

Abstract
RAD51 recombinase plays a critical role in homologous recombination and DNA damage repair. Here we showed that expression of RAD51 is frequently upregulated in lung cancer tumors compared with normal tissues and is associated with poor survival (hazard ratio (HR) = 2, P = 0.0009). Systematic investigation of lung cancer cell lines revealed higher expression of RAD51 in KRAS mutant (MT) cells compared to wildtype (WT) cells. We further showed that MT KRAS, but not WT KRAS, played a critical role in RAD51 overexpression via MYC. Moreover, our results revealed that KRAS MT cells are highly dependent on RAD51 for survival and depletion of RAD51 resulted in enhanced DNA double strand breaks, defective colony formation and cell death. Together, our results suggest that mutant KRAS promotes RAD51 expression to enhance DNA damage repair and lung cancer cell survival, suggesting that RAD51 may be an effective therapeutic target to overcome chemo/radioresistance in KRAS mutant cancers.
Keywords: DNA damage repair, KRAS, Lung cancer, MYC, RAD51
INTRODUCTION
Lung cancer is the most common malignancy and leading cause of cancer-related death worldwide. Mutational activation of KRAS is among the most common oncogenic events in lung cancers (1–3). Predominantly occur in lung adenocarcinomas, KRAS mutations are associated with poor survival, accelerated metastatic progression, shortened time to relapse and resistance to multiple drugs (1–4). Despite recent advances in understanding the molecular mechanisms and personalized therapy, the oncogenic roles of MT KRAS oncogene in lung cancer are still unclear, yet effective therapies targeting KRAS remain to be developed (5).
RAD51 is an evolutionarily conserved recombinase which plays a critical role in homologous recombination (HR) repair of double-strand DNA breaks (DSBs) (6). RAD51 expression could be induced upon DNA damage stimuli and translocate between cytoplasm and nucleus (7). Disruption of RAD51 activity by mutations result in increased sensitivity to DNA damaging agents that create DSBs (8). Overexpression of RAD51 has been observed in several cancer types, such as breast (9) and pancreatic cancers (10, 11) and is associated with enhanced tumor progression and drug resistance. It has also been reported that elevated expression of RAD51 confers radioresistance and reduced survival in gliomas (12). Remarkably, KRAS-MT colorectal cancer cells are highly dependent on RAD51 for survival (13). Given the high frequency of KRAS mutations in lung adenocarcinomas and the putative link between KRAS and RAD51, we ask whether overexpression of RAD51 contributes to the unfavorable prognosis of lung cancer and the differentially response between patients bearing WT and MT KRAS.
To this end, using data from TCGA project, we first analyzed RAD51 expression and its association with prognostic survival in large cohorts of lung adenocarcinomas patients. We further performed a systematic analysis of RAD51 expression in numerous lung cancer cell lines with WT or MT KRAS. These data, together with further functional studies, revealed MT KRAS-dependent upregulation of RAD51, which is essential for DNA damage repair and survival of KRAS MT cells. Our results provided evidence in support of the role of RAD51 in lung cancer tumorigenesis and drug resistance, and suggested that RAD51 might serve as a potential biomarker and therapeutic target for KRAS MT lung cancer.
RESULTS
High expression of RAD51 is associated with poor survival
To examine the expression of RAD51 in lung adenocarcinomas, we analyzed RNA-seq data from large cohorts of patients from the TCGA project. We firstly examined the expression of RAD51 in lung adenocarcinomas tumors (n = 483) and normal lung tissues (n = 347). As shown in Fig. 1A, our results revealed that RAD51 expression is significantly elevated in tumor samples compared with normal tissues (P < 0.05), consistent with its role in promoting tumor development. We then asked whether higher expression of RAD51 is associated with advanced disease stages. As shown in Fig. 1B, the stage-dependent analysis suggested that RAD51 expression was lowest in stage I tumors, and was remarkably enhanced in advanced-stage tumors (P = 0.0005). In addition, we investigated the association between RAD51 expression and overall patient survival. Survival analysis using Kaplan-Meier and log rank test revealed poorer overall survival in lung adenocarcinomas patients with high RAD51 expression (Upper-Quartile, n = 120) as compared to patients with low RAD51 expression (Lower-Quartile, n = 120) (hazard ratio (HR) = 2, P = 0.0009). Together, our data showed that expression of RAD51 protein is enhanced in lung cancer cells and is particularly associated with advanced disease stages and poor survival, supporting its putative oncogenic role in lung adenocarcinomas.
Fig. 1.
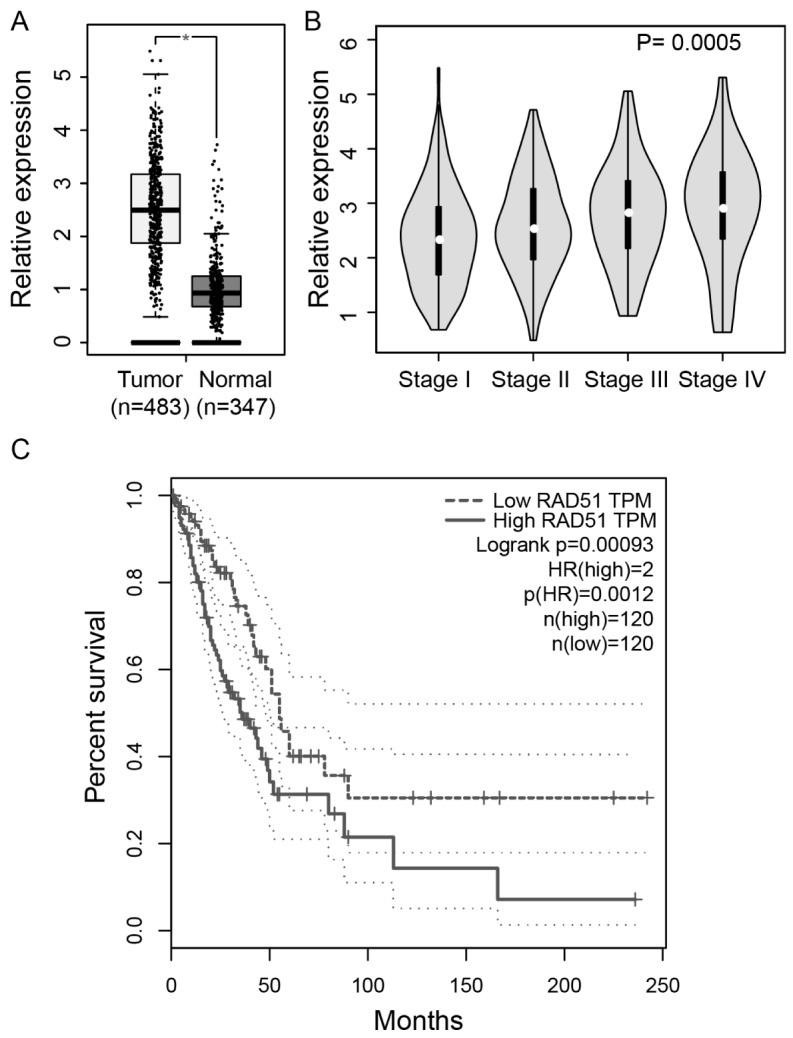
Association between RAD51 expression and survival. (A) Expression levels of RAD51 in 483 lung adenocarcinomas tumor samples and 347 normal tissues. *P < 0.05. (B) Expression levels of RAD51 in lung adenocarcinomas tumor samples stratified by tumor stages. (C) Kaplan–Meier analysis of lung adenocarcinoma-specific overall survival of patients with high and low expression of RAD51. Upper-Quartile (n = 120) and Lower-Quartile (n = 120) patients were used for the analysis. Dotted line indicates 95% confidence interval. Hazard ratio (HR) and P-value are shown.
Enhanced expression of RAD51 in KRAS mutant lung cancer cell lines
KRAS mutations are the most frequently oncogene aberrations in lung adenocarcinoma patients and is associated with therapeutic resistance and poor survival. It has been shown that DNA damage repair by homologous recombination (HR) pathway was enhanced in KRAS MT colorectal cancer cells through hyperactivation of c-MYC (13). We thus asked whether enhanced expression of RAD51 protein is associated with KRAS mutation in lung cancer cells. To this end, we used realtime PCR to investigate RAD51 expression in normal lung fibroblasts (n = 2) and lung cancer cell lines bearing wildtype (WT, n = 7) or mutant KRAS (MT, n = 8) (Fig. 2A). As shown in Fig. 2B, compared to normal lung fibroblasts, higher expression of RAD51 was observed in lung cancer cell lines with either WT KRAS or MT KRAS. In particular, KRAS MT cells exhibited even higher expression of RAD51 compared to KRAS WT cells (P < 0.05). The results were further validated by western blot (Fig. 3C, D), indicating that MT KRAS is associated with overexpression of RAD51 in lung cancer cells.
Fig. 2.

High expression of RAD51 in KRAS mutant cells. (A) Realtime PCR analysis of RAD51 expression levels in normal lung fibroblasts and lung cancer cell lines containing WT or MT KRAS. Data represents Mean ± SEM of three replicates. (B) Summary of data in (A), each dot indicates mean expression of RAD51 in one cell line. (C) Western blot analysis of RAD51 in KRAS WT and MT cell lines. (D) Quantification of data in (C), each dot indicates protein expression of RAD51 in one cell line. *P < 0.05.
Fig. 3.

Mutant KRAS induces overexpression of RAD51. (A) Western blot analysis of RAD51 expression upon KRAS knockdown in KRAS MT cell lines A549 and LU99A, or KRAS WT cell lines NCIH2023 and NCIH3122 cells. Two different siRNAs targeting KRAS were used. (B) Western blot analysis of RAD51 expression upon transient expression of FLAG-tagged WT KRAS or MT KRAS in A549 cells and NCIH3122 cells. (C) Western blot analysis of RAD51 expression upon MYC knockdown in A549 cells. Two different siRNAs targeting MYC were used. (D) Western blot analysis of RAD51 expression upon MYC or control knockdown in combination with overexpression of FLAG-tagged MT KRAS or control in A549 cells.
Mutant KRAS drives overexpression of RAD51
We next asked whether overexpression of RAD51 is directly linked to MT KRAS in lung cancer cells. To this end, we used siRNAs to knockdown KRAS in two KRAS MT (A549 and LU99A) and two WT cell lines (NCIH2023 and NCIH3122) and examined changes in RAD51 expression. As shown in Fig. 3A, our results revealed that depletion of KRAS resulted in remarkable reduction of RAD51 expression in KRAS MT cells but not in KRAS WT cells. Furthermore, A549 cells and NCIH3122 cells were transfected with empty vector or vectors expression FLAG-tagged, WT KRAS (FLAG-KRASWT) or MT KRAS bearing the G13D oncogenic mutation (FLAG-KRASG13D). As shown in Fig. 3B, western blot analysis revealed equal amount of WT and MT KRAS expression following transfection. However, overexpression of KRASG13D, but not KRASWT, resulted in significantly elevated RAD51 expression in both cell lines (all P < 0.01, Fig. 3A), and a more dramatic effect was observed in NCIH3122 cells, probably because they have a WT KRAS background and low expression of endogenous RAD51. In contrast, signaling pathways inducing RAD51 expression might be readily activated by endogenous MT KRAS in A549 cells, and the effect of exogenous MT KRAS might thus not be as strong as in KRAS WT cells. These results are consistent with the MT, but not the WT, KRAS being important in inducing overexpression of RAD51 in lung cancer cells. The intimate association of KRAS mutation and RAD51 overexpression thus provide a putative mechanism underlying the role of KRAS mutations in hyperactivating homologous recombination repair (14, 15).
Previous studies suggested that KRAS mutation is associated with hyperactivation of MYC, which plays a critical role in regulating expression of genes essential for KRAS-driven cancer (16–18).
We thus examined the role of MYC in regulating RAD51 expression in KRAS MT lung cancer cells. As shown in Fig. 3C, MYC knockdown results in downregulation of RAD51 in KRAS MT A549 cells. In addition, as shown in Fig. 3D, MYC knockdown abolished RAD51 upregulation induced by transient overexpression of MT KRAS. These data supported the hypothesis that MT KRAS may affect the DNA repair process through upregulation of RAD51 via MYC in lung adenocarcinomas cells, consistent with its role as an independent risk factor for radioresistance and unfavorable prognosis.
KRAS mutant cells are more dependent on RAD51 for survival
To examine the function of RAD51 in regulating DNA damage checkpoint recovery and lung cancer survival, we used shRNAs to obtain stable knockdown of RAD51 in NCIH3122 and A549 cells. As shown in Fig. 4A, western blot showed efficient knockdown of RAD51 in both cell lines. Remarkably, RAD51 knockdown resulted in accumulation of histone H2AX phosphorylation (γH2AX), a sensitive molecular marker of DNA damage, in both cell lines, indicating that RAD51 is essential for repairing of DNA double-strand breaks (Fig. 4A). A more dramatic accumulation of γH2AX was observed in A549 cells compared to NCIH3122, indicative of a more essential role of RAD51 in KRAS mutant cells. Furthermore, as shown in Fig. 4A, our results revealed that knockdown of RAD51 selectively induced cleavage of PARP1, a hallmark of apoptotic cell death. We thus further examined apoptosis and cell viability by colony formation assay and flow cytometry (Fig. 4B, C). Consistent with the western blot results, RAD51-depleted A549 cells exhibited more dramatic decrease in colony-forming ability and increase in apoptotic cell population than NCIH3122 with RAD51 knockdown. We thus suggested that KRAS MT lung cancer cells are highly dependent on RAD51 for DNA damage repair and survival, providing important insights into the development of novel therapies to counteract the oncogenic effects of KRAS.in facilitating tumor cells to survive the DNA damage inflicted by chemotherapy agents.
Fig. 4.

KRAS mutant cells are more dependent on RAD51 for survival. (A) Western blot of RAD51, γH2AX and PARP1 upon stable knockdown of RAD51 in A549 or NCIH3122 cells. (B) Colony formation assay of A549 or NCIH3122 cells with stable knockdown of RAD51. Data represents Mean ± SEM of three replicates. **P < 0.01. (C) Flow cytometry analysis of A549 or NCIH3122 cells with stable knockdown of RAD51 stained with Annexin V/PI. The percentage of apoptotic cells are shown.
DISCUSSION
Activating mutations of KRAS are frequently associated with some of the deadliest forms of cancer, however, the need for developing efficient KRAS inhibitors remain unmet (2, 3, 14). Accordingly, underlying the molecular mechanisms of oncogenic KRAS signaling and drug response KRAS in lung cancer patients could help to identify novel targeted agents and combinations that may allow a tailored patient management. In the current study, we reported overexpression of RAD51 driven by mutant KRAS in lung cancer cells. In addition, we provided evidence that KRAS mutant lung cancer cells are more dependent on RAD51 for DNA damage repair and survival than wildtype cells. We thus suggest that the KRAS-RAD51 cascade could be used as a negative prognostic factor for unfavorable survival and resistance to chemo/radiotherapies. This may provide important insights into personalized therapies in lung cancer management.
Previous studies suggested that oncogenic KRAS mutations induce DNA damage and genotoxic stress (13, 19). Consistent with this notion, KRAS mutant tumor cells always contain pre-malignant lesions (e.g., mutations or deletions in ATM, Chk2, and p53) that inactivate DNA damage response and cell cycle checkpoint (20–23), which normally lead to cellular senescence and/or cell death. According, deregulation of canonical DNA damage response signaling, as well as repression of damage-induced checkpoint, might be critical prerequisites for malignant transformation. In this regard, RAD51 might function as a key regulator that suppresses KRAS oncogene-induced genotoxic stress and/or allow fast repair of the damaged DNA to avoid activation of the canonical DNA damage response, which is deleterious to the tumor cells. Consistent with this notion, our results revealed an increase in double strand breaks marked by γH2AX accumulation following RAD51 depletion. We thus hypothesized that high expression of RAD51 driven by mutant KRAS might at least partially contribute to drug resistance in lung cancer cells. Accordingly, targeting RAD51, and probably other unknown druggable targets in the same signaling cascade, could provide novel therapeutic interventions to enhance the efficiency of conventional and targeted chemotherapeutic regimes for treatment of mutant KRAS-driven lung cancers (24, 25).
One interesting question is how mutant KRAS induces expression of RAD51. Previous studies identified MYC as a critical downstream effector that contributes to the malignant transformation of KRAS-driven cancers (17, 26). Remarkably, as an important transcription factor, MYC plays a critical role regulating expression of multiple DNA damage repair genes, including RAD51, XRCC2, BRCA1, BRCA2, DNA-PKcs, XRCC4 and KU70 (27). Chromatin immunoprecipitation studies revealed that MYC directly binds to RAD51 promoter (27), and the expression levels of MYC of RAD51 are highly correlated in a panel of cancer cell lines. In addition, it has been shown that forced expression of MYC in resulted in RAD51 upregulation in colorectal cancer cells (13). Accordingly, MYC-RAD51 signaling may be a potential therapeutic target to sensitize cancer cells to DNA damage or to prevent genomic instability.
Together, our data revealed a mechanistic link between mutant KRAS and RAD51 overexpression, which in turn promotes DNA damage repair and survival in lung cancer cells. These data thus provide insights into a promising therapeutic vulnerability for the otherwise nondruggable KRAS mutations, which warrants further clinic investigation in lung cancer patients.
MATERIALS AND METHODS
Cell culture
KRAS wildtype lung cancer cell lines (NCIH3122, NCIH2023, HCC827, NCIH1755, NCIH1299, NCIH1781 and NCIH1563) and KRAS wildtype lung cancer cell lines (CORL23, LU65, NCIH460, NCIH1734, NCIH358, SW900, A549 and LU99A) were cultured in RPMI medium (Gibco Co., Gaithersburg, MD, USA) containing penicillin/streptomycin and 15% heat-inactivated FBS (Gibco Co., Gaithersburg, MD, USA), as recommended by ATCC, at 37°C, 5% CO2, and 95% humidity. Normal lung fibroblast cells (HEL-299 and MRC-5) were cultured in EMEM medium (Gibco, Gaithersburg, MD, USA) containing penicillin/streptomycin and 10% FBS.
Western blot
Whole cell extracts were prepared using lysate buffer containing 10 mM TrisHCl, pH7.4, 150 mM NaCl,1 mM EDTA, 10% glycerol, 1% Triton X100, 40 mM NaVO4, 0.1% SDS, and 1x protease inhibitors (Roche, Indianapolis, IN, USA). The following antibodies were used in this study: anti-RAD51 (Abcam Co., Milton, Cambridge, UK), anti-FLAG (Sigma-Aldrich Co., St. Louis, MO, USA), anti-GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-γH2AX (Cell Signaling Co., Danvers, MA, USA). Western blot signals were detected by enhanced chemiluminescent reagent (Promega Co., Madison, WI, USA).
Realtime PCR
Total RNAs were isolated from different lung cancer cell lines using Trizol reagent (Thermo Fisher Scientific Co., Waltham, MA, USA). Reverse transcription was performed using using M-MLV reverse transcriptase as per manufacturer’s instructions (Promega Co., Madison, WI, USA). Realtime PCR was carried out using SYBRⓇ Green (Thermo Fisher Co., Chelmsford, MA) in an Applied Biosystems QuantStudio 6 thermal cycler (Applied Biosystems Co., Waltham, MA, USA). Reaction profiles were set up as follows: initial denaturation at 95°C for 10 min followed by 40 cycles of 95°C for 15 s, 63°C for 1 min, and 72°C for 30 s. Relative transcript amounts were calculated using the threshold cycle method (ΔΔCT) using GAPDH as the internal control. The following primers were used: RAD51_forward, 5′-TCTCTGGCAGTGATGTCCTGGA-3′; RAD51_reverse, 5′-TAAAGGGCGGTGGCACTGTCTA-3′; GAPDH_forward, 5′-CTGTTGCTGTAGCCAAATTCGT-3′; GAPDH_reverse, 5′-ACC CACTCCTCCACCTTTGAC-3′.
Overexpression and knockdown
The human wild-type KRAS mRNA sequence was cloned from the pCMV6-Myc-DDK-tagged-KRAS vector (OriGene Technologies Co., Rockville, MD, USA) into the 3XFLAG-CMV vector to create FLAG-KRAS constructs. G13D mutation was generated by site-directed mutagenesis kit (Qiagen Co., Hilden, Germany). Small interfering RNAs (siRNA) targeting KRAS, MYC or scrambled control siRNAs were purchased from Dharmacon Co. (Lafayette, CO, USA). KRAS expression vectors or siRNAs were transfected into the cells using the Lipofectamine2000 transfection reagent (Thermo Fisher Scientific Co., Waltham, MA, USA) according to the manufacturer’s instructions. For stable knockdown of RAD51, lung cancer cells were transfected with the SMARTvector Lentiviral vectors encoding RAD51 small hairpin RNA (shRNAs) or scramble control shRNAs (Dharmacon Co., Lafayette, CO, USA). Single clones were selected using puromycin selection and validated by western-blot. Sequences of siRNAs were:
KRAS_1_sense: 5′-GUGCAAUGAAGGGACCAGUA-3′, KRAS_1_antisense: 5′-UACUGGUCCCUCAUUGCAC-3′; KRAS_2_sense: 5′-GCAAGUAGUAAUUGAUGGA-3′, KRAS_1_antisense: 5′-UCCAUCAAUUACUACUUGC-3′; MYC_1_sense: 5′-GCGA CGAGGAGGAGAACUUCUACCA-3′, MYC_1_antisense: 5′-U GGUAGAAGUUCUCCUCCUCGUCGCAG-3′; MYC_2_sense: 5′-UUUGUGUUUCAACUGUUCUCGUCGU-3′, MYC_2_antisense: 5′-ACGACGAGAACAGUUGAAACACAAA-3′, Control: _sense: 5′-UUCUCCGAACGUGUCACGU-3′, control_antisense: 5′-ACGUGACACGUUCGGAGAA-3′. The target sequence of RAD51 shRNA: 5′-GAAGGAAAGGCCATGTACATTG-3′ and the scrambled target sequence: 5′-TTCTCCGAACGTGTCACGT-3′.
Colony formation assays and viability
The stable RAD51 knockdown lung cells were seeded in semisolid agar medium (2,000 cells/well) in a 6-well plate in triplicate. After 15 days, colonies were stained with crystal violet (Sigma-Aldrich, St. Louis, MO, USA) and counted using an inverted microscope. The apoptotic rates of the lung cancer cells were examined using flow cytometry and Annexin V/PI staining kit (Thermo Fisher Scientific, Waltham, MA, USA).
Gene expression and Kaplan–Meier survival analysis
Expression of RAD51 in lung cancer tumors, its association with disease stages, and the impact on prognostic survival of patients (Kaplan–Meier survival analysis) were analyzed using GEPIA (http://gepia.cancer-pku.cn/) (28), a web-based interactive toolkit for cancer gene expression profiling datasets with default parameters.
Statistical analysis
Western blot quantification was performed using ImageJ 1.51 (NIH). Statistical analyses were performed using GRAPHPAD PRISM v6.0 (GraphPad Software, LaJolla, CA, USA). Student’s t-tests and the P-values were calculated as indicated in figure legends. Asterisks indicate significant differences (*P < 0.05; **P < 0.01; ***P < 0.001).
ACKNOWLEDGEMENTS
This study was supported by Foundation of Science and Technology Department of Jiangxi Province (GJJ160266).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Blandin Knight S, Crosbie PA, Balata H, Chudziak J, Hussell T, Dive C. Progress and prospects of early detection in lung cancer. Open Biol. 2017;7:170070. doi: 10.1098/rsob.170070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaufman J, Stinchcombe TE. Treatment of KRAS-Mutant Non-Small Cell Lung Cancer: The End of the Beginning for Targeted Therapies. JAMA. 2017;317:1835–1837. doi: 10.1001/jama.2017.3436. [DOI] [PubMed] [Google Scholar]
- 3.Matikas A, Mistriotis D, Georgoulias V, Kotsakis A. Targeting KRAS mutated non-small cell lung cancer: A history of failures and a future of hope for a diverse entity. Crit Rev Oncol Hematol. 2017;110:1–12. doi: 10.1016/j.critrevonc.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res. 2015;21:1819–1827. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 6.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zafar F, Seidler SB, Kronenberg A, Schild D, Wiese C. Homologous recombination contributes to the repair of DNA double-strand breaks induced by high-energy iron ions. Radiat Res. 2010;173:27–39. doi: 10.1667/RR1910.1. [DOI] [PubMed] [Google Scholar]
- 8.Su F, Mukherjee S, Yang Y, et al. Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep. 2014;9:1387–1401. doi: 10.1016/j.celrep.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alshareeda AT, Negm OH, Aleskandarany MA, et al. Clinical and biological significance of RAD51 expression in breast cancer: a key DNA damage response protein. Breast Cancer Res Treat. 2016;159:41–53. doi: 10.1007/s10549-016-3915-8. [DOI] [PubMed] [Google Scholar]
- 10.Nagathihalli NS, Nagaraju G. RAD51 as a potential biomarker and therapeutic target for pancreatic cancer. Biochim Biophys Acta. 20111816:209–218. doi: 10.1016/j.bbcan.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Maacke H, Jost K, Opitz S, et al. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19:2791–2795. doi: 10.1038/sj.onc.1203578. [DOI] [PubMed] [Google Scholar]
- 12.Balbous A, Cortes U, Guilloteau K, et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer. 2016;16:604. doi: 10.1186/s12885-016-2647-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalimutho M, Bain AL, Mukherjee B, et al. Enhanced dependency of KRAS-mutant colorectal cancer cells on RAD51-dependent homologous recombination repair identified from genetic interactions in Saccharomyces cerevisiae. Mol Oncol. 2017;11:470–490. doi: 10.1002/1878-0261.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roman M, Baraibar I, Lopez I, et al. KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol Cancer. 2018;17:33. doi: 10.1186/s12943-018-0789-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eder S, Arndt A, Lamkowski A, et al. Baseline MAPK signaling activity confers intrinsic radioresistance to KRAS-mutant colorectal carcinoma cells by rapid upregulation of heterogeneous nuclear ribonucleoprotein K (hnRNP K) Cancer Lett. 2017;385:160–167. doi: 10.1016/j.canlet.2016.10.027. [DOI] [PubMed] [Google Scholar]
- 16.Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22:2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ischenko I, Zhi J, Hayman MJ, Petrenko O. KRAS-dependent suppression of MYC enhances the sensitivity of cancer cells to cytotoxic agents. Oncotarget. 2017;8:17995–18009. doi: 10.18632/oncotarget.14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perera D, Venkitaraman AR. Oncogenic KRAS triggers MAPK-dependent errors in mitosis and MYC-dependent sensitivity to anti-mitotic agents. Sci Rep. 2016;6:29741. doi: 10.1038/srep29741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grabocka E, Commisso C, Bar-Sagi D. Molecular pathways: targeting the dependence of mutant RAS cancers on the DNA damage response. Clin Cancer Res. 2015;21:1243–1247. doi: 10.1158/1078-0432.CCR-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dietlein F, Kalb B, Jokic M, et al. A Synergistic Interaction between Chk1- and MK2 Inhibitors in KRAS-Mutant Cancer. Cell. 2015;162:146–159. doi: 10.1016/j.cell.2015.05.053. [DOI] [PubMed] [Google Scholar]
- 21.Zhao L, Pu X, Ye Y, Lu C, Chang JY, Wu X. Association between genetic variants in DNA double-strand break repair pathways and risk of radiation therapy-induced pneumonitis and esophagitis in non-small cell lung cancer. Cancers (Basel) 2016;8:E23. doi: 10.3390/cancers8020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scarbrough PM, Weber RP, Iversen ES, et al. A cross-cancer genetic association analysis of the DNA repair and DNA damage signaling pathways for lung, ovary, prostate, breast, and colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2016;25:193–200. doi: 10.1158/1055-9965.EPI-15-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothschild SI, Gautschi O, Lara PN, Jr, Mack PC, Gandara DR. Biomarkers of DNA repair and related pathways: significance in non-small cell lung cancer. Curr Opin Oncol. 2011;23:150–157. doi: 10.1097/CCO.0b013e328341ee38. [DOI] [PubMed] [Google Scholar]
- 24.O’Grady S, Finn SP, Cuffe S, Richard DJ, O’Byrne KJ, Barr MP. The role of DNA repair pathways in cisplatin resistant lung cancer. Cancer Treat Rev. 2014;40:1161–1170. doi: 10.1016/j.ctrv.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 25.Chen P, Li J, Chen YC, et al. The functional status of DNA repair pathways determines the sensitization effect to cisplatin in non-small cell lung cancer cells. Cell Oncol (Dordr) 2016;39:511–522. doi: 10.1007/s13402-016-0291-7. [DOI] [PubMed] [Google Scholar]
- 26.Soucek L, Whitfield JR, Sodir NM, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504–513. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luoto KR, Meng AX, Wasylishen AR, et al. Tumor cell kill by c-MYC depletion: role of MYC-regulated genes that control DNA double-strand break repair. Cancer Res. 2010;70:8748–8759. doi: 10.1158/0008-5472.CAN-10-0944. [DOI] [PubMed] [Google Scholar]
- 28.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]