of genetic information in both DNA strands. While unrepaired DSBs may result in cell death, their inaccurate repair can lead to mutagenesis and inappropriate chromosomal translocations (Ranjha et al, 2018). Cells possess two main mechanisms for DSB repair. This includes end‐joining pathways including Ku‐dependent non‐homologous end‐joining (NHEJ) and Ku‐independent microhomology‐mediated end‐joining (MMEJ), which are functional in any phase of the cell cycle and do not require a DNA template (Ranjha et al, 2018). The second mechanism is template‐dependent homologous recombination (HR), which is generally restricted to the S‐G2 phase of the cell cycle. The sister chromatid serves as an HR template in most cases in vegetative cells, explaining why recombination can only function in the cell cycle phases when sister chromatids are available (Ranjha et al, 2018). This is achieved by a regulatory mechanism involving cyclin‐dependent kinases (CDKs), which phosphorylate key factors that function in the first step of the HR pathway termed DNA end resection (Huertas et al, 2008; Huertas & Jackson, 2009). Resection involves nucleolytic degradation of the 5′‐terminated DNA strands at DSBs, leading to 3′ overhangs that are essential for the downstream steps in the recombination pathway. While limited DNA end resection may be involved in MMEJ, DNA that underwent extended resection is no longer ligatable and therefore unsuitable for end‐joining. The decision whether and to which extent to resect DNA ends therefore regulates the pathway choice in DSB repair (Ranjha et al, 2018).
The MRE11‐RAD50‐NBS1 (MRN, in human cells) or Mre11‐Rad50‐Xrs2 (MRX, in budding yeast) complex has multiple key evolutionarily conserved functions to initiate and coordinate the repair of DSBs (Paull, 2010; Stracker & Petrini, 2011). This includes roles in both end‐joining and HR pathways (Moore & Haber, 1996; Carney et al, 1998; Paull & Gellert, 1998, 1999; de Jager et al, 2001; Rass et al, 2009). Additionally, MRN promotes DNA end tethering and is required to signal the presence of DSBs via the ATM kinase (Carney et al, 1998; Stewart et al, 1999; de Jager et al, 2001; Usui et al, 2001; Lee & Paull, 2005; Williams et al, 2008). This is achieved by a conserved interaction between the MRN subunit NBS1 (Xrs2 in Saccharomyces cerevisiae) and ATM (Tel1 in yeast). ATM in turn phosphorylates hundreds of protein targets that regulate the response to DSBs. Beyond ATM, NBS1 also interacts with MDC1, which binds phosphorylated proteins, including H2AX (γH2AX), to amplify the DNA damage signaling beyond the vicinity of the DSB (Goldberg et al, 2003). In recombination, the MRN complex has a direct role in DNA end resection mediated by the MRE11 nuclease in the S‐G2 phase of the cell cycle. MRE11 is likely the first nuclease to resect DSBs, which is particularly important for the processing of breaks with secondary DNA structures or protein adducts such as stalled topoisomerases or Ku (Shibata et al, 2014; Anand et al, 2016; Deshpande et al, 2016; Wang et al, 2017; Reginato et al, 2018). The current models posit that resection by MRE11 is initiated by endonucleolytic DNA cleavage internal to the DSB past the protein block, followed by 3′→5′ exonucleolytic degradation back toward the DNA end (Keeney & Kleckner, 1995; Neale et al, 2005; Garcia et al, 2011; Cannavo & Cejka, 2014; Shibata et al, 2014). The endonucleolytic cleavage sites also represent entry sites for processive DNA end resection nucleases that function downstream of MRN in the 5′→3′ direction, including EXO1 and DNA2 (Gravel et al, 2008; Mimitou & Symington, 2008; Zhu et al, 2008; Nimonkar et al, 2011). The MRN nuclease furthermore likely functions to promote MMEJ independently of the cell cycle, which to date remains very poorly defined (Ma et al, 2003; Rahal et al, 2010; Taylor et al, 2010; Truong et al, 2013; Deng et al, 2014; Sharma et al, 2015).
The processing of protein‐blocked DSBs is an evolutionarily conserved capacity of MRE11 homologues and their co‐factors. In bacteria, the MRE11‐RAD50‐like SbcC‐SbcD complex endonucleolytically cleaves DNA near blocked DNA ends (Connelly et al, 2003). Eukaryotic cells possess a third member of the MRE11‐RAD50 complex, NBS1/Xrs2, as well as CtIP/Sae2, which function as co‐factors of the MRE11 nuclease in resection (Carney et al, 1998; Sartori et al, 2007). In S. cerevisiae, Sae2 phosphorylated by CDK and other kinases has a critical function to promote the Mre11‐Rad50 endonuclease near protein blocks (Huertas et al, 2008; Cannavo & Cejka, 2014; Wang et al, 2017; Reginato et al, 2018), while Xrs2 per se is not essential for the DNA clipping reaction in vitro (Oh et al, 2016), but may have a stimulatory function (Wang et al, 2017). It has been demonstrated that Xrs2 is responsible for the nuclear import of the MRX complex (Carney et al, 1998; Tsukamoto et al, 2005). When this function was bypassed by placing the nuclear localization signal on Mre11, Xrs2 became partially dispensable for the Mre11‐Rad50‐dependent functions in resection also in vivo (Oh et al, 2016). In contrast, Xrs2 per se was strictly required for the DNA damage signaling function of the MRX complex (Oh et al, 2016). In humans, both CtIP, when phosphorylated by CDK, and NBS1 promote the MRE11‐RAD50 endonuclease (Anand et al, 2016; Deshpande et al, 2016). The comparatively stronger requirement for NBS1 in human cells with respect to budding yeast likely reflects the need for more complex regulatory mechanisms to control the MRE11 nuclease in high eukaryotes. How NBS1 performs this function, however, remains poorly characterized. Beyond DSB processing, unscheduled DNA degradation by MRE11 at stalled DNA replication forks may be responsible for the toxicity associated with defects in BRCA1 or BRCA2, further highlighting the importance to understand how MRE11 nuclease is regulated in human cells (Schlacher et al, 2011; Ray Chaudhuri et al, 2016; Feng & Jasin, 2017; Mijic et al, 2017).
Human NBS1 consists of 754 amino acids. The N‐terminus contains a forkhead‐associated domain (FHA) and tandem BRCA1 C‐terminal (BRCT) motifs (BRCT1 and BRCT2), which bind phosphorylated proteins, including CtIP and MDC1 (Spycher et al, 2008; Williams et al, 2009; Wang et al, 2013). At its C‐terminal part, NBS1 contains ATM and MRE11 interaction sites, as well as three potential nuclear localization signals that promote the nuclear import of MRN (Carney et al, 1998; Nakada et al, 2003; Falck et al, 2005; Tsukamoto et al, 2005; You et al, 2005). The FHA domain of S. cerevisiae Xrs2 binds phosphorylated Sae2, although this capacity appears to be partially dispensable for DNA end resection in vitro and in vivo (Liang et al, 2015; Oh et al, 2016). Structural and biochemical characterization of MRN in Schizosaccharomyces pombe revealed that it also binds phosphorylated Ctp1, an ortholog of CtIP/Sae2 in S. pombe, through the FHA motif of Nbs1 (Lloyd et al, 2009; Williams et al, 2009). A point mutation that affects this interaction resulted in hypersensitivity to ionizing radiation and camptothecin, as well as impaired Ctp1 enrichment at DSBs (Williams et al, 2009). Similarly in human cells, CtIP phosphorylated by CDK at multiple sites in the center of the protein binds the FHA‐BRCT domains of NBS1, which is important for DNA end resection (Wang et al, 2013). However, this CDK‐dependent phosphorylation was also important for the subsequent modification of T859 by ATM that is required for resection in vivo. Phosphomimetic T859E could partially bypass the requirement for the phosphorylation of CtIP at the CDK sites at the center of the protein that mediate its interaction with the FHA‐BRCT domains of NBS1. This raised questions whether modification of these central CDK sites is required for resection per se, or only serves as a platform to help phosphorylate CtIP at T859 (Wang et al, 2013). Furthermore, mutations in FHA‐BRCT abrogate the interaction of NBS1 with MDC1, which may have an indirect effect on resection as a result of disrupted signaling or MRN recruitment (Hari et al, 2010). Because of the multiple functions of the MRN complex and potential pleiotropic phenotypes associated with NBS1 defects, the interpretation of cell‐based resection assays with NBS1 variants is challenging.
Here, we primarily employ in vitro reconstituted reactions to define the function of NBS1 in DNA end resection by MRN‐CtIP. We show that both FHA and BRCT domains of NBS1 promote resection by MRE11 through interactions with phosphorylated CtIP. When NBS1 senses that CtIP is phosphorylated, it promotes resection by a mechanism that is dependent on its interaction with MRE11. This is in agreement with a recent study showing that an NBS1 fragment containing the MRE11 binding site but not the FHA‐BRCT domains rescues the inviability of NBS1‐deficient mouse embryonic fibroblasts (Kim et al, 2017). Importantly, we identify an NBS1‐independent DNA cleavage activity of MRE11‐RAD50 and CtIP. Although less efficient than the resection capacity of the MRN‐CtIP holocomplex, the NBS1‐independent activity is promoted by CtIP but surprisingly does not strictly require its phosphorylation. Accordingly, we find limited CtIP‐ and MRE11‐dependent but NBS1‐independent DSB resection activity in vivo. These results suggest that a mechanism that interferes with the function of NBS1 might allow limited resection in the absence of CDK‐dependent modification of CtIP, which might be relevant for understanding MRE11 nuclease functions in G1.
Results
NBS1 in trans promotes endonucleolytic cleavage by MRE11‐RAD50 and phosphorylated CtIP
Previously, we showed that phosphorylated CtIP (pCtIP) promotes the clipping of the 5′‐terminated DNA strand near protein‐blocked DSBs by the MRE11 nuclease within the MRE11‐RAD50‐NBS1 (MRN) complex (Anand et al, 2016). This reaction is believed to initiate DNA end resection by MRN. The dsDNA clipping efficiency of MRE11‐RAD50 (MR) was strongly reduced compared to MRN, showing that NBS1 has an important function to stimulate this activity (Anand et al, 2016; Deshpande et al, 2016). To define the function of NBS1 in the regulation of the MRE11 nuclease, we purified NBS1, as well as MR, MRN, and pCtIP from baculovirus‐infected Spodoptera frugiperda 9 (Sf9) insect cells (Fig EV1A–D).
Figure EV1. Preparation and analysis of NBS1 constructs.
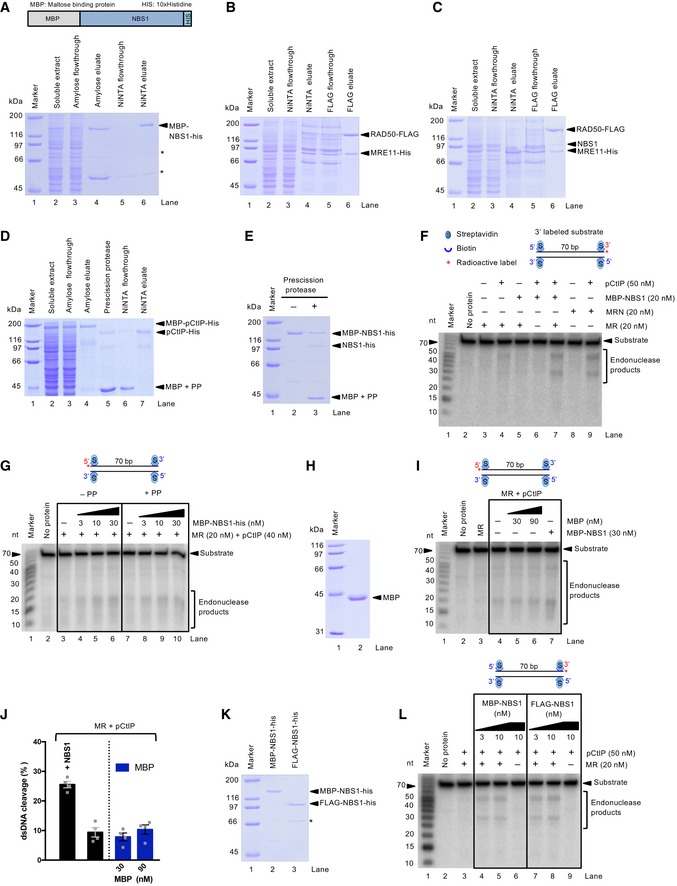
- A representative 10% polyacrylamide gel showing purification of recombinant MBP‐NBS1‐his. The gel was stained with Coomassie brilliant blue. Amylose flowthrough and eluate, flowthrough and eluate from amylose resin; Ni‐NTA flowthrough and eluate, flowthrough and eluate from nickel‐nitrilotriacetic acid (Ni‐NTA). * indicates truncated products.
- Samples from a representative purification of the MR complex were analyzed by 10% polyacrylamide gel electrophoresis. The gel was stained with Coomassie brilliant blue. FLAG flowthrough and eluate, flowthrough and eluate from anti‐FLAG affinity resin.
- Samples from a representative purification of the MRN complex analyzed by 10% polyacrylamide gel electrophoresis.
- Samples from a representative purification of phosphorylated CtIP (expressed and purified with phosphatase inhibitors) analyzed by 10% polyacrylamide gel electrophoresis. MBP, maltose binding protein; PP, PreScission protease.
- Polyacrylamide gel stained with Coomassie brilliant blue showing the partial cleavage of MBP‐NBS1‐his by PreScission protease (PP). The PP recognition site is in between the MBP tag and NBS1. 2 μg recombinant MBP‐NBS1‐his was incubated for 1 h at 4°C with 1 μg of PreScission protease.
- Nuclease assays as in Fig 1A but with 3′‐end‐labeled 70‐bp dsDNA.
- Nuclease assays with MR and pCtIP and either uncleaved (left panel) or partially cleaved MBP‐NBS1 (right panel, ˜ 50% cleaved) with PreScission protease. 5′‐end‐labeled 70‐bp‐long dsDNA was used as a substrate.
- Polyacrylamide gel stained with Coomassie brilliant showing purified recombinant MBP tag.
- Nuclease assays with MR, pCtIP, MBP‐NBS1, or MBP, as indicated. 5′‐end‐labeled 70‐bp‐long dsDNA was used as a substrate. MBP tag does not affect the nuclease reactions.
- Quantitation of assays such as in (I). Averages shown; n = 3; error bars, SEM.
- Polyacrylamide gel stained with Coomassie brilliant showing purified recombinant MBP‐NBS1‐his and FLAG‐NBS1‐his. * indicates truncated product.
- Nuclease assays with MR and pCtIP and either MBP‐NBS1‐his (left panel) or FLAG‐NBS1 (right panel). 3′‐end‐labeled 70‐bp‐long dsDNA was used a substrate.
NBS1 was purified by affinity chromatography by utilizing maltose binding protein (MBP) and 10× histidine (his) tags, located at the N‐ and C‐termini, respectively. As the removal of the MBP tag by PreScission protease cleavage was not very efficient (Fig EV1E), the MBP tag was retained during the final purification (Fig EV1A). MBP‐NBS1‐his added in trans promoted dsDNA clipping by MR and pCtIP (Fig 1A and B). This was almost as efficient as DNA cleavage by pCtIP and MRN purified as a complex, where NBS1 was untagged (Figs 1A and B, and EV1F). Therefore, the affinity tags did not notably impair the stimulatory function of NBS1 on dsDNA clipping by MR and pCtIP in vitro. To further confirm this, we note that treatment of MBP‐NBS1 with PreScission protease, which cleaves ~ 50% of the MBP tag off NBS1, did not affect dsDNA clipping efficiency (Fig EV1E and G). The MBP tag, expressed separately, did not affect the cleavage capacity of MR and pCtIP (Fig EV1H–J). Finally, FLAG‐NBS1‐his construct promoted DNA cleavage similarly as MBP‐NBS1‐his (Fig EV1K and L). In summary, we conclude that MBP‐NBS1‐his (hereafter NBS1 for brevity) added in trans promotes the capacity of MR and pCtIP ensemble to clip 5′‐terminated DNA in the vicinity of protein blocks.
Figure 1. NBS1 in trans with MR and pCtIP cleaves DNA similarly as MRN‐pCtIP.
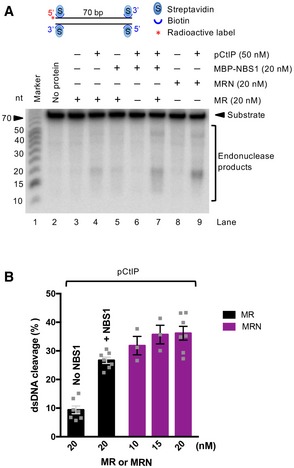
- A representative nuclease assay with MR, MBP‐NBS1‐his (denoted MBP‐NBS1), MRN, and pCtIP on 5′‐end‐labeled 70‐bp dsDNA with all ends blocked with streptavidin. Samples were separated on 15% denaturing polyacrylamide gel.
- Quantitation of nuclease assays such as in (A). Averages shown; n ≥ 3; error bars, SEM.
FHA and BRCT domains of NBS1 are important for the processing of protein‐blocked DSBs, while its interaction with MRE11 is essential
NBS1 mediates physical interactions of the MRN complex with ATM, BRCA1, BLM, MDC1, CtIP, and possibly other factors (Carney et al, 1998; Stewart et al, 1999; Wang et al, 2000, 2013; Goldberg et al, 2003; Chen et al, 2008; Spycher et al, 2008; Williams et al, 2008; Hari et al, 2010). Out of these, the interactions with CtIP and MDC1 are at least in part phosphorylation‐dependent. To specifically define the functional interaction of NBS1 with MR and pCtIP in resection, we employed our in vitro reconstituted system (Anand et al, 2016). To this point, we prepared NBS1 variants lacking particular domains or carrying mutations specifically affecting known interactions with the MR and pCtIP ensemble (Figs 2A and EV2A), and assayed them in nuclease assays to determine their effect on the MRE11 endonuclease.
Figure 2. FHA and BRCT domains of NBS1 are important for MR‐pCtIP stimulation while MRE11‐NBS1 interaction is essential.
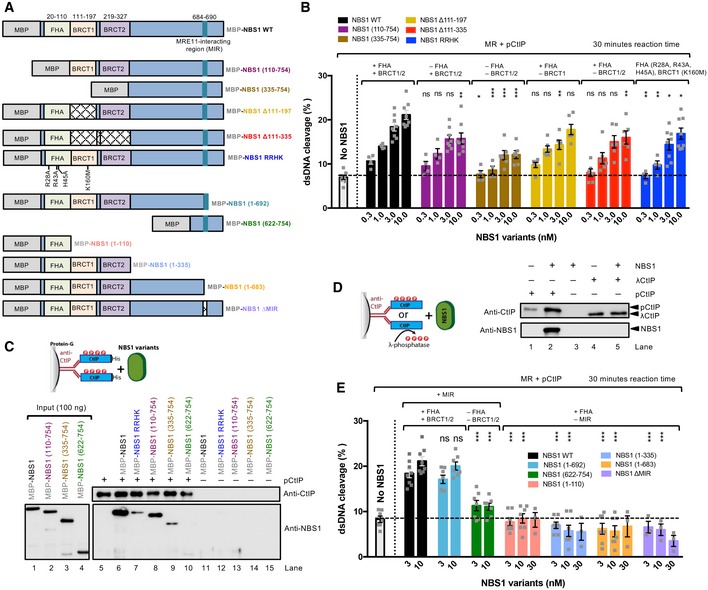
- A schematic representation of purified recombinant wild‐type NBS1 and variants. All constructs were MBP tagged at the N‐ and his‐tagged at the C‐terminus.
- The effect of mutations in FHA and/or BRCT domains of NBS1 on the nuclease of MR and pCtIP. Quantitation of experiments such as shown in Fig EV2D. Averages shown; n ≥ 4, error bars, SEM. Statistical significance denotations represent the analysis between wild‐type full‐length NBS1 (in black) and the corresponding concentrations of the NBS1 variants; ns (P > 0.05, not significant), *(P < 0.05), **(P < 0.01), ***(P < 0.001), two‐tailed t‐test.
- Analysis of pCtIP binding to NBS1 variants. Anti‐CtIP antibody was immobilized on protein G agarose, bound to pCtIP, and tested for interactions with the indicated NBS1 variants (see cartoon at the top). Western blot of eluates (right part) was performed with anti‐CtIP and anti‐NBS1 antibodies. Left part (input) indicates that the anti‐NBS1 antibody recognizes the NBS1 constructs to a similar level.
- Analysis of phosphorylated CtIP (pCtIP, mock‐treated) or λ phosphatase‐treated CtIP (λCtIP) interactions with NBS1. The CtIP variants were immobilized on protein G agarose and incubated with NBS1 (see cartoon on the left). The Western blot was performed with anti‐CtIP and anti‐NBS1 antibodies.
- The effect of the MRE11 interaction region (MIR) of NBS1 on the nuclease of MR and pCtIP. Quantitation of experiments such as shown in Fig EV2E. Averages shown, n ≥ 3, error bars, SEM. The quantitation of wild‐type NBS1 (NBS1 WT, in black) is the same as in panel (B) and is shown again for reference. Statistical significance denotations represent the analysis between wild‐type full‐length NBS1 (in black) and the corresponding concentrations of the NBS1 variants; ns (P > 0.05, not significant), ***(P < 0.001), two‐tailed t‐test.
Figure EV2. Stimulation of MR‐pCtIP by NBS1.
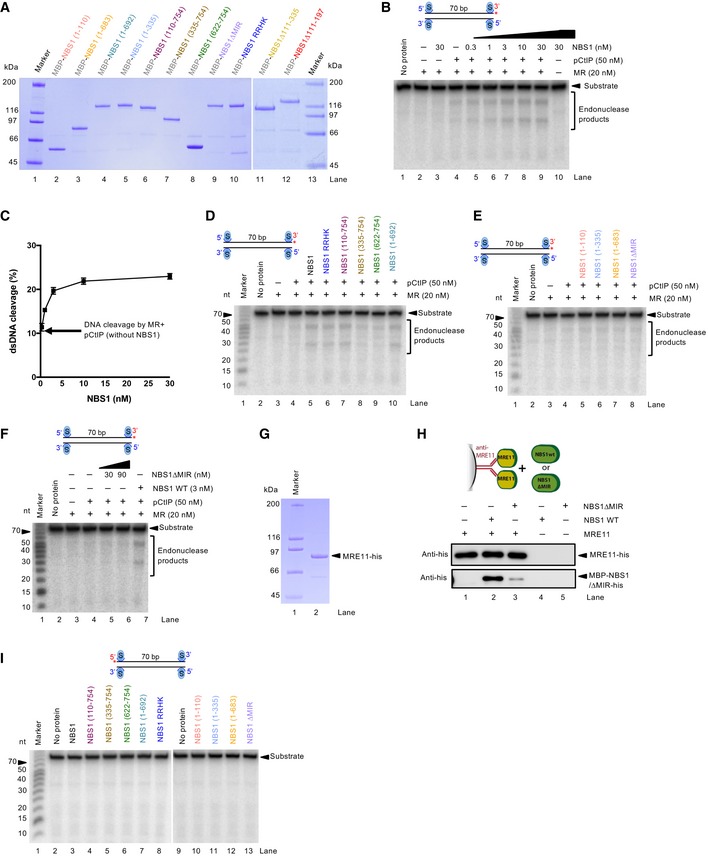
- A representative polyacrylamide gel (4–15%) stained with Coomassie brilliant blue showing purified NBS1 variants. All constructs were MBP tagged at the N‐ and his‐tagged at the C‐terminus.
- A representative nuclease assay with MR, pCtIP, and various concentrations of MBP‐NBS1‐his (labeled NBS1) on 3′‐end‐labeled dsDNA.
- Quantitation of experiments such as shown in (B). Averages shown; n = 5; error bars, SEM.
- Representative nuclease assays with MR, pCtIP, and various MBP‐NBS1‐his fragments (10 nM) containing MRE11 interaction region (MIR), using 3′‐end‐labeled dsDNA as a substrate.
- Representative nuclease assays with MR, pCtIP, and various NBS1 fragments (10 nM) lacking MRE11 interaction region (MIR) on 3′‐end‐labeled dsDNA.
- Representative nuclease assays with MR, pCtIP, and various concentrations of MBP‐NBS1∆MIR, using 3′‐end‐labeled dsDNA as a substrate.
- A representative 10% polyacrylamide gel stained with Coomassie brilliant blue showing purified recombinant MRE11‐his.
- Interaction of MRE11 with NBS1. MRE11 was immobilized on antibody‐coupled protein G agarose and incubated with MBP‐NBS1‐his or MBP‐NBS1∆MIR‐his (see cartoon). Bound proteins were analyzed by Western blot with anti‐his antibodies.
- Nuclease assay with NBS1 variants, showing that these proteins alone do not possess any nuclease activity.
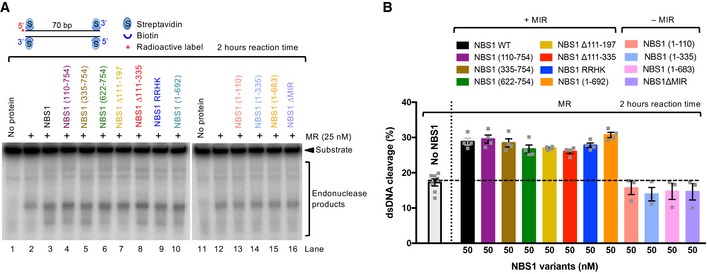
We first varied the concentrations of wild‐type NBS1 in reactions with MR and pCtIP. While weak NBS1‐independent DNA cleavage was observed, we found that less than equimolar concentrations of NBS1, compared to the MR complex, were sufficient for maximal DNA clipping activity (Fig EV2B and C). We next tested various NBS1 fragments lacking FHA, BRCT, or both domains (Figs 2B and EV2D). As phosphorylation of CtIP is required for resection (Huertas & Jackson, 2009), and the MRN complex primarily binds phosphorylated CtIP via the FHA and BRCT domains of NBS1 (Fig 2C and D; Wang et al, 2013; Williams et al, 2009), a strong defect in resection was anticipated. As shown in Fig 2B, a deletion of either FHA or BRCT domain in NBS1 had only a minor effect on resection in vitro, while elimination of both domains significantly reduced, but did not eliminate resection. Similarly to the elimination of both FHA and BRCT domains, a stronger inhibitory effect was observed with the NBS1 RRHK variant, carrying point mutations in FHA‐BRCT (R28A, R43A, and H45A in FHA and K160M in BRCT1, Fig 2A and B; Wang et al, 2013). The RRHK mutations decreased the phosphorylation‐dependent interaction of NBS1 with phosphorylated MDC1 (Hari et al, 2010), as well as with pCtIP (Fig 2C), in agreement with a dramatic DNA end resection defect in vivo, as scored by the single‐strand annealing reporter assay (Wang et al, 2013). Despite the strong physical interaction between the FHA and BRCT domains of NBS1 and pCtIP (Fig 2C), which is dependent on CtIP phosphorylation (Fig 2D), we conclude that both domains of NBS1 are important, but not essential for the DNA clipping activity in vitro together with MR and pCtIP. Furthermore, NBS1 (335–754), which lacks FHA‐BRCT but contains a central linker region, exhibited similar stimulatory activity to NBS1 (622–754) lacking the central region (Fig 2B and E). This result revealed that the central NBS1 region (residues 335–621) is largely dispensable for MR‐ and pCtIP‐mediated resection, despite this region mediated residual interaction with pCtIP (Fig 2C).
It has been demonstrated that the MRE11‐RAD50 complex directly interacts with NBS1 via the MRE11 interaction region (MIR) within the C‐terminal part of NBS1, with the most important motif located between residues 684–690 of NBS1 (Desai‐Mehta et al, 2001; Schiller et al, 2012; Kim et al, 2017). This interaction facilitates MRN entry into the nucleus as only NBS1 contains the nuclear localization sequence. Therefore, in vivo experiments with mutated MIR of NBS1 cannot easily distinguish effects related to impaired nuclear entry from direct effects on the biochemical activities of the MR complex. Using our in vitro system, where any effects on nuclear import are irrelevant, we observed that in contrast to the FHA and BRCT domains, the MRE11 interaction region in NBS1 was absolutely essential for the stimulatory function of NBS1 on the MRE11‐RAD50 endonuclease in conjunction with pCtIP (Figs 2E and EV2E). Specifically, the NBS1 (1–692) fragment lacking the very C‐terminal region of NBS1, but possessing MIR, exhibited similar activity as full‐length NBS1 (Figs 2E and EV2D). In contrast, the NBS1 (1–683) mutant lacking nine residues comprising MIR (residues 684–692) completely lost its stimulatory activity (Figs 2E and EV2E). Likewise, the internal deletion of MIR (residues 684–690) totally abolished NBS1 function in DNA clipping, even at high concentrations (Figs 2E, and EV2E and F). In accord with previous studies (Desai‐Mehta et al, 2001; Schiller et al, 2012), we observed a dramatic reduction of the MRE11 and NBS1 physical interaction when NBS1 ∆MIR variant was used in pulldown experiments instead of wild‐type NBS1 (Fig EV2G and H). None of the NBS1 variants used here had any DNA cleavage activity, as expected (Fig EV2I). In summary, the NBS1‐dependent interaction with MRE11‐RAD50, more than that with pCtIP, is important for the endonuclease activity of the MRE11‐RAD50 and pCtIP nuclease ensemble.
In the absence of pCtIP, NBS1 promotes MR cleavage independently of its FHA and BRCT domains
Recently, it has been shown that NBS1 alone is capable of stimulating the endonucleolytic activity of MR on protein‐blocked dsDNA, independently of CtIP (or pCtIP; Deshpande et al, 2016). Although CtIP‐independent DNA end resection does not likely occur in vivo (Sartori et al, 2007), we used the in vitro assay to learn more about the function of NBS1. To this point, we employed our NBS1 mutants in an MR‐dependent nuclease assay with streptavidin‐blocked dsDNA. In contrast to the assays that included pCtIP, the reactions were incubated for 2 h instead of 30 min to compensate for the lower cleavage efficacy in the absence of pCtIP. We observed that all NBS1 fragments containing MIR stimulated the clipping activity of MR almost indistinguishably, irrespectively of FHA and BRCT domains (Fig 3A and B). Notably, one of these constructs, the short C‐terminal NBS1 fragment containing 133 residues (NBS1 622–754), stimulated the endonucleolytic cleavage to the same extent as full‐length wild‐type NBS1. Conversely, NBS1 fragments lacking MIR did not at all promote MR (Fig 3A and B). Very similar results were obtained using circular ssDNA as a substrate, which forms a variety of secondary DNA structures (Fig EV3A and B). However, as noted previously, we believe that the cleavage of dsDNA near protein blocks is a better model for resection taking place in cells (Fig EV3C–G; Anand et al, 2016; Chen et al, 2005; Deshpande et al, 2014). In summary, we show that in the absence of pCtIP, the phosphoprotein binding domains of NBS1 become dispensable for resection in vitro. Our data support a model for resection in wild‐type cells, where the FHA‐BRCT domains of NBS1 promote resection by first binding phosphorylated CtIP, and NBS1 in turn activates the MR complex by interacting with MRE11.
Figure 3. Without pCtIP, NBS1 promotes the MR endonuclease independently of its FHA and BRCT domains.
- Nuclease assays with MR and various NBS1 fragments (50 nM) on oligonucleotide‐based 5′‐end‐labeled dsDNA. The reaction buffer contained 60 mM NaCl and reactions were incubated for 2 h.
- Quantitation of experiments such as shown in (A). Averages shown; n ≥ 3, error bars, SEM.
Figure EV3. Stimulation of MR by NBS1.
- Nuclease assays with MR and various NBS1 fragments (50 nM) on M13 ssDNA. The reaction products were separated on agarose gels (1.1%). The reaction buffer contained 60 mM NaCl and reactions were incubated for 2 h.
- Quantitation of experiments such as shown in (A). Averages shown; n = 3, error bars, SEM.
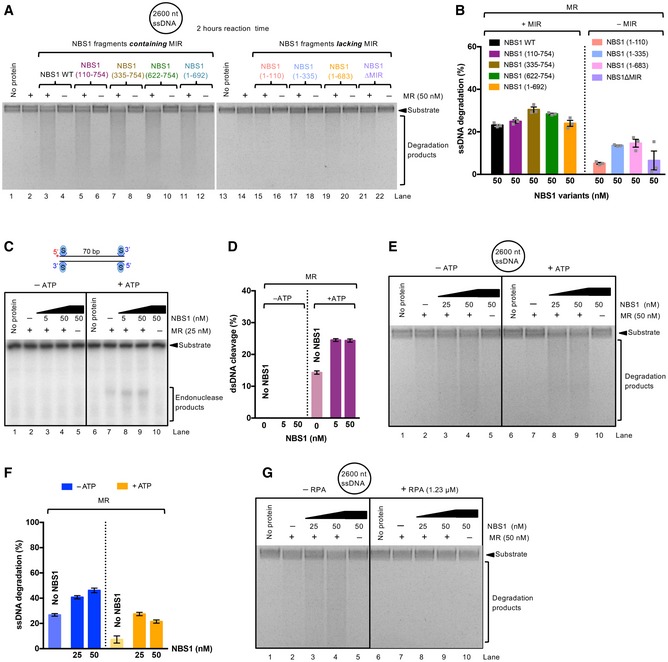
- Nuclease assay with MR and MBP‐NBS1‐his (denoted NBS1), with and without ATP on 5′‐end‐labeled dsDNA. The reaction buffer contained 60 mM NaCl and reactions were incubated for 2 h. The DNA cleavage reaction requires ATP, in agreement with observations that ATPase‐deficient RAD50 mutants are incapable of resection in vivo.
- Quantitation of experiments such as shown in (C). Averages shown, n = 2, error bars, range.
- Nuclease assays with MR and MBP‐NBS1‐his (denoted NBS1), with and without ATP, using M13 ssDNA as a substrate. The reaction buffer contained 60 mM NaCl and reactions were incubated for 2 h. The reactions were separated on 1.1% agarose gel. ATP did not stimulate M13 ssDNA cleavage.
- Quantitation of experiments such as shown in (E). Averages shown; n = 2, error bars, range.
- Nuclease assay with MR and MBP‐NBS1 as in panel (E), but with or without RPA. RPA inhibits M13 ssDNA cleavage (this panel) and ATP is partially dispensable (see panels E and F). In cells, RPA is ubiquitous and RAD50 ATPase‐deficient mutants are incapable of resection. Therefore, we believe that the M13‐based assays do not accurately mimic the reaction occurring in vivo, and that the clipping of dsDNA (see Fig 3A and B) is a better model.
BRCT and FHA domains of NBS1 are important but not absolutely essential for resection in vivo
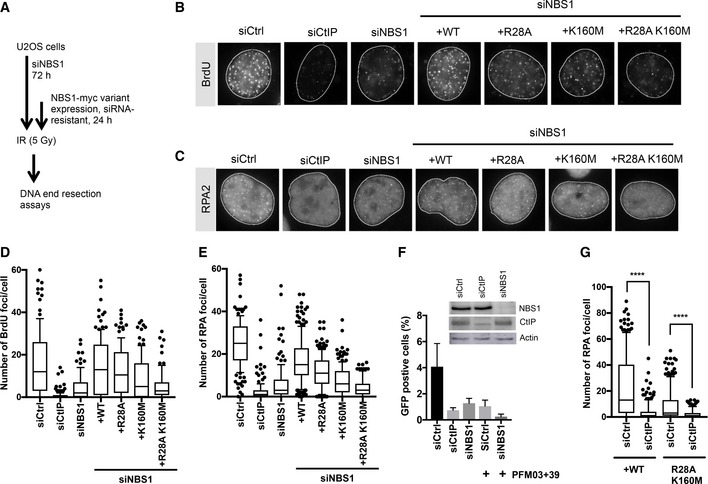
To verify the physiological significance of our data, we first designed an NBS1 complementation system in U2OS cells to define the requirement of the FHA and BRCT domains of NBS1 for DNA end resection in vivo. The expression of endogenous wild‐type NBS1 was suppressed with siRNA, and siRNA‐resistant myc‐tagged NBS1 variants were expressed from a construct containing doxycycline‐inducible promoter stably integrated into chromosomal DNA (Fig 4A). Downregulation of endogenous NBS1 led to cellular sensitivity to ionizing radiation, retention of MRE11 in the cytoplasm, and a DNA end resection deficiency, as anticipated (Figs 4B–E and EV4A–D). Expression of wild‐type myc‐tagged NBS1 overcame these defects, verifying the functionality of the complementation system (Figs 4B–E and EV4A–D).
Figure 4. The function of NBS1 in DNA end resection in vivo .
- A schematic representation of the NBS1 knockdown‐rescue experiments in U2OS cells.
- In vivo resection assay using BrdU staining under native conditions to directly detect ssDNA at sites of DSBs. Shown are representative images.
- In vivo resection assay using RPA2 staining as a marker for ssDNA present at sites of DSBs. Shown are representative images.
- Quantitation of the experiment shown in (B). At least 100 cells were assessed per condition. The horizontal line represents a median, the box spans from 25th to 75th percentile, and the whiskers from 10th to 90th percentile.
- Quantitation of the experiment shown in (C). At least 100 cells were assessed per condition. The horizontal line represents a median, the box spans from 25th to 75th percentile, and the whiskers from 10th to 90th percentile.
- Quantitation of the single‐strand annealing (SSA) reporter assay in U2OS cells. All cells were treated with DMSO; where indicated, both PFM03 (50 μM) and PFM39 (50 μM) MRE11 inhibitors were added. Averages shown; n = 4, error bars, SEM. Insert, Western blot analysis of NBS1 and CtIP protein levels upon depletion with siRNA.
- Quantitation of resection activity in NBS1∆N cells complemented with wild‐type (WT) or NBS1 R28A K160M mutant and either mock‐depleted (siCTRL) or depleted of CtIP (siCtIP). At least 300 cells were assessed per condition. The horizontal line represents a median, the box spans from 25th to 75th percentile, and the whiskers from 10th to 90th percentile. Statistical significance was determined using ordinary one‐way ANOVA with Tukey's multiple comparison test, ****(P < 0.0001).
Figure EV4. Cellular function of NBS1 in DNA end resection.
- Western blot of Flp‐In T‐REx NBS1‐myc expressing cells. Expression of recombinant NBS1 was induced for 24 h with doxycycline (DOX) prior to extract preparation.
- Clonogenic survival assay showing that expression of siRNA‐resistant recombinant NBS1 rescues the radiosensitivity of NBS1‐depleted cells. Averages shown; n = 3, error bars, SD.
- Immunofluorescence experiment showing efficient depletion of NBS1 by siRNA and cytoplasmic localization of MRE11 in the NBS1‐depleted cells. Expression of myc‐tagged NBS1 restored MRE11 nuclear localization.
- Western blot showing efficient depletion of NBS1 and CtIP by siRNA transfection and expression of siRNA‐resistant myc‐tagged recombinant wt NBS1 and variants.
- NBS1 R28A and K160M mutations impair the interaction between NBS1 and CtIP in cell extracts. Flag‐CtIP and NBS1‐myc variants were overexpressed in 293‐T cells, where indicated. Anti‐FLAG affinity resin was used to pull down FLAG‐CtIP and interacting partners.
- Western blot showing the downregulation of CtIP and NBS1 upon siRNA treatment in the HR reporter assays in (G).
- HR repair reporter assay after CtIP and NBS1 depletion. Bars and error bars represent mean and SD of four independent experiments. Statistical significance was tested by unpaired t‐test (*P = 0.02).
- Western blot of extracts prepared from wild‐type U2OS cells, NBS1ΔN cells, and NBS1ΔN cells stably expressing mNeonGreen (mNG)‐tagged wild‐type (WT) and R28A K160M NBS1 mutant full‐length NBS1, either mock‐depleted (siCTRL) or depleted of CtIP (siCtIP) by siRNA. Note the presence of a presumably hypomorphic 40 kDa C‐terminal fragment of NBS1 (NBS1hm) in the NBS1ΔN cell line.
- Immunofluorescence of NBS1∆N U2OS cells stably expressing mNeonGreen (mNG)‐tagged wild‐type (WT) and R28A K160M mutant full‐length NBS1. Note that MRE11 nuclear localization is partially impaired in NBS1∆N cells, which is fully rescued by heterologous expression of NBS1 wt and R28A K160M mutant.
- Quantitation of resection activity as scored by RPA foci in wild‐type U2OS cells and NBS1∆N cells. At least 260 cells were assessed per condition. The horizontal line represents a median, the box spans from 25th to 75th percentile, and the whiskers from 10th to 90th percentile.
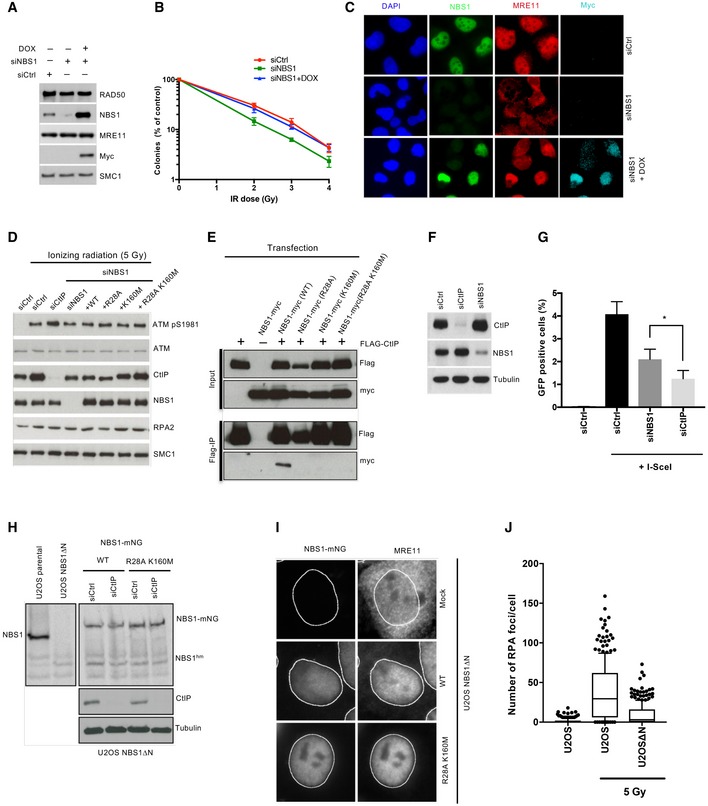
To test for the involvement of the FHA and BRCT domains of NBS1 in resection, we generated cell lines expressing wild‐type NBS1, NBS1 R28A (a point mutation in the FHA domain), NBS1 K160M (a point mutation in the BRCT1 domain), or the NBS1 R28A K160M double mutant. The respective point mutations in the FHA and BRCT domains affect their phosphorylation‐dependent protein binding function (Cerosaletti & Concannon, 2003; Hari et al, 2010; Wang et al, 2013), and disrupted the interaction with CtIP in cell extracts (Fig EV4E). The cell lines were treated with siNBS1 for 72 h to knock down endogenous NBS1 (Fig 4A). For the last 24 h, the expression of the complementing NBS1 variant was induced by the addition of doxycycline. Cells were then irradiated (5 Gy) and DNA end resection was scored by automated counting of BrdU foci in ssDNA under non‐denaturing conditions or by counting of RPA2 foci (Fig 4B–E). Mutations in either FHA or BRCT domains led to an intermediate defect in DNA end resection in both resection assays. The NBS1 R28A K160M double mutant led to a more severe, but not complete abrogation of resection, comparable to NBS1 depletion (Fig 4B–E). Both FHA and BRCT domains thus individually promote, but are not absolutely essential, for the DNA end resection function of NBS1, which is in full agreement with our biochemical analysis.
Depletion of CtIP brought about a more severe defect than that of NBS1, and nearly completely eliminated BrdU or RPA2 foci upon IR (Fig 4B–E). This result confirmed that CtIP is essential for almost all DNA end resection activity, as observed previously by others (Sartori et al, 2007). Furthermore, the depletion of NBS1 brought about lesser defects compared to CtIP depletion in homologous recombination as scored by a DR‐GFP reporter assay (Fig EV4F and G). Finally, depletion of NBS1 did not completely eliminate single‐strand annealing (SSA; Fig 4F), which is a good readout for extensive DNA end resection (Stark et al, 2004). We also note that the residual SSA activity observed in the reporter assay without NBS1 was MRE11 dependent (Fig 4F).
The depletion of NBS1 by siRNA may not be complete and may thus explain the residual resection we observed. To this point, using CRISPR/Cas9, we constructed a U2OS hypomorphic NBS1ΔN cell line that expresses low levels of a C‐terminal fragment of NBS1, which completely lacks the FHA and BRCT domains (Fig EV4H). Similarly as in mouse embryonic fibroblasts, where a C‐terminal fragment of NBS1 rescued viability and ATM activation (Kim et al, 2017), the NBS1ΔN cells were viable and grew normally in the absence of exogenous DNA damage. MRE11 was partially retained in the cytoplasm in this cell line, most likely owing to the low expression level of the hypomorphic NBS1 allele that may not be sufficient to fully localize MRE11 in the nucleus (Fig EV4I). However, stable expression of recombinant wild‐type or NBS1 R28A K160M (C‐terminally tagged with the monomeric GFP analogue mNeonGreen) fully restored MRE11 nuclear localization (Fig EV4I). DNA end resection in the NBS1ΔN cell line was similarly compromised as in the cells depleted from NBS1 by siRNA (Fig EV4J). Consistent with our siRNA‐based complementation system, stable expression of wild‐type NBS1 in the NBS1ΔN cell line efficiently restored DNA end resection activity, whereas expression of the NBS1 R28A K160M variant did not, indicating that even though MRE11 nuclear localization was restored by NBS1 R28A K160M, full resection activity was not (Fig 4G). Interestingly, both in wild‐type and NBS1 R28A K160M expressing cells, the residual resection activity was dependent on CtIP (Fig 4G), thus suggesting that CtIP can stimulate resection independently of NBS1 also in vivo. Together, we demonstrated that limited CtIP‐dependent DNA end resection in vivo can occur without the involvement of FHA‐BRCT domains of NBS1.
Phosphorylation of CtIP is partially dispensable for stimulation of MR in the absence of NBS1
NBS1 specifically binds phosphorylated CtIP (Fig 2D), and phosphorylation of CtIP by CDKs is key for stimulating the MRN endonuclease activity (Anand et al, 2016). Therefore, the observation that limited CtIP‐dependent resection can occur without the FHA‐BRCT domains of NBS1 raised the question whether phosphorylation of CtIP is required under these conditions. In Figs 2B, and EV2B and C, we observed weak NBS1‐independent resection in vitro. To better define the mechanism of this reaction, we modified our biochemical assay conditions and employed the extended reaction incubation time (2 h) to improve the detection limit. As shown in Fig 5A and B, we observed that pCtIP stimulated dsDNA cleavage by MR in the absence of NBS1 in a concentration‐dependent manner. Our previous experiments established NBS1 as a reader of CtIP phosphorylation, and the NBS1‐independent reaction thus identifies a separate mechanism for CtIP‐facilitated stimulation of MR. Next, we set out to test whether phosphorylation of CtIP was required. We treated or mock‐treated pCtIP with λ phosphatase. As shown previously (Anand et al, 2016), we observed that only pCtIP (i.e., mock‐treated), but not λCtIP, stimulated DNA cleavage by MRN in a concentration‐dependent manner (Fig 5C–E). In contrast, we observed a robust stimulation of MR‐dependent DNA clipping by both phosphorylated and λ phosphatase‐treated CtIP (Fig 5C–E). Very similar results were obtained in kinetic assays: with MRN, pCtIP was ~ 7‐fold more efficient in promoting the DNA cleavage compared to λCtIP; with MR, the difference was only ~ 1.5‐fold (Figs 5F and G, and EV5A). Therefore, NBS1 appears to restrict DNA cleavage by MR to conditions when CtIP is phosphorylated. Without NBS1, although less efficient, the MR‐dependent DNA cleavage is more promiscuous and can occur without CtIP phosphorylation.
Figure 5. Phosphorylation of CtIP is partially dispensable for MR stimulation in the absence of NBS1.
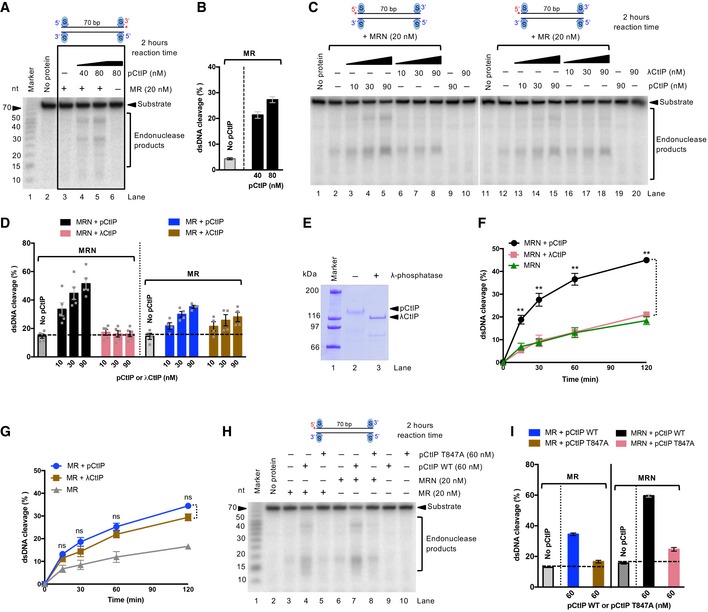
- Nuclease assays with MR and pCtIP on 3′‐end‐labeled dsDNA, incubated for 2 h.
- Quantitation of experiments such as shown in (A). Averages shown; n = 2; error bars, range.
- Nuclease assays with MRN, MR, and various concentrations of pCtIP (mock‐treated) or λCtIP on 5′‐end‐labeled dsDNA, incubated for 2 h.
- Quantitation of experiments such as shown in (C). Averages shown; n = 3; error bars, SEM.
- Phosphorylated CtIP was mock‐incubated (pCtIP, lane 2) or treated with λ phosphatase (λCtIP, lane 3). The proteins were separated on a 10% polyacrylamide gel and stained with Coomassie brilliant blue.
- Quantitation of kinetic nuclease assays with MRN and pCtIP (mock‐treated) or λCtIP. Averages shown; n = 3; error bars, SEM. Statistical denotations, **(P < 0.01), two‐tailed t‐test.
- Quantitation of nuclease assays as in (F), but with MR complex instead of MRN. Averages shown; n = 3; error bars, SEM. Statistical denotations, ns (P > 0.05), two‐tailed t‐test.
- Nuclease assays with MR, MRN, pCtIP, or pCtIP T847A, as indicated, on 5′‐end‐labeled dsDNA. Reactions were incubated for 2 h.
- Quantitation of experiments such as shown in (H). Averages shown; n = 4; error bars, SEM.
Figure EV5. Function of CtIP phosphorylation in MR stimulation with or without NBS1.
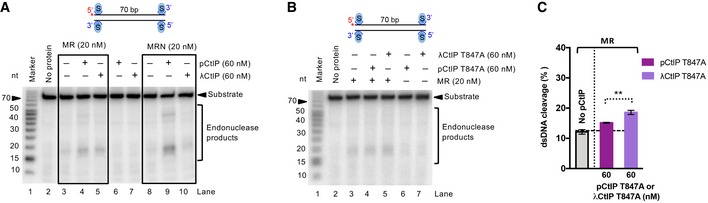
- Representative nuclease assays with MR, MRN, and pCtIP or λCtIP using 5′‐end‐labeled dsDNA. Quantitation of these and similar experiments is shown in Fig 5F and G. Reactions were incubated for 2 h.
- Nuclease assays with MR and pCtIP T847A or λCtIP T847A on 5′‐end‐labeled dsDNA. Reactions were incubated for 2 h.
- Quantitation of experiments such as shown in (B). Averages shown; n = 4; error bars, SEM. Statistical denotations, **(P < 0.01), two‐tailed t‐test.
One of the key residues in CtIP that needs to be phosphorylated by CDKs to allow DNA end resection during S‐phase is T847, which maps to a conserved region that is equivalent to S267 of S. cerevisiae Sae2 (Huertas et al, 2008; Huertas & Jackson, 2009). Non‐phosphorylatable substitution of T847 with alanine, resulting in T847A mutation, renders pCtIP incapable to promote DNA end resection (Huertas & Jackson, 2009; Anand et al, 2016). Unexpectedly, we observed that the pCtIP T847A mutant was incapable to promote both MR and MRN (Fig 5H and I). This result suggested that the T847A variant is likely structurally impaired, beyond being non‐phosphorylatable at the key CDK consensus site. Alternatively, the failure of pCtIP T847A to promote MR, in contrast to λCtIP, may stem from the presence of other phosphorylated residues in pCtIP T847A that have an inhibitory function together with MR. To test for this scenario, we treated pCtIP T847A with λ phosphatase, and observed a minor, yet statistically significant, rescue of DNA clipping by MR (Fig EV5B and C). As wild‐type, dephosphorylated λCtIP was still more efficient in promoting the MR endonuclease than dephosphorylated λCtIP T847A, the T847A substitution in pCtIP likely brings about structural changes within the conserved CtIP region that affect CtIP activity beyond mimicking a non‐phosphorylated threonine.
Together, our results demonstrate that NBS1, as CtIP, is a co‐factor that stimulates the DNA cleavage efficacy of the MR complex. Reactions with MRN require CtIP phosphorylation and lead to extensive DNA end resection activity that is required for HR and SSA. In contrast, in the absence of NBS1, the MR‐dependent cleavage is more restricted, and CtIP phosphorylation is partially dispensable for the DNA cleavage activity.
The physical interaction of MRE11 and RAD50 with CtIP does not require CtIP phosphorylation
Our observation that CtIP was able to stimulate the DNA cleavage capacity of the MR complex prompted us to define physical interactions between MRE11, RAD50, and phosphorylated or non‐phosphorylated CtIP. CtIP is known to interact with all subunits of the MRN complex (Sartori et al, 2007; Chen et al, 2008; Williams et al, 2009; Yuan & Chen, 2009); in accord, we detected direct interactions between pCtIP/CtIP and both MRE11 and RAD50 (Fig 6A and B). While phosphorylation of CtIP was absolutely required for its interaction with NBS1 (Fig 2D), it was not the case for MRE11 and RAD50. In contrast, λ phosphatase‐treated CtIP interacted with both MRE11 and RAD50 even better than pCtIP, suggesting that CtIP phosphorylation may interfere with the interaction with the isolated MRE11 and RAD50 subunits (Fig 6A and B). We cannot exclude that the apparent increase in the protein–protein interactions was due to aggregation of non‐phosphorylated CtIP, or that our preparation of pCtIP contained phosphorylated residues that are not normally modified in vivo, which could inhibit the interaction with MRE11 and RAD50. Nevertheless, NBS1 was mostly responsible for the interaction between the MRN complex and pCtIP, and phosphorylation of CtIP was indispensable for this interaction (Fig 6C). The interaction between MRN and pCtIP was not affected by the presence of ATP and/or DNA (Fig EV6A). Finally, NBS1 largely mediated the interaction of the MRN complex even when DNA bound (Fig EV6B). In summary, the interactions of MRE11 and RAD50 with CtIP did not require CtIP phosphorylation. These data are in agreement with the results from the nuclease assays, which indicated that CtIP phosphorylation was not strictly required to promote the nuclease of the MR complex.
Figure 6. CtIP phosphorylation is dispensable for its interaction with MRE11 and RAD50.
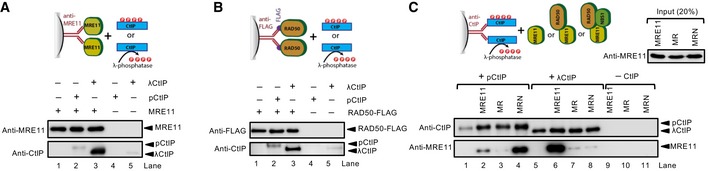
- Interaction between MRE11 and CtIP. MRE11 was immobilized on antibody‐coupled protein G agarose and incubated with pCtIP or λCtIP (see cartoon). Western blot analysis of bound proteins was performed with anti‐MRE11 and anti‐CtIP antibodies.
- Interaction between RAD50 and CtIP. RAD50‐FLAG was immobilized on anti‐FLAG affinity resin and incubated with pCtIP and λCtIP (see cartoon). Western blot analysis of bound proteins was performed with anti‐FLAG and anti‐CtIP antibodies.
- Interaction between MRE11, MR, or MRN and pCtIP or λCtIP, as indicated. Phosphorylated (pCtIP) or λ phosphatase‐treated CtIP (λCtIP) were immobilized on antibody‐coupled protein G agarose and incubated with MRE11, MR, or MRN complexes (see cartoon). Bound proteins were analyzed by Western blot using anti‐CtIP and anti‐MRE11 antibodies.
Figure EV6. Analysis of physical interactions in the presence of DNA .
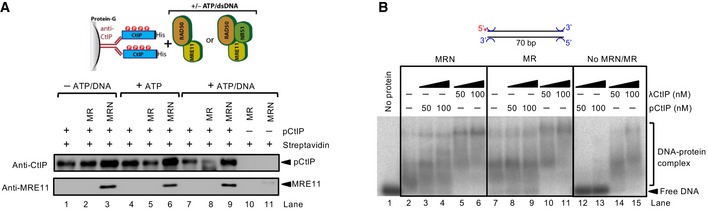
- Interaction assays as in Fig 6C, but with ATP (2 mM ATP, 10 mM magnesium acetate, no EDTA) or ATP/DNA (2 mM ATP, 10 mM magnesium acetate, no EDTA, 20 nM dsDNA resulting from annealing PC210 and PC211 oligonucleotides, in molecules). No ATP/DNA reactions contained 10 mM magnesium acetate and lacked EDTA.
- Electrophoretic mobility shift assays with MR, MRN, and either pCtIP or λCtIP. The substrate was 5′‐end‐labeled dsDNA that was not blocked with streptavidin. The reaction buffer was the same as in nuclease assays, except pyruvate kinase and phosphoenolpyruvate were omitted. The binding reactions were carried out on ice for 15 min and separated by 0.6% agarose at 4°C.
NBS1 is dispensable for DNA cleavage opposite to a strand discontinuity
To learn more about the involvement of NBS1 in the DNA clipping reaction, we employed a substrate that contains a pre‐existing nick in the strand opposite to the anticipated cleavage site, which was shown to increase the efficacy of DNA cleavage (Deshpande et al, 2016). To this point, we performed nuclease analyses with both nicked and non‐nicked DNA substrates, which had phosphorothioate bonds at the 3′ ends to prevent exonucleolytic DNA degradation by MRE11 (or MR/MRN). We observed that MRE11 alone had no endonuclease activity on any of the substrates tested, with or without pCtIP, as expected (Fig 7A and B, lanes 3 and 6). NBS1 promoted the cleavage of the non‐nicked DNA substrate by MR, and pCtIP was essential (Fig 7A). In contrast, NBS1 was dispensable when nicked DNA substrate was used (Fig 7B). Under these conditions, pCtIP provided a moderate stimulatory effect, with or without NBS1 (Fig 7B). The same results were obtained in kinetic assays (Fig 7C and D). The endonuclease activity was promoted by a nick or a gap in the top strand, as a substrate lacking the top left oligonucleotide was not endonucleolytically cleaved by MR‐pCtIP or MRN‐pCtIP (Fig EV7). Together, our results suggested that NBS1, and to a lesser extent pCtIP, is dispensable for the endonucleolytic DNA cleavage in case of a pre‐existing nick opposite to the cleavage site.
Figure 7. NBS1 is not required for DNA cleavage opposite to a nick.
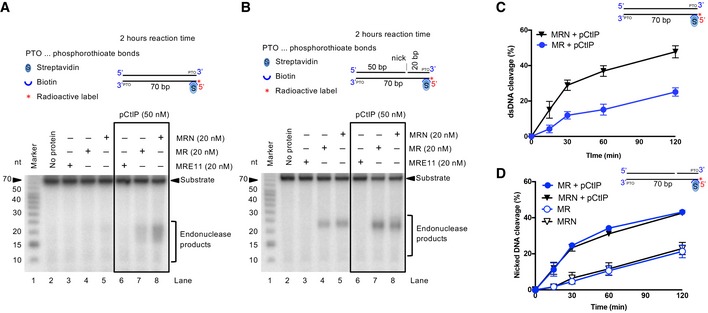
- Representative nuclease assays with MRE11, MR, MRN, and pCtIP, as indicated, on 5′‐end‐labeled dsDNA with a single streptavidin‐blocked end (S). The substrate contained five phosphorothioate bonds (PTO) at both 3′‐ends to prevent exonucleolytic DNA degradation. Reactions were incubated for 2 h.
- Representative nuclease assays as in (A), but with a substrate containing a nick in the top oligonucleotide (20 nt away from the 3′‐end, see cartoon).
- Quantitation of experiments such as shown in (A), with pCtIP. Averages shown; n ≥ 4; error bars, SEM.
- Quantitation of experiments such as shown in (B), with and without pCtIP. Averages shown; n = 3; error bars, SEM.
Figure EV7. MRN and pCtIP do not efficiently cleave overhanged DNA.
Nuclease assays as in Fig 7B, but with a substrate missing the top left oligonucleotide.
Discussion
The MRE11 nuclease plays critical functions at DSBs as well as at stalled, collapsed, or reversed replication forks (Schlacher et al, 2011; Stracker & Petrini, 2011; Mijic et al, 2017). Scheduled activation of the MRE11 nuclease is important for both homologous recombination and microhomology‐mediated end‐joining DSB repair pathways. In contrast, inappropriate MRE11 nuclease may result in illegitimate recombination leading to genome rearrangements or excessive DNA degradation at challenged replication forks, leading to cell lethality in certain genetic backgrounds (Schlacher et al, 2011). Therefore, the activity of the MRE11 nuclease must be finely controlled and restricted to situations when it is appropriate for genome stability maintenance. MRE11 is a 3′→5′ exonuclease on its own (Paull & Gellert, 1998). In complex with RAD50 and other co‐factors, MRE11 can cleave DNA endonucleolytically (Paull & Gellert, 1998, 1999). It is believed that the processing of DSBs or reversed DNA replication forks is initiated by the endonucleolytic cleavage of the 5′‐terminated DNA strand by MRE11 and co‐factors (Neale et al, 2005; Garcia et al, 2011; Cannavo & Cejka, 2014; Lemacon et al, 2017). Eukaryotic cells possess two key regulators of the MR nuclease complex: CtIP/Sae2 and NBS1/Xrs2. In yeast, phosphorylated Sae2 was required for the endonucleolytic DNA cleavage by MR, while the lack of Xrs2 had a more moderate effect (Oh et al, 2016). In humans, both NBS1 and phosphorylated CtIP promote the activity of the MR endonuclease, revealing that NBS1 is a regulator of MRE11 that appears more important in high eukaryotes (Anand et al, 2016; Deshpande et al, 2016). In this study, we define the mechanism by which NBS1 regulates the MRE11 endonuclease using primarily a reconstituted in vitro system.
We show that NBS1 functions as a sensor of CtIP phosphorylation, which allows MRE11 nuclease activation only when CtIP is phosphorylated (Fig 8A–C). The FHA and BRCT are highly conserved phosphoprotein binding domains, which mediate the physical interaction of NBS1 with phosphorylated CtIP and both these domains promote resection in vitro as well as in vivo. Once NBS1 senses that CtIP is phosphorylated, it activates the MR endonuclease, which is dependent on a direct physical interaction between NBS1 (residues 684–692) and MRE11 (Fig 8A). We found that the FHA and BRCT domains of NBS1 are important, but not absolutely essential for the MR‐ and pCtIP‐dependent cleavage of dsDNA near protein blocks. More than the FHA‐BRCT domains, the short region within the C‐terminus of NBS1 that interacts with MRE11 is absolutely required for NBS1 to promote the MR complex in conjunction with pCtIP. In support of this model, we observed residual NBS1‐dependent, but pCtIP‐independent nuclease activity of the MR complex in vitro (Fig 8C). Without CtIP, NBS1 still promotes DNA cleavage by MR. In this case, the FHA‐BRCT domains of NBS1 were entirely dispensable, while the MRE11 interaction region was required. Nevertheless, while this in vitro reaction informs us about the underlying mechanism, we believe that CtIP‐independent resection does not occur in vivo, as depletion of CtIP entirely eliminated resection in cells in all assays tested (Fig 4; Sartori et al, 2007).
Figure 8. A model for MRN and CtIP functions in endonucleolytic DNA cleavage.
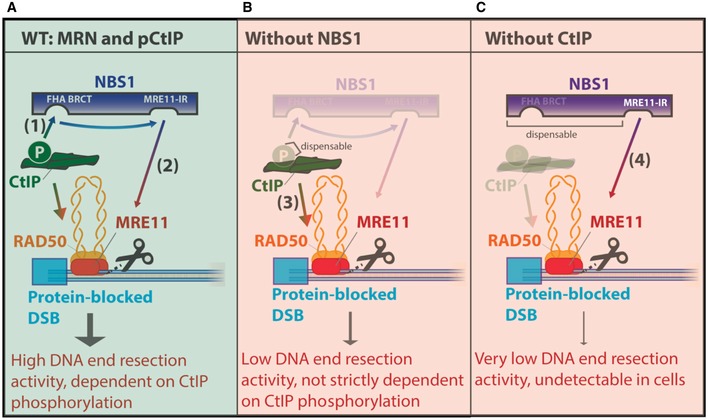
- When phosphorylated CtIP and MRN are present (in S‐G2), CtIP phosphorylation is detected by the FHA and BRCT domains of NBS1 (1), which in turn promotes DNA cleavage by MR via direct interaction with MRE11, mediated by the MRE11 interaction region of NBS1 (2). This results in maximal DNA end resection activity compatible with homologous recombination.
- In the absence of NBS1 function (see text), CtIP promotes the nuclease of MR in a reaction that is partly independent of its phosphorylation (3). This results in low resection activity of MR, which may be sufficient for limited resection in G1.
- In the absence of CtIP, NBS1, through its MRE11 interaction region, but independently of its FHA and BRCT domains, promotes the MR endonuclease (4). This activity is likely very limited and therefore unlikely to be physiologically relevant.
Next, we observed that in the absence of NBS1, CtIP still promotes the endonuclease of MR, albeit to a lesser extent. This functional interaction was observed in vitro (Fig 5), as well as in cells upon NBS1 depletion or gene editing that eliminated the FHA and BRCT domains of NBS1 (Fig 4). Strikingly, CtIP phosphorylation was not strictly required in the absence of NBS1 (Figs 5C–G and 8B). While these data are in agreement with the function of NBS1 as a sensor of CtIP phosphorylation, the result was nevertheless surprising, as it revealed fundamental differences between the budding yeast and human systems. In yeast, Sae2 phosphorylation regulates its physical interaction with RAD50 (Cannavo et al, 2018), in addition to Xrs2 (Liang et al, 2015). Even in the absence of Xrs2, Sae2 phosphorylation is still fully required for the stimulation of the Mre11‐Rad50 endonuclease. Using human proteins instead, we found that the phosphorylation of CtIP was largely but not completely dispensable for the activation of MR in the absence of NBS1. This result might help explain how CtIP promotes limited resection in G1. It is well established that CDK‐ and ATM‐dependent phosphorylation of CtIP is necessary for the activation of extended resection and hence homologous recombination in S‐G2 (Huertas et al, 2008; Huertas & Jackson, 2009; Wang et al, 2013). Nevertheless, CtIP likely activates MRE11 in MMEJ, which is not restricted to S‐G2 and requires only minimal resection (Truong et al, 2013; Averbeck et al, 2014; Sharma et al, 2015; Dutta et al, 2017). In G1, CtIP is phosphorylated at S327, which mediates its interaction with BRCA1, but the G1 form of CtIP will likely lack other phosphorylation modifications that are present in S‐G2 (Yu & Chen, 2004; Biehs et al, 2017). We speculate that this form of CtIP could promote the MR complex in case the NBS1 function is inhibited. However, we were unable to test this hypothesis directly, as a CtIP variant that is non‐phosphorylatable at the key CDK site at T847 appears to be impaired beyond mimicking a non‐phosphorylated residue (Fig 5H and I).
While MRE11 and RAD50 form a constitutively interacting dimer of dimers complex, there are conflicting reports about the stoichiometry of NBS1 within the MRN complex (Trujillo et al, 1998; Paull & Gellert, 1999; Schiller et al, 2012). Furthermore, it has been reported that the physical interaction of NBS1 with MR may be differentially regulated in G1 vs. S‐G2, upon DNA damage or at various subcellular locations, such as at telomeres (Zhu et al, 2000; Zhou et al, 2017; Limbo et al, 2018). Specifically, the MR complex was shown to localize to DSBs in the absence of NBS1 (Limbo et al, 2018), and NBS1 co‐localized with MR to telomeres in S but not during interphase (Zhu et al, 2000). NBS1 also shows slower kinetics of recruitment to DSBs compared to MRE11 (Haince et al, 2008). Finally, we show here that DNA end resection was strongly reduced but not completely eliminated in the absence of NBS1 in cells, despite the reduced nuclear import of MR, showing that NBS1 may not be an obligate component of the resection machinery under all conditions. Our data suggest that a reduction of the MR interaction with NBS1 may allow the complex to respond to non‐phosphorylated or differently phosphorylated CtIP. It is additionally possible that the function of NBS1 as a sensor of CtIP phosphorylation might be regulated by posttranslational modifications of NBS1 (which is heavily phosphorylated) and/or by other proteins that could regulate NBS1 function. More research is needed to fully understand the mechanisms of resection in G1, and the involvement of MRN and CtIP proteins in this context. Together, our results demonstrate that the MRE11‐RAD50 endonuclease is on its own restricted and becomes fully functional in the presence of NBS1 and phosphorylated CtIP. By regulating these co‐factors through multiple mechanisms, cells can fine‐tune the function of the MR endonuclease to optimally promote genome stability in various physiological contexts.
Material and Methods
Cloning, expression, and purification of recombinant proteins
Recombinant MRN, MR, and phosphorylated CtIP were expressed in insect Spodoptera frugiperda 9 (Sf9) cells and purified by affinity and ion exchange chromatography as described previously (Anand et al, 2016, 2018). MBP‐NBS1‐his and its variants, as well as MRE11‐his and FLAG‐NBS1‐his, were expressed in Sf9 cells and purified as described in detail in the Appendix Supplementary Methods. To prepare the pFB‐MBP‐NBS1‐his expression construct, NBS1 was amplified from pTP36 (a kind gift from Tanya Paull, University of Texas) by PCR using primers NBS1_F and NBS1_R (Appendix Tables S1 and S2). The PCR product was digested with NheI and XmaI (New England Biolabs) and cloned into corresponding sites of pFB‐MBP‐MLH3‐his (Ranjha et al, 2014). The preparation of other NBS1 variant expression constructs is described in the Appendix Supplementary Methods. To prepare FLAG‐NBS1‐his, a NBS1 sequence that was codon‐optimized for expression in Sf9 cells (NBS1co) was purchased from Synbio Technologies and cloned into BamHI‐XhoI sites of pFB‐MBP‐NBS1‐his to create pFB‐FLAG‐NBS1co‐his. To prepare pFB‐MBP‐MRE11‐his, the MRE11 gene was amplified by PCR from pTP17 (a kind gift from Tanya Paull, University of Texas) using primers HMRE11‐FO and HMRE11‐RE (Appendix Tables S1 and S2). The amplified product was digested with NheI and XmaI, and cloned into the same sites in pFB‐MBP‐Sgs1‐his plasmid (Cejka & Kowalczykowski, 2010); the sequence of Sgs1 was removed during this step. The scheme and sequences of oligonucleotides used for cloning of the NBS1 fragments and all other constructs in this study are listed in Appendix Tables S1 and S2. Where indicated, 1.2 μg pCtIP was dephosphorylated in 20 μl reactions with 200 units of λ phosphatase (λ‐PP, New England Biolabs) in 1× PMP buffer (New England Biolabs) supplemented with 1 mM magnesium chloride for 15 min at 30°C. For “mock” control, λ‐PP was excluded from the reaction.
Nuclease assays
Unless indicated otherwise, nuclease assays (15 μl volume) were carried out in nuclease buffer containing 25 mM Tris–HCl pH 7.5, 5 mM magnesium acetate, 1 mM manganese acetate, 1 mM dithiothreitol (DTT), 1 mM ATP, 0.25 mg/ml BSA (New England Biolabs), 1 mM phosphoenolpyruvate (Sigma), 80 U/ml pyruvate kinase (Sigma), and 1 nM oligonucleotide‐based DNA substrate (in molecules). Unless specified otherwise, the final NaCl concentration in the reactions was adjusted to 15 mM, accounting for salt brought to the reactions with protein storage or dilution buffer. The reactions were supplemented with 15 nM streptavidin (Sigma) and incubated for 5 min at room temperature to block the biotinylated end(s) of the DNA substrates. The recombinant proteins were then added to the reactions on ice and samples were incubated at 37°C for 30 min, unless indicated otherwise. Reactions were stopped by adding 0.5 μl of 0.5 M ethylenediaminetetraacetic (EDTA) and 1 μl Proteinase K (19 mg/ml, Roche), and incubated at 50°C for 30 min. Finally, 15 μl loading buffer (5% formamide, 20 mM EDTA, bromophenol blue) was added to all samples and the products were separated on 15% polyacrylamide denaturing urea gels (19:1 acrylamide‐bisacrylamide, Bio‐Rad), as described (Pinto et al, 2018). The gels were fixed in fixing solution (40% methanol, 10% acetic acid, 5% glycerol) for 30 min at room temperature and dried on a 3MM Chr paper (Whatman). The dried gels were exposed to storage phosphor screen (GE Healthcare) and scanned by a Typhoon Phosphor Imager (FLA 9500, GE Healthcare). Nuclease assays with circular ssDNA substrate (M13, 100 ng per reaction, New England Biolabs) were carried out similarly except the reaction buffer contained 60 mM NaCl and incubation time was extended to 2 h. The reaction products were separated on 1.1% agarose gels, which were stained with GelRed (1:20,000, Biotium) after completion of electrophoresis for 45 min. The gels were imaged with gel imager (Ingenius3, Syngene).
Preparation of oligonucleotide‐based DNA substrates
All oligonucleotides were purified by polyacrylamide gel electrophoresis and purchased from Eurogentec. The labeling of oligonucleotides at the 5′‐end was carried out by T4 polynucleotide kinase (New England Biolabs) and [γ‐32P] ATP (Perkin Elmer). The labeling of oligonucleotides at the 3′‐end was carried out by terminal deoxynucleotidyl transferase (New England Biolabs) and [α‐32P] cordycepin 5′ triphosphate (Perkin Elmer), as described (Pinto et al, 2018). To prepare quadruple blocked 70‐bp‐long DNA substrate, PC210 and PC211 oligonucleotides were used, as described previously (Cannavo & Cejka, 2014). To prepare single‐blocked non‐nicked substrate (Fig 7A), PC216C_5XSS (GATGCATGAGGTGGAGTACGCGCCCGGGGAGCCCAAGGGCACGCCCTGGCACCCGCACCGCGGCA*C*T*T*A*C) was labeled at 5′‐end and annealed with PC216_5XSS (GTAAGTGCCGCGGTGCGGGTGCCAGGGCGTGCCCTTGGGCTCCCCGGGCGCGTACTCCACCTCAT*G*C*A*T*C), the bold T represents the biotin modification and * represent phosphorothioate linkages between nucleotides. To prepare the single‐blocked nicked substrate (Fig 7B), PC216C_5XSS labeled at 5′‐end was annealed with PC216_50nt (GTAAGTGCCGCGGTGCGGGTGCCAGGGCGTGCCCTTGGGCTCCCCGGGCG) and PC216_20nt_5XSS (CGTACTCCACCTCAT*G*C*A*T*C). To prepare substrate used in Fig EV7, PC216C_5XSS labeled at 5′‐end was annealed to PC216_20nt_5XSS.
Protein interaction assays
To test for interactions between recombinant proteins, the “bait” proteins were first immobilized on a resin [either M2 anti‐FLAG affinity gel (Sigma) or protein G agarose (Invitrogen) coupled to specific antibodies]. The resin was then incubated with “prey” proteins in binding buffer, washed, and bound proteins were analyzed by Western blotting. As a negative control, prey proteins were incubated with resin without bait proteins.
Specifically, to detect the interaction between pCtIP and NBS1 variants (Fig 2C and D), 1.5 μg anti‐CtIP antibody (Active Motif, 61141) was captured on 10 μl protein G beads upon incubation in 150 μl PBS‐T pH 7.4 (1.37 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 0.1% Tween‐20) for 40 min at room temperature with continuous rotation. Next, 1 μg recombinant CtIP (mock‐treated pCtIP or dephosphorylated λCtIP) was added and incubated in 60 μl binding buffer (25 mM Tris–HCl pH 7.5, 1 mM DTT, 3 mM EDTA, 50 mM NaCl, 0.2 mg/ml BSA) for 1 h at 4°C with continuous rotation. The resin with the immobilized antibody was washed two times with 150 μl wash buffer (25 mM Tris–HCl pH 7.5, 1 mM DTT, 3 mM EDTA, 80 mM NaCl, 0.05% Triton X), and incubated with 320 nM NBS1 variants in 60 μl binding buffer for 1 h at 4°C with continuous rotation. The resin was then washed three times with 150 μl wash buffer. The proteins were eluted by incubation of the washed resin for 3 min at 95°C in 60 μl SDS buffer (50 mM Tris–HCl pH 6.8, 1.6% sodium dodecyl sulfate, 100 mM DTT, 10% glycerol, 0.01% bromophenol blue). CtIP and NBS1 variants were detected by Western blot using anti‐CtIP (Active Motif, 61141, 1:1,000) and anti‐NBS1 (Novus Biologicals, NB100‐143, 1:1,000) antibodies. In assays shown in Figs 6C and EV6A, 90 nM MRE11, MR, or MRN was incubated with immobilized λCtIP or pCtIP as described above. MRE11 was detected with anti‐MRE11 antibody (Abcam, ab214, 1:1,000).
To detect the interaction between MRE11‐his and MBP‐NBS1‐his/MBP‐NBS1 ∆MIR‐his, or between MRE11‐his and pCtIP/λCtIP as shown in Figs EV2H and 6A, respectively, 1.4 μg MRE11‐his was immobilized on beads and coupled to anti‐MRE11 antibody (Abcam, ab214). After washing, 200 nM MBP‐NBS1‐his/MBP‐NBS1∆MIR‐his or 330 nM pCtIP/λCtIP was incubated with the MRE11‐coated resin in a 60 μl reaction in binding buffer as described above. For the assay shown in Fig EV2H, both MRE11‐his and MBP‐NBS1‐his/MBP‐NBS1 ∆MIR‐his were detected by anti‐his antibody (MBL, M091‐3, 1:1,000) by Western blotting.
For the interaction assay shown in Fig 6B, 50 ml Sf9 cells were infected with RAD50‐FLAG baculovirus. Cells were lysed and RAD50‐FLAG (~ 300 ng) was immobilized on 30 μl anti‐FLAG M2 affinity gel. Next, in a 150 μl reaction, 330 nM pCtIP/λCtIP was added and incubated in binding buffer for 1 h at 4°C with continuous rotation. Beads were washed four times with 300 μl wash buffer, and proteins were eluted with wash buffer supplemented with 3× FLAG peptide (150 ng/μl, Sigma). Bound RAD50‐FLAG was detected by Western blotting using anti‐FLAG antibody (Sigma, F3165, 1:2,000).
For FLAG co‐IP experiments (Fig EV4E), 293‐T cells were co‐transfected with FLAG‐CtIP expression constructs (Sartori et al, 2007) and NBS1‐myc expression constructs (Hari et al, 2010). Immunoprecipitations were done using anti‐FLAG M2 affinity beads (Sigma, A2220). Western blots were probed with rabbit anti‐FLAG (Sigma, F7425) and mouse anti‐myc (GeneTex, 9E10, GTX80249) antibodies.
DR‐GFP and SSA reporter assays
For DR‐GFP HR repair reporter assays, DR‐GFP U2OS cells (a gift from J. Stark) were plated at 20,000 cells/cm2 in 6‐well plates. After 24 h, cells were transfected with control siRNA or siRNA against CtIP (5′‐GCUAAACAGGAACGAAUCTTdTdT‐3′) and NBS1 (5′‐GGAGGAAGAUGUCAAUGUUTTdTdT‐3′), respectively. Forty‐eight hours after siRNA transfection, cells were transfected with I‐SceI expression plasmid (pCBASce). Four hours after transfection, the medium was replaced and a second transfection with siRNA oligonucleotides was carried out. Forty‐eight hours after I‐SceI transfection, cells were analyzed for GFP expression by flow cytometry on a LSRII Fortessa flow cytometer (BD). For the SSA reporter assays, 0.4 × 105 SA‐GFP U2OS cells were seeded in 0.5 ml antibiotic‐free media onto 24‐well plates with 5 pmol siRNA (siCTRL, 5′‐TGGTTTACATGTCGACTAA; siCtIP, 5′‐GCTAAAACAGGAACGAATC; siNBS1, 5′‐GGAGGAAGATGTCAATGTT) and 1.8 μl RNAiMAX (Invitrogen) in 100 μl Opti‐MEM (Gibco) and cultured overnight (20 h). Following RNAi treatment, the cells were transfected with 0.4 μg I‐SceI (pCBASce) and 0.2 μg empty vector (pcDNA3.1) with Lipofectamine 2000 (Invitrogen) in 100 μl Opti‐MEM and 0.5 ml antibiotic‐free media for 3 h. After the I‐SceI transfection, cells were washed and treated with MRE11 inhibitors 50 μM PFM03 and 50 μM PFM39 (a kind gift from Davide Moiani and John Tainer, MD Anderson Cancer Center; Shibata et al, 2014) or mock‐treated with DMSO and cultured for 3 days. GFP‐positive cells were quantified on an LSRFortessa flow cytometer (BD).
Generation of siRNA‐based NBS1 complementation system in U2OS cells
To generate the siRNA‐based complementation system for NBS1, the Flp‐In™ T‐REx™ system (Thermo Fisher Scientific) was used according to the manufacturer's protocols. Briefly, myc‐tagged full‐length NBS1 cDNAs (wild‐type and mutants) were sub‐cloned from retroviral expression constructs (Hari et al, 2010) and ligated into the pcDNA5/FRT/TO vector. Silent mutations were introduced in the siRNA target sequence (see below) by site‐directed mutagenesis using the following mutagenesis primer (sense): 5′‐GTTCAAAAACAGGAGGAAGACGTGAACGTTAGAAAAAGGCCAAGG‐3′. A Flp‐In T‐REx U2OS cell line (gift from D. Durocher) was cultured in Dulbecco's modified Eagle medium (DMEM), supplemented with 10% fetal calf serum (FCS), 2 mM l‐glutamine, and penicillin‐streptomycin antibiotics under standard cell culture conditions in a CO2 incubator (37°C; 5% CO2). NBS1‐myc harboring vectors and the Flp‐In recombinase expression plasmid pOG44 were mixed in a 1:9 ratio and transfected into the Flp‐In T‐REx U2OS cells. Stable clones were established by hygromycin B (250 μg/ml) and blasticidin (10 μg/ml) selection and were characterized for inducible NBS1‐myc expression by immunofluorescence and Western blotting.
Generation of the NBS1∆N cell line
The NBS1∆N cell line was generated according to a protocol published by the Zhang laboratory (Ran et al, 2013). The sgRNA target sequence (5′‐GCGTTGAGTACGTTGTTGGA‐3′) was cloned into the PX330‐U6‐hSpCas9 vector and verified by sequencing. The targeting vector was then transfected into U2OS cells, and clonal cell lines were isolated by dilution in 96‐well plates. Single clones were analyzed for NBS1 expression by immunofluorescence. Several clones without detectable NBS1 expression were isolated and one of them was further characterized (data will be published elsewhere). Frameshift‐inducing insertion/deletion (indel) mutations in all of the three NBS1 alleles present in U2OS cells were verified by Sanger sequencing of a PCR‐amplified genomic fragment that was cloned in the pCR II Blunt TOPO plasmid (sequences will be published elsewhere), indicating that the FHA‐BRCT domain coding region was eliminated. Western blotting, however, revealed the presence of weakly expressed ~ 40 kDa protein that is recognized by a monoclonal antibody raised against full‐length NBS1 (GeneTex, clone 1D7, GTX70224), which was sensitive to downregulation of the NBS1 mRNA by siRNA transfection, thus suggesting that this cell line expresses low levels of hypomorphic C‐terminal fragment of NBS1 that is produced by internal translation initiation within the NBS1 mRNA using an open reading frame generated by one of the indel mutations, similar to the NBS1 hypomorphic alleles expressed in cell lines derived from NBS patients (Maser et al, 2001).
In vivo resection assay
U2OS cell lines were grown on coverslips for 24 h prior to treatment. When indicated, cells were transfected with siRNAs against NBS1 or CtIP (siRNA sequence indicated in the previous section where the generation of siRNA‐based NBS1 complementation system was explained) and incubated for 72 h. For heterologous expression of full‐length recombinant myc‐tagged NBS1, doxycycline (1 μg/ml) was added 24 h prior to the experiment. For BrdU staining under native conditions, BrdU (10 μg/ml) was also added 24 h prior to the experiment. Cells were exposed to 5 Gy of IR and then incubated for 3 h at 37°C. The medium was removed, and cells were washed with PBS, followed by a brief pre‐extraction on ice in CSK buffer (100 mM PIPES, pH 7.0; 100 mM NaCl; 300 mM sucrose; 3 mM MgCl2; 0.4% Triton X‐100). Cells were then fixed in 4% paraformaldehyde, washed, and prepared for immunofluorescence analysis (see below).
Immunofluorescence
Cells were grown on glass coverslips and fixed with either ice‐cold methanol for 10 min (for staining with NBS1 and MRE11 antibodies), or with 4% buffered formaldehyde (for staining with all other antibodies) for 15 min at room temperature, and subsequently permeabilized for 5 min in PBS containing 0.2% Triton X‐100. Following 1 h of blocking in blocking buffer (10% FBS, 3% BSA in PBS), primary antibody incubations were performed at room temperature for 2 h. Coverslips were washed three times with PBS, and secondary antibody incubations were performed for 1 h at room temperature in the dark. After washing with PBS for three times, coverslips were mounted on glass microscope slides with Vectrashield mounting medium containing 0.5 μg/ml 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI). Widefield image acquisition was done on either a Leica DMI6000B inverted microscope equipped with a 63× Apochromat oil immersion objective (NA 1.3) using Leica's LAS‐AF software or on a Zeiss AxioObserver Z1 widefield microscope, equipped with a Lumencor SpectraX illumination system and a 63×, 1.4‐NA, i‐plan apochromat oil immersion objective, using Zeiss’ ZEN (blue edition) software. Images were analyzed using Fiji software (Schindelin et al, 2012). Counting of BrdU foci or RPA2 foci was done with a Fiji macro written for automatic image analysis. The following antibodies were used at the indicated dilutions: rabbit anti‐NBS1 (Novus Biologicals, NB100‐143, 1:500), affinity purified sheep anti‐MRE11 (Goldberg et al, 2003, 1:1,000), mouse anti‐myc (GeneTex, 9E10, GTX80249, 1:250), mouse anti‐BrdU (Amersham/GE Healthcare, BU‐1, RPN202, 1:100), and mouse anti‐RPA2 (Abcam, 9H8, ab2175, 1:250).
Western blotting of total cell extracts
SDS–PAGE and Western blotting were performed using total extracts prepared from cultured cells following standard procedures. The following antibodies were used at the indicated dilutions: rabbit anti‐ATMpS1981 (Eptimics, YE070901r, 1:5,000), rabbit anti‐ATM (Calbiochem, PC‐116, 1:250), mouse anti‐CtIP (Santa Cruz, sc‐271339, 1:250), mouse anti‐NBS1 (GeneTex, 1D7, GTX70224, 1:500), mouse anti‐RPA2 (Abcam, 9H8, ab2175, 1:500), rabbit anti‐SMC1 (Abcam, ab9262, 1:500), mouse anti‐RAD50 (GeneTex, 13B3, GTX70228, 1:1,000), mouse anti‐MRE11 (Abcam, 12D7, ab214, 1:500), mouse anti‐myc (GeneTex, 9E10, GTX80249, 1:250), and mouse anti‐tubulin (Sigma, DM1A, T6199, 1:2,000). For Western blot shown in Fig 4F, the following antibodies were used: rabbit anti‐NBS1 (Novus Biologicals, NB100 143, 1:1,000), mouse anti‐CtIP (Active Motif, 61141, 1:1,000), and mouse anti‐β actin (Abcam, ab8227, 1:1,000).
Clonogenic survival assay
U2OS Flp‐In T‐REx cell lines were transfected with siNBS1 for 48 h and then plated at low density in medium supplemented with 0.5 μg/ml doxycycline. 24 h later, the plates were exposed to the indicated IR doses, followed by incubation for 12–14 days. Colonies were fixed and stained with Coomassie brilliant blue, and the number of surviving colonies for each cell was counted. The percentage of surviving colonies for each cell line was calculated using the plating efficiencies of the non‐irradiated cells as a reference. Survival experiments were carried out in biological triplicates.
Author contributions
RA performed all biochemical experiments. LR performed the experiment shown in Fig EV6B. AJ, SMH, and DB performed cell biology experiments. All authors designed the experiments and analyzed the data. RA, PC, and MS wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Dr. Jeremy Stark (City of Hope), Dr. Xiohua Wu (Scripps), Dr. Davide Moiani and Dr. John Tainer (MD Anderson Cancer Center), Dr. Tanya Paull (UT Austin), A. Sartori (University of Zurich), and Dan Durocher (Lunenfeld‐Tanenbaum Research Institute) for cell lines, plasmids, and reagents. We thank members of the Cejka (S. Halder, G. Reginato, E. Cannavo, I. Ceppi, A. Acharya) and Stucki laboratories for comments on the manuscript. This work was supported by the Swiss National Science Foundation grants 31003A_144284 to MS, 31003A_175444 to PC, and European Research Council grant 681630 to PC.
The EMBO Journal (2019) 38: e101005
References
- Anand R, Ranjha L, Cannavo E, Cejka P (2016) Phosphorylated CtIP functions as a co‐factor of the MRE11‐RAD50‐NBS1 endonuclease in DNA end resection. Mol Cell 64: 940–950 [DOI] [PubMed] [Google Scholar]
- Anand R, Pinto C, Cejka P (2018) Methods to study DNA end resection I: recombinant protein purification. Methods Enzymol 600: 25–66 [DOI] [PubMed] [Google Scholar]
- Averbeck NB, Ringel O, Herrlitz M, Jakob B, Durante M, Taucher‐Scholz G (2014) DNA end resection is needed for the repair of complex lesions in G1‐phase human cells. Cell Cycle 13: 2509–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biehs R, Steinlage M, Barton O, Juhasz S, Kunzel J, Spies J, Shibata A, Jeggo PA, Lobrich M (2017) DNA double‐strand break resection occurs during non‐homologous end joining in G1 but is distinct from resection during homologous recombination. Mol Cell 65: 671–684.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P (2014) Sae2 promotes dsDNA endonuclease activity within Mre11‐Rad50‐Xrs2 to resect DNA breaks. Nature 514: 122–125 [DOI] [PubMed] [Google Scholar]
- Cannavo E, Johnson D, Andres SN, Kissling VM, Reinert JK, Garcia V, Erie DA, Hess D, Thoma NH, Enchev RI, Peter M, Williams RS, Neale MJ, Cejka P (2018) Regulatory control of DNA end resection by Sae2 phosphorylation. Nat Commun 9: 4016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR III, Hays L, Morgan WF, Petrini JH (1998) The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double‐strand break repair to the cellular DNA damage response. Cell 93: 477–486 [DOI] [PubMed] [Google Scholar]
- Cejka P, Kowalczykowski SC (2010) The full‐length Saccharomyces cerevisiae Sgs1 protein is a vigorous DNA helicase that preferentially unwinds holliday junctions. J Biol Chem 285: 8290–8301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerosaletti KM, Concannon P (2003) Nibrin forkhead‐associated domain and breast cancer C‐terminal domain are both required for nuclear focus formation and phosphorylation. J Biol Chem 278: 21944–21951 [DOI] [PubMed] [Google Scholar]
- Chen L, Trujillo KM, Van Komen S, Roh DH, Krejci L, Lewis LK, Resnick MA, Sung P, Tomkinson AE (2005) Effect of amino acid substitutions in the rad50 ATP binding domain on DNA double strand break repair in yeast. J Biol Chem 280: 2620–2627 [DOI] [PubMed] [Google Scholar]
- Chen L, Nievera CJ, Lee AY, Wu X (2008) Cell cycle‐dependent complex formation of BRCA1.CtIP.MRN is important for DNA double‐strand break repair. J Biol Chem 283: 7713–7720 [DOI] [PubMed] [Google Scholar]
- Connelly JC, de Leau ES, Leach DR (2003) Nucleolytic processing of a protein‐bound DNA end by the E. coli SbcCD (MR) complex. DNA Repair (Amst) 2: 795–807 [DOI] [PubMed] [Google Scholar]
- Deng SK, Gibb B, de Almeida MJ, Greene EC, Symington LS (2014) RPA antagonizes microhomology‐mediated repair of DNA double‐strand breaks. Nat Struct Mol Biol 21: 405–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai‐Mehta A, Cerosaletti KM, Concannon P (2001) Distinct functional domains of nibrin mediate Mre11 binding, focus formation, and nuclear localization. Mol Cell Biol 21: 2184–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande RA, Williams GJ, Limbo O, Williams RS, Kuhnlein J, Lee JH, Classen S, Guenther G, Russell P, Tainer JA, Paull TT (2014) ATP‐driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J 33: 482–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande RA, Lee JH, Arora S, Paull TT (2016) Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein‐DNA adducts. Mol Cell 64: 593–606 [DOI] [PubMed] [Google Scholar]
- Dutta A, Eckelmann B, Adhikari S, Ahmed KM, Sengupta S, Pandey A, Hegde PM, Tsai MS, Tainer JA, Weinfeld M, Hegde ML, Mitra S (2017) Microhomology‐mediated end joining is activated in irradiated human cells due to phosphorylation‐dependent formation of the XRCC1 repair complex. Nucleic Acids Res 45: 2585–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA‐PKcs to sites of DNA damage. Nature 434: 605–611 [DOI] [PubMed] [Google Scholar]
- Feng W, Jasin M (2017) BRCA2 suppresses replication stress‐induced mitotic and G1 abnormalities through homologous recombination. Nat Commun 8: 525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V, Phelps SE, Gray S, Neale MJ (2011) Bidirectional resection of DNA double‐strand breaks by Mre11 and Exo1. Nature 479: 241–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP (2003) MDC1 is required for the intra‐S‐phase DNA damage checkpoint. Nature 421: 952–956 [DOI] [PubMed] [Google Scholar]
- Gravel S, Chapman JR, Magill C, Jackson SP (2008) DNA helicases Sgs1 and BLM promote DNA double‐strand break resection. Genes Dev 22: 2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, Poirier GG (2008) PARP1‐dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem 283: 1197–1208 [DOI] [PubMed] [Google Scholar]
- Hari FJ, Spycher C, Jungmichel S, Pavic L, Stucki M (2010) A divalent FHA/BRCT‐binding mechanism couples the MRE11‐RAD50‐NBS1 complex to damaged chromatin. EMBO Rep 11: 387–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Cortes‐Ledesma F, Sartori AA, Aguilera A, Jackson SP (2008) CDK targets Sae2 to control DNA‐end resection and homologous recombination. Nature 455: 689–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Jackson SP (2009) Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 284: 9558–9565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C (2001) Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell 8: 1129–1135 [DOI] [PubMed] [Google Scholar]
- Keeney S, Kleckner N (1995) Covalent protein‐DNA complexes at the 5′ strand termini of meiosis‐specific double‐strand breaks in yeast. Proc Natl Acad Sci USA 92: 11274–11278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Grosbart M, Anand R, Wyman C, Cejka P, Petrini JH (2017) The Mre11‐Nbs1 interface is essential for viability and tumor suppression. Cell Rep 18: 496–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Paull TT (2005) ATM activation by DNA double‐strand breaks through the Mre11‐Rad50‐Nbs1 complex. Science 308: 551–554 [DOI] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, Vindigni A (2017) MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81‐dependent fork rescue in BRCA2‐deficient cells. Nat Commun 8: 860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Suhandynata RT, Zhou H (2015) Phosphorylation of Sae2 mediates forkhead‐associated (FHA) domain‐specific interaction and regulates its DNA repair function. J Biol Chem 290: 10751–10763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbo O, Yamada Y, Russell P (2018) Mre11‐Rad50‐dependent activity of ATM/Tel1 at DNA breaks and telomeres in the absence of Nbs1. Mol Biol Cell 29: 1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd J, Chapman JR, Clapperton JA, Haire LF, Hartsuiker E, Li J, Carr AM, Jackson SP, Smerdon SJ (2009) A supramodular FHA/BRCT‐repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell 139: 100–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JL, Kim EM, Haber JE, Lee SE (2003) Yeast Mre11 and Rad1 proteins define a Ku‐independent mechanism to repair double‐strand breaks lacking overlapping end sequences. Mol Cell Biol 23: 8820–8828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser RS, Zinkel R, Petrini JH (2001) An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat Genet 27: 417–421 [DOI] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, Lopes M (2017) Replication fork reversal triggers fork degradation in BRCA2‐defective cells. Nat Commun 8: 859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double‐strand break processing. Nature 455: 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Haber JE (1996) Cell cycle and genetic requirements of two pathways of nonhomologous end‐joining repair of double‐strand breaks in Saccharomyces cerevisiae . Mol Cell Biol 16: 2164–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Matsumoto K, Sugimoto K (2003) ATM‐related Tel1 associates with double‐strand breaks through an Xrs2‐dependent mechanism. Genes Dev 17: 1957–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale MJ, Pan J, Keeney S (2005) Endonucleolytic processing of covalent protein‐linked DNA double‐strand breaks. Nature 436: 1053–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC (2011) BLM‐DNA2‐RPA‐MRN and EXO1‐BLM‐RPA‐MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25: 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Al‐Zain A, Cannavo E, Cejka P, Symington LS (2016) Xrs2 dependent and independent functions of the Mre11‐Rad50 complex. Mol Cell 64: 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Gellert M (1998) The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double‐strand breaks. Mol Cell 1: 969–979 [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M (1999) Nbs1 potentiates ATP‐driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev 13: 1276–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT (2010) Making the best of the loose ends: Mre11/Rad50 complexes and Sae2 promote DNA double‐strand break resection. DNA Repair (Amst) 9: 1283–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto C, Anand R, Cejka P (2018) Methods to study DNA end resection II: biochemical reconstitution assays. Methods Enzymol 600: 67–106 [DOI] [PubMed] [Google Scholar]
- Rahal EA, Henricksen LA, Li Y, Williams RS, Tainer JA, Dixon K (2010) ATM regulates Mre11‐dependent DNA end‐degradation and microhomology‐mediated end joining. Cell Cycle 9: 2866–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjha L, Anand R, Cejka P (2014) The Saccharomyces cerevisiae Mlh1‐Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions. J Biol Chem 289: 5674–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjha L, Howard SM, Cejka P (2018) Main steps in DNA double‐strand break repair: an introduction to homologous recombination and related processes. Chromosoma 127: 187–214 [DOI] [PubMed] [Google Scholar]
- Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS (2009) Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol 16: 819–824 [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB et al (2016) Replication fork stability confers chemoresistance in BRCA‐deficient cells. Nature 535: 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginato G, Cannavo E, Cejka P (2018) Physiological protein blocks direct the Mre11‐Rad50‐Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev 31: 2325–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP (2007) Human CtIP promotes DNA end resection. Nature 450: 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller CB, Lammens K, Guerini I, Coordes B, Feldmann H, Schlauderer F, Mockel C, Schele A, Strasser K, Jackson SP, Hopfner KP (2012) Structure of Mre11‐Nbs1 complex yields insights into ataxia‐telangiectasia‐like disease mutations and DNA damage signaling. Nat Struct Mol Biol 19: 693–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC (2015) Homology and enzymatic requirements of microhomology‐dependent alternative end joining. Cell Death Dis 6: e1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum‐Fikkert S, Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, Neale MJ, Bristow RG, Masson JY, Wyman C, Jeggo PA, Tainer JA (2014) DNA double‐strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53: 7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spycher C, Miller ES, Townsend K, Pavic L, Morrice NA, Janscak P, Stewart GS, Stucki M (2008) Constitutive phosphorylation of MDC1 physically links the MRE11‐RAD50‐NBS1 complex to damaged chromatin. J Cell Biol 181: 227–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M (2004) Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol 24: 9305–9316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM (1999) The DNA double‐strand break repair gene hMRE11 is mutated in individuals with an ataxia‐telangiectasia‐like disorder. Cell 99: 577–587 [DOI] [PubMed] [Google Scholar]
- Stracker TH, Petrini JH (2011) The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 12: 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EM, Cecillon SM, Bonis A, Chapman JR, Povirk LF, Lindsay HD (2010) The Mre11/Rad50/Nbs1 complex functions in resection‐based DNA end joining in Xenopus laevis . Nucleic Acids Res 38: 441–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo KM, Yuan SS, Lee EY, Sung P (1998) Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J Biol Chem 273: 21447–21450 [DOI] [PubMed] [Google Scholar]
- Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, Wu X (2013) Microhomology‐mediated end joining and homologous recombination share the initial end resection step to repair DNA double‐strand breaks in mammalian cells. Proc Natl Acad Sci USA 110: 7720–7725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto Y, Mitsuoka C, Terasawa M, Ogawa H, Ogawa T (2005) Xrs2p regulates Mre11p translocation to the nucleus and plays a role in telomere elongation and meiotic recombination. Mol Biol Cell 16: 597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Ogawa H, Petrini JH (2001) A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 7: 1255–1266 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J (2000) BASC, a super complex of BRCA1‐associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14: 927–939 [PMC free article] [PubMed] [Google Scholar]
- Wang H, Shi LZ, Wong CC, Han X, Hwang PY, Truong LN, Zhu Q, Shao Z, Chen DJ, Berns MW, Yates JR III, Chen L, Wu X (2013) The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR‐mediated double‐strand break repair. PLoS Genet 9: e1003277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Daley JM, Kwon Y, Krasner DS, Sung P (2017) Plasticity of the Mre11‐Rad50‐Xrs2‐Sae2 nuclease ensemble in the processing of DNA‐bound obstacles. Genes Dev 31: 2331–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA (2008) Mre11 dimers coordinate DNA end bridging and nuclease processing in double‐strand‐break repair. Cell 135: 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JN, Iwasaki H, Russell P, Tainer JA (2009) Nbs1 flexibly tethers Ctp1 and Mre11‐Rad50 to coordinate DNA double‐strand break processing and repair. Cell 139: 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Chahwan C, Bailis J, Hunter T, Russell P (2005) ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol 25: 5363–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Chen J (2004) DNA damage‐induced cell cycle checkpoint control requires CtIP, a phosphorylation‐dependent binding partner of BRCA1 C‐terminal domains. Mol Cell Biol 24: 9478–9486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Chen J (2009) N terminus of CtIP is critical for homologous recombination‐mediated double‐strand break repair. J Biol Chem 284: 31746–31752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Kawamura K, Yanagihara H, Kobayashi J, Zhang‐Akiyama QM (2017) NBS1 is regulated by two kind of mechanisms: ATM‐dependent complex formation with MRE11 and RAD50, and cell cycle‐dependent degradation of protein. J Radiat Res 58: 487–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XD, Kuster B, Mann M, Petrini JH, de Lange T (2000) Cell‐cycle‐regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet 25: 347–352 [DOI] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double‐strand break ends. Cell 134: 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File