

Abstract
Hydrogen sulfide (H2S) is an endogenous gaseous signaling molecule that plays important roles in the cardiovascular system. In our previous studies, we demonstrated that H2S regulates lipid metabolism. In the present study, we aimed to explore the mechanisms through which H2 regulates lipid metabolism in HepG2 cells in vitro. Treatment of the HepG2 cells with H2S inhibited the expression of proprotein convertase subtilisin/kexin type 9 (PCSK9) and increased the level of low-density lipoprotein receptor (LDLR) in a time- and dose-dependent manner. The knockdown of PCSK9 by siRNA effectively increased the levels of LDLR and 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate-labeled LDL (DiI-LDL) uptake in the H2S-treated HepG2 cells. Furthermore, the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)-sterol regulatory element binding proteins 2 (SREBP-2) signaling pathway was confirmed to be involved in H2S-regulated PCSK9 expression. Notably, the HepG2 cells were incubated with 30% serum and DiI-LDL for 24 h, and the results revealed that H2S increased lipid uptake, but caused no increase in lipid accumulation. On the whole, the findings of this study demonstrate that H2S is involved in the regulation of lipid metabolism in HepG2 cells through the regulation of the expression of PCSK9 via the PI3K/Akt-SREBP-2 signaling pathway. To the very best of our knowledge, this study is the first to report that H2S can regulate the expression of PCSK9.
Keywords: hydrogen sulfide, lipid metabolism, proprotein convertase subtilisin/kexin type 9, low-density lipoprotein receptor, phosphoinositide 3-kinase/protein kinase B, sterol regulatory element-binding protein 2, HepG2 cells
Introduction
Hydrogen sulfide (H2S), an endogenous gaseous signaling molecule, performs important regulatory functions, such as vascular toning, leukocyte adhesion and smooth muscle cell proliferation, in the cardiovascular system. Recently, Li et al demonstrated that H2S reduces atherosclerosis by regulating lipid metabolism (1). Exogenous H2S can mitigate fatty liver and reduce triglyceride and total cholesterol levels in obese mice (2). Cytathiohine β-synthase (a key enzyme for the biosynthesis of H2S) knockout mice have been reported to exhibit higher low-density lipoprotein cholesterol (LDL-C) and lower plasma levels of high-density lipoprotein cholesterol than normal mice (3). Additionally, patients with atherosclerosis exhibit reduced serum H2S concentrations (4). However, the precise mechanisms through which H2S regulates lipid metabolism remain unclear.
It is well established that proprotein convertase subtilisin/kexin type 9 (PCSK9) plays an important role in lipid metabolism and atherosclerosis (5). PCSK9 alters LDL-C concentrations by promoting hepatic low-density lipoprotein receptor (LDLR) degradation and decreasing the hepatic clearance of plasma LDL-C levels (6). Numerous factors regulate PCSK9 expression. For example, statin treatment leads to an increased transcription of PCSK9 (7), in contrast to berberine treatment, which decreases PCSK9 expression (8). However, to date, whether H2S is involved in the regulation of hepatic PCSK9 expression remains unknown.
In the present study, we thus aimed to examine the effects of H2S on the expression of PCSK9 in HepG2 cells and to elucidate the mechanisms through which H2S regulates lipid metabolism.
Materials and methods
Reagents
Sodium hydrosulfide (NaHS) and Oil Red O were purchased from Sigma-Aldrich (St. Louis, MO, USA). Specific monoclonal anti-PCSK9 (cat. no. 55206), anti-LDLR (cat. no. 66414), anti-SREBP-2 (cat. no. 557037) and anti-SREBP-1-c (cat. no. 66875) antibodies were purchased from Proteintech (Rosemont, IL, USA). Specific monoclonal anti-protein kinase B (Akt) antibody (cat. no. BM4400) was purchased from Wuhan Boshide Bioengineering Co. (Wuhan, China). Specific monoclonal anti-phosphorylated Akt antibody (cat. no. YM3621) was purchased from ImmunoWay Biotechnology (Plano, TX, USA). Anti-β-actin antibody (cat. no. LCA01) was purchased from Aijia Biological Technology Co., Ltd. (Changsha, China). Cy3-conjugated goat anti-rabbit IgG secondary antibody (cat. no. E031620) was purchased from EarthOx Life Sciences (Millbrae, CA, USA). Dulbecco's modified Eagle's medium (DMEM) was purchased from HyClone (Logan, UT, USA). Fetal bovine serum (FBS) was purchased from Hangzhou Sijiqing Biocompany (Hangzhou, China). The Hoechst Staining kit was purchased from Beyotime Institute of Biotechnology (Jiangsu, China). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate-labeled LDL (DiI-LDL) was purchased from Haoyuan Biological Technology Co., Ltd. (Guangzhou, China). siRNAs were synthesized by Ruibo Biotechnology Co., Ltd. (Guangzhou, China).
Cells and cell culture
The HepG2 human liver cancer cell line was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and maintained in tissue culture bottles with DMEM supplemented with 10% heat-inactivated FBS at 37°C under an atmosphere of 5% CO2 and 95% air. The cell culture medium was replaced with fresh medium 3 times a week.
Oil Red O staining protocol
Oil Red O staining was used to quantify intracytoplasmic lipid droplets. The HepG2 cells were seeded in a 6-well plate covered with coverslips. After the cells had become adherent, they were incubated for 12 h at 37°C in serum-free medium, treated with various concentrations (0, 50, 100 and 200 µmol/l) of NaHS (a H2S donor) for 0.5 h, and subsequently incubated with 30% FBS for 24 h. The HepG2 cells were first fixed with 4% paraformaldehyde for 30 min at room temperature, and washed 3 times with distilled water. Subsequently, the cells were stained with Oil Red O at room temperature for 15 min. The cells were then washed 3 times with distilled water, and nuclear counterstaining was performed with hematoxylin and eosin (Beyotime Institute of Biotechnology, Jiangsu, China) staining for 30 sec at room temperature. Lipid droplets were observed by light microscopy (Olympus, Tokyo, Japan) and then imaged using Image Pro-Plus 6.0 software (Media Cybernetics, Rockville, MD, USA).
DiI-LDL uptake assay
The HepG2 cells were seeded in 24-well plates. Following treatment with various concentrations of NaHS (0, 50, 100 and 200 µmol/l) for 24 h, the cells were rinsed twice with phosphate-buffered saline (PBS). Subsequently, the cells were incubated with 25 µg/ml DiI-LDL for 4 h at 37°C, followed by fixation in 3% paraformaldehyde for 20 min. The nuclei were counterstained with Hoechst 33258 (250 nM; Beyotime Institute of Biotechnology) for 5 min at room temperature. Immunofluorescence was visualized using a Nikon TE2000-U fluorescence microscope (Nikon, Tokyo, Japan).
Immunofluorescence assay
The HepG2 cells were plated in 24-well plates. Following treatment with various concentrations of NaHS (0, 50, 100 and 200 µmol/l) for 24 h, the cells were rinsed 3 times with PBS, fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.5% Triton X-100 in PBS for 30 min, and incubated with anti-LDLR antibody (1:1,000) overnight at 4°C. The cells were rinsed 3 times for 5 min with PBS, followed by incubation with the appropriate fluorophore-conjugated secondary antibody (SA012; 1:1,000; Aijia Biological Technology Co., Ltd.) for 2 h. The nuclei were counterstained with Hoechst 33258 (250 nM; Beyotime Institute of Biotechnology). Immunofluorescence was visualized using a Nikon TE2000-U fluorescence microscope (Nikon).
Western blot analysis
The cells were washed twice with PBS and incubated on ice with cell lysis buffer and phenylmethanesulfonyl fluoride for 30 min, followed by cell lysate collection and centrifugation at 10,000 x g for 10 min at 4°C. The BCA protein assay kit was used for the quantification of total protein in cell lysates (Beyotime Institute of Biotechnology) according to the manufacturer's instructions. Proteins were separated by 12% SDS-PAGE and electrotransferred onto a polyvinylidene fluoride membrane. The membrane was incubated in a blocking buffer (0.1% Tween-20, 150 mM NaCl, 20 mM Tris base, pH 7.6 and 5% non-fat milk) for 2 h at 37°C and subsequently with the appropriate primary antibodies (PCSK9, 1:400; LDLR, 1:1,000; SREBP-2, 1:5,000; SREBP-1c, 1:2,000; Akt, 1:400; phosphorylated Akt, 1:2,000; and β-actin, 1:1,000) in blocking buffer at 4°C overnight. The membranes were then incubated with anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibodies (SA009 and SA001; 1:1,000; Aijia Biological Technology Co., Ltd.) for 2 h. Proteins were visualized by chemiluminescence using AuraECL (Aijia Biological Technology Co., Ltd.) following the manufacturer's instructions. Image Pro-Plus 6.0 software (Media Cybernetics) was used to analyze relative protein band density.
siRNA transfection
The PCSK9 mRNA sequence was retrieved from GenBank (no. NM 199253). The target sequences of PCSK9 were as follows: siPCSK9 sense strand, 5′-GGC AGA GAC UGA UCC ACU UdT dT-3′ and siPCSK9 antisense strand, 5′-AAG UGG AUC AGU CUCU GCC TdT d-3′. The sequences of the negative control siRNA were as follows: Sense strand, 5′-UAU AGC UGU CUC GAG CAA GdT dT-3′ and antisense strand, 5′-CUU GCU CGA GAC AGC UAU ATd Td-3′. The HepG2 cells were seeded in 6-well plates. siRNA transfection was performed using riboFECT ™CP (Ruibo Biotechnology Co., Ltd.) following the manufacturer's instructions. Cells were harvested for the subsequent experiments at 48 h following transfection.
Statistical analysis
Quantitative data are presented as the means ± standard error of the mean, with the significance determined by a Student's t-test or one-way analysis of variance with Tukey's multiple comparisons post hoc test. The results were plotted using GraphPad Prism 5.0 software. Differences between datasets were considered statistically significant at P<0.05.
Results
H2S inhibits PCSK9 expression in HepG2 cells
To elucidate the role of H2S in PCSK9 expression, we first incubated the HepG2 cells with various concentrations of NaHS (0, 50, 100 and 200 µmol/l) for 24 h. PCSK9 protein expression was detected by western blot analysis. As shown in Fig. 1, H2S inhibited PCSK9 expression in the HepG2 cells in a concentration-dependent manner. Subsequently, we incubated the HepG2 cells with 200 µmol/l NaHS for the indicated periods of time (0, 6, 12 and 24 h). As shown in Fig. 2, H2S inhibited PCSK9 expression in the HepG2 cells in a time-dependent manner.
Figure 1.
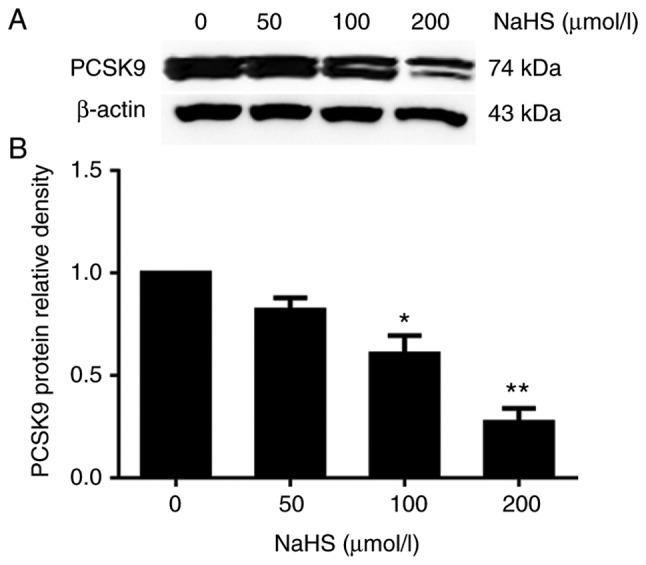
H2S inhibits PCSK9 expression in HepG2 cells in a concentration-dependent manner. HepG2 cells were treated with various concentrations of NaHS (0, 50, 100 and 200 µmol/l). (A and B) The protein levels of PCSK9 were detected by western blot analysis (*P<0.05 and **P<0.01 vs. the 0 µmol/l NaHS group) (n=3). H2S, hydrogen sulfide; NaHS, sodium hydrosulfide; PCSK9, proprotein convertase subtilisin/kexin type 9.
Figure 2.
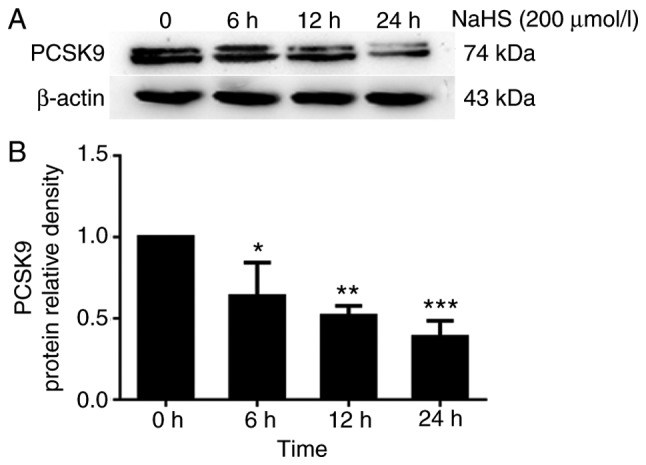
H2S inhibits PCSK9 expression in HepG2 cells in a time-dependent manner. HepG2 cells were treated with 200 µmol/l NaHS for the indicated periods of time (0, 6, 12 and 24 h). (A and B) The protein levels of PCSK9 were detected by western blot analysis (*P<0.05, **P<0.01 and ***P<0.001 vs. the 0 h NaHS group) (n=3). H2S, hydrogen sulfide; NaHS, sodium hydrosul-fide; PCSK9, proprotein convertase subtilisin/kexin type 9.
H2S inhibits the expression of PCSK9, upregulating the expression of LDLR and increase lipid uptake in HepG2 cells
The above-mentioned results indicated that H2S inhibited PCSK9 expression. Thus, the LDLR levels may be increased in the HepG2 cells. To determine whether H2S inhibits the expression of PCSK9 to influence the expression of LDLR and lipid uptake, we first examined the effects of H2S on LDLR expression in the HepG2 cells. The HepG2 cells were incubated with various concentrations of NaHS (0, 50, 100 and 200 µmol/l) for 24 h. LDLR expression was detected by western blot analysis and immunofluorescence assay. As shown in Fig. 3, H2S upregulated LDLR expression in the HepG2 cells in a concentration-dependent manner. In addition, as shown in Fig. 4, red fluorescence intensity increased with the increasing NaHS concentrations. This finding indicates that H2S upregulates the LDLR protein levels.
Figure 3.
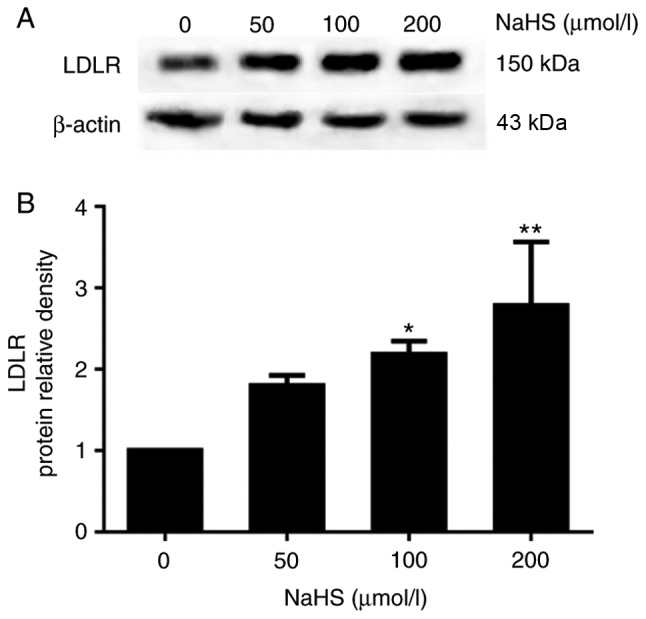
H2S upregulates LDLR expression in HepG2 cells in a concentration-dependent manner. HepG2 cells were incubated with various concentrations of NaHS (0, 50, 100 and 200 µmol/l) for 24 h. (A and B) LDLR expression was detected by western blot analysis (*P<0.05 and **P<0.01 vs. the 0 µmol/l NaHS group) (n=3). H2S, hydrogen sulfide; NaHS, sodium hydrosulfide; LDLR, low-density lipoprotein receptor.
Figure 4.

H2S increases the number of LDLRs on the cell membrane in a concentration-dependent manner (magnification, ×400). LDLR expression was detected by immunofluorescence assays. As shown in the figure, the red fluorescence intensity increased with the increasing NaHS concentrations. H2S, hydrogen sulfide; NaHS, sodium hydrosulfide; LDLR, low-density lipoprotein receptor.
It is well known that PCSK9 can promote LDLR degradation (9). Thus, to determine whether H2S upregulates LDLR expression by inhibiting PCSK9 expression, we first used RNA interference to suppress PCSK9 expression, and LDLR expression was detected. The results of western blot analysis revealed that PCSK9 inhibition was maintained following transfection for 48 h (Fig. 5A and B). Subsequently, DiI-LDL was incubated with the HepG2 cells for 4 h, and DiI-LDL uptake by the HepG2 cells was detected. The uptake of DiI-LDL at 37°C represented the cell surface-bound DiI-LDL. As shown in Figs. 5 and 6, compared with the cells treated with 200 µmol/l NaHS, those that were transfected with PCSK9 siRNA exhibited an increased LDLR expression and increased lipid uptake. This result indicated that H2S reduced the effects of PCSK9 on LDLR degradation by inhibiting PCSK9 expression, thereby increasing lipid uptake by HepG2 cells.
Figure 5.

PCSK9 siRNA reduces PCSK9 expression, which in turn increases the LDLR levels. (A-F) The protein levels of PCSK9 and LDLR were detected by western blot analysis (*P<0.05 and **P<0.01 vs. the control group; ***P<0.001 vs. the control siRNA group; #P<0.05, PCSK9 siRNA+200 µmol/l NaHS group vs. control siRNA + 200 µmol/l NaHS group) (n=3). NaHS, sodium hydrosulfide; LDLR, low-density lipoprotein receptor; PCSK9, proprotein conver-tase subtilisin/kexin type 9.
Figure 6.

PCSK9 siRNA increases the uptake of DiI-LDL by HepG2 cells (magnification, ×400). DiI-LDL was incubated with the HepG2 cells for 4 h, and the DiI-LDL uptake by HepG2 cells was detected. PCSK9, proprotein convertase subtilisin/kexin type 9; DiI-LDL, 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocar-bocyanine perchlorate-labeled LDL.
H2S regulates the expression of PCSK9 by affecting the PI3K/Akt-SREBP-2 signaling pathway in HepG2 cells
It has been reported that SREBP-2 may upregulate PCSK9 expression (10). Thus, to explore the potential mechanisms underlying the H2S regulation if PCSK9 expression, we investigated whether H2S regulates PCSK9 expression by regulating SREBP-2 expression. Following treatment with various concentrations of NaHS (0, 50, 100 and 200 µmol/l) for 24 h, SREBP-1c and SREBP-2 expression was detected by western blot analysis. As shown in Fig. 7, SREBP-1c expression increased and that of SREBP-2 decreased with the increasing NaHS concentration.
Figure 7.

The mechanisms underlying the regulation of PCSK9 expression by H2S. Effects of incubation with various concentrations of NaHS for 24 h on the expression of SREBPs in HepG2 cells. (A-C) The protein levels of SREBPs were detected by western blot analysis (*P<0.05 and **P<0.01 vs. the 0 µmol/l NaHS group) (n=3). PCSK9, proprotein convertase subtilisin/kexin type 9; NaHS, sodium hydrosulfide; SREBPs, sterol regulatory element-binding proteins.
PI3K/Akt is known to regulate the expression of SREBPs (11). Thus, in this study, we treated the HepG2 cells with various concentrations of NaHS (0, 50, 100 and 200 µmol/l) for 24 h, and phosphorylated Akt protein expression was detected by western blot analysis. As shown in Fig. 8, phosphorylated Akt protein expression increased with the increasing NaHS concentration. Following treatment with 200 µmol/l NaHS, the level of Akt phosphorylation increased, and SREBP-2 and PCSK9 expression decreased accordingly (Fig. 9).
Figure 8.

Effects of various concentrations of NaHS on the expression of p-Akt for 24 h in HepG2 cells. (A and B) The protein levels of phosphorylated Akt protein were detected by western blot analysis (*P<0.05 vs. the 0 µmol/l NaHS group) (n=3). NaHS, sodium hydrosulfide; Akt, protein kinase B.
Figure 9.

Effects of H2S on the expression of p-Akt, Akt, SREBP-2 and PCSK9 in HepG2 cells. Following treatment with 200 µmol/l NaHS, the level of Akt phosphorylation was increased, and SREBP-2 and PCSK9 expression decreased accordingly. (A-D) The protein levels of phosphorylated Akt, SREBP-2 and PCSK9 were detected by western blot analysis (*P<0.05 and **P<0.01 vs. the control group) (n=3). Akt, protein kinase B; NaHS, sodium hydrosulfide; SREBP-2, sterol regulatory element-binding protein 2; PCSK9, proprotein convertase subtilisin/kexin type 9.
H2S reduces lipid accumulation in HepG2 cells
The above-mentioned results indicated that H2S increased lipid uptake. Therefore, we hypothesized that lipid accumulation may be increased in the HepG2 cells. To explore whether H2S influences lipid accumulation in the HepG2 cells, the HepG2 cells were incubated with culture medium supplemented with 30% FBS for 24 h, and intracellular lipids were detected by Oil Red O staining. Surprisingly, as shown in Fig. 10, lipid accumulation in the HepG2 cells exhibited no increase, but exhibited a decrease with the increasing H2S concentration. Subsequently, DiI-LDL was incubated with the HepG2 cells for 24 h, and intracellular red fluorescence was also reduced, suggesting the reduced lipid accumulation in HepG2 cells (Fig. 11). The above-mentioned results indicate that H2S can inhibit lipid accumulation in HepG2 cells induced by high serum concentrations.
Figure 10.
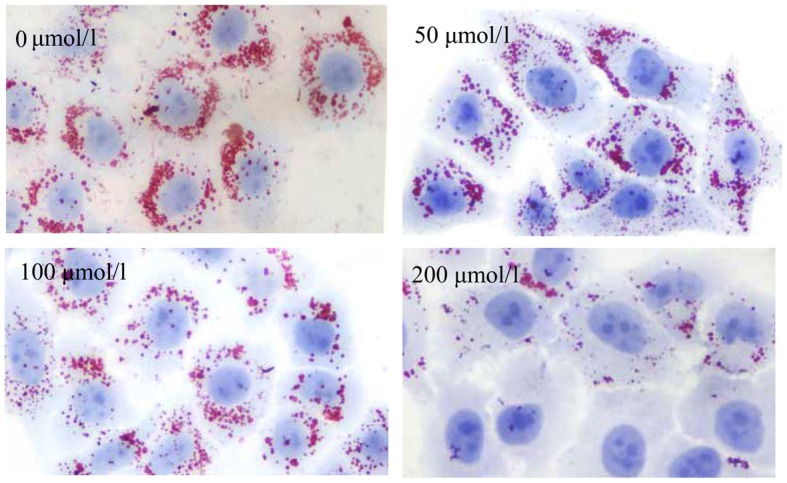
H2S reduces lipid accumulation in HepG2 cells. Following treatment with 30% fetal bovine serum, lipid accumulation was detected by Oil Red O in HepG2 cells (magnification, ×400). As shown in the figure, lipid accumulation in HepG2 cells did not increase with increasing H2S concentration and was reduced with the increasing H2S concentrations.
Figure 11.

H2S reduces lipid accumulation detected by DiI-LDL in HepG2 cells (magnification, ×400). DiI-LDL was incubated with HepG2 cells for 24 h, and intracellular red fluorescence was also reduced, implying that lipid accumulation in HepG2 cells was reduced. H2S, hydrogen sulfide; DiI-LDL, 1,1′-diocta-decyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate-labeled LDL.
Taken together, these results indicated that H2S downregulated the expression of PCSK9 in a time- and concentration-dependent manner, thereby regulating HepG2 lipid metabolism (Fig. 12).
Figure 12.
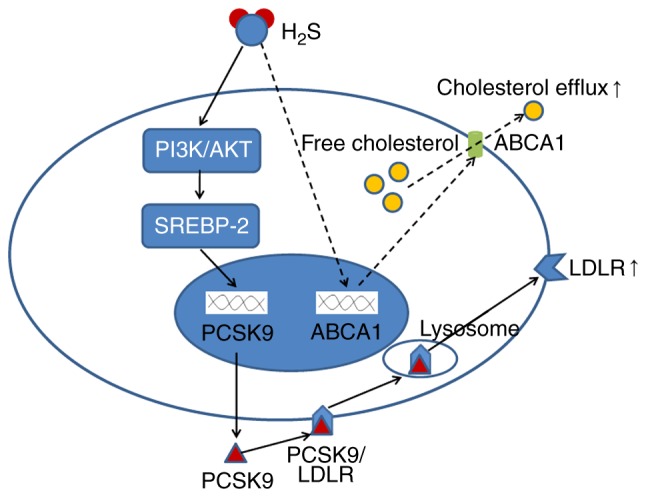
Schematic diagram of the regulatory effects of H2S on PCSK9 expression and lipid metabolism. Solid lines show the pathway of H2S regulating the LDLR, which is concluded by the research results of this study. The dotted line is speculative as to why H2S increases LDLR, but does not increase the intracellular lipid content of HepG2 cells. H2S, hydrogen sulfide; PCSK9, proprotein convertase subtilisin/kexin type 9; LDLR, low-density lipoprotein receptor.
Discussion
H2S inhibits PCSK9 expression to regulate lipid metabolism in HepG2 cells
H2S has emerged as an important cardiovascular signaling molecule similar to nitric oxide and carbon monoxide (12). H2S carries out a wide range of physiological and pathological functions in the cardiovascular system, including maintaining endothelial homeostasis (13), inhibiting the proliferation of vascular smooth muscle cells (14), and regulating lipid metabolism (15). However, the precise mechanisms through which H2S carries out its functions remain unknown. In this study, we found that H2S inhibited the expression of PCSK9 and increased lipid uptake by HepG2 cells.
The PCSK9 gene was discovered in 2003 (16); it plays an important role in lipid metabolism (17). PCSK9 accelerates the degradation of hepatic LDLR and prevents its recycling to the cell surface, which reduces lipid removal from the liver and increases serum lipid levels, thus promoting atherogenesis (18). PCSK9 mutations cause autosomal dominant hypercholesterolemia (19). Numerous factors, such as statins (20) and berberine (21), regulate the expression of PCSK9. This study demonstrated that H2S downregulated the expression of PCSK9 in HepG2 cells in a concentration- and time-dependent manner. To the very best of our knowledge, this study is the first to report that H2S can regulate the expression of PCSK9.
Previous studies have confirmed that PCSK9 regulates cellular lipid metabolism by degrading LDLR on the cell surface (22,23). The overexpression of PCSK9 has been shown to significantly increase LDLR degradation in HepG2 cells, thus reducing LDLR expression (24). In this study, H2S treatment upregulated the expression of LDLR by decreasing the expression of PCSK9 in HepG2 cells. The expression of LDLR further increased after the expression of PCSK9 was interfered with by siRNA. Consistent with this result, following incubation with DiI-LDL for 4 h, DiI-LDL uptake by HepG2 cells increased in the 200 µmol/l NaHS and PCSK9 siRNA groups compared with that in the 200 µmol/l NaHS group. This result indicates that H2S reduces the expression of PCSK9, thereby inhibiting the degradation of LDLR by PCSK9, and increasing the expression of LDLR on the cell surface to enhance lipid uptake. In addition, the highest concentration of NaHS (200 µmol/l) used in the treatment of HepG2 cells was confirmed by previous experiments, and this concentration does not cause toxic effects on cells (25).
PI3K/Akt-SREBP-2 is involved in the regulation of PCSK9 and LDLR expression by H2S
SREBPs play an important role in lipid metabolism by regulating the expression of lipid metabolism-related genes (26). SREBP-1c primarily enhances the synthesis of fatty acids and the transcription of metabolic enzymes, while SREBP-2 primarily regulates the transcription of cholesterol-metabolizing enzymes (27). SREBP-1c and SREBP-2 are the predominant isomers in the liver (27). The promoter region of PCSK9 contains a sterol-regulatory element, which implies that PCSK9 transcription depends on sterols (28). Dubuc et al demonstrated that PCSK9 is a target gene of SREBP-2 (29), whereas SREBP-2 may participate in lipid metabolism by regulating PCSK9 expression. In this study, following treatment with various concentrations of H2S, the expression of SREBP-1c and SREBP-2 was detected in HepG2 cells. The results revealed that the expression of SREBP-1c increased, while that of SREBP-2 decreased with the increasing NaHS concentration. This opposite result may be due to the fact that SREBP-1c primarily enhances the synthesis of fatty acids and triglycerides, while SREBP-2 preferentially synthesizes cholesterol. Chae et al demonstrated that LDLR was increased by downregulating the expression of hepatic PCSK9 via SREBP-2 (30). In this study, H2S inhibited the expression of SREBP-2 to inhibit the expression of PCSK9 and reduced the degradation of LDLR by PCSK9 to increase the LDLR level on the cell surface, consistent with the findings in the study by Chae et al (30).
PI3K/Akt can upregulate SREBP-2 expression and participate in lipid metabolism regulation (31). In this study, following treatment with various concentrations of NaHS, the levels of phosphorylated Akt were detected in the HepG2 cells. The results revealed that H2S increased the content of phosphorylated Akt protein to activate the PI3K/Akt signaling pathway, consistent with the findings of the study by Zheng et al (32). The increased Akt phosphorylation resulted in the decreased expression of SREBP-2. As a positive transcriptional regulator of PCSK9, the inhibition of SREBP-2 by phosphorylated Akt also downregulated the expression of PCSK9. The above-mentioned results indicate that PI3K/Akt-SREBP-2 is involved in the regulation of PCSK9 and LDLR expression by H2S.
Notably, this study demonstrated that H2S inhibited PCSK9 expression to reduce LDLR degradation at the HepG2 cell membrane to increase lipid uptake in the HepG2 cells. However, following incubation with 30% FBS or DiI-LDL, lipid accumulation in the HepG2 cells exhibited no increase, but instead decreased. This result suggests that H2S may also promote lipid efflux in addition to promoting lipid uptake through the SREBP-2 PCSK9/LDLR pathways in HepG2 cells. Gong et al (33) reported that H2S upregulated ATP-binding cassette transporter A1 (ABCA1) expression, whereas ABCA1 played an important role in promoting intracellular cholesterol efflux, which may explain why H2S increases lipid uptake, but causes no increase in lipid accumulation in HepG2 cells.
Notably, H2S can affect pH and Na+, but whether pH or Na+ affects lipid metabolism in HepG2 cells remains unclear. Therefore, further studies are required to determine the effects of pH or Na+ on lipid metabolism in HepG2 cells and whether other liver cancer cells have the same effect as HepG2.
In conclusion, the findings of this study demonstrate that H2S downregulates the expression of PCSK9 in a time- and concentration-dependent manner, thereby regulating HepG2 lipid metabolism (Fig. 12). To the very best of our knowledge, this study is the first to report that H2S can regulate the expression of PCSK9. Our findings provide new insight into the understanding of H2S-regulated lipid metabolism and anti-atherosclerosis. More importantly, the findings of this study suggest that H2S may be a novel candidate as PCSK9 inhibitor in addition to PCSK9 monoclonal antibodies and may provide a novel method with which to discover novel PCSK9 inhibitors (34).
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the National Natural Science Foundation of China (no. 81370376), the Natural Science Foundation of Hunan province (nos. 2018JJ2343 and 2015JJ4097), the Key Project of the Educational Department of Hu-nan Province (no. 15A137), the Visiting Scholar Foundation of Key Laboratory of Biorheological Science and Technology (Chongqing University), Ministry of Education (nos. CQKLBST-2014-002 and CQKLBST-2015-004), and the first-class discipline in Hunan Province, and Undergraduate Innovative Experiment Project (no. 2018XJXZ369).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
LSL and MMW contributed to the conception and design of the experiment. JX, XQB, LL, MZ, JP and QX performed the experiments and collected the data. ZR, ZSJ, ZHT, HYW and MMW analyzed the data and helped interpret the results. JX, XQB and LL wrote the manuscript. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Li D, Xiong Q, Peng J, Hu B, Li W, Zhu Y, Shen X. Hydrogen Sulfide upregulates the expression of ATP-binding cassette transporter A1 via promoting nuclear translocation of PPARα. Int J Mol Sci. 2016;17:E635. doi: 10.3390/ijms17050635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu D, Zheng N, Qi K, Cheng H, Sun Z, Gao B, Zhang Y, Pang W, Huangfu C, Ji S, et al. Exogenous hydrogen sulfide mitigates the fatty liver in obese mice through improving lipid metabolism and antioxidant potential. Med Gas Res. 2015;5:1. doi: 10.1186/s13618-014-0022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H, Mani S, Wu L, Fu M, Shuang T, Xu C, Wang R. The interaction of estrogen and CSE/H2S pathway in the development of atherosclerosis. Am J Physiol Heart Circ Physiol. 2017;312:H406–H414. doi: 10.1152/ajpheart.00245.2016. [DOI] [PubMed] [Google Scholar]
- 4.Gao L, Xu Z, Yin Z, Chen K, Wang C, Zhang H. Association of hydrogen sulfide with alterations of monocyte chemokine receptors, CCR2 and CX3CR1 in patients with coronary artery disease. Inflamm Res. 2015;64:627–635. doi: 10.1007/s00011-015-0844-7. [DOI] [PubMed] [Google Scholar]
- 5.Lin XL, Xiao LL, Tang ZH, Jiang ZS, Liu MH. Role of PCSK9 in lipid metabolism and atherosclerosis. Biomed Pharmacother. 2018;104:36–44. doi: 10.1016/j.biopha.2018.05.024. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro MD, Fazio S. PCSK9 and atherosclerosis-lipids and beyond. J Atheroscler Thromb. 2017;24:462–472. doi: 10.5551/jat.RV17003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shahreyar M, Salem SA, Nayyar M, George LK, Garg N, Koshy SKG. Hyperlipidemia: Management with proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors. J Am Board Fam Med. 2018;31:628–634. doi: 10.3122/jabfm.2018.04.170447. [DOI] [PubMed] [Google Scholar]
- 8.Ochin CC, Garelnabi M. Berberine encapsulated PLGA-PEG nanoparticles modulate PCSK-9 in HepG2 Cells. Cardiovasc Hematol Disord Drug Targets. 2018;18:61–70. doi: 10.2174/1871529X18666180201130340. [DOI] [PubMed] [Google Scholar]
- 9.Tavori H, Rashid S, Fazio S. On the function and homeostasis of PCSK9: Reciprocal interaction with LDLR and additional lipid effects. Atherosclerosis. 2015;238:264–270. doi: 10.1016/j.atherosclerosis.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res. 2008;49:399–409. doi: 10.1194/jlr.M700443-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: Cell signaling meets lipid metabolism. Trends Endocrinol Metab. 2010;21:268–276. doi: 10.1016/j.tem.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Cao X, Ding L, Xie ZZ, Yang Y, Whiteman M, Moore PK, Bian JS. A review of hydrogen sulfide synthesis, metabolism, and measurement: Is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxid Redox Signal. 2018 doi: 10.1089/ars.2017.7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altaany Z, Moccia F, Munaron L, Mancardi D, Wang R. Hydrogen sulfide and endothelial dysfunction: Relationship with nitric oxide. Curr Med Chem. 2014;21:3646–3661. doi: 10.2174/0929867321666140706142930. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Wang X, Liang X, Wu J, Dong S, Li H, Jin M, Sun D, Zhang W, Zhong X. Inhibition of hydrogen sulfide on the proliferation of vascular smooth muscle cells involved in the modulation of calcium sensing receptor in high homocysteine. Exp Cell Res. 2016;347:184–191. doi: 10.1016/j.yexcr.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Cheung SH, Lau JYW. Hydrogen sulfide mediates atheroprotection against oxidative stress via S-sulfhydration. PLoS One. 2018;13:e0194176. doi: 10.1371/journal.pone.0194176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 17.Singh A, Davidson M. Update on PCSK9 therapies for the treatment of dyslipidemia. Expert Rev Endocrinol Metab. 2016;11:87–95. doi: 10.1586/17446651.2016.1131120. [DOI] [PubMed] [Google Scholar]
- 18.Shapiro MD, Tavori H, Fazio S. PCSK9: From basic science discoveries to clinical trials. Circ Res. 2018;122:1420–1438. doi: 10.1161/CIRCRESAHA.118.311227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elbitar S, Susan-Resiga D, Ghaleb Y, El Khoury P, Peloso G, Stitziel N, Rabès JP, Carreau V, Hamelin J, Ben-Djoudi-Ouadda A, et al. New sequencing technologies help revealing unexpected mutations in autosomal dominant hypercholesterolemia. Sci Rep. 2018;8:1943. doi: 10.1038/s41598-018-20281-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reiss AB, Shah N, Muhieddine D, Zhen J, Yudkevich J, Kasselman LJ, DeLeon J. PCSK9 in cholesterol metabolism: From bench to bedside. Clin Sci (Lond) 2018;132:1135–1153. doi: 10.1042/CS20180190. [DOI] [PubMed] [Google Scholar]
- 21.Spigoni V, Aldigeri R, Antonini M, Micheli MM, Fantuzzi F, Fratter A, Pellizzato M, Derlindati E, Zavaroni I, Bonadonna RC, Dei Cas A. Effects of a new nutraceutical formulation (berberine, red yeast rice and chitosan) on non-HDL cholesterol levels in individuals with dyslipidemia: Results from a randomized, double blind, placebo-controlled study. Int J Mol Sci. 2017;18:E1498. doi: 10.3390/ijms18071498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poirier S, Hamouda HA, Villeneuve L, Demers A, Mayer G. Trafficking dynamics of PCSK9-induced LDLR degradation: Focus on human PCSK9 mutations and C-terminal domain. PLoS One. 2016;11:e0157230. doi: 10.1371/journal.pone.0157230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tavori H, Giunzioni I, Predazzi IM, Plubell D, Shivinsky A, Miles J, Devay RM, Liang H, Rashid S, Linton MF, Fazio S. Human PCSK9 promotes hepatic lipogenesis and atherosclerosis development via apoE- and LDLR-mediated mechanisms. Cardiovasc Res. 2016;110:268–278. doi: 10.1093/cvr/cvw053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Anderson NN, Ho YK, Hammer RE, Horton JD. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116:2995–3005. doi: 10.1172/JCI29383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qu K, Liu YM, He XL, Zhang H, Zhang K, Peng J, Tang YL, Yu XH, Zeng JF, Lei JJ, et al. H2 S inhibits apo(a) expression and secretion through PKCα/FXR and Akt/HNF4α pathways in HepG2 cells. Cell Biol Int. 2016;40:906–916. doi: 10.1002/cbin.10632. [DOI] [PubMed] [Google Scholar]
- 26.Park SW, Moon YA, Horton JD. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem. 2004;279:50630–50638. doi: 10.1074/jbc.M410077200. [DOI] [PubMed] [Google Scholar]
- 27.Horton JD, Goldstein JL, Brown MS. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI0215593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lebeau P, Al-Hashimi A, Sood S, Lhoták Š, Yu P, Gyulay G, Paré G, Chen SR, Trigatti B, Prat A, et al. Endoplasmic reticulum stress and Ca2+ depletion differentially modulate the sterol regulatory protein PCSK9 to control lipid metabolism. J Biol Chem. 2017;292:1510–1523. doi: 10.1074/jbc.M116.744235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, Prat A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–1459. doi: 10.1161/01.ATV.0000134621.14315.43. [DOI] [PubMed] [Google Scholar]
- 30.Chae HS, You BH, Kim DY, Lee H, Ko HW, Ko HJ, Choi YH, Choi SS, Chin YW. Sauchinone controls hepatic cholesterol homeostasis by the negative regulation of PCSK9 transcriptional network. Sci Rep. 2018;8:6737. doi: 10.1038/s41598-018-24935-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang J, Chen S, Cai D, Bian D, Wang F. Long noncoding RNA lncARSR promotes hepatic cholesterol biosynthesis via modulating Akt/SREBP-2/HMGCR pathway. Life Sci. 2018;203:48–53. doi: 10.1016/j.lfs.2018.04.028. [DOI] [PubMed] [Google Scholar]
- 32.Zheng D, Chen Z, Chen J, Zhuang X, Feng J, Li J. Exogenous hydrogen sulfide exerts proliferation, anti-apoptosis, migration effects and accelerates cell cycle progression in multiple myeloma cells via activating the Akt pathway. Oncol Rep. 2016;36:1909–1916. doi: 10.3892/or.2016.5014. [DOI] [PubMed] [Google Scholar]
- 33.Gong D, Cheng HP, Xie W, Zhang M, Liu D, Lan G, Huang C, Zhao ZW, Chen LY, Yao F, et al. Cystathionine γ-lyase(CSE)/hydrogen sulfide system is regulated by miR-216a and influences cholesterol efflux in macrophages via the PI3K/AKT/ABCA1 pathway. Biochem Biophys Res Commun. 2016;470:107–116. doi: 10.1016/j.bbrc.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 34.He NY, Li Q, Wu CY, Ren Z, Gao Y, Pan LH, Wang MM, Wen HY, Jiang ZS, Tang ZH, Liu LS. Lowering serum lipids via PCSK9-targeting drugs: Current advances and future perspectives. Acta Pharmacol Sin. 2017;38:301–311. doi: 10.1038/aps.2016.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.