

SUMMARY
Cortical hyperexcitability is a hallmark of fragile X syndrome (FXS). In the Fmr1 knockout (KO) mouse model of FXS, cortical hyperexcitability is linked to sensory hypersensitivity and seizure susceptibility. It remains unclear why homeostatic mechanisms fail to prevent such activity. Homeostatic intrinsic plasticity (HIP) adjusts membrane excitability through regulation of ion channels to maintain activity levels following activity perturbation. Despite the critical role of HIP in the maturation of excitability, it has not been examined in FXS. Here, we demonstrate that HIP does not operate normally in a disease model, FXS. HIP was either lost or exaggerated in two distinct neuronal populations from Fmr1 KO cortical cultures. In addition, we have identified a mechanism for homeostatic intrinsic plasticity. Compromising HIP function during development could leave cortical neurons in the FXS nervous system vulnerable to hyperexcitability.
Graphical Abstract
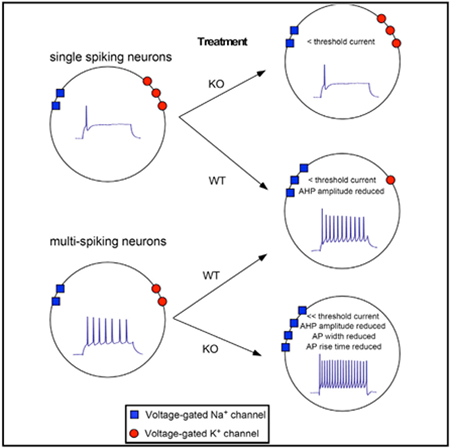
In Brief
Fragile X syndrome (FXS) is characterized by cortical hyperexcitability, but the mechanisms driving hyperexcitability are poorly understood. Homeostatic intrinsic plasticity (HIP) regulates ion channel function to maintain appropriate activity levels. Bülow et al. show that HIP is functionally altered in FXS neurons, which may leave cortical neurons vulnerable to hyperexcitability.
INTRODUCTION
Fragile X syndrome (FXS) is a neurodevelopmental disorder that is the leading monogenetic cause of autism and inherited form of intellectual disability (Bassell and Warren, 2008). Patients with FXS do not express the fragile X mental retardation protein (FMRP) and suffer from a diversity of debilitating symptoms, including sensory hypersensitivity, seizures, and sleep disturbances (Contractor et al., 2015). The mouse model of FXS, the Fmr1 knockout (KO), is hypersensitive to sensory stimuli and displays increased seizure susceptibility (Contractor et al., 2015). Many studies in vitro and in vivo have now demonstrated that the Fmr1 KO cortex exhibits hyperexcitable features (Gibson et al., 2008; Gonçalves et al., 2013; Gross et al., 2015; Zhang et al., 2014). A wide variety of mechanisms underlying cortical hyperexcitability have been reported, including alterations in synaptic transmission and ion channel expression and function (Kalmbach et al., 2015; Ronesi et al., 2012). Despite significant progress in this field, the hyperexcitable features and the underlying mechanisms reported are often heterogeneous if not contradictory. Although it is clear that the mechanisms underlying cortical hyperexcitability in FXS are complicated and multifactorial, recognition of a guiding principle driving the expression of these altered mechanisms would be transformative.
One set of mechanisms that is known to be important for ensuring appropriate levels of excitability is referred to as homeostatic plasticity (Davis, 2013; Desai, 2003; Turrigiano, 2011). Homeostatic mechanisms are expressed in the nervous system to maintain an appropriate level of neuronal activity despite ongoing challenges to the network. Homeostatic plasticity is thought to be particularly important during early development, a period characterized by vast challenges to neuronal activity due to increases in cell size, the transition of GABA from excitatory to inhibitory, and changes in ion channel expression (Marder and Goaillard, 2006; Wenner, 2014). Despite these major alterations, neuronal and network function is remarkably stable, and homeostatic mechanisms are thought to be especially important in this process (Marder, 2011; Marder and Goaillard, 2006). The hyperexcitability associated with FXS suggests that homeostatic mechanisms are either incapable of preventing increased excitability or that the loss of FMRP directly impairs homeostatic mechanisms. Recent work suggests that one type of homeostatic plasticity, synaptic scaling, fails in the Fmr1 KO hippocampus (Sarti et al., 2013; Soden and Chen, 2010).
Although less studied, homeostatic regulation of intrinsic membrane excitability may be even more influential in setting network excitability than synaptic scaling (Karmarkar and Buonomano, 2006; Marder, 2011; Wilhelm et al., 2009). This homeostatic intrinsic plasticity (HIP) regulates the expression of voltage-gated ion channels to alter membrane excitability and thereby maintain certain features of neural function such as firing rate (Desai, 2003; Desai et al., 1999; Turrigiano et al., 1995). Interestingly, FMRP has previously been shown to regulate a subset of ion channels, some of which are known to be regulated by HIP (Contractor et al., 2015; Desai et al., 1999; Turrigiano et al., 1995; Wilhelm et al., 2009). In FXS, the loss of FMRP could significantly impair the function of HIP leading to dysregulated excitability levels. We therefore examined the function of HIP in the Fmr1 KO. We hypothesized that HIP was either absent or functioned inappropriately in Fmr1 KO cortical neurons, which contributed to hyperexcitability. In order to test this idea, we used primary cultures of mouse cortical Fmr1 KO neurons and assessed the function of HIP following prolonged activity deprivation. In summary, we find that HIP does not operate appropriately in the KO but that the alterations observed in these neurons depend on the specific cell population. These results show that a fundamental neuronal mechanism implicated in maintaining neural activity levels through development is not working normally in Fmr1 KO cortical neurons.
RESULTS
Baseline Intrinsic Membrane Excitability Is Not Enhanced in Fmr1 KO Primary Cortical Excitatory Neurons
We examined if cultured Fmr1 KO cortical neurons displayed hyperexcitability at baseline by measuring action potential (AP) threshold (the current and voltage necessary to trigger an AP) and firing frequency. We recorded from large, pyramidal-shaped neurons (presumed excitatory) at days in vitro (DIV) 10–15. We found that AP threshold (Figures 1A and 1B), and the number of spikes per current step (shown as “spike number frequency” in Hz) were no different between genotypes (Figure 1C). Furthermore, we measured no differences in passive membrane properties (Table S1). The observation that these electrophysiologically assessed properties were similar in wild-type (WT) and KO cells raised the possibility that hyperexcitability may not emerge in cortical Fmr1 KO neurons until later stages of development.
Figure 1. Fmr1 KO Neurons Do Not Display Differences in Baseline Intrinsic Excitability Levels or Homeostatic Intrinsic Plasticity.
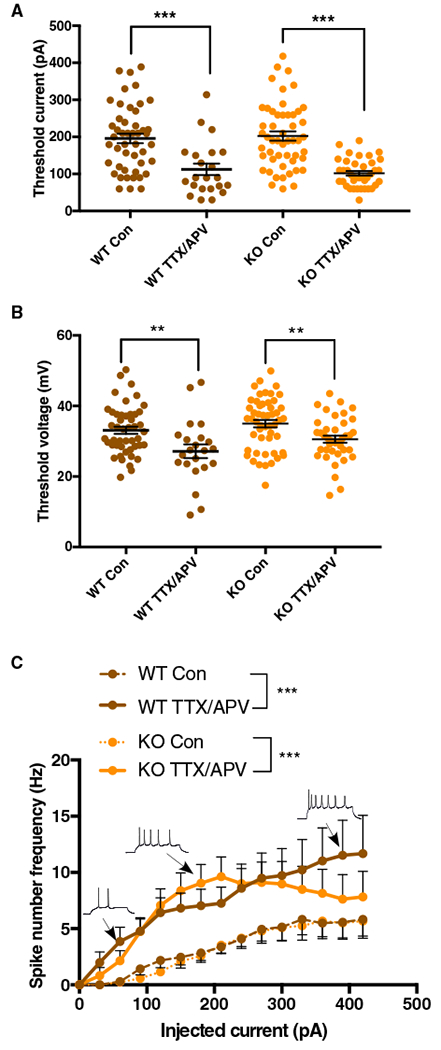
(A and B) Action potential threshold current (A) and voltage (B) in control and TTX/APV conditions (two-way ANOVA [α = 0.05]: threshold current, F[1, 155] = 53.6; Bonferroni: p < 0.001; threshold voltage, F[1, 156] = 18.31; Bonferroni: p < 0.01).
(C) Measurement of spike number frequency in control and TTX/APV conditions (GLM [alpha = 0.05], three-way interaction [coefficient = 0.199, SE = 0.042, Z value = 4.658, p < 0.001)].
Post hoc significance testing was done by first calculating the grand mean of number of spikes for each cell (across all steps) and then comparing those means using the Wilcoxon nonparametric test (alpha = 0.05) (WT: W = 226, p = 0.0001; KO: W = 552, p = 0.0003). Error bars indicate SEM. WT control, n = 47 cells/12 litters; WT TTX/APV, n = 23/5; KO control, n = 51/13; KO TTX/APV, n = 39/10. Con, control. See also Table S1. **p < 0.01 and ***p < 0.001.
Some Features of Homeostatic Intrinsic Plasticity Are Intact in Cortical Fmr1 KO Neurons
Previous studies have shown an absence of synaptic scaling in hippocampal Fmr1 KO neurons following activity deprivation with TTX (Na+ channel blocker) and APV (NMDA receptor blocker) (Sarti et al., 2013; Soden and Chen, 2010). We therefore used this pharmacological approach to test the hypothesis that HIP was altered as well. For purposes of clarity we use the following terminology: WT or KO Con (untreated cortical cultures/control) and WT T/A or KO T/A (cortical cultures treated for 48 h with TTX and APV). As expected, we found that AP threshold was reduced in WT T/A (Figures 1A and 1B), and the number of spikes generated in response to increasing current injections was increased (Figure 1C). Surprisingly, the compensatory adjustments following TTX/APV treatment were no different between WT and Fmr1 KO cultures (Figure 1). These initial findings appear to suggest that HIP is acting in a normal fashion in cortical Fmr1 KO neurons.
A Subset of Cortical Fmr1 KO Neurons Displays Loss of HIP
Although many aspects of excitability appeared to be similar between WT Con and KO Con neurons, we noted that following chronic activity deprivation there were striking differences in the proportions of single-spiking neurons (i.e., cells that spike only once at all steps; Figure 2A) and multi-spiking neurons (i.e., cells that spike more than once during the step protocol; Figure 2A). Approximately 50% of the WT Con neurons exhibited a single-spiking (SS) profile, while almost all WT T/A neurons were multi-spiking (MS; Figure 2A). This conversion from SS neurons to a MS phenotype represents a previously unrecognized mechanism of HIP. Strikingly, this conversion from SS to MS neurons failed to occur in the Fmr1 KO cultures, where the proportion of SS neurons was statistically indistinguishable between KO Con and KO T/A sister cultures (Figure 2A). Although our total n for the WT T/A was lower than other conditions, power analysis demonstrated that our n was sufficient to identify the true proportions of SS and MS neurons (see STAR Methods for power analysis; power = 0.98). Interestingly, although SS Fmr1 KO neurons were unable to convert to the MS phenotype, we did observe a non-significant reduction in the threshold current of KO T/A SS compared with KO Con SS (Figure 2B2), which was due to a significant increase in input resistance (Table S2).
Figure 2. Cortical Fmr1 KO Neurons Display Cell Population-Specific Alterations in Homeostatic Intrinsic Plasticity.
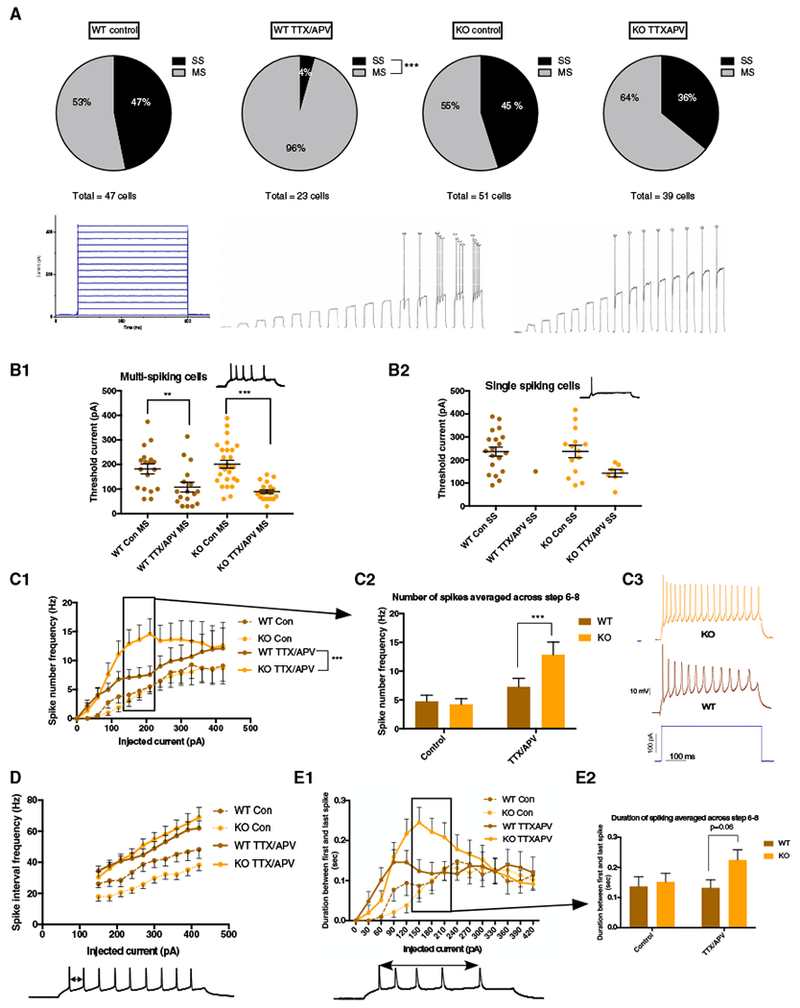
(A) Top: WT neurons significantly reduced the number of SS cells following TTX/APV (proportion Z test [two-tailed, α = 0.05] p < 0.0001), while KO SS cells failed to convert. Bottom: current injection protocol and example traces from multi-spiking and single-spiking neurons across all 15 current steps.
(B1) Reduced threshold current in MS cells following TTX/APV (two-way ANOVA [α = 0.05]: F[1, 97] = 34.84; Bonferroni: WT: p = 0.01, KO: p < 0.0001).
(B2) Trend toward reduced threshold in SS cells following TTTX/APV (two-way ANOVA [α = 0.05]: F[1, 54] = 3.32; Bonferroni: p = 0.07).
(C1 and C2) Spike number frequency in control and TTX/APV MS cells (GLM [α = 0.05], three-way interaction among drug, current, and genotype [coefficient = 0.235, SE = 0.045, Z value = 5.21, p < 0.001)]. Wilcoxon post hoc significance testing was done by combining spike number frequency data from steps 6, 7, and 8 and found significant differences in these current steps between MS WT and KO after TTX/APV (W = 3, 131, p = 0.0007).
(C3) Example traces from TTX/APV-treated WT and KO MS cells.
(D) Spike interval frequency in control and TTX/APV MS cells.
(E1 and E2) TTX/APV KO MS cells display trends toward longer duration of spiking during steps 6–8 compared with WT (two-way ANOVA [α = 0.05]: interaction effect F(1, 77) = 1.56; Bonferroni: p = 0.06).
Error bars indicate SEM. Con, control; MS, multi-spiking; SS, single-spiking. Number of cells in each condition: WT control (MS = 25 cells, SS = 22 cells, 12 litters), WT TTX/APV (MS = 22 cells, SS = 1 cell, 5 litters), KO control (MS = 28 cells, SS = 23 cells, 13 litters), and KO TTX/APV (MS = 25 cells, SS = 14 cells, 10 litters). See also Figure S1 and Tables S1–S3. **p < 0.01 and ***p < 0.001.
A Subset of Cortical Fmr1 KO Neurons Displays Exaggerated HIP
The inappropriate maintenance of a large SS population following TTX/APV in the Fmr1 KO compared with WT cultures significantly affected our analysis of the spike number frequency in response to increasing current injections (Figure 1C). To better compare multi-spiking populations between genotypes, we analyzed the number of spikes generated per step from only MS neurons. Applying this approach, we observed a significantly larger increase of spike number in KO T/A compared with WT T/A through steps 6, 7, and 8 (150–210 pA) (Figures 2C1–2C3). The populations of WT T/A MS cells and KO T/A MS cells appeared to be distinct. KO T/A MS neurons were likely to be derived from cells that were multi-spiking before treatment, as SS cells failed to convert to the MS phenotype. However, the WT T/A MS cells were likely to be derived from cells that were either MS or SS. We considered the possibility that KO T/A MS cells expressed more spiking than in WT T/A MS cells because the SS cells that converted to MS cells in the WT had smaller spike numbers. Several observations argue against such a possibility (Figure S1): (1) we did not see any evidence of a bimodal distribution of firing frequency in WT T/A MS cells (Figure S1A), (2) the proportion of cells firing at ≥ 10Hz on steps 6–8 (150–210 pA) was far larger for KO T/A MS cells compared with WT T/A MS cells (WT T/A MS versus KO T/A MS: step 6, 9% versus 48%; step 7, 14% versus 40%; step 8, 18% versus 40%; Figures S1A and S1B), and finally, (3) we noted that for steps 4–8, the two or three cells with the highest firing rates were always KO T/A MS cells (Figure S1A).
Two results became clear when we measured the spike time interval between the first two spikes at each of the depolarizing steps (shown as “spike interval frequency” in Hz; Figure 2D). First, we noticed that spike interval frequency was lower in KO Con MS cells compared with the WT Con MS cells (Figure 2D). Although this was true for the interval between the first two spikes, overall spike interval frequency was not different between WT and KO controls as the number of spikes (Figure 2C1) and period of spiking (duration between first and last spike; Figure 2E1) were nearly identical. The results contribute to the observation that KO cells were not more excitable than the WT at baseline. Second, although spike number frequency was higher in the KO T/A MS cells compared with the WT T/A MS cells (Figures 2C1–2C3), we did not observe any differences in spike interval frequency in these populations (Figure 2D). By assessing the duration between the first and last spike generated per step, KO T/A MS cells fired more APs than WT T/A MS cells by firing for a longer period rather than at a faster rate (Figures 2E1 and 2E2). However, because of the slower spike interval frequency in KO Con MS cells, the spike interval frequency was increased to a greater extent following TTX/APV treatment in the KO compared with WT multi-spikers, consistent with exaggerated HIP in these cells. Finally, we found a significant reduction in threshold current for both WT T/A MS cells and KO T/A MS cells compared with their untreated counterparts (Figure 2B1).
Homeostatic Synaptic Scaling in Cortical Fmr1 KO Neurons
One of our initial motivations for studying homeostatic plasticity in the Fmr1 KO cortex were the findings that AMPAergic and GABAergic synaptic scaling were absent in the Fmr1 KO hippocampus following TTX/APV treatment (Sarti et al., 2013; Soden and Chen, 2010). However, it remained unknown whether scaling was perturbed in different regions of the Fmr1 KO brain. We therefore tested whether scaling was also impaired in cortical Fmr1 KO neurons. Unexpectedly, we found that AMPAergic synaptic up-scaling was intact in KO T/A cells. We observed a similar increase in the miniature excitatory postsynaptic current (mEPSC) amplitudes (Figure 3A1) and a clear rightward shift in the distribution of mEPSC amplitudes (Figure 3A2). This shift in the distribution of both WT T/A and KO T/A mEPSC amplitudes exhibited a multiplicative relationship to untreated control amplitudes, consistent with synaptic scaling (see STAR Methods; Figures 3A2 and S2).
Figure 3. Synaptic Scaling in Excitatory Cortical Fmr1 KO Neurons.
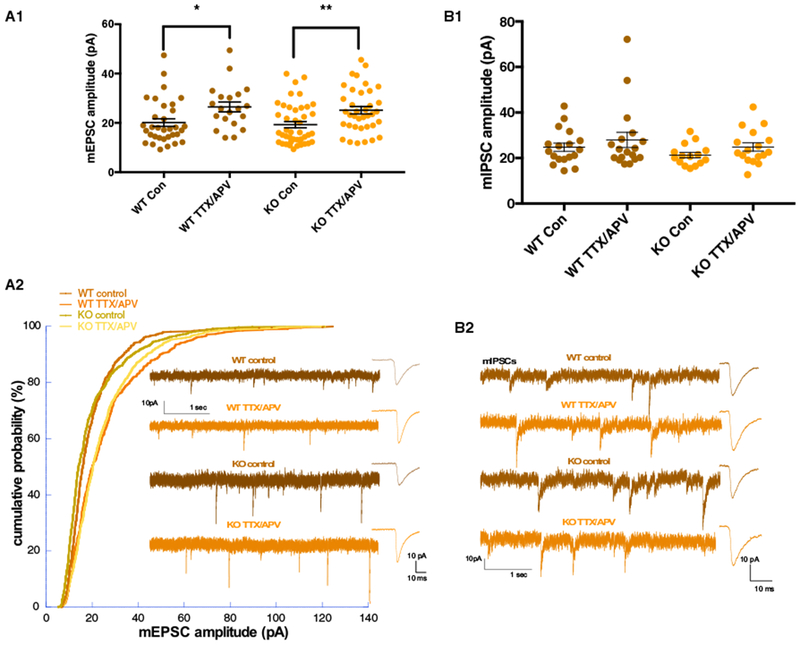
(A1) mEPSC amplitude in control and TTX/APV conditions (two-way ANOVA [α = 0.05]: F(1, 29) = 0.027; Bonferroni: WT control versus TTX/APV: p = 0.023, KO control versus TTX/APV: p = 0.007). WT control n = 33 cells, 13 litters; WT TTX/APV n = 20 cells, 8 litters; KO control n = 43 cells, 15 litters; and KO TTX/APV n = 37 cells, 11 litters.
(A2) After TTX/APV, both WT and KO cells Kolmogorov-Smirnov test shows a significant shift in their distribution compared with control conditions. After downscaling (WT scaled by 0.82, KO scaled by 0.75), the treated conditions were not significantly different from control. WT and KO TTX/APV distributions are not different.
(B1 and B2) No differences were observed in mIPSC amplitudes before or after treatment in either genotype (two-way ANOVA [α = 0.05]). WT control n = 18 cells, 6 litters; WT TTX/APV n = 18 cells, 5 litters; KO control n = 15 cells, 5 litters; and KO TTX/APV n = 17 cells, 4 litters.
Error bars indicate SEM. Con, control. See also Figures S2 and S3. *p < 0.05 and **p < 0.01.
TTX/APV treatment did not trigger GABAergic down-scaling in cortical WT or KO neurons. There were no differences in miniature inhibitory postsynaptic current (mIPSC) amplitudes among any of the four conditions (Figures 3B1 and 3B2). We also found no differences in mEPSC or mIPSC frequency (Figure S3).
Thus, homeostatic synaptic scaling was no different in WT and Fmr1 KO cortical neurons and therefore would be unlikely to contribute to cortical hyperexcitability. These findings further focus our attention on the loss and gain of HIP function as a mechanism underlying cortical hyperexcitability.
Reduction of AMPA Receptor-Mediated Transmission Also Triggers Altered HIP in Cortical Fmr1 KO Neurons
In order to test HIP function in WT and KO cultures in response to a distinct and less severe activity perturbation, we treated cultures with the AMPA receptor blocker NBQX. We found that NBQX treatment produced a non-significant trend toward a reduction in the proportion of SS neurons in WT cultures, but this reduction was less apparent in Fmr1 KO cultures (Figure 4A). Furthermore, chronic AMPAR blockade led to a significant increase in spike interval frequency of MS KO neurons that was absent in the WT (Figures 4B1 and 4B2). Specifically, Fmr1 KO neurons fired a second AP more quickly. We did not observe an obvious difference in spike number frequency between genotypes after NBQX, although the KO cells did appear to have a stronger increase than WT cells in the early steps (Figure 4C). In short, we found that Fmr1 KO neurons displayed functionally similar alterations in HIP function after either NBQX or TTX/APV treatment, but the phenotypic changes of NBQX were less pronounced than TTX/APV, and in some cases qualitatively distinct. Surprisingly, NBQX did not trigger reductions in threshold current or threshold voltage (Figures S4A and S4B), did not increase mEPSC amplitude or frequency (Figures S4C and S4D), and also did not change mIPSC amplitude or frequency (Figures S4E and S4F). Overall, these results demonstrate that altered HIP function is expressed in response to multiple pharmacological types of activity deprivation.
Figure 4. Reduction of AMPA Receptor-Mediated Transmission Also Triggers Altered HIP in Cortical Fmr1 KO Neurons.
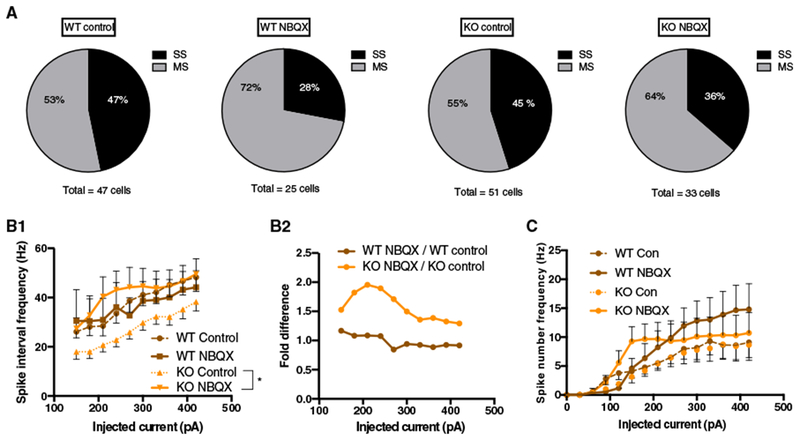
(A) NBQX triggered a non-significant trend toward a conversion from SS to MS phenotype in WT (proportion Z test [two tailed, α = 0.05]: p = 0.17).
(B1) Spike interval frequency in control and NBQX MS cells (GLM [α = 0.05], two-way interaction between drug and genotype [beta = 22.7, SE = 10.64, df = 115, t = 2.135, p = 0.035]).
(B2) MS Fmr1 KO neurons display a greater fold increase in the spike interval frequency.
(C) Spike number frequency in control and NBQX MS conditions.
Error bars indicate SEM. Note that values for WT control and KO control are the same as plotted in Figure 1. Con, control; MS, multi-spiking; SS, single-spiking. Number of cells in each condition: WT control (MS = 25 cells, SS = 22 cells, 12 litters), WT NBQX (MS = 18 cells, SS = 7 cells, 6 litters), KO control (MS = 28 cells, SS = 23 cells, 13 litters), KO NBQX (MS = 21 cells, SS = 12 cells, 7 litters). See also Figure S4 and Tables S1–S3. *p < 0.05.
Activity Perturbation Alters AP Parameters
Previous work has demonstrated that specific ion channel conductances mediate distinct components of the AP waveform (Figure 5A) (Bean, 2007). Thus, to estimate how activity perturbation (TTX/APV or NBQX) may have triggered different changes in ion channel populations, a comprehensive analysis of AP parameters was conducted. We start with the first AP generated during the first step that exhibited an AP (called “threshold AP”; Figure 5A) and then assess the first APs generated during the current steps expressing the overcompensation in KO T/A MS cells (steps 6–8, i.e., 150–210 pA).
Figure 5. Activity Perturbation Triggers Different Changes in Action Potential Parameters Depending on Cell Population, Treatment, and Genotype.
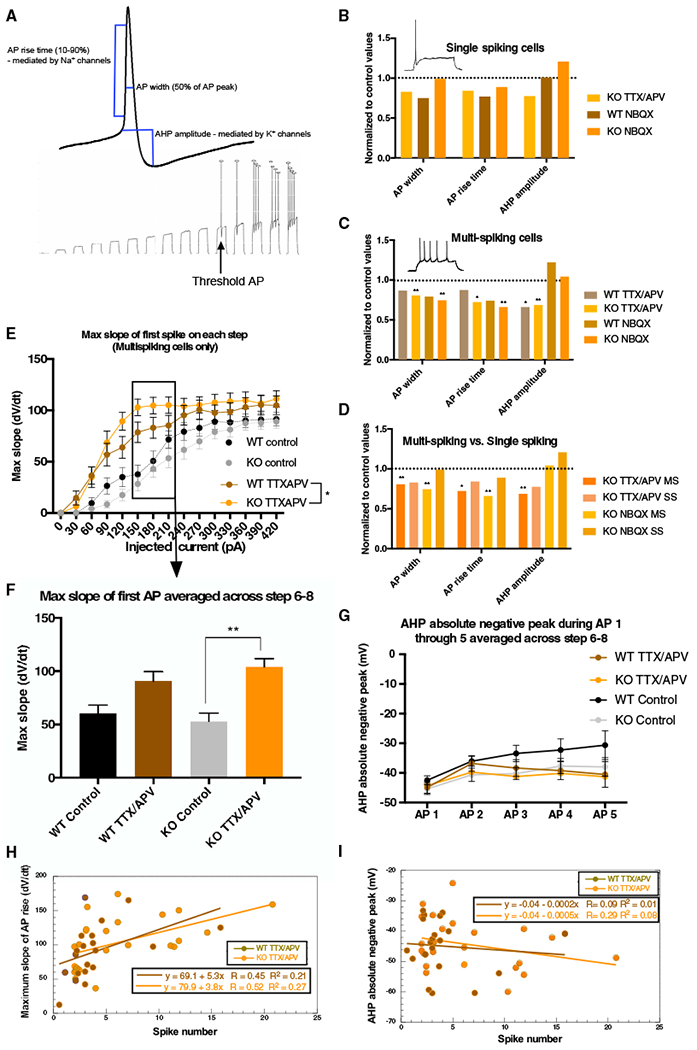
(A) Top: model of measured AP parameters; bottom: “AP threshold” example trace.
(B) Treated non-converting KO SS neurons do not significantly reduce AP parameters.
(C) Treated MS KO neurons show larger changes in AP parameters. NBQX and TTX/APV trigger reductions in AP width and rise time but only in the MS KO (AP width: F[2, 123] = 8.06, KO TTX/APV p = 0.006, WT TTX/APV p = 0.39, KO NBQX p = 0.0057, WT NBQX p = 0.084; rise time: F[2, 123] = 7.45, TTX/APV p = 0.026, WT TTX/APV p = 0.92, KO NBQX p = 0.005, WT NBQX p = 0.14). MS WT and KO reduced AHP amplitude after TTX/APV (F[2, 118] = 16.33, KO TTX/APV p = 0.0006, WT p = 0.012).
(D) TTX/APV treated MS KO neurons display larger changes in AP parameters than treated, non-converting KO SS cells.
(E and F) TTX/APV triggers increased maximum slope of the AP rising phase in both WT MS and KO MS cells during current steps 6–8 (E), but the increase only reaches significance in the KO MS cells following treatment (F) (treatment effect: F[1, 83] = 22.58, p = 0.006).
(G) AHP absolute negative peak in control and TTX/APV MS cells at steps 6–8.
(H and I) Maximum slope (H), but not (I) AHP amplitude, correlates with spike number frequency in both WT and KO MS cells after TTX/APV (Pearson correlation test [α = 0.05], maximum slope versus spike number frequency: KO TTX/APV: correlation coefficient = 0.51, p = 0.009; WT TTX/APV: correlation coefficient = 0.46, p = 0.04).
All statistical tests for AP parameters are two-way ANOVA (α = 0.05) with Bonferroni. Error bars indicate SEM. MS, multi-spiking; SS, single-spiking. Number of cells in each condition: WT control (MS = 25 cells, SS = 22 cells, 12 litters), WT TTX/APV (MS = 22 cells, SS = 1 cell, 5 litters), WT NBQX (MS = 18 cells, SS = 7 cells, 6 litters), KO control (MS = 28 cells, SS = 23 cells, 13 litters), KO TTX/APV (MS = 25 cells, SS = 14 cells, 10 litters), KO NBQX (MS = 21 cells, SS = 12 cells, 7 litters). See also Figures S5 and S6 and Tables S2 and S3. *p < 0.05 and **p < 0.01.
Interestingly, we found that KO Con APs had significant increases in rise time and afterhyperpolarization (AHP) amplitude compared with WT Con APs, consistent with the idea that loss of FMRP alters the function of several ion channel populations (Figure S5). In contrast, we observed no differences between single-spiking or multi-spiking cells in threshold AP parameters (Figure S5) or passive properties (Tables S2 and S3). Thus, despite significant differences in their functional output (firing single versus multiple spikes), these cell populations appear otherwise similar in AP waveform.
As described below, differences in AP parameters were observed in MS neurons in treated and control conditions. We considered the possibility that the failure of HIP to convert KO SS neurons into MS neurons would be associated with weak or no changes in AP parameters. Consistent with this idea, following treatment (TTX/APV or NBQX), KO SS neurons did not show significant changes in any of the parameters of the threshold AP compared with KO Con SS cells, although trends were observed (Figures 5B and S6B). Furthermore, these changes were less prominent in SS compared with MS KO cells (Figure 5D). The results are consistent with impaired regulation of ion channel function in non-converting Fmr1 KO cells.
In the case of exaggerated HIP in which KO T/A MS cells had greater increases in spike number frequency compared with WT T/A MS cells, we hypothesized that we would observe larger changes in AP parameters (i.e., ion channel conductances) in KO T/A MS neurons. Indeed, we found that KO T/A MS cells displayed significantly larger treatment-dependent reductions in threshold AP width and rise time than WT T/A MS cells (Figures 5C and S6A). Although AHP amplitude of the threshold AP was significantly reduced in treated KO MS cells, it was also reduced in treated WT MS cells (Figures 5C and S6A). These results suggest that Na+ channels rather than K+ channels might be involved in the expression of exaggerated HIP. Consistent with the idea that Na+ channels were important in the overcompensation of spike number frequency in steps 6–8 (Figure 2C1), we observed an overcompensation of the maximum slope in these same steps for KO T/A MS cells compared with WT T/A MS cells (Figures 5E and 5F). On the other hand, when we measured the AHP amplitude of the first five APs (averaged across step 6–8), we saw no differences across genotypes or conditions in the MS cells at any point in the spike train (Figure 5G). This result suggests that reduction in AHP amplitude, and thus K+-channel conductance associated with the AP, is not critical in driving the overcompensation.
Next, we correlated slope of the rising phase of the AP or AHP amplitude with spike number frequency in each cell. Interestingly, when we correlated maximum slope with spike number frequency for steps 6–8, we observed significant correlations for both WT T/A MS cells and KO T/A MS cells, consistent with the idea that rise time, and presumably Na+ channels, are associated with determining the number of spikes a cell generates (Figure 5H). In contrast, when we correlated AHP amplitude (steps 6–8), we did not observe correlations with spike number frequency (Figure 5I).
In summary, our results suggest that (1) Fmr1 KO MS cells display larger changes in Na+ channel function and that this difference likely contributes to an exaggerated HIP, and, conversely, (2) SS Fmr1 KO neurons display reduced changes in AP parameters and thus underlying ion channel conductances. Together, the observed changes in AP waveform provide indications of some of the channels that could be involved in the loss and gain of homeostatic regulation observed in the KO.
DISCUSSION
Compensatory Shift in WT Neurons from Single- to Multi-spiking Phenotype Fails in the Fmr1 KO
Several previous studies have shown that activity perturbations can trigger changes in ion channel conductances that lead to homeostatic compensations in some aspect of membrane excitability (Desai et al., 1999; Turrigiano et al., 1994; Wilhelm et al., 2009). Here we describe a distinct form of HIP in which chronic activity perturbation converts single-spiking neurons into multi-spiking neurons, which should elevate the excitability of the network. Strikingly, this conversion fails to occur in Fmr1 KO neurons.
What are the mechanisms underlying this conversion in the WT, and why might it fail in the KO? WT SS cells convert to a multi-spiking phenotype through what must be a change in channel conductances that favor multiple spike generation, while these changes would not be replicated in the KO. Although SS Fmr1 KO neurons fail to convert to MS neurons, they do show some compensatory trends such as reductions in threshold (Figure 2B2), rise time, and AHP amplitude (Figure 5B), but these changes may not be sufficient for conversion. Alternatively, the channels mediating the conversion could be separate from those shaping the AP waveform. Although we do not currently know which channels are involved in the conversion from single- to multi-spiking neurons, potential candidates include the delayed rectifier K+ channels, slow K+ conductance (M-current), and Na+ persistent inward currents (NaPICs). Each of these conductances have been shown to shape firing pattern (Bean, 2007). In these scenarios, KO SS neurons would fail to alter these channels sufficiently, thus preventing the conversion to a MS phenotype. Importantly, FMRP has been shown to regulate or associate with the delayed rectifier potassium channel Kv1.1 and slow-activating potassium channel Kv7.2, and the NaPIC (Contractor et al., 2015; Darnell et al., 2011; Deng and Klyachko, 2016; Kalmbach et al., 2015). In addition, Kv1.1 and the slow-activating K+ channel kv7.2 have also recently been shown to be targets of HIP (Kuba et al., 2015; Lee et al., 2015). Overall our results suggest that FMRP is necessary for the WT conversion from SS to MS neurons.
Compensatory Increase in the Firing Frequency of WT Multi-spiking Cells Is Exaggerated in the Fmr1 KO
Following activity deprivation, MS WT cells increase their overall excitability, and this is associated with a significant reduction in the AHP amplitude as well as trends toward reduced AP rise time and AP width (Figures 5C and S6A), suggesting that alterations in Na+ and K+ conductances could facilitate increased excitability. Interestingly, treated MS Fmr1 KO neurons increase their spike number frequency to an even greater extent, and this was associated with a significant increase in the maximum slope of the rising phase of the AP during the steps when we observed the overcompensation (Figures 5E and 5F). Moreover, rise time, but not AHP amplitude, correlated with spike number frequency (Figures 5H and 5I), suggesting that treatment-induced changes in Na+ channels could contribute to the increase in spike number frequency and that the changes in Na+ channel function were greater in the KO. Previous literature has shown that FMRP interacts with Nav1.2 and Nav 1.6 at the level of the mRNA (Darnell et al., 2011), and both transient and persistent Na+ conductances are increased in mature cortical Fmr1 KO neurons (Deng and Klyachko, 2016; Routh et al., 2017). One possibility is that HIP could trigger increased Na+ channel function in KO MS neurons because of a loss of FMRP translation suppression. This exaggerated plasticity is reminiscent of the overactive mGluR pathway mediating LTD in the Fmr1 KO hippocampus (Huber et al., 2002; Osterweil et al., 2010). Alternatively, impairments in HIP may be attributed to loss of FMRP’s translation-independent interactions (Contractor et al., 2015). Our result introduces a potentially novel mechanism whereby an impaired homeostatic intrinsic plasticity drives alterations in ion channel function that lead to the development of cortical hyperexcitability in the Fmr1 KO.
Development of Hyperexcitability in Cortical Fmr1 KO Neurons
Several studies have demonstrated various measures of hyperexcitability in mature cortical Fmr1 KO neurons (Gibson et al., 2008; Gonçalves et al., 2013; Gross et al., 2015; Zhang et al., 2014), which have been linked to the loss of FMRP regulation of ion channels at the level of the mRNA (Gross et al., 2011; Lee et al., 2011; Strumbos et al., 2010) or via direct protein-protein interactions (Brown et al., 2010; Deng et al., 2013). Recent reports have demonstrated a gradual development of hyperexcitability in the Fmr1 KO rat visual cortex in vivo and in FXS patient-derived human induced pluripotent stem cells in vitro (Berzhanskaya et al., 2016; Liu et al., 2018). These results are consistent with the observation that symptoms associated with cortical hyperexcitability (seizures, sensory hypersensitivity) do not emerge until a few years into FXS development (Hagerman et al., 2017). These findings support the idea that hyperexcitability is not caused by the changes in ion channel function that are directly produced by the loss of FMRP but rather indirect compensations to these initial changes (Brager et al., 2012). Previous studies have shown in WT neurons that altering the function of a particular ion channel leads to compensatory changes in the function of other channel types (MacLean et al., 2005; Pratt and Aizenman, 2007).
Our findings, in what are likely immature cortical cultures (derived from ~P1 cortices) support the idea that KO neurons compensate for the direct effects of FMRP loss early in development. First, there was no sign of baseline hyperexcitability in Fmr1 KO neurons (Figure 1), despite the observation that certain AP parameters were significantly altered, suggesting changes in ion channel function (Figure S5). Second, we found that certain features of HIP were intact as chronic activity blockade produced similar compensatory changes in AP threshold in WT and KO cultures (Figures 1A, 1B, 2B1, and 2B2). Finally, differences in WT Con and KO Con spike interval frequency (Figure 2D) and AP rise time and AHP amplitude (Figures S5B and S5C) suggested that if anything the KO cells were hypoexcitable. This could represent a compensation to higher activity levels that occur at an even earlier stage in the KO cultures.
If various compensations in early development of the Fmr1 KO network are intact, then how does hyperexcitability emerge later in development? Homeostatic mechanisms will be particularly important in defining the mature constellation of ion channels and synaptic receptors because this plasticity regulates their expression through development in order to maintain some aspect of neuronal function (Marder and Goaillard, 2006; O’Leary et al., 2014). Recent work has demonstrated that virtually identical activity patterns can be achieved through highly distinct combinations of ion channel and receptor combinations (parameter solutions) (Marder and Goaillard, 2006; Prinz et al., 2004). In the Fmr1 KO, the constellation of ion channels and receptors that are homeostatically regulated will have been altered by the loss of FMRP. The loss of FMRP could lead to parameter solutions that maintain most aspects of excitability at early developmental stages. However, as the network is repeatedly challenged through the natural process of development, or experimentally by activity blockade (TTX/APV or NBQX), HIP may drive the cells into parameter solutions that are less successful in maintaining appropriate excitability. If continued developmental challenges lead to a subset of neurons that express very high firing frequencies, as occured following activity blockade, we might expect that this would contribute to the emergence of hyperexcitability of the Fmr1 KO cortical network. As development progresses, HIP may not be able to prevent hyperexcitability, because of developmental challenges that are out of the range of HIP adjustments or because homeostatic capacity is reduced in later development.
Region-Specific Differences in Homeostatic Impairments
Previous work has suggested that the loss of FMRP is complicated in that loss of this protein produces distinct deficits in a very specific fashion depending on the tissue and cell type (Contractor et al., 2015). Earlier work has shown that the Fmr1 KO can exhibit cell type-specific changes that can even be found among different cells within the same layer of cortex (Kalmbach et al., 2015). Our results are consistent with this complexity in that two different cell populations display opposing alterations in HIP function.
We have also made the surprising finding that cortical Fmr1 KO neurons show intact AMPAergic up-scaling (Figure 3). This result is consistent with region-specific differences because previous reports suggest that scaling is impaired in hippocampal Fmr1 KO neurons (Sarti et al., 2013; Soden and Chen, 2010). In fact, region-specific deficits in plasticity mechanisms have been reported previously in the Fmr1 KO (Brager et al., 2012; Zhang et al., 2014). Alternatively, it is possible that scaling deficits are strain specific (Dobkin et al., 2000; Ranson et al., 2012). Although we used Fmr1 KO mice bred on the C57BL/6 background, Lu Chen’s group used the FVB strain (Soden and Chen, 2010). Moving forward, it will be important to recognize that plasticity mechanisms in FXS may be expressed differently in different cell types or regions of the nervous system.
Conclusions
We have shown that homeostatic intrinsic plasticity, a fundamental neuronal mechanism driving nervous system development, does not operate normally in Fmr1 KO cortical neurons. We discovered a form of HIP in single-spiking WT neurons and find that this plasticity was lost in the Fmr1 KO. Conversely, Fmr1 KO multi-spiking neurons displayed an exaggerated form of HIP. Our findings that HIP is altered in the KO distinctly in different cells could provide an explanation for subtle or contradictory results that have been reported. This work motivates further studies to investigate how loss of FMRP leads to impaired HIP, and how altered HIP function may contribute to the emergence of cortical hyperexcitability in FXS. Future studies on underlying mechanisms of alterations in HIP could provide a platform for understanding the development of hyperexcitability and could suggest therapeutic strategies for manipulating HIP’s contribution to the development of hyperactive networks.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Wenner (pwenner@emory.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
FMR1HET females (backcrossed on C57BL6 background) were crossed with WT C57BL6 males (Jackson Laboratory) to generate litters of pups with mixed genotypes (Fmr1 KO, Fmr1HET or wild-type (WT)). Thus, for all experiments, Fmr1 KO male pups were compared to their WT littermate control. We performed PCR to identify genotypes as described previously (Gross et al., 2015; Muddashetty et al., 2007) on postnatal day 0 – 2 (P0-P2). The mice were housed in a 12 hr light/dark cycle and the animal protocol was approved by the Institutional Animal Care and Use Committees at Emory University.
Primary cortical neuronal cultures
Cerebral cortices were dissected and cultured from genotyped WT and Fmr1 KO pups on P0-P3. The cortices were enzymatically dissociated using trypsin (Thermo Fisher Scientific; 10 min), mechanically dissociated in Minimum Essential Media (MEM; Fisher) supplemented with 10% Fetal Bovine Serum (FBS; Hyclone) and stained to assess viability using Trypan Blue (Sigma). The neurons were plated on coverslips (Matsunami Inc., 22mm) coated with FBS, poly-l-lysine (Sigma) and laminin (Sigma). A total of 50,000 neurons were plated as a ‘spot’ on the center of the coverslip to create a small, high density network. The neurons were cultured in standard growth medium (glial conditioned neurobasal (Fisher) supplemented with glutamax (GIBCO) and B27 (Invitrogen)), and half of the media was exchanged 2-3 times a week until experimental treatments began. No antibiotics or antimycotics were used. The cultures were maintained in an incubator regulated at 37 C, 5% CO2 and 95% relative humidity (culturing protocol is similar to (Fong et al., 2015)).
METHOD DETAILS
Pharmacology and whole cell recordings
Drugs were used in the following concentrations (in μM): TTX, 1 (Tocris); APV, 100 (Tocris); NBQX, 10 (Tocris). Drugs were added to fresh standard growth medium and added to the cultures by a complete media change. Control cultures had a simultaneous complete media change but without drugs. Cultures were randomly assigned to each treatment group (control, TTX/APV, NBQX). Whole cell recordings were performed on all groups (control, TTX/APV, NBQX) on the same day for each genotype.
Large, pyramidal shaped neurons were selected for whole cell recordings during a continuous perfusion of artificial cerebrospinal fluid (aCSF) containing (in mM): 126 NaCl, 3 KCl, 2 CaCl2, 1.5 MgSO4, 1 NaH2PO4, 25 HEPES, and 25 D-glucose (pH 7.4) (Fong et al., 2015). For intrinsic excitability and AMPAergic mEPSC recordings, the internal solution contained the following (in mM): 94 K-gluconate, 10 NaCl, 36 KCl, 1.1 EGTA, 10 HEPES, 1 MgCl, 0.1 CaCl, 1 ATP-salt, 0.5 GTP (pH 7.3). To isolate AMPAergic mEPSCs the external solution was supplemented with 1 μM TTX, 20 μM gabazine and 100 μM APV, and the recordings were done in voltage-clamp at −70 mV. For GABAergic mIPSCs, recordings the internal solution contained the following (in mM): 136 CsCl, 1.1 EGTA, 10 HEPES, 0.1 CaCl, 0.3 MgCl, 3 Mg-ATP, 0.5 Na-GTP (pH 7.3). To isolate GABAergic mIPSCs the external solution was supplemented with 1 μM TTX, 20 μM NBQX and 100 μM APV, and the recordings were done in voltage-clamp at −70 mV. mPSCs were recorded using an AxoClamp 2B amplifier. Pipette resistances ranged from 7-12 MΩ. All data was collected at 5, 10 or 20 kHz sampling rate and analyzed blind to genotype and treatment using Axograph Software. The mPSCs with amplitudes less than 5 pA and cells with less than 20 mPSCs were excluded from analysis. Intrinsic excitability measurements were done in a drug-free external solution in current clamp. A15 current step (500 ms long injection; 0-420 pA) protocol with 30 pA step-increments was used to assess AP threshold and firing frequency. The current injection protocol was repeated 3 times for each cell, and the average value was used for analysis. Cells with a resting membrane potential less than −55 mV were excluded from analysis. For intrinsic excitability experiments, we calculated the number of APs generated during the current pulse, and measured spike interval frequency as 1 s divided by the interval between the onset of the 1st and 2nd AP at each current step. For spike time interval, we did not analyze steps if any of the conditions had an n below 7 (led to exclusion of current step 1-5 in all conditions (control, TTX/APV and NBQX)). All analysis was done blind to genotype and treatment, and the cultures were assigned randomly to treatment conditions.
AP parameter analysis
Analysis of “threshold AP” (AP) waveform parameters was carried out offline using Axograph software. Analysis was done blind to genotype and treatment. We performed the analysis of the first spike generated during the current step protocol and measured the following parameters: AP peak (measured from AP threshold to peak), AP rise time (10 to 90% of AP peak), AP width (measured at 50% of AP peak), after hyperpolarization (AHP) amplitude (the deepest negative peak immediately following the AP peak compared to the threshold voltage). Measurement of maximum slope of the AP rising phase across all steps was carried out in MATLAB using custom scripts to identify the maximum change in voltage over time (dV/dt). Measurement of AHP amplitudes of spike trains during step 6-8 was carried out in MATLAB using in-house constructed programs to identify the maximum negative peak following the peak of the AP (measured in voltage). Duration of spiking was measured in MATLAB using custom scripts which identified the time between the first and last AP generated during each step.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance tests were done in GraphPad Prism v7, XLSTAT or RStudio. All data are expressed as mean ± SEM. Ns are reported in the figure legends or on the figures. All statistical information (incl. tolerated alpha error, statistical test and p value (adjusted for multiple comparisons where applicable)) is reported in the figure legends.
mEPSC and mIPSC amplitudes were compared in Prism using 2-way ANOVA (alpha = 0.05) with post hoc multiple comparisons tests with Bonferroni corrections. mEPSC amplitude distributions were compared using the Kolmogorov-Smirnov test (XLSTAT) with alpha = 0.05.
Threshold current and threshold voltage datasets were compared in Prism using 2-way ANOVA (alpha = 0.05) with post hoc multiple comparison tests with Bonferroni corrections.
Spike number frequency (Figures 1C and 2C1) and spike interval frequency (Figure 4B1) datasets were fit using a generalized linear mixed model holding drug (control and treatment (either TTX/APV or NBQX)) and genotype (WT and KO) as fixed effects, with current steps as a random effect (glmer (Figures 1C and 2C) or lmer (Figure 4B), lme4 package v1.1-18-1, R vs3.5.1). We used a poisson distribution to fit the spike number frequency datasets (Figures 1C and 2C) and a Gaussian distribution to fit the spike interval frequency dataset (Figure 4B). For Figures 1C and 2C we performed post hoc significance using Wilcoxon nonparametric test (alpha = 0.05). Since the difference in spike number frequency in Figure 2C was clearly restricted to a small current range, we decided to focus on the steps that showed the largest fold difference in spike number (step 6, 7, 8) for any further comparisons. Thus, for our post hoc test of spike number frequency we averaged spike number frequency across step 6, 7, 8 and compared this combined data with a Wilcoxon test.
The proportions of single versus multi-spiking cells were compared using z-test for two proportions (two-tailed) (XLSTAT) with alpha = 0.05. We determined the power of our proportion results (Figure 2A only) by performing a custom Monte Carlo simulation in R (alpha = 0.05, two-tailed). We created a random distribution of numbers based on our observed proportion of WT TTX/APV SS cells (= 0.043) and total N of the WT TTX/APV group (= 23) and simulated that dataset 1000×. For each of the simulated datasets, we ran a proportion test to test whether there was a significant difference in the proportions generated by the simulation. Finally, we calculated the fraction of simulated datasets with a p value < 0.05 to calculate the power of our experiment. With this Monte Carlo simulation and proportion analysis we achieved a power of 0.98.
To compare duration of spiking (Figures 2E1 and 2E2) we averaged duration of spiking across the steps we hypothesized would show significant differences (step 6, 7, 8) due to our observation of exaggeration of HIP during these steps. We performed the duration of spiking comparison in Prism using 2-way ANOVA (alpha = 0.05) with post hoc multiple comparisons using Bonferroni corrections as the data was continuous.
“Threshold AP” waveform parameters were analyzed in Prism using 2-way ANOVA (alpha = 0.05) with post hoc multiple comparisons using Bonferroni corrections. To compare the maximum slope of the AP rising phase we averaged data across step 6, 7, 8 as these were the steps where we hypothesized we would identify a difference due to our observation of exaggerated HIP during these steps. We performed the max slope comparison in Prism using 2-way ANOVA (alpha = 0.05) with post hoc multiple comparisons using Bonferroni corrections, as data was continuous.
The significance of the correlations of max slope and spike number frequency were tested in Rstudio using Pearson’s test (cor.test, ggpubr package version 0.2, Rvs3.5.1)
DATA AND SOFTWARE AVAILABILITY
MATLAB custom script for measuring max slope of AP rising phase, AHP absolute negative peak, and duration of spiking, as well as R custom script for Monte Carlo simulation can be assessed though links provided in the Key Resource Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tetrodotoxin (TTX) | Tocris | Cat#: 1078 |
| D-(-)-2-Amino-5-phosphonopentanoic acid (APV) | Tocris | Cat#: 0106 |
| 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) | Tocris | Cat#: 0373 |
| 6-Imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide (Gabazine) | Tocris | Cat#: 1262 |
| Experimental Models: Organisms/Strains | ||
| Experimental mouse: Fmr1 KO C57BL/6 | Bred in house | N/A |
| Experimental mouse: WT C57BL/6 | Bred in house | N/A |
| Breeder mouse: Fmr1 het C57BL/6 female | Bred in house | N/A |
| Breeder mouse: WT C57BL/6 male | Jackson Laboratory | N/A |
| Software and Algorithms | ||
| Prism - 2-way ANOVAs | GraphPad Software | Version 7 |
| MATLAB – measurement of max slope of AP rising phase, AHP negative peak, and duration of spiking | MathWorks | Version R2017b https://github.com/pernillebuelow/Scripts/blob/master/Pernille_CC_Step_Analysis.m |
| RStudio – generalized linear mixed models, Wilcoxon post hoc significance tests, correlation statistics, Monte Carlo simulation of proportions, power analyses | RStudio | Version 1.1.463 https://github.com/pernillebuelow/Scripts/blob/master/Proportions_R_script.R |
| Axograph – measurement and analysis of all intrinsic excitability and synaptic transmission data, measurement of all “threshold AP” waveform parameters | Axograph X | Version 1.7.0 |
| Excel – proportion statistics (z-test), KS tests | Microsoft | Version 16.18 |
Supplementary Material
Highlights.
Activity block normally drives conversion of single- to multi-spiking neurons
Fmr1 knockout (KO) neurons fail to convert single- to multi-spiking neurons
Multi-spiking Fmr1 KO neurons display exaggerated compensatory increase in spiking
Altered homeostatic intrinsic plasticity may contribute to hyperexcitability in FXS
ACKNOWLEDGMENTS
We sincerely thank Drs. Ming-fai Fong, Nina Gross, and Mark Rich for their comments on the manuscript and Dr. Robert Liu for insightful suggestions on the study. This work was supported by grants R01MH109026 from the National Institute of Mental Health (G.J.B.) and R01NS065992 from the National Institute of Neurological Disorders and Stroke (P.W.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.01.035.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bassell GJ, and Warren ST (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP (2007). The action potential in mammalian central neurons. Nat. Rev. Neurosci 8, 451–465. [DOI] [PubMed] [Google Scholar]
- Berzhanskaya J, Phillips MA, Shen J, and Colonnese MT (2016). Sensory hypo-excitability in a rat model of fetal development in fragile X syndrome. Sci. Rep 6, 30769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH, Akhavan AR, and Johnston D (2012). Impaired dendritic expression and plasticity of h-channels in thefmrl (-/y) mouse model offragile X syndrome. Cell Rep 1, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D, and Kaczmarek LK (2010). Fragile X mental retardation protein controls gating ofthe sodium-activated potassium channel Slack. Nat. Neurosci 13, 819–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, and Portera-Cailliau C (2015). Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW (2013). Homeostatic signaling and the stabilization of neural function. Neuron 80, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, and Klyachko VA (2016). Increased persistent sodium current causes neuronal hyperexcitability in the entorhinal cortex of Fmr1 knockout mice. Cell Rep 16, 3157–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS, and Klyachko VA (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS (2003). Homeostatic plasticity in the CNS: synaptic and intrinsic forms. J. Physiol. Paris 97, 391–402. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, and Turrigiano GG (1999). Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat. Neurosci 2, 515–520. [DOI] [PubMed] [Google Scholar]
- Dobkin C, Rabe A, Dumas R, El Idrissi A, Haubenstock H, and Brown WT (2000). Fmr1 knockout mouse has a distinctive strain-specific learning impairment. Neuroscience 100, 423–429. [DOI] [PubMed] [Google Scholar]
- Fong MF, Newman JP, Potter SM, and Wenner P (2015). Upward synaptic scaling is dependent on neurotransmission rather than spiking. Nat. Commun 6, 6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA, and Huber KM (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model offragileXsyndrome. J. Neurophysiol 100, 2615–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves JT, Anstey JE, Golshani P, and Portera-Cailliau C (2013). Circuit level defects in the developing neocortex of Fragile X mice. Nat. Neurosci 16, 903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Yao X, Pong DL, Jeromin A, and Bassell GJ (2011). Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J. Neurosci 31, 5693–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Chang CW, Kelly SM, Bhattacharya A, McBride SM, Danielson SW, Jiang MQ, Chan CB, Ye K, Gibson JR, et al. (2015). Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep 11, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB Jr., Moine H, Kooy RF, Tassone F, Gantois I, Sonenberg N, Mandel JL, and Hagerman PJ (2017). Fragile X syndrome. Nat. Rev. Dis. Primers 3, 17065. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, and Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U S A99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach BE, Johnston D, and Brager DH (2015). Cell-type specific channelopathies in the prefrontal cortex of the fmr1-/y mouse model of fragile X syndrome. eNeuro 2, ENEURO.0114-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmarkar UR, and Buonomano DV (2006). Different forms of homeostatic plasticity are engaged with distinct temporal profiles. Eur. J. Neurosci 23, 1575–1584. [DOI] [PubMed] [Google Scholar]
- Kuba H, Yamada R, Ishiguro G, and Adachi R (2015). Redistribution of Kv1 and Kv7 enhances neuronal excitability during structural axon initial segment plasticity. Nat. Commun 6, 8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Ge WP, Huang W, He Y, Wang GX, Rowson-Baldwin A, Smith SJ, Jan YN, and Jan LY (2011). Bidirectional regulation ofdendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 72, 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Royston SE, Vest MO, Ley DJ, Lee S, Bolton EC, and Chung HJ (2015). N-methyl-D-aspartate receptors mediate activity-dependent down-regulation of potassium channel genes during the expression of homeostatic intrinsic plasticity. Mol. Brain 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J, Hnisz D, Li CH, Yuan B, Xu C, et al. (2018). Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 172, 979–992. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Goeritz ML, Casey R, Oliva R, Guckenheimer J, and Harris-Warrick RM (2005). Activity-independent coregulation of IA and Ih in rhythmically active neurons. J. Neurophysiol 94, 3601–3617. [DOI] [PubMed] [Google Scholar]
- Marder E (2011). Variability, compensation, and modulation in neurons and circuits. Proc. Natl. Acad. Sci. U S A 108 (Suppl 3), 15542–15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, and Goaillard JM (2006). Variability, compensation and homeo-stasis in neuron and network function. Nat. Rev. Neurosci 7, 563–574. [DOI] [PubMed] [Google Scholar]
- Muddashetty RS, Kelic S, Gross C, Xu M, and Bassell GJ (2007). Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci 27, 5338–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary T, Williams AH, Franci A, and Marder E (2014). Cell types, network homeostasis, and pathological compensation from a biologically plausible ion channel expression model. Neuron 82, 809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, and Bear MF (2010). Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci 30, 15616–15627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, and Aizenman CD (2007). Homeostatic regulation of intrinsic excitability and synaptic transmission in a developing visual circuit. J. Neurosci 27, 8268–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz AA, Bucher D, and Marder E (2004). Similar network activity from disparate circuit parameters. Nat. Neurosci 7, 1345–1352. [DOI] [PubMed] [Google Scholar]
- Ranson A, Cheetham CE, Fox K, and Sengpiel F (2012). Homeostatic plasticity mechanisms are required for juvenile, but not adult, ocular dominance plasticity. Proc. Natl. Acad. Sci. U S A 109, 1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, and Huber KM (2012). Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci 15, 431–440, S431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routh BN, Rathour RK, Baumgardner ME, Kalmbach BE, Johnston D, and Brager DH (2017). Increased transient Na+ conductance and action potential output in layer 2/3 prefrontal cortex neurons of the fmr1-/y mouse. J. Physiol 595, 4431–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarti F, Zhang Z, Schroeder J, and Chen L (2013). Rapid suppression of inhibitory synaptic transmission by retinoic acid. J. Neurosci 33, 11440–11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soden ME, and Chen L (2010). Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J. Neurosci 30, 16910–16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumbos JG, Brown MR, Kronengold J, Polley DB, and Kaczmarek LK (2010). Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J. Neurosci 30, 10263–10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G (2011). Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu. Rev. Neurosci 34, 89–103. [DOI] [PubMed] [Google Scholar]
- Turrigiano G, Abbott LF, and Marder E (1994). Activity-dependent changes in the intrinsic properties of cultured neurons. Science 264, 974–977. [DOI] [PubMed] [Google Scholar]
- Turrigiano G, LeMasson G, and Marder E (1995). Selective regulation of current densities underlies spontaneous changes in the activity of cultured neurons. J. Neurosci 15, 3640–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner P (2014). Homeostatic synaptic plasticity in developing spinal networks driven by excitatory GABAergic currents. Neuropharmacology 78, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm JC, Rich MM, and Wenner P (2009). Compensatory changes in cellular excitability, not synaptic scaling, contribute to homeostatic recovery of embryonic network activity. Proc. Natl. Acad. Sci. U S A 106, 6760–6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Bonnan A, Bony G, Ferezou I, Pietropaolo S, Ginger M, Sans N, Rossier J, Oostra B, LeMasson G, and Frick A (2014). Dendritic channelopathies contributeto neocortical and sensory hyperexcitability in Fmr1(-/y) mice. Nat. Neurosci 17, 1701–1709. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MATLAB custom script for measuring max slope of AP rising phase, AHP absolute negative peak, and duration of spiking, as well as R custom script for Monte Carlo simulation can be assessed though links provided in the Key Resource Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tetrodotoxin (TTX) | Tocris | Cat#: 1078 |
| D-(-)-2-Amino-5-phosphonopentanoic acid (APV) | Tocris | Cat#: 0106 |
| 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) | Tocris | Cat#: 0373 |
| 6-Imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide (Gabazine) | Tocris | Cat#: 1262 |
| Experimental Models: Organisms/Strains | ||
| Experimental mouse: Fmr1 KO C57BL/6 | Bred in house | N/A |
| Experimental mouse: WT C57BL/6 | Bred in house | N/A |
| Breeder mouse: Fmr1 het C57BL/6 female | Bred in house | N/A |
| Breeder mouse: WT C57BL/6 male | Jackson Laboratory | N/A |
| Software and Algorithms | ||
| Prism - 2-way ANOVAs | GraphPad Software | Version 7 |
| MATLAB – measurement of max slope of AP rising phase, AHP negative peak, and duration of spiking | MathWorks | Version R2017b https://github.com/pernillebuelow/Scripts/blob/master/Pernille_CC_Step_Analysis.m |
| RStudio – generalized linear mixed models, Wilcoxon post hoc significance tests, correlation statistics, Monte Carlo simulation of proportions, power analyses | RStudio | Version 1.1.463 https://github.com/pernillebuelow/Scripts/blob/master/Proportions_R_script.R |
| Axograph – measurement and analysis of all intrinsic excitability and synaptic transmission data, measurement of all “threshold AP” waveform parameters | Axograph X | Version 1.7.0 |
| Excel – proportion statistics (z-test), KS tests | Microsoft | Version 16.18 |