

Abstract
Biocompatible chemical protein cleavage methods have been long-sought to replace enzymatic cleavages, but have yet to be realized. Here, we report the development of the SNAC-tag (Sequence-specific Nickel Assisted Cleavage) to achieve sequence-specific chemical protein cleavage under biocompatible conditions with comparable efficiency to enzymes. We demonstrate that the SNAC-tag can be inserted before both water-soluble and membrane proteins to achieve fusion protein cleavage, even when enzymatic cleavages fail.
Protein cleavage is an integral step of recombinant protein expression and purification processes. Different tags are often fused to the N-terminus or C-terminus of target proteins to improve expression yield, solubility, folding or purification.1,2 However, after the target protein is purified, removal of these tags is often desired. Currently, the only strategy to remove fusion tags under biocompatible conditions is enzymatic cleavage using proteases, including Tobacco Etch Virus protease, thrombin protease.3 Unfortunately, the enzymes added often need to be removed, adding one more purification step. Enzymes can also be prohibitively expensive, especially when producing proteins at large scales. Moreover, enzymatic cleavages often fail, and this is especially common for membrane proteins when the enzyme recognition site is proximal to the hydrophobic domain. Finally, detergents and denaturants, which are widely used to solubilize membrane proteins or proteins expressed in inclusion bodies, inactivate common proteolytic enzymes used to cleave tags3.
One approach to circumvent enzymatic cleavage and its drawbacks is chemical cleavage. Cyanogen bromide has been used for chemical protein cleavages. However, under harsh conditions with low sequence specificity (recognizing single Met residue).4 Metal ions including Pd2+, Cu2+, Ni2+ have also been explored to this end.5–8 Though use of Pd2+ and Cu2+ has major obstacles, Ni2+ has shown more potential. Bal and coworkers found Ni2+ could cleave the sequence pattern –XSXHZ– prior to the Ser residue.7 The best optimized sequence was inserted in a mini-protein SPI-2 to show cleavage can indeed progress.9,10 However, the reaction requires heating to 50 °C for ~20 hours,9,10 which makes this method impractically harsh for most proteins.
To find better methods for biocompatible chemical protein cleavage, we turned to substrate phage display.11 We built a hexapeptide substrate phage library between a N-terminal AviTag12 and M13 phage pIII protein (in Fig. 1a). We performed four rounds of selection (Supplementary Fig. 1) in the presence of different metal ions with increasingly stringent cleavage conditions (shorter incubation, lower temperature). Final output phage libraries were sequenced and we readily identified a consensus sequence pattern for cleavage using Ni2+ ion (Fig. 1b).
Fig. 1. Phage selection and optimization of best Ni2+ cleavage sequence.
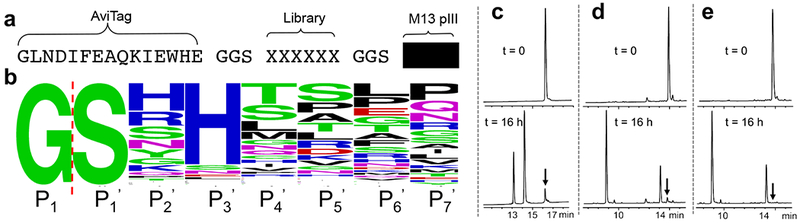
a. Substrate phage construct. b. Sequence frequency plot of Ni2+ cleavage selection, red dashed line indicates cleavage site between P1 and P1’ positions, P2’ to P7’ correspond to the randomized positions in Fig. 1a. Panels c-e. Evaluation of the cleavage of synthetic peptides (black arrow indicates uncleaved peptide). c. The most frequently observed phage selected peptide sequence YFLGGSHHTDLPGGSRRLFY; d. optimized peptide YFLPGSRHWG; e. best optimized peptide YFLPGSHHWG (the Arg for His substitution is based on the sequence logo in Fig. 1b). All peptides contain C-terminal carboxamides. The cleavage conditions were: peptide 0.2 mM, 0.1 M CHES, pH 8.2, 1 mM NiCl2, 22 °C, 16 hours. The mass of uncleaved and cleaved peptides were measured using MALDI-TOF shown in Supplementary Fig. 5.
This sequence pattern agrees with previous reports where Ser and His are very effective at positions P1’ and P3’.13 We found a small but significant preference for His at P2’ and Thr at P4’. We picked the two most enriched sequences and made the corresponding peptides to test the cleavage efficiency by HPLC. The most frequently observed sequence (–GSHHTDLP–) could achieve 90% cleavage at room temperature at pH 8.2 within 18 hours (Fig. 1c).
We found Gly at P1 position is critical for high cleavage efficiency. A Gly to Ala replacement leads to a dramatically reduced cleavage rate (Supplementary Fig. 2). This can be rationalized through the proposed cleavage mechanism (Supplementary Fig. 1), involving Ni2+-assisted N-to-O acyl shift of the P1 carbonyl to the P1’ Ser side chain.14 The resulting ester intermediate is then cleaved during the rate-limiting ester hydrolysis,7 and hence is sensitive to steric effects that slows hydrolysis for residues other than Gly.15,16 We therefore introduced a Gly at position P1 of a previously reported cleavage sequence7 to give -GSRHW-. This peptide performed slightly better than the best phage selected sequence (Fig. 1c-d). We further confirmed that His at P2’ position (-GSHHW-) indeed performs better than Arg (-GSRHW-) when embedded in a short peptide construct (Fig. 1d-e). We then did a cleavage comparison without or with the critical Gly insertion on the miniprotein SPI-2 (Supplementary Fig. 3).9 With the critical Gly insertion, the cleavage progressed to more than 90% yield (22 °C, 16 hours). As expected, absence of the Gly gave only 15% cleavage yield under the same conditions. These data together designate the resulting minimal sequence –GSHHW– as the SNAC-tag, which was generated from combined efforts of our phage selection, validation using synthetic peptides and previous reports.7
We also examined the effect of buffers and exogenous nucleophiles on the rate of the cleavage reaction. We found CHES and HEPES buffers performed the best (Supplementary Fig. 4), weak nucleophile acetone oxime can accelerate cleavage slightly without inducing non-specific cleavages (Supplementary Fig. 4).
Encouraged by the activity of the SNAC-tag, in peptides and a small protein, we sought to examine its utility as a cleavable tag in a number of difficult cases where fusion-tag enzymatic cleavage was very inefficient. We first examined its utility in conjunction with a HisTag (6xHis) fused to the N-terminus of a water-soluble α-helical bundle protein, HB2225. Between the HisTag and HB2225, a cleavage site was included: either TEV (-ENLYFQS-, Fig. 2a) or a SNAC-tag (–GSHHW–, Fig. 2b). Cleavage by TEV protease under standard conditions was mostly incomplete (Fig. 2a). However, cleavage of the SNAC-tag containing construct yielded ~80% cleavage into the desired product (Fig. 2b) with good reagents compatibility (Fig. 2c). This result demonstrates that the SNAC-tag can indeed achieve biocompatible cleavage in full-sized globular proteins when enzymatic cleavage fails.
Fig. 2. SNAC-tag cleavage in fusion proteins.
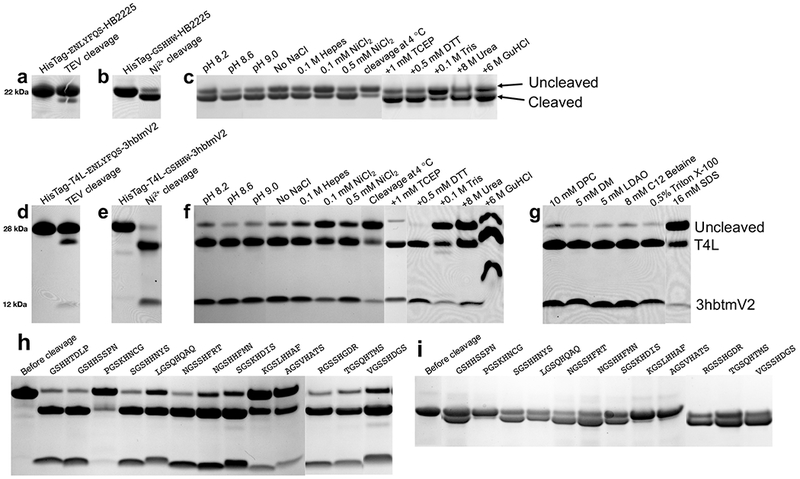
a. TEV protease cleavage of HisTag-ENLYFQS-HB2225 construct, cleavage conditions: protein 1 mg/mL, 50 mM Tris, pH 8.0, 0.5 mM EDTA, 1 mM DTT, 0.25 M NaCl, 22 °C, 16 hours, TEV protease 0.04 mg/mL. b. Ni2+ cleavage of HisTag-GSHHW-HB2225 construct, cleavage conditions: protein 1 mg/mL, 1.0 mM NiCl2, 0.1 M CHES, 0.1 M acetone oxime, 0.1 M NaCl, pH 8.2, 22 °C, 16 hours. c. Cleavage compatibility test for Ni2+ cleavage of HisTag-GSHHW-HB2225. d. TEV protease cleavage of HisTag-T4L-ENLYFQS-3hbtmV2 construct, cleavage conditions: protein 1 mg/mL, 50 mM Tris, pH 8.0, 0.5 mM EDTA, 1mM DTT, 0.25 M NaCl, 22 °C, 5 mM DPC, 16 hours, TEV 0.04 mg/mL. e. Ni2+ cleavage of HisTag-T4L-GSHHW-3hbtmV2 construct, cleavage conditions: protein 1 mg/mL, 1.0 mM NiCl2 0.1 M CHES, 0.1 M acetone oxime, 0.1 M NaCl, pH 8.2, 22 °C, 5 mM DPC, 16 hours. f. Cleavage compatibility test for Ni2+ cleavage of HisTag-T4L-GSHHW-3hbtmV2. g. Ni2+ cleavage of HisTag-T4L-GSHHW-3hbtmV2 in different detergents. h. Cleavage of the membrane protein HisTag-T4L-XXXXXXXX-3hbtmV2. Lane 1 shows protein before cleavage, lanes 2 to 14 shows cleavage using difference sequences from phage selections, the sequence inserted for each lane is shown on top of the lane. i. Cleavage of the globular protein construct HisTag-XXXXXXXX-HB2225, lane 1 shows the position of the protein before cleavage, lane 2 to lane 13 shows cleavage using difference sequences from phage selection, the sequence inserted for each lane is shown on top of the lane. Cleavage percentages were quantified based on cleaved and uncleaved protein bands in each individual lane in Imagelab 5.2.1 and is summarized in Supplement Table 1, 2. Full gels are shown in Supplementary Fig. 6.
We next explored the utility of SNAC-tag for membrane protein cleavage. In particular, we sought to cleave a construct consisting of a designed membrane protein 3hbtmV2 fused to T4 lysozyme via a TEV-cleavable linker. After screening a number of constructs, we found only a very low level of cleavage with TEV (<20%) (Fig. 2d). By contrast, replacing the TEV substrate sequence with the SNAC-tag (–GSHHW–) at the same site, resulted in >90% cleavage (Fig. 2e). This demonstrates SNAC-tag is perfectly compatible with cleavage of membrane proteins and detergent micelles. The SNAC-tag cleavage was further tested in two more water soluble proteins and one more membrane protein, all cleavages achieved >80% completion at pH 8.2, 22 °C, 18 hours (Supplementary Fig. 7). In another T4L fusion membrane protein (T4L-PL5) where thrombin cleavage was met with difficulty (Supplementary Fig. 8). We inserted SNAC-tag to replace thrombin cleavage sequence and shown SNAC-tag can be effectively cleaved (Supplementary Fig. 8).
We investigated the effect of different buffer and detergent conditions on the SNAC-tag cleavages (Fig. 2c, Fig. 2f-g). Importantly, cleavage was fully compatible with common mild detergents, although the reaction was partially inhibited by SDS (Sodium dodecyl sulfate) (Fig. 2g). Gratifyingly, strong denaturing conditions including 6 M guanidinium chloride or 8 M Urea are also compatible, though with decreased efficiency. Lower concentrations up to 2 M of guanidinium chloride or Urea do not obviously affect cleavage efficiency (Supplementary Fig. 9). The SNAC-tag is most efficiently cleaved at pH 8.6, with a small decrease in yield at lower (8.2) and higher (9.0) pH. Removal of salt has little effect on T4L-3hbtmV2 cleavage, and only a modest effect for HisTag-HB2225 cleavage. We also found that HEPES and CHES buffers perform comparably while the weakly Ni2+ chelating Tris buffer slows cleavage rate. Ni2+ concentration at 1 mM seems to be necessary for efficient cleavage. Low levels of reducing agent (1 mM TCEP, 0.5 mM DTT) are compatible, and in fact cleavage of a Cys-containing peptide shows negligible oxidation (Supplementary Fig. 10). For proteins that require low temperature handling, cleavage can occur at 4 °C albeit at a significantly slower rate than at room temperature.
We studied the potential for cleaving SNAC-tag from proteins bound to Ni2+-NTA (nitrilotriacetic acid) resins, via a HisTag. One miligram of HisTag-T4L-GSHHW-3hbtmV2 was loaded onto a bed volume of 0.5 mL Ni-NTA resins. After exchange into cleavage buffer, 1 mM NiCl2 was added and the reaction was incubated overnight at 22 °C (Supplementary Fig. 11). On-resin cleavages proceeded nearly to completion, comparable to the efficiency observed in solution. No HisTag containing protein was eluted off the beads during cleavage, nor did we observe protein cleavage while SNAC-tag containing proteins were purified using Ni-NTA resins. Thus, SNAC-tag cleavage is compatible with Ni-NTA purifications and on-resin cleavages.10 However, significant scale-up of protein loading for on-resin cleavage (50 mg protein per mL bed volume Ni-NTA resin) resulted in visible protein precipitation as the cleavage proceeds, suggesting loading as an important variable to consider for on-resin applications.
Knowing that Gly at P1 position plays a key role in the rate of hydrolysis, we generated a second targeted phage library (–X1X2X3GSX4HX5X6X7–) to further explore the influences of nearby residues on cleavage efficiency. Four more rounds of selection were performed and the final output library was sequenced. We ranked each particular sequence observed in the output library (normalized to the control library: the same selection protocol in the absence of Ni2+) shown in Supplementary Table 2. Guided by this analysis, we then picked 11 distinct sequences from this selection and the top two sequences from the initial phage selection to replace the TEV substrate site in HisTag-HB2225 and in HisTag-T4L-3hbtmV2 for Ni2+ cleavage. We confirmed that several of these SNAC-tag variants yielded more than 80% cleavage under our standard conditions (Fig. 2h). For the water-soluble HisTag-HB2225, efficiency is slightly lower (Fig. 2i). This result indicates that a number of diverse sequence combinations can be used to achieve high cleavage efficiency. All the high-ranking cleavage sequences are listed in the Supplementary Table 2. To achieve high cleavage efficiency, Gly at P1, Ser at P1’ and His P3’ seems to be mandatory, while hydrophobic residues at P2’ are not favored. For the sequences listed in Fig. 2h-i and Supplementary Table 2, the specific sequences seem to be important to achieve high cleavage efficiency, though we didn’t investigate further to find the minimal sequence for each individual sequence. Furthermore, cleavage requires correct geometry of the Ni2+ complex (Supplementary Fig. 1), and is only likely achieved in poorly structured protein regions. 9,14,17
We also systematically evaluated the effect of the surrounding residues by screening hundreds of peptides at five nearby positions (Supplementary Table 3-4). Better cleavage peptides than –GSHHW– were identified. However, they didn’t perform better when inserted in proteins (Supplementary Fig. 12).
In summary, we develop the SNAC-tag as a new chemical protein cleavage strategy. The SNAC-tag can be inserted into both water soluble and membrane proteins to achieve biocompatible protein cleavage in a sequence-specific manner with efficiency comparable to enzymes. The method is particularly attractive as it only leaves a small Gly residue at the C-terminus of the released protein, when it is used as a C-terminal tag. Additionally, this approach circumvents the demands of subsequent enzyme removal. Critically, we show cases where the SNAC-tag can succeed when enzymatic cleavage fails. We suggest using –GSHHW– as a general SNAC-tag although –GSRHW– is also efficiently cleaved. Cleavage conditions are: protein concentration ~1 mg/mL, 1 mM NiCl2, 0.1 M CHES buffer, 0.1 M acetone oxime, pH 8.6, incubating at room temperature, 0.1-0.5 M NaCl. For specific sequence requirements and additional cleavage details, consult the methods. We expect the SNAC-tag to greatly reduce the cost and labor of recombinant protein production at both the laboratory and industrial scale and to find broad applications throughout medicine, basic biological research, and biotechnology.
Methods
Reagents.
All reagents were used without further treatment. Fmoc-protected amino acids were purchased from GL Biochem. 2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU), trifluoroacetic acid (TFA), and hydroxybenzotriazole hydrate (HOBt) were purchased from Chem-Impex International. 4-methylpiperidine were purchased from Acros Organics. Rink Amide-ChemMatrix resin (0.5 mmol/g loading) was purchased from Biotage. All other reagents including tris(2-carboxyethyl)phosphine hydrochloride (TCEP), DL-Dithiothreitol (DTT), N,N-Dimethyldodecylamine N-oxide (LDAO), N-Dodecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate (C12 Betaine), Triton X100 were purchased from Sigma-Aldrich. Fos-choline-12 (DPC), Decyl β-D-maltopyranoside (DM) were purchased from Anatrace. Sodium dodecyl sulfate (SDS) was purchased from Calbiochem, IPTG (isopropyl β-D-1-thiogalactopyranoside) was purchased from Genesee Scientific Corporation.
Protein Gels and DNA Gels.
Precast NuPAGE 4-12% Bis-Tris polyacrylamide protein gels were purchased from Invitrogen. Protein gels were run at 200 V for 35 minutes in MES buffer, gels were then washed 9 times with warm water, stained with coomassie blue for 4 hours. It was then destained using tap water for 16 hours before we imaged them on Bio-Rad ChemiDoc MP imaging System. All protein bands were analyzed and quantified using Imagelab 5.2.1. Agarose DNA gels were made in house using 1% UltraPure Agarose 1000 in TAE buffer with 0.01% EZ-Vision (Amresco), DNA gels were run at 150 V for 25 minutes and visualized under UV.
Reverse-phase HPLC and Mass Spectrometry.
Analytical reverse-phase HPLC analyses were performed on an Agilent 1100 series HPLC system using a Phenomenex Kinetex 2.6 μm C18, 50 × 2.1 mm column. Chromatographic separations were obtained using a linear gradient of 1-61% acetonitrile (with 0.08% TFA) in water (with 0.1% TFA) over 10 min, 1-41% acetonitrile (with 0.08% TFA) in water (with 0.1% TFA) over 20 min, or 1-41% acetonitrile (with 0.08% TFA) in water (with 0.1% TFA) over 40 min with column at room temperature. Flow rates were controlled at 0.35 mL/min. Peptide detection was based on UV absorption at 220 nm, and mass spectrometry data were obtained using Shimadzu AXIMA Performance MALDI-TOF spectrometer in a reflectron mode with α-Cyano-4-hydroxycinnamic acid as the matrix.
Preparative HPLC.
Products from solid-phase peptide synthesis were purified using a Phenomenex Jupiter 5.0 μm C4, 250 × 10.0 mm column. A shallow gradient of acetonitrile (with 0.08% TFA) versus water (with 0.1% TFA) was designed for each peptide based on its elution characteristics. Flow rates were controlled at 5 mL/min. Fractions containing the desired pure peptide were identified by analytical HPLC and mass spectrometry, then combined and lyophilized.
Solid-phase peptide synthesis (SPPS).
All peptides were synthesized at 20 μmol scale using a Biotage Syro II parallel peptide synthesizer or at 0.1 mM scale using a Biotage Initiator+ Alstra peptide synthesizer. Rink Amide-ChemMatrix resin (Biotage, 0.5 mmol/g loading) was used for the synthesis. A typical SPPS reaction cycle includes Fmoc deprotection, washing, coupling, and post-coupling washing steps. The deprotection was carried out for 5 min at room temperature with 20% 4-methylpiperidine in dimethylformamide (DMF). A standard double coupling was done for 8 min at room temperature or 5 min under microwave with 5 equivalents Fmoc-protected amino acids, 4.98 equivalents HCTU, and 10 equivalents DIEA (relative to the amino groups on resin) in DMF at a final concentration of 0.125 M amino acids.
Phage library construction.
All phages were propagated in the E. coli strains TG1 (Lucigen). E. coli CJ236 was purchased from Lucigen. The recombinantly expressed BirA enzyme used for biotinylating AviTag was a gift from the Wells lab at UCSF. Phagemid vector pCES1 was obtained from Craik lab at UCSF. Pierce streptavidin coated high capacity plates were purchased from ThermoFisher Scientific. The AviTag-displaying M13 phage libraries were constructed in phagemid vector pCES1. AviTag-TEV sequence was first fused to the N-terminal of pIII protein in pCES1 vector using ApaL1 and Not1 restriction sites affording protein sequence as MGLNDIFEAQKIEWHEGGSENLYFQGGSAAAHHHHHHGAAEQKLISEEDLNG-, Single strand M13 DNA was purified using the M13 DNA kit from Qiagen. For library construction, we followed the protocol by Chen et al.18 Phage libraries were constructed using an oligonucleotide encoding the reverse complement of protein sequence –IEWHEGGSXXXXXXGGSAAAHHH- using primer 5’-TGATGATGATGTGCGGCCGCACTACCACCMNNMNNMNNMNNMNNMNNGCTGCCGCCTTCATGCCATTCAAT-3’ for AviTag-XXXXXX- library, or using 5’-TGATGATGATGTGCGGCCGCACTACCACCMNNMNNMNNATGMNNACTACCMNNMNNMNNACCACCGCTGCCGCCTTCATGCCATTCAAT-3’ for AviTag-XXXGSXHXXX-library (codon M = A, C; N = A, C, T, G). Double-stranded DNA was generated according to Sidhu et al. 18,19 and electroporated into TG1. Library size of 4×109 were generated and amplified for 15 h at 37 °C in 250 mL 2xYT broth containing AMP (30 mg/mL) and Kan (15 mg/mL final). For phage biotinylation, purified phages (4 × 1012 cfu) were resuspended in 0.5 mL of 10 mM Tris, pH 8.0 to exchange buffer (10 mM Tris, pH 8.0) three times using 30 kDa MWCO tube. After buffer exchange, we added 40 uL water, 80 uL 2× biotinylation buffer (0.1 M Tris, 10 mM MgCl2, 2 mM Biotin, pH 8.0) to wash the tube and get the solution to eppendorf tube. 20 uL water, and 20 uL 2× biotinylation buffer was added to the centrifugation tube again to rinse one more time. We then added ~6 uL of 0.1 M ATP, 2 uL of BirA enzyme (3U/uL) to the ~200 uL phage solution, vortexed gently to mix well and leave at 4 °C overnight for biotinylation. Anti-biotin western blot was then performed to confirm phages were biotinylated.
Substrate phage selection experiments.
Four rounds of substrate selection were conducted (Supplementary Fig. 1). In round one, streptavidin-coated ELISA plate was blocked with 2% BSA for 30 min and washed three times with phosphate-buffered saline containing 0.1 % Tween 20 (PBST) buffer. The plate was then coated with ~ 109 phages (one well) by gently shaking the phages for 2 hours. The phages were washed out of the wells, well washed with PBST × 6, batch wash with PBST (0.3 mL) for 3 × 20 min. Cleavage buffer containing 2 mM NiCl2, 0.1 M Tris, pH 9.0 was then added at 37 ˚C for 3 hours. Cleaved phages were amplified overnight in 2xYT (30 mL) at 37 ˚C with shaking. The second, third and fourth rounds of selection were performed by repeating the procedure above with shorten the cleavage time to 30 min as well as lowering the cleavage temperature from 37 °C to room temperature. Over different rounds of selection, output phage titer is always around 3×108 cfu. The final output phages were sequenced either by picking colonies or doing deep sequencing.
We also ran a control selection along with the cleavage selections. In the control selection, all selection procedures were kept the same except in the elution step (cleavage step), which lacked added Ni2+ in the Tris buffer. The purpose of performing control selection was to estimate sequence enrichment due to differences in growth rate or background-binding for individual displayed peptides.
Protein expression, purification and cleavage.
Initial HB2225 and 3hbtmV2 genes were introduced into a pET-28a vector (Novagen) using Gibson assembly, their sequences were confirmed (Genewiz San Francisco), and were then transformed and expressed in One Shot® BL21(DE3) Chemically Competent E. coli (ThermoFisher Scientific). Different cleavage peptide sequences (Supplementary Table 5) were then introduced into the pET-28a vector containing the HB2225 and 3hbtmV2 genes following Gibson assembly protocol (New England Biolabs).
Protein sequences are listed as below:
-
6×His-ENLYFQS-HB2225:
MGSSHHHHHHSSGENLYFQSHMDYLRELLKLELQAIKQYEKLRQTGDELVQAFQRLREIFDKGDDDSLEQVLEEIEELIQKHRQLASELPKLELQAIKQYREALEYVKLPVLAKILEDEEKHIEWLEEAAKQGDQWVQLFQRFREAIDKGDKDSLEQLLEELEQALQKIRELARLTRKILEDEEKHIEWLETILG
-
6×His-GSHHW-HB2225:
MGSSHHHHHHSSGGSHHWSHMDYLRELLKLELQAIKQYEKLRQTGDELVQAFQRLREIFDKGDDDSLEQVLEEIEELIQKHRQLASELPKLELQAIKQYREALEYVKLPVLAKILEDEEKHIEWLEEAAKQGDQWVQLFQRFREAIDKGDKDSLEQLLEELEQALQKIRELARLTRKILEDEEKHIEWLETILG
-
6×His-T4L-ENLYFQG-3hbtmV2:
MGSSHHHHHHSSGLVPRGSHMGNIFEMLRIDEGLRLKIYKDTEGYYTIGIGHLLTKSPSLNAAKSELDKAIGRNTNGVITKDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRAALINMVFQMGETGVAGFTNSLRMLQQKRWDEAAVNLAKSRWYNQTPNRAKRVITTFRTGTWDAYAAGGSGSTENLYFQSNSPDLEAWLLFIMLLTNALIFLAQKWVRETRDGDEAWKKIFLATFALNLLXXXXXXXXXXXXXXXXXXXXXXXXXTLAFNLLTILLGYKTIEGR (de novo designed membrane protein, full sequence to be released in a separate paper)
-
6×His-T4L-GSHHW-3hbtmV2:
MGSSHHHHHHSSGLVPRGSHMGNIFEMLRIDEGLRLKIYKDTEGYYTIGIGHLLTKSPSLNAAKSELDKAIGRNTNGVITKDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRAALINMVFQMGETGVAGFTNSLRMLQQKRWDEAAVNLAKSRWYNQTPNRAKRVITTFRTGTWDAYAAGGSGSTEGSHHWNSPDLEAWLLFIMLLTNALIFLAQKWVRETRDGDEAWKKIFLATFALNLLXXXXXXXXXXXXXXXXXXXXXXXXXTLAFNLLTILLGYKTIEGR (de novo designed membrane protein, full sequence to be released in a separate paper)
For expressing HB2225 constructs, cells were grown in LB broth (15 μg/mL Kan), induced at 0.6-0.8 OD600 with 1 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) and incubated for 4 hours at 37 °C before harvesting. Cell pellets were resuspended in buffer containing 50 mM Tris (pH 8.0), 300 mM NaCl, 2% glycerol and lysed by sonication, and cell debris was subsequently removed by centrifugation (18000 rpm and 4 °C for 30 min). The soluble cell lysate was purified with Ni-NTA affinity chromatography.
For expressing 3hbtmV2 constructs, cells were grown in LB broth (15 μg/mL Kan), induced at 0.6-0.8 OD600 with 0.4 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) and incubated overnight at 18 °C before harvesting. Cell pellets were resuspended in buffer containing 50 mM Tris (pH 8.0), 300 mM NaCl, 2% glycerol, 5 mM DPC and lysed by sonication, and cell debris was subsequently removed by centrifugation (18000 rpm and 4 °C for 30 min). The soluble cell lysate was purified with Ni-NTA affinity chromatography.
For cleaving HB2225 constructs, proteins were first exchanged into buffer containing 0.1 M CHES, 0.1 M Acetone oxime, 0.1 M NaCl at appropriate pH, 1 mM NiCl2 was then added, the solution was mixed well and left at room temperature (~22 °C) for cleavage to proceed without shaking or stirring. Imidazole from Ni-NTA elution needs to be removed completely prior cleavage. For cleaving 3hbtmV2 constructs, procedure is essentially the same as HB2225 except appropriate detergents needs to be added to the cleavage solutions. Cleavage efficiency were quantified using Imagelab 5.2.1 on Bio-Rad ChemiDoc MP imaging System. A time course cleavage of HisTag-T4L-GSHHW-3hbtmV2 and HisTag-GSHHW-HB2225 are shown in Supplementary Fig. 13.
TEV protease was purchased from Sigma-Aldrich, TEV cleavage was carried out under standard conditions: protein 1 mg/mL, 50 mM Tris, pH 8.0, 0.5 mM EDTA, 1 mM DTT, 0.25 M NaCl, 22 °C, 5 mM DPC, 16 hours, TEV 0.04 mg/mL. We also tried TEV cleavage at 34 °C for HisTag-T4L-ENLYFQS-3hbtmV2 and HisTag-ENLYFQS-HB2225, no obvious difference was observed from 22 °C cleavage. We ran a positive control of TEV protease cleavage on a different protein that could be efficiently cleaved by TEV protease to show TEV protease we obtained indeed has good quality (Supplementary Fig. 14).
Thrombin was purchased from EMD Milipore. Cleavage conditions: 50 mM Tris, pH 8.4, 150 mM NaCl, 0.5 mM CaCl2, 5 mM DPC, 22 °C, 16 hours. One thrombin unit is defined as that amount of enzyme required to cleave 1 mg of a test protein when incubated in standard digest buffer at 20°C for 16 hours.
Peptide cleavage screening:
As an alternative approach to explore nearby amino acid influence on cleavage efficiency, we made ~100 peptides (Supplementary Table 3) varying the amino acid identity individually around the cleavage site in the peptide WLX1X2SX3HX4X5. Peptides cleavage were performed under cleavage conditions described above in Fig. 1, cleavage yields were quantified using Agilent HPLC online system by integrating peak area. The sum of the extinction coefficient for the two product peaks was assumed to be the same as the initial peptide. We found position X5 has minimum impact on cleavage; position X4 has strong preference for Trp; position X3 strongly prefer His; for position X2, His, Asn, perform the best, followed by Lys, Arg; position X1 prefer Pro. We then further picked ~30 sequence combinations (Supplementary Table 4) and tested the cleavage efficiency. We found two sequence combinations (-PGSHHW- -HNSHHW-) indeed gave better cleavage efficiency (Supplementary Fig. 12). But when we then inserted these two sequences individually into the globular protein and membrane protein construct we tested previously. Unfortunately, they didn’t perform better (Supplementary Fig. 12). Thus, for searching best protein cleavage sequences, peptide models didn’t seem to be the best system. Protein based selections such as phage display is probably a more reliable way as we did here. Another thing to point out is the amino acid preference at X2 position didn’t fully hold up in a different peptide system (Supplementary Table 4), so we think Gly is still the best amino acid to use for best cleavage at X2 position.
A few tips for running cleavage reactions:
For peptide cleavage tests, lyophilized peptides were dissolved in cleavage buffer (0.1 M CHES, 0.1 M NaCl, 0.1 M acetone oxime, pH 8.2 or otherwise noted in the context) at 0.2 mM concentration, 1 mM NiCl2 was then added for the reaction to proceed without stirring or shaking.
We tested the compatibility of single unpaired cysteine with cleavage conditions in a peptide sample (Supplementary Fig. 10) and found no obvious Cys oxidation was observed during the cleavage process. In cases where cysteine oxidation is observed, 1-2 mM TCEP could be added to keep cleavage under reducing conditions to minimize oxidation while keeping cleavage rate essentially the same.
For the cleavage of short peptides, pH does make a big difference, cleavage rate increase about two-fold from 8.2 to pH 8.6, and from pH 8.6 to pH 9.0 (Supplementary Fig. 15). However, this trend of rate increase didn’t hold for the two protein cleavage examples, we therefore recommend doing cleavage at pH 8.6 as it performs slightly better on the two protein examples shown in Fig. 2.
For some proteins, adding 1 mM NiCl2 could cause protein precipitation. Under this situation, adding 0.5 M GuHCl to protein solution prior mixing protein with Ni2+ could dramatically reduce protein precipitation without affecting cleavage efficiency (Supplementary Fig. 9), since most of the proteins cannot be denatured with this diluted GuHCl concentration, we recommend using 0.5 M GuHCl when protein precipitation occurs. Alternatively, 1 mM TCEP could be added to protein solution prior adding NiCl2, since TCEP binds with Ni2+ weakly and could minimize non-specific metal mediated protein-protein interactions preventing protein precipitation. Other weak Ni2+ ligand could potentially be explored to achieve the same result. The detailed protocol for SNAC-tag cleavage has been published on Protocol Exchange.20
Data Availability
The data that support the findings of this study are available from the corresponding author upon appropriate request.
Supplementary Material
Acknowledgements.
We thank the Wells lab at UCSF for generously providing BirA enzymes, Alex Martinko and Sam Pollock for helping carrying out phage biotinylation reactions. We thank the Craik lab at UCSF for providing the pCES1 phagemid vector, Natalia Sevillano for helping building phage libraries, Matthew Ravalin for helping operating multichannel peptide synthesizer. This work was supported in part by Grants 5R35GM122603-02 from the NIH (to W.F.D.).
Footnotes
Competing interests. The authors declare no conflict of interests.
References
- 1.Young CL, Britton ZT & Robinson AS Biotechnol. J 7, 620–34 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Kimple ME, Brill AL & Pasker RL Curr. Protoc. Protein Sci. 73, Unit 9.9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waugh DS Protein Expr. Purif. 80, 283–93 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gross E & Witkop BJ Am. Chem. Soc 83, 1510–1511 (1961). [Google Scholar]
- 5.Parac TN & Kostic NM J. Am. Chem. Soc 118, 51–58 (1996). [Google Scholar]
- 6.Dutca LM, Ko KS, Pohl NL & Kostic NM Inorg. Chem 44, 5141–5146 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Krezel A et al. J. Am. Chem. Soc. 132, 3355–3366 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Allen G & Campbell RO Int. J. Pept. Protein Res. 48, 265–73 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Kopera E, Belczyk-Ciesielska A & Bal W PLoS One 7, e36350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kopera E et al. PLoS One 9, e106936 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matthews DJ . & Wells JA Science 260, 1113–1117 (1993). [DOI] [PubMed] [Google Scholar]
- 12.Fairhead M & Howarth M Methods Mol. Biol. 1266, 171–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krezel A, Mylonas M, Kopera E & Bal W Acta Biochimica Polonica 53, 721–727 (2006). [PubMed] [Google Scholar]
- 14.Kopera E et al. Inorg. Chem 49, 6636–6645 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Hackeng TM, Griffin JH & Dawson PE Proc. Natl. Acad. Sci. USA 96, 10068–10073 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hay RW, Porter LJ & Morris PJ Aust. J. Chem 19, 1197–1205 (1966). [Google Scholar]
- 17.Wezynfeld NE, Fraczyk T & Bal W Coordination Chemistry Reviews 327, 166–187 (2016). [Google Scholar]
- 18.Chen G & Sidhu SS Methods Mol Biol 1131, 113–131 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Sidhu SS, Lowman HB, Cunningham BC & Wells JA Methods Enzymol 328, 333–363 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Dang B & DeGrado WF Protocol Exchange (2019) DOI: 10.1038/protex.2019.013. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon appropriate request.