

Abstract
Endothelial dysfunction-induced lipid retention is an early feature of atherosclerotic lesion formation. Apoptosis of vascular smooth muscle cells (VSMCs) is one of the major modulating factors of atherogenesis, which accelerates atherosclerosis progression by causing plaque destabilization and rupture. However, the mechanism underlying VSMC apoptosis mediated by endothelial dysfunction in relation to atherosclerosis remains elusive. In this study, we reveal differential expression of several genes related to lipid retention and apoptosis, in conjunction with atherosclerosis, by utilizing a genetic mouse model of endothelial nitric oxide synthase (eNOS) deficiency manifesting endothelial dysfunction. Moreover, eNOS deficiency led to the enhanced susceptibility against pro-apoptotic insult in VSMCs. In particular, the expression of aggrecan, a major proteoglycan, was elevated in aortic tissue of eNOS deficient mice compared to wild type mice, and administration of aggrecan induced apoptosis in VSMCs. This suggests that eNOS deficiency may elevate aggrecan expression, which promotes apoptosis in VSMC, thereby contributing to atherosclerosis progression. These results may facilitate the development of novel approaches for improving the diagnosis or treatment of atherosclerosis.
Keywords: Aggrecan, Apoptosis, Atherosclerosis, Endothelial nitric oxide synthase, Vascular smooth muscle cells
INTRODUCTION
Endothelial dysfunction is a pathophysiological condition characterized by diminished production of vasodilating factors, mainly nitric oxide (NO), and an increase in endothelium-derived vasoconstrictors (1). NO produced by endothelial nitric oxide synthase (eNOS) in endothelial cells (ECs) plays an important role in proper endothelium functions by regulating vascular homeostasis and the functional endothelium, and acts as an anti-thrombotic and anti-inflammatory barrier for the vascular wall (1). The maintenance of endothelium function is, thus, critical for the protection of the cardiovascular system against atherosclerosis (2–4), myocardial ischemia (5), and unstable angina (6). In this context, endothelium dysfunction is known to be an early feature of atherosclerotic lesion formation and progression (7).
Atherosclerosis is a representative disease, showing progressive and chronic inflammatory pathology of the arterial intima, and it is the leading cause of death in developed countries (8). Progression of this disease is characterized by real-life clinical events of the rupture and thrombosis of atherosclerotic plaques that consist of macrophages, ECs, vascular smooth muscle cells (VSMCs), and different collagen types (8, 9). In the case of vessel damage, VSMCs change from the contractile into the proliferative type, to repair the vascular wall. However, in atherosclerosis, VSMCs become aberrantly regulated and this promotes extracellular matrix formation in plaque areas (10–12). The major atherogenic factors are known to be lipid dysfunction, cell death, arterial lipid accumulation, and inflammatory responses in directing the initiation and development of a prominent feature of advanced atherosclerotic lesion (10, 13, 14). In particular, apoptosis of VSMCs may cause plaque destabilization and rupture by inducing loss of collagen and matrix, accumulation of cell debris, and intimal inflammation, thereby reinforcing atherosclerosis progression (10). This is because the tensile strength of the protective cap mainly depends on structural properties determined by the number of VSMCs and the collagen which they produce (15). VSMC apoptosis may also promote calcification, become procoagulant both locally and systemically, and provoke inflammation (16), further accelerating atherosclerosis progression in later stage. However, the mechanism underlying eNOS deficiency-mediated induction of VSMC apoptosis in relation to atherosclerosis remains elusive.
In this study, we aimed to investigate the differential gene expression profiles associated with atherosclerosis in aortic tissues using a genetic model of eNOS deficiency, and identified differentially expressed genes related to the pathogenesis of atherosclerosis. We found that eNOS deficiency led to an increase in aggrecan expression, which promotes apoptosis in VSMCs, indicating that NO-deficiency-mediated apoptosis of VSMCs may be involved in the pathological pathways of atherosclerosis. Understanding the changes in gene expression induced by eNOS deficiency could reveal molecular mechanism in atherosclerosis and facilitate the development of novel approaches for improving the diagnosis or treatment of the disease.
RESULTS
Genetic deletion of eNOS increases apoptosis by altering apoptosis-related gene expression in murine aortic tissue
Although previous studies report that eNOS protects against atherosclerosis while eNOS deficiency leads to increased atherosclerosis in a mouse model (4, 17), gene expression changes associated with atherosclerosis in eNOS−/− mice have not been investigated. Therefore, we performed Affymetrix Gene-Chip microarray gene expression profiling of aortic tissue isolated from wild type (WT) and eNOS−/− mice to analyze differentially expressed genes in eNOS deficient mouse manifesting endothelial dysfunction. We found that the expression of genes related to the pathogenesis of atherosclerosis was elevated in eNOS−/− mice. These genes include fibronectin 1 (FN1; response to stress, adhesion and extracellular molecules (ECM)), connective tissue growth factor (CTGF; adhesion, ECM and cell growth), and thrombospondin 4 (THBS4; adhesion and ECM) (Supplementary Table 1).
Evidence suggests that apoptosis in atherosclerotic lesions is responsible for prominent atherogenesis (10, 13, 14). Therefore, we then examined differential apoptosis-related gene expression profiles of WT and eNOS−/− mice, and found that the expressions of Tnfrsf11b (tumor necrosis factor receptor superfamily, member 11b) and Dapk1 (death associated protein kinase 1) were dysregulated in the eNOS−/− aorta (Supplementary Table 1). In order to verify the microarray results, we performed qPCR for Tnfrsf11b and Dapk1 using total RNA extracted from WT and eNOS−/− aortas. Being consistent with microarray expression profiling data, these genes were found to be upregulated in the eNOS−/− aorta compared with those of WT (Fig. 1C).
Fig. 1.
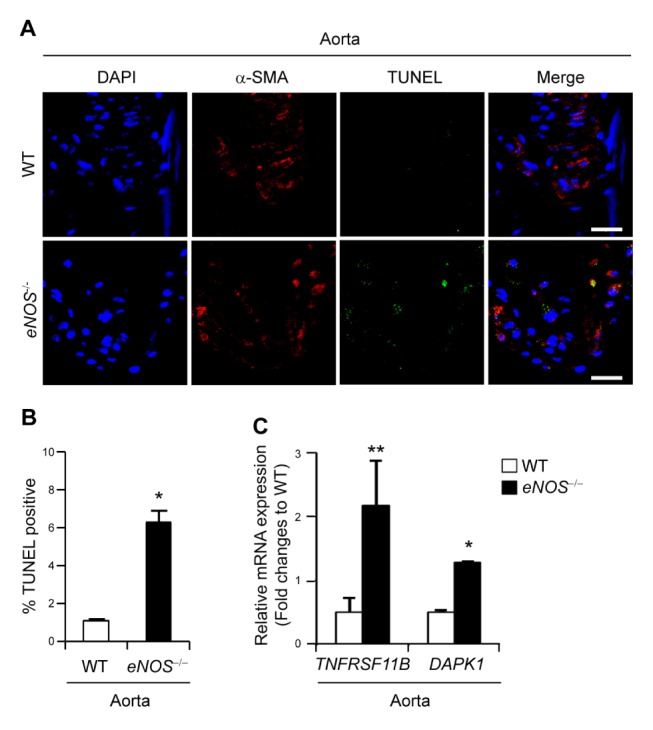
Increased apoptosis in the aortic sinus from eNOS−/− mice. (A) Representative images of α-SMA (stained in red) and TUNEL staining showing apoptotic cells (stained in green) in the aortic sinus from wild type (WT) and eNOS−/− mice. Scale bars, 20 μm. (B) The number of TUNEL-positive cells in the aortic sinus of WT and eNOS−/− mice (mean ± SD). *P < 0.05 compared to WT control. (C) Validation of microarray experiment by quantitative real-time PCR for apoptosis-related genes including TNFRSF11B and DAPK1 in eNOS−/− relative to WT controls. *P < 0.05 and **P < 0.005 compared to WT control. P values were calculated using Mann-Whitney test (Fig. B) or Kruskal-Wallis test (Fig. C).
We next evaluated apoptosis in aortic tissue isolated from WT and eNOS−/− mice. TUNEL assay revealed that eNOS−/− mice aortic tissue is sensitized to undergo apoptosis compared to that of WT, as shown by increased TUNEL-positive cells which co-stained against α-SMA, the representative marker of VSMC (Fig. 1A and 1B).
Increased apoptosis in eNOS−/− VSMCs
Cell death is an important factor for plaque rupture in atherosclerosis (10, 13). In particular, VSMC apoptosis in atherosclerotic plaque could be detrimental to plaque stability and increase the risk for thrombosis (10, 14). We observed an increased rate of apoptosis in aortic tissue from eNOS−/− mouse (Fig. 1) and thus investigated whether eNOS deficiency leads to induction of apoptosis in VSMCs utilizing TUNEL assay. While WT VSMCs showed negligible evidence of apoptosis, eNOS−/− VSMCs showed presence of apoptosis as indicated by TUNEL positive staining (Fig. 2A). Followed by serum deprivation, apoptosis was further increased in eNOS−/− VSMCs, as shown by increased TUNEL positive staining (Fig. 2A and 2B). To confirm eNOS deficiency-induced apoptosis, VSMCs were also subjected to Annexin V/propidium iodide (PI) staining and analyzed by flow cytometry. We observed enhanced apoptosis in eNOS−/− VSMCs, as shown by 14.9% annexin V positive cells, whereas it was only 3.4% in WT VSMC (Fig. 2C). eNOS−/− VSMCs exhibited a marked increase (4.4 fold; P < 0.05) compared with WT VSMCs in the apoptotic cell fraction assessed by FACs analysis of an annexin V staining assay (Fig. 2D), suggesting that VSMC apoptosis was promoted by eNOS deficiency.
Fig. 2.
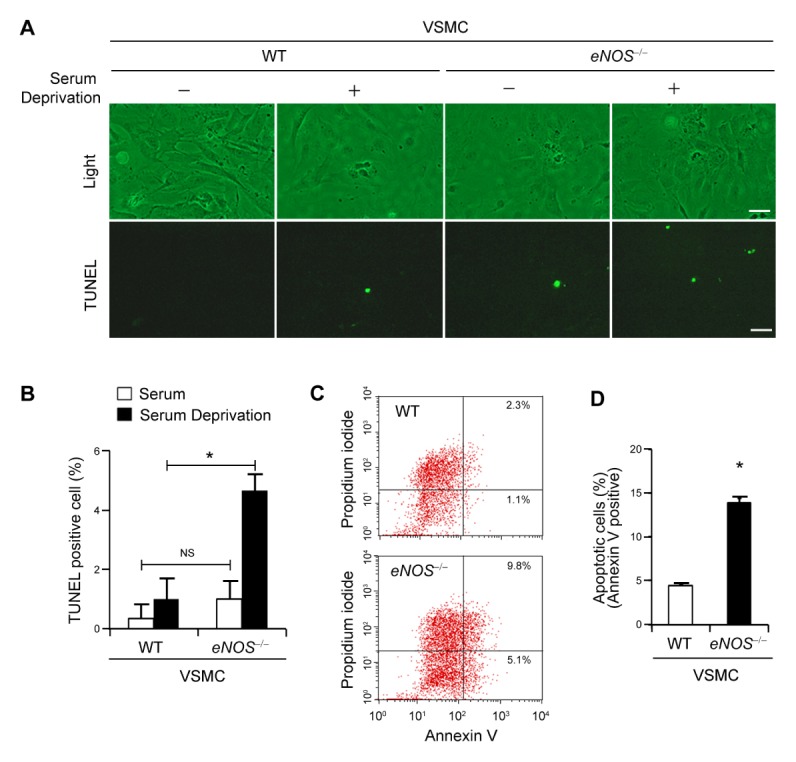
Enhanced apoptosis in eNOS−/− vascular smooth muscle cells (VSMCs). (A) Representative light microscopy (upper panels) and images of TUNEL staining (lower panels) showing apoptotic cells (stained in green) in VSMCs from WT and eNOS−/− mice in the presence or absence of serum deprivation. Scale bars, 100 μm. (B) The percentage of apoptosis was calculated as percent of TUNEL-positive cells out of total cells (mean ± SD). NS, not significant; *P < 0.05 compared to WT VSMCs incubated without serum. (C) Flow cytometry analysis of annexin-V and propidium iodide (PI) staining of apoptotic cells in VSMCs from WT and eNOS−/− mice. (D) Quantification of annexin-V positive apoptotic cells (annexin-V positive and PI negative + annexin-V and PI double positive cells). *P < 0.05 compared to WT VSMCs. P values were calculated using Kruskal-Wallis test (Fig. B) or Mann-Whitney test (Fig. D).
eNOS deficiency leads to elevated expression of aggrecan which induces apoptosis in VSMCs
Previous reports showed that deposition of proteoglycan is selectively enhanced in the intima of advanced atherosclerotic lesions (18, 19), and aggrecan induces apoptosis in chondrocytes and murine thymoma cells (20, 21). Thus, we next determined whether eNOS deficiency leads to accumulation of aggrecan in aortic tissue and, in turn, aggrecan induces apoptosis in VSMC. The major lipoprotein-binding proteoglycans, aggrecan (Acan), biglycan (Bgn), and versican (Vcan) showed higher mRNA expressions in eNOS−/− mice compared to those of WT in microarray expression profiling data (Fig. 3A). In particular, aggrecan mRNA expression was significantly higher in eNOS−/− mice and these results were confirmed using qPCR (Fig. 3B). Aggrecan protein expression was higher in aortic sinus tissue from eNOS deficient mouse compared with that of WT mouse, as evidenced by immunoblot assay (Fig. 3C) and increased aggrecan positive staining (Fig. 3D).
Fig. 3.
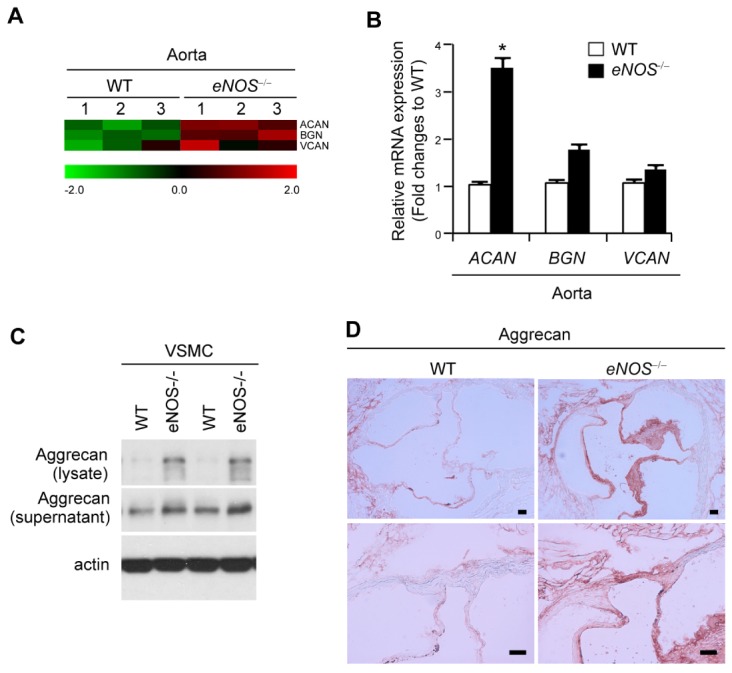
Elevated expression of aggrecan in aorta from eNOS−/− mouse. (A) Gene expression profiles of remodeling-associated genes. Transcripts that are upregulated and downregulated are shown in red and green, respectively. The columns represent the aorta samples from WT or eNOS−/− mice. (B) The levels of mRNA expression of aggrecan (Acan), biglycan (Bgn), and versican (Vcan) were compared in aorta from WT and eNOS−/− mice using qPCR. *P < 0.05 compared to WT control. P values were calculated using Kruskal-Wallis test. (C) Protein expression of aggrecan and β-actin were detected by western blotting in VSMCs. (D) Representative photographs of immunohistochemistry staining of aggrecan in aortic sinus sections from WT and eNOS−/− mice. Scale bars, 100 μm.
Enhanced apoptosis by aggrecan and NO-mediated aggrecan gene expression in eNOS−/− VSMCs
The administration of aggrecan modestly induced, at a concentration of 1 μg/ml in the presence of serum, whereas upon serum withdrawal aggrecan promoted VSMC apoptosis in a dose-dependent manner, as shown by TUNEL staining (Fig. 4A). Annexin V/PI staining also revealed the dose-dependent increase in apoptosis in response to aggrecan treatment in these cells (Fig. 4B). Treatment of VSMCs with DETA-NO, a NO donor, induced a dose-dependent decrease of gene expression of aggrecan (Fig. 4C). In contrast, L-NAME, an NOS inhibitor, increased aggrecan mRNA in a dose-dependent manner (Fig. 4D). Moreover, DETA-NO treatment decreased the number of apoptotic VSMCs (Fig. 4E). Altogether, these results suggest that eNOS deficiency may induce expression of aggrecan, which in turn promotes apoptosis in VSMC, thereby triggering atherosclerosis.
Fig. 4.

Induction of apoptosis by aggrecan and NO-mediated gene expression of aggrecan in VSMCs. (A) VSMCs from WT mice were treated with various concentrations of aggrecan, followed by induction of apoptosis with serum deprivation. The percentage of TUNEL-positive cells was determined using TUNEL staining. (B) Quantification of annexin-V positive apoptotic cells in VSMC treated with indicated concentrations of aggrecan. *P < 0.05 and **P < 0.005 compared to vehicle control. (C, D) VSMCs were treated with the indicated concentrations of DETA-NO (C) or L-NAME (D) and relative gene expression of ACAN was quantitated using qPCR. GAPDH was used as an internal control (mean ± SD). (E) VSMCs were treated with DETA-NO, followed by induction of apoptosis with serum deprivation. The percentage of TUNEL-positive cells was determined using TUNEL staining. *P < 0.05 and **P < 0.005 compared to vehicle control. P values were calculated using Kruskal-Wallis test.
DISCUSSION
To investigate the contribution of endothelial dysfunction in pathological conditions of atherosclerosis, we used a genetic model of eNOS deficiency and showed that several genes associated with lipid retention and apoptosis, implicated in the pathogenesis of atherosclerosis, were differentially expressed in eNOS−/− mouse, demonstrating the involvement of endothelium dysfunction in pathological pathways deriving atherosclerosis.
In atherosclerotic aorta of apoE-KO mouse, the expression level of a disintegrin and metalloprotease with thrombospondin motifs-5 (ADAMTS-5) was markedly reduced, which cleaves aggrecan, one of the major lipoprotein-binding proteoglycans (22). Such results raise the possibility that aggrecan may be accumulated in atherosclerotic plaques. Indeed, we found that aortic sinus tissue from eNOS deficient mice possessing atherosclerotic regions expressed higher amounts of aggrecan than WT mice (Fig. 3). In line with this result, selective deposition of proteoglycan such as aggrecan, biglycan, and veriscan was enhanced in the intima of advanced atherosclerotic lesions (18, 19) and apolipoprotein B-containing lipoproteins are trapped by these proteoglycans, initiating atherosclerosis in the arterial intima through macrophage accumulation and lipid core formation (22).
It has been reported that aggrecan induces apoptosis in chondrocytes (20) and murine thymoma cell line expressing human Fas (21). As VSMC apoptosis may cause plaque destabilization and rupture, accelerating atherosclerosis progression (10, 15), we determined whether aggrecan affects apoptosis in VSMC. We found aggrecan-mediated induction of apoptosis in VSMC (Fig. 4) for the first time as far as we know. This result suggests that VSMC apoptosis mediated by elevated aggrecan may be associated with atherosclerosis progression in eNOS deficient mice. In this context, Stevens et al. reported that NO enhances aggrecan degradation by inducing aggrecanase activity at posttranscriptional level (23), suggesting that NO deficiency is likely to induce accumulation of aggrecan protein. Nonetheless, we found that eNOS deficiency increases expression of aggrecan mRNA as evidenced by microarray data profiling and qPCR, as well as aggrecan protein as shown by IHC in animal model (Fig. 3). Taken together, these results suggest that eNOS deficiency may induce elevated expression of aggrecan which promotes apoptosis in VSMC, thereby triggering atherosclerosis.
We revealed several unrecognized genes potentially implicated in the pathogenesis of apoptosis and lipid retention, as well as atherosclerosis, by utilizing a genetic model of eNOS deficiency. We showed that eNOS deficiency may induce elevated expression of aggrecan in VSMCs, which promotes apoptosis and subsequent progression of atherosclerosis. This suggests that amplified VSMC apoptosis mediated by elevated level of aggrecan may be involved in pathological pathways of atherosclerosis due to endothelial dysfunction. These results may shed light on the mechanisms by which endothelium dysfunction contributes to the progression of atherosclerosis. Future research unraveling the regulation of aggrecan-mediated apoptosis in the cardiovascular system could provide new therapeutic avenues for prevention or control of atherosclerosis.
MATERIALS AND METHODS
Materials
Monoclonal anti-β-actin antibody (Ab) were purchased from Sigma–Aldrich (St Louis, MO, USA). Recombinant aggrecan was obtained from R and D (Minneapolis, MN, USA). DAPI (4′,6-diamidino-2-phenylindole) was obtained from Invitrogen (Carlsbad, CA, USA). Donkey FITC-conjugated anti-mouse IgG was purchased from Jackson Immunoresearch (West Grove, PA, USA). Anti-aggrecan polyclonal antibody was purchased from Abcam (Cambridge, MA, USA).
Cell culture and animals
eNOS−/− mice were purchased from Jackson laboratories (Bar Harbor, ME, USA). All animal experiments were performed according to protocols approved by the Institutional Committee for Use and Care of Laboratory animals. The studies with mice were conducted according to the protocol granted by the Ethics Committee of Ulsan University (Seoul, Korea) and conformed to the Guide for the Care and Use of Laboratory Animals published by NIH. The application form included a statement guaranteeing strict observation to the animal’s rights. Female mice were maintained in a pathogen free facility with 12 hr light/dark cycle. Murine VSMCs were isolated from the aorta by enzyme isolation and maintained in Dulbecco modified eagle medium (DMEM, Hyclone, South Logan, UT, USA) supplemented with 10% fetal bovine serum (Hyclone), penicillin (100 U · ml−1, Life Technologies, Carlsbad, CA, USA), and streptomycin sulfate (Life Technologies) (24).
Immunofluorescence staining and confocal microscopy
The animals were anesthetized and perfused with PBS, pH 7.4. The hearts were dissected, embedded in OCT compound (Sakura Finetek, Torrance, CA, USA), and snap-frozen in liquid nitrogen. Serial 5 μm sections were taken through the aortic sinus. Immunohistochemistry (IHC) was carried out utilizing the EnVisionTM G|2 Doublestain System and rabbit/mouse (DAB+/Permanent Red) in accordance to the manufacturer’s protocol (Dako, Carpinteria, CA, USA). Sections were fixed with paraformaldehyde and then permeabilized with 0.25% Triton X-100 in PBS. After being blocked with dual endogenous enzyme-blocking reagent, sections were incubated with primary antibody against aggrecan for 30 minutes at RT, and the polymer/HRP system was applied for detection. Corresponding rabbit sera were used as negative controls. Sections were stained with 4′,6-diamidino-2-phenylindole (DAPI) and α-smooth muscle actin (α-SMA). Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end-labeling (TUNEL) staining for apoptotic nuclei was performed using In Situ Cell Death Detection Kit (Roche, Germany) in accordance to the manufacturer’s instructions. Digital images were acquired using a confocal laser-scanning microscope (LSM 710, Carl Zeiss).
RNA isolation
Total RNA was extracted from the aorta of WT and eNOS−/− mice using RNeasy Lipid Tissue Kit (QIAGEN, Hilden, Germany). RNA quality was assessed by Agilent 2100 bioanalyzer using the RNA 6000 Nano Chip (Agilent Technologies, Amstelveen, Netherlands), and its quantity was determined by ND-1000 Spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE, USA).
Gene Expression Profiling (Microarray)
Global gene expression analyses of aorta specimens from WT and eNOS−/− mice were performed using Affymetrix GeneChipⓇ Mouse Gene 2.0 ST Array (Affymetrix, Santa Clara, CA, USA). Sample preparation was performed according to the instructions and recommendations provided by the manufacturer. In brief, 300 ng of total RNA from each sample was converted to double-strand cDNA using a random hexamer incorporating a T7 promoter. Amplified RNA was generated from the double-stranded cDNA template through an in vitro transcription reaction and purified with the Affymetrix sample cleanup module. cDNA was regenerated through random-primed reverse transcription using a dNTP mix which contained dUTP. The cDNA was then fragmented by UDG and APE 1 restriction endonucleases, and end-labeled by terminal transferase reaction incorporating a biotinylated dideoxynucleotide. Fragmented end-labeled cDNA was hybridized to the GeneChipⓇ Mouse Gene 2.0 ST arrays for 17 hr at 45°C as described in the Gene Chip Whole Transcript Sense Target Labeling Assay Manual (Affymetrix). After hybridization, the chips were stained, washed in a Genechip Fluidics Station 450 (Affymetrix) and scanned using a Genechip Array scanner 3000 7G (Affymetrix) (25).
Data processing and analysis
The intensity values of CEL files were normalized to remove bias between the samples, using the Robust Multi-Average algorithm implemented in the Affymetrix Expression Console software version 1.3.1. The normalized data were imported into the statistical programming environment R (Version3.0.2) for further analysis, such as density and MA plots with tools available from the Bioconductor Project (http://www.bioconductor.org). In order to classify the co-expression gene groups which have similar expression patterns, hierarchical clustering analysis was performed with the Multi Experiment Viewer software version 4.4 (http://www.tm4.org). The web-based tool Database for Annotation, Visualization, and Integrated Discovery was used to perform the biological interpretation of the differentially expressed genes. Genes were classified based on the information of the gene function in Gene ontology and KEGG pathway databases (http://david.abcc.ncifcrf.gov/home.jsp). The complete data set is available with NCBI GEO accession number GSE123855.
Quantitative real time PCR (qPCR)
Quantitative real-time PCR was performed to validate the microarray experiment with the same RNA used on the microarray, using a Power SYBR Green 1-Step Kit and the ABI 7000 Real Time PCR System (Applied Biosystems, Carlsbad, CA, USA) following manufacturer’s instructions. Gene expression was normalized to GAPDH gene and the quantity of target gene was calculated according to the comparative CT method.
Western blot assay
Cells were lysed, and protein lysates were resolved on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and electophoretically transferred to a polyvinyli-dene difluoride membrane (Bio-Rad, Hercules, CA, USA). Nonspecific interactions were blocked using 5% BSA solution in Tris–buffered saline (20 mM Tris/HCl, pH 7.6, 150 mM NaCl, and 0.1% Triton X-100) for 1 hr and membranes were then incubated with the indicated primary antibodies overnight at 4°C. Membranes were incubated with the appropriate secondary antibodies conjugated with horseradish peroxidase and immunoreactivity was detected by the use of an enhanced chemiluminescence detection kit (Millipore, Billerica, MA, USA).
TUNEL assay
Apoptosis of VSMCs from WT and eNOS−/− mice were induced with serum deprivation. VSMCs were also treated with various concentrations of aggrecan in the presence or absence of serum and TUNEL staining for apoptotic nuclei was performed.
Annexin V/propidium iodide (PI) staining
Apoptosis was detected by annexin-V and propidium iodide (PI) staining using the FITC Annexin-V Apoptosis Detection Kit I (BD Pharmingen, San Diego, CA, USA). Briefly, VSMCs were washed twice in cold PBS, and resuspended in 1× Annexin-V binding buffer (10 mM HEPES/NaOH, pH 7.4; 140 mM NaCl; 2.5 mM CaCl2). Cells were then stained with Annexin-V-FITC and PI, and incubated for 15 minutes at 4°C in the dark and analyzed with a FACScalibur flow cytometer and analyzed in CellQuest software.
Statistical analysis
All quantitative experiments were performed at least in triplicate and the data values are presented as the mean ± standard deviation. Mann-Whitney test (Fig. 1B and Fig. 2D) or Kruskal-Wallis test (Fig. 1C, 2B, 3B and 4) were used to determine significance. P value of less than 0.05 was considered statistically significant.
Supplementary Information
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea (NRF) MRC grant (2018R1A5A2020732) funded by the Korean government (MSIT) and by grant (2018-524) from the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 2.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 3.Mooradian DL, Hutsell TC, Keefer LK. Nitric oxide (NO) donor molecules: effect of NO release rate on vascular smooth muscle cell proliferation in vitro. J Cardiovasc Pharmacol. 1995;25:674–678. doi: 10.1097/00005344-199504000-00023. [DOI] [PubMed] [Google Scholar]
- 4.Ponnuswamy P, Schröttle A, Ostermeier E, et al. eNOS protects from atherosclerosis despite relevant superoxide production by the enzyme in apoE mice. PLoS One. 2012;7:e30193. doi: 10.1371/journal.pone.0030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flammer AJ, Anderson T, Celermajer DS, et al. The assessment of endothelial function: from research into clinical practice. Circulation. 2012;126:753–767. doi: 10.1161/CIRCULATIONAHA.112.093245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bogaty P, Hackett D, Davies G, Maseri A. Vasoreactivity of the culprit lesion in unstable angina. Circulation. 1994;90:5–11. doi: 10.1161/01.CIR.90.1.5. [DOI] [PubMed] [Google Scholar]
- 7.Cooke JP, Singer AH, Tsao P, Zera P, Rowan RA, Billingham ME. Antiatherogenic effects of L-arginine in the hypercholesterolemic rabbit. J Clin Invest. 1992;90:1168–1172. doi: 10.1172/JCI115937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 10.Clarke MC, Figg N, Maguire JJ, et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 11.Klouche M, Peri G, Knabbe C, et al. Modified atherogenic lipoproteins induce expression of pentraxin-3 by human vascular smooth muscle cells. Atherosclerosis. 2004;175:221–228. doi: 10.1016/j.atherosclerosis.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 12.Zhang MJ, Zhou Y, Chen L, et al. Impaired SIRT1 promotes the migration of vascular smooth muscle cell-derived foam cells. Histochem Cell Biol. 2016;146:33–43. doi: 10.1007/s00418-016-1408-9. [DOI] [PubMed] [Google Scholar]
- 13.Kockx MM, Herman AG. Apoptosis in atherosclerosis: beneficial or detrimental? Cardiovasc Res. 2000;45:736–746. doi: 10.1016/S0008-6363(99)00235-7. [DOI] [PubMed] [Google Scholar]
- 14.Martinet W, Kockx MM. Apoptosis in atherosclerosis: focus on oxidized lipids and inflammation. Curr Opin Lipidol. 2001;12:535–541. doi: 10.1097/00041433-200110000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Martinet W, De Meyer GR. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res. 2009;104:304–317. doi: 10.1161/CIRCRESAHA.108.188318. [DOI] [PubMed] [Google Scholar]
- 16.Littlewood TD, Bennett MR. Apoptotic cell death in atherosclerosis. Curr Opin Lipidol. 2003;14:469–475. doi: 10.1097/00041433-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Kuhlencordt PJ, Gyurko R, Han F, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 18.Strom A, Ahlqvist E, Franzen A, Heinegard D, Hultgardh-Nilsson A. Extracellular matrix components in atherosclerotic arteries of Apo E/LDL receptor deficient mice: an immunohistochemical study. Histol Histopathol. 2004;19:337–347. doi: 10.14670/HH-19.337. [DOI] [PubMed] [Google Scholar]
- 19.Talusan P, Bedri S, Yang S, et al. Analysis of intimal proteoglycans in atherosclerosis-prone and atherosclerosis-resistant human arteries by mass spectrometry. Mol Cell Proteomics. 2005;4:1350–1357. doi: 10.1074/mcp.M500088-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao L, Yang BB. Chondrocyte apoptosis induced by aggrecan G1 domain as a result of decreased cell adhesion. Exp Cell Res. 1999;246:527–537. doi: 10.1006/excr.1998.4335. [DOI] [PubMed] [Google Scholar]
- 21.Fujimoto T, Kawashima H, Tanaka T, et al. CD44 binds a chondroitin sulfate proteoglycan, aggrecan. Int Immunol. 2001;13:359–366. doi: 10.1093/intimm/13.3.359. [DOI] [PubMed] [Google Scholar]
- 22.Didangelos A, Mayr U, Monaco C, Mayr M. Novel role of ADAMTS-5 protein in proteoglycan turnover and lipoprotein retention in atherosclerosis. J Biol Chem. 2012;287:19341–19345. doi: 10.1074/jbc.C112.350785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stevens AL, Wheeler CA, Tannenbaum SR, Grodzinsky AJ. Nitric oxide enhances aggrecan degradation by aggrecanase in response to TNF-alpha but not IL-1beta treatment at a post-transcriptional level in bovine cartilage explants. Osteoarthritis Cartilage. 2008;16:489–497. doi: 10.1016/j.joca.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross R, Glomset J, Kariya B, Harker L. A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro. Proc Natl Acad Sci U S A. 1974;71:1207–1210. doi: 10.1073/pnas.71.4.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.