

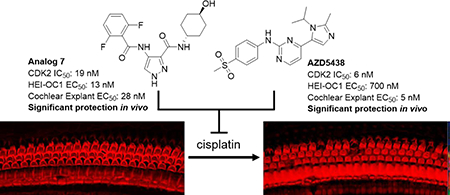
Abstract
There are currently no FDA-approved therapies to prevent the hearing loss associated with the usage of cisplatin in chemotherapeutic regimens. We recently demonstrated that the pharmacologic inhibition with kenpaullone or genetic deletion of CDK2 preserved hearing function in animal models treated with cisplatin, which suggests that CDK2 is a promising therapeutic target to prevent cisplatin-induced ototoxicity. In this study, we identified two lead compounds, AT7519 and AZD5438, from a focused library screen of 187 CDK2 inhibitors, performed in an immortalized cell line derived from neonatal mouse cochleae treated with cisplatin. Moreover, we screened 36 analogs of AT7519 and identified analog 7, which exhibited an improved therapeutic index. When delivered locally, analog 7 and AZD5438 both provided significant protection against cisplatin-induced ototoxicity in mice. Thus, we have identified two additional compounds that prevent cisplatin-induced ototoxicity in vivo, and provided further evidence that CDK2 is a druggable target for treating cisplatin-induced ototoxicity.
Graphical Abstract
INTRODUCTION
Cisplatin is a chemotherapeutic agent that is widely used for treating malignant neoplasms, and it is included on the World Health Organization’s list of essential medicines. Unfortunately, its use is often associated with potentially debilitating ototoxicity in approximately 63% of patients.1 This ototoxicity results from damage to cochlear structures such as the outer hair cells, the spiral ganglion nerve fibers, and the stria vascularis.2 The mechanism by which cisplatin induces cytotoxicity in auditory cells is not completely understood, but it is widely accepted that the generation of reactive oxygen species (ROS) by cisplatin and its ability to alkylate DNA are key contributors.3 Compounds such as sodium thiosulfate,4 N-acetylcysteine,5 D-methionine,6 amifostine,7 and dexamethasone,8 can act through the direct inactivation of cisplatin through chelation, serve as antioxidants, strengthen cellular antioxidant defenses, and/or regulate cytokines. However, none of these compounds have exhibited complete hearing protection across all frequencies in clinical trials, and none have been approved by the FDA.9
Protein kinases are attractive therapeutic targets due to their role in cellular signaling pathways which regulate key cellular functions, such as metabolism and cell cycle progression, and many kinases have been investigated for their role in ototoxic pathways.10,11 For instance, organic cation transporter 2 (OCT2) has been identified as one the primary methods by which cisplatin enters inner-ear hair cells and its inhibition or down regulation by kinase inhibitors has been shown to significantly reduce platinum drug uptake and toxicity.12,13,14 In another example, the Src-protein tyrosine kinase inhibitor, KX1–004, provided significant protection against cisplatin-induced ototoxicity in rats, without interfering the antitumor activity of cisplatin.15 Lastly, the glycogen synthase kinase 3 beta (GSK3B) inhibitors, SB 216763 and LiCl, were reported to inhibit cisplatin-induced apoptosis, and thereby prevent cisplatin-induced ototoxicity in rats.16
We recently reported that the pan-kinase inhibitor kenpaullone provided significant protection against cisplatin-induced ototoxicity in zebrafish and, when delivered by transtympanic injection, in mice and rats.17 Mechanistically, kenpaullone was shown to directly inhibit cyclin-dependent kinase 2 (CDK2) and thereby attenuate cisplatin-induced mitochondrial ROS production and caspase 3/7–mediated cell death.17 The same study also showed that CDK2-deficient mice treated with cisplatin exhibited no hearing loss. These results demonstrated that CDK2 is a promising therapeutic target for future drug development.
In the present study, we sought to identify additional CDK2 inhibitors that protect against cisplatin-induced ototoxicity and possess desirable drug-like properties. From a screen of 187 additional CDK2 inhibitors in HEI-OC1 cells treated with cisplatin, we identified two hits, namely AT7519 and AZD5438. Our structure-activity relation (SAR) studies around AT7519 and AZD5438 led to the discovery of the lead compound designated analog 7. Analog 7 and AZD5438 exhibited high potency (5–28 nM) in cochlear explants and effectively protected against cisplatin-induced ototoxicity in mice, thus expanding the repertoire of CDK2 inhibitors that protect against ototoxicity caused by cisplatin.
RESULTS
Identification of AZD5438 and AT7519 as Protective Agents Against Cisplatin Cytotoxicity.
We screened a focused library of 187 CDK2 inhibitors by performing dose-response analyses for potency and toxicity in HEI-OC1 cells as previously described (Table S1).18 Cell death from cisplatin was quantified by means of the luminescent product generated from the specific cleavage of a caspase-3/7 substrate. The caspase-3/7 activity in cells treated with cisplatin alone was considered to be 100%, and the activity in cells not treated with cisplatin was considered to be 0%. Of the compounds tested, 36 had an EC50 of less than 5 μM; and of those compounds, eight were further evaluated ex vivo in P3 mouse cochlear explants treated with cisplatin (Table 1). Cochleae were dissected from P3 mice and cultured with the test compounds and cisplatin (150 μM) in an incubator. After 24 h, the explants were fixed with 4% PFA (paraformaldehyde), stained with phalloidin, and imaged by confocal microscopy. The OHCs of the explants treated with test compounds and cisplatin were counted per 160 μm and compared to the counts of the untreated explants (considered complete protection) and the explants treated with cisplatin only (considered no protection). AT7519 and AZD5438 were the most potent compounds in tests on cochlear explants, and they exhibited substantially greater potency when compared with kenpaullone. In addition, both AT7519 and AZD5438 have been investigated in clinical trials and are safe for use and well-tolerated in humans. Therefore, we pursued further investigations of AT7519 and AZD5438 as lead compounds in the study.
Table 1.
Top Protective Compounds in HEI-OC1 Cell Assay and Cochlear Explants
| Name | HEI-OC1 EC50 (μM) | Explant EC50 (μM) |
|---|---|---|
| AZD5438 | 0.700 | 0.005 |
| AT7519 | 0.380 | 0.025a |
| Kenpaullone | 0.349 | 0.150b |
| BIO | 0.192 | 0.570c |
| CDK2 inhibitor II | 0.127 | 1.000b |
| Olomoucine II | 0.595 | 3.000b |
| SU9516 | 1.594 | 6.000c |
| AZD1080 | 3.634 | 15.000c |
EC50 could not be determined; maximal protection was observed at 25 nM.
Report by Teitz et al.17
Estimated EC50 based on 2–3 tested dose concentrations.
Generation and Study of an AT7519 Analog Library.
AT7519 is a potent inhibitor of CDK2 that has been evaluated in phase I and phase II clinical trials for treating relapsed/refractory chronic lymphocytic leukemia and mantle cell lymphoma and has been shown to be well tolerated at plasma concentrations higher than those obtained with the established biologically active doses.19,20 Although AT7519 at a concentration of 25 nM provided excellent protection in cochlear explant cultures treated with cisplatin, a loss of activity was observed at higher doses and an EC50 could not be determined (Figure 1). Nevertheless, because of the impressive activity of AT7519 in cochlear explants and its amendable scaffold, we selected AT7519 as a lead compound to improve upon its efficacy and therapeutic index.
Figure 1.

Dose response and morphology of explants treated with AT7519 and cisplatin. (A) The chemical structure of AT7519. (B) The dose response of AT7519 in P3 cochlear explants of mice treated with cisplatin (150 μM). OHC counts for media alone were considered as complete protection from cisplatin-induced toxicity and OHC counts for cisplatin alone were considered as no protection. Number of explants tested indicated within each bar. Cell counts for explants treated with cisplatin + AT7519 (25 nM) were significantly different from explants treated with cisplatin alone (*P < 0.05). A dosing curve could not be generated because of a loss of efficacy at higher doses.
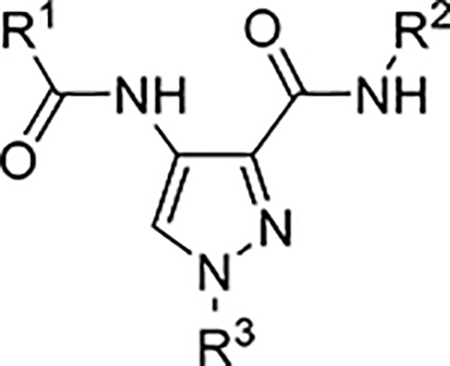
Many analogs of AT7519 have been described in the literature. Accordingly, we generated a small library of known AT7519 analogs from which to identify new analogs with improved efficacy and potency over AT7519 for protection against cisplatin-induced toxicity. Analogs 1–13 have been previously reported and were synthesized according to the procedures previously described by Wyatt et al.21 Compounds 14–36 were obtained from commercial sources. The compounds were screened with our HEI-OC1 cell assay under the conditions described previously (Table 2 and Table S2).18 CDK2 activity has been reported for analogs 1–13, and was measured for analogs 14–36 at concentrations of 1 μM and 10 μM. Of the latter group of 23 compounds, only analog 14 exhibited CDK2 inhibition activity in our assay, and dose response curve was generated (IC50 = 760.2 nM).21 We observed no correlation between efficacy in the HEI-OC1 cell assay and CDK2 activity for any of the compounds tested. Lastly, the permeability and aqueous solubility for each compound was determined (Table S2). The permeability was assessed by a parallel artificial membrane permeability assay (PAMPA). No significant correlation was observed between efficacy in the HEI-OC1 cell assay and cell permeability or aqueous solubility.
Table 2.
Cellular Activity of Selected Analogs of AT7519
 | |||||
|---|---|---|---|---|---|
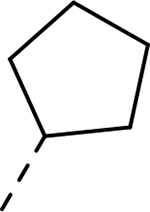
| Compound | R1 | R2 | R3 | HEI-OC1 EC50 (μM) | CDK2 IC50 (μM) |
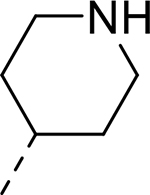
| AT7519 | 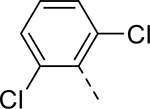 |
 |
H | 0.380 | 0.047a |
| 1 | 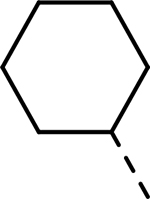 |
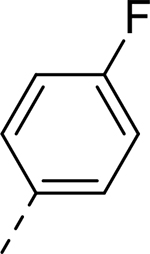 |
H | >32.219 | 0.090a |
| 2 | 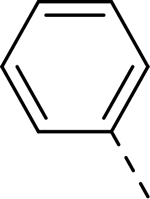 |
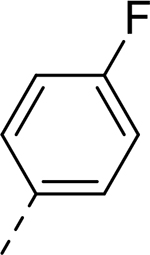 |
H | >33.208 | 0.140a |
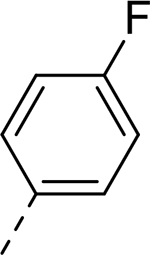
| 3 | 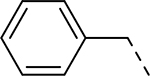 |
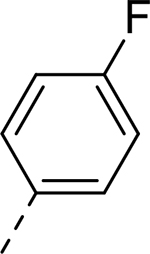 |
H | >32.714 | 1.600a |
| 4 | 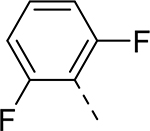 |
 |
H | 0.507 | 0.003a |
| 5 | 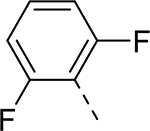 |
 |
H | 0.044 | 0.025a |
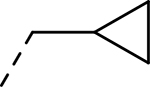
| 6 | 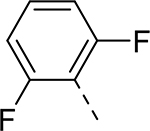 |
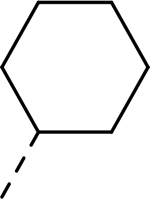 |
H | >27.260 | 0.012a |
| 7 | 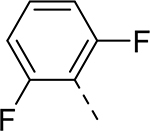 |
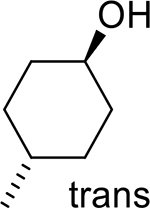 |
H | 0.013 | 0.019a |
| 8 | 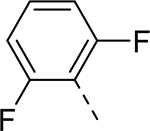 |
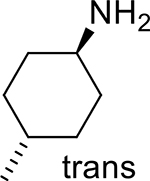 |
H | >33.457 | 0.038a |
| 9 | 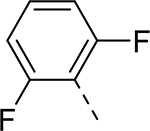 |
 |
H | >37.918 | 0.044a |
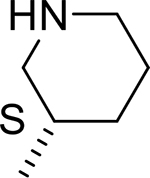
| 10 | 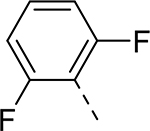 |
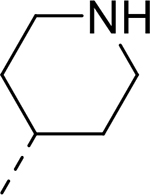 |
H | 22.463 | 0.140a |
| 11 | 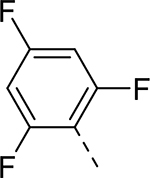 |
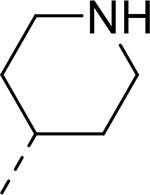 |
H | >30.855 | 0.750a |
| 12 | 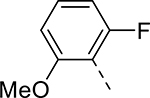 |
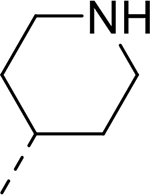 |
H | >37.1747 | 0.035a |
| 13 | 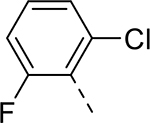 |
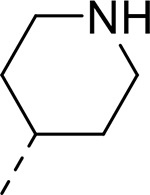 |
H | 2.606 | 0.110a |
| 14 | 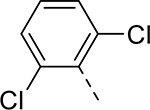 |
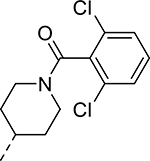 |
H | 0.459 | 760.2b |
| 15 | 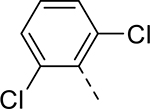 |
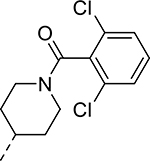 |
Me | >49.442 | >10b |
| 16 | 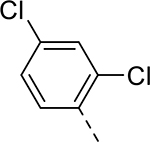 |
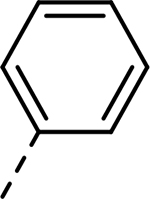 |
Et | 31.149 | >10b |
| 17 | 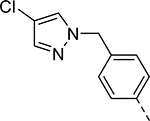 |
 |
Et | 5.039 | >10b |
| 18 | 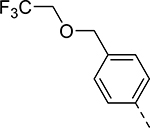 |
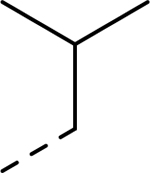 |
Et | 6.753 | >10b |
Reported by Wyatt et al.21
From this work.
The analogs that proved most potent in the cellular assay, i.e., analogs 4, 5, 7, and 14, were each evaluated at a concentration of 150 nM in cochlear explants treated with cisplatin (Figure 2A). Only analog 7 provided significant protection against cisplatin toxicity in cochlear explants from P3 mice. A dose-response study of analog 7, performed in cochlear explants, demonstrated that the analog exhibits potency comparable to that of AT7519 and maintains the level of protection at higher doses in explants treated with cisplatin (Figure 2B and 2C), whereas AT7519 does not do so (Figure 1B).
Figure 2.

Efficacy of top analogs in cochlear explants treated with cisplatin. (A) Outer hair-cell counts for cochlear explants treated with medium alone, cisplatin alone, cisplatin + analog 4, cisplatin + analog 5, cisplatin + analog 7, or cisplatin + analog 14. All compounds were tested at a concentration of 150 nM. OHC counts for media alone were considered as complete protection from cisplatin-induced toxicity and OHC counts for cisplatin alone were considered as no protection. Number of explants tested indicated within each bar. Cell counts for explants treated with cisplatin + analog 7 (150 nM) were significantly different from explants treated with cisplatin alone (*P < 0.05). (B) The chemical structure of analog 7 and its EC50 in cochlear explants treated with cisplatin. (C) The dose response of analog 7 in P3 cochlear explants from mice.
Analog 7 Protects Against Cisplatin-induced Ototoxicity in Mice.
Because of its superior potency in the HEI-OC1 cells and in cochlear explant culture, analog 7 was selected for in vivo study in adult FVB mice. To minimize the risk of systemic toxicity, 5 μL of the compound was administered to each left mouse ear by transtympanic injection, which is a well-established method of drug delivery for treating middle- and inner-ear diseases.22,23 Before the compound was administered, the auditory brainstem responses (ABRs) of the mice were evaluated to determine their baseline hearing thresholds at 8, 16, and 32 kHz. In the absence of cisplatin, all tested doses of analog 7 were nontoxic in the ears of mice at 15 days after injection, and a dose concentration of 50 μM was selected for the study of protection against cisplatin-induced ototoxicity.
Cisplatin (10 mg/kg) was administered by intraperitoneal injection (i.p.) 1 h after transtympanic delivery of the analog or vehicle alone (DMSO) (recipients were blinded to the individuals who performed the hearing tests until after the tests were completed), and hearing thresholds were measured by recording ABRs after 15 days. Mouse ears treated with DMSO alone exhibited substantial threshold elevations (due to cisplatin) that were similar to those previously reported in studies using FVB mice.12 In comparison, mice treated with analog 7 exhibited significantly reduced ABR threshold elevations at 16 kHz and 32 kHz, with an average threshold reduction of 10 dB at either frequency (Figure 3).
Figure 3.

Effect of analog 7 on cisplatin-induced ototoxicity when administered 1 h before cisplatin administration. P30 FVB mice received analog 7 (50 μM) in saline with 0.5% DMSO (5 μL) by transtympanic injection of the left ear. The ABR of the left ear of each mouse was recorded before treatment and again 15 days after. The data in the figure represent the mean change in threshold shifts before and after treatment for each group. The threshold shifts at 16 and 32 kHz in mice treated with analog 7 + cisplatin were significantly different from those in mice treated with vehicle alone and cisplatin (16 kHz, P = 0.0075; 32 kHz, P = 0.0288) or with vehicle alone and no cisplatin (32 kHz, P = 0.0235). Results are presented as the mean ± SEM. *P < 0.05 and **P < 0.01 by the paired 2-tailed t-test.
Morphologic analysis revealed that cisplatin induced the loss of outer hair cells (OHCs) in the middle basal turn (the 32-kHz region) of mice that received vehicle alone (Figure 4). Substantially less OHC loss was observed in the middle basal turn of mice that received cisplatin plus analog 7 than in the corresponding region of the mice that were treated with cisplatin alone. These morphologic data, along with the OHC counts, demonstrate that analog 7, when delivered transtympanically, prevents cisplatin-induced hearing loss in adult FVB mice.
Figure 4.

Morphology of cochleae treated with analog 7 (50 μM) and cisplatin. (A) OHC counts per 160 μm in the 32-kHz region of the organ of Corti. Results are presented as the mean ± SEM. (B) The morphology of the 32-kHz region of the organ of Corti. The sections are stained with phalloidin (red), DAPI (blue), and an antibody to parvalbumin (green).
AZD5438 Protects Against Cisplatin-Induced Ototoxicity in Mice.
AZD5438 is another hit identified from our focused library of 187 CDK2 inhibitors by screening in HEI-OC1 cells. Notably, AZD5438 is an orally bioavailable inhibitor of CDK1, CDK2, and CDK9 that has been used in solid-tumor therapy and was safe and well tolerated in patients on a weekly dosing schedule in a phase I study.24,25 In cochlear explant cultures treated with cisplatin, AZD5438 was the most potent of the CDK2 inhibitors tested in our study (EC50 = 5 nM) (Figure 5). As with AT7519, we designed a small library of AZD5438 analogs in order to identify additional compounds with activity against cisplatin-induced ototoxicity. We acquired nine analogs from commercial sources (Table S3); however, none of these analogs exhibited activity comparable to that of AZD5438 in terms of potency in HEI-OC1 cells treated with cisplatin. Therefore, AZD5438 was selected for in vivo study in adult FVB mice to assess its effectiveness at protecting against cisplatin-induced ototoxicity.
Figure 5.

(A) The chemical structure of AZD5438 and its EC50 in cochlear explants treated with cisplatin. (B) The dose-response and morphology of P3 cochlear explants of mice treated with AZD5438 and cisplatin. OHC counts for media alone were considered as complete protection from cisplatin-induced toxicity and OHC counts for cisplatin alone were considered as no protection.
When AZD5438 was delivered transtympanically to adult FVB mice at dose concentrations of 2 μM, 10 μM, or 50 μM, we observed no toxicity after 15 days. Consequently, a dose concentration of 50 μM was selected for further study with cisplatin. To determine whether AZD5438 protected against cisplatin-induced ototoxicity in vivo, we administered AZD5438 to mice by transtympanic injection 1 h before cisplatin administration (i.p.) and again 24 h afterwards. As before, we evaluated hearing loss by comparing ABR thresholds before treatment and 15 days after treatment. Compared to the group of mice treated with the vehicle and cisplatin, the group that received AZD5438 and cisplatin had significantly reduced ABR threshold shifts at 32 kHz (14 dB) (Figure 6). In addition, we observed no significant difference in the hearing thresholds of mice that received AZD5438 with cisplatin and mice that received the vehicle alone. These results demonstrate that AZD5438 is fully protective against cisplatin ototoxicity in mice.
Figure 6.

Effect of AZD5438 on cisplatin-induced ototoxicity in adult FVB mice when dosed 1 h before cisplatin administration and again 24 h after cisplatin administration. P30 FVB mice received AZD5438 (50 μM) in saline with 0.5% DMSO (5 μL) by transtympanic injection of the left ear. The ABR of the left ear of each mouse was recorded before treatment and again 15 days after treatment. The data in the figure represent the mean ABR threshold shifts before and after treatment for each group. The threshold shifts at 32 kHz for mice treated with the analog AZD5438 + cisplatin were significantly different from those for mice treated with cisplatin alone (32 kHz, P = 0.002). Results are presented as the mean ± S.E.M. **P < 0.01 by the paired 2-tailed t-test.
Morphologic analysis revealed that cisplatin primarily caused the loss of OHCs in the middle basal cochlear turn (the 32-kHz region) of mice that received vehicle alone (Figure 7). In comparison, the mice that received AZD5438 had significantly less OHC loss overall and virtually no OHC loss in the 32-kHz region. Together, these data suggest that AZD5438 provides full protection against cisplatin-induced ototoxicity in vivo at both the functional and morphologic levels
Figure 7.

Morphologic analysis of cochleae treated with AZD5438 and cisplatin. (A) OHC counts per 160 μm in the 32-kHz region of the organ of Corti, showing a significant difference between the OHC counts in mice treated with AZD5438 + cisplatin and mice treated with cisplatin alone (P = 0.0075). (B) The morphology of the 32-kHz region of the organ of Corti. The sections are stained with phalloidin (red), DAPI (blue), and an antibody to parvalbumin (green). Results are presented as the mean ± SEM. **P < 0.01 by the paired 2-tailed t-test.
Kinome Profiling of AZD5438, AT7519, and Analog 7.
To compare the kinase specificity of our top compounds, AZD5438, AT7519, and analog 7 were screened at a concentration of 1 μM against an assay panel of 468 kinases by the KINOMEscan™ Profiling Service provided by DiscoverX (Figure 8 and Table S4). All three compounds screened had the most hits in the CMGC kinase group. Based on their selectivity scores at <35% of the control, AT7519 and its analog exhibited better selectivity when compared with AZD5438 (the selectivity scores were 0.07, 0.08, and 0.18, respectively) (Figure 8 and Table S5). The results were compared to a previously reported kinome of kenpaullone.26 AZD5438, AT7519, and analog 7 were more potent kinase inhibitors than kenpaullone, and all four compounds had significant overlaps in their activity for GSK3A, GSK3B, and members of the CDK family (CDK2, 3, and 7). These results support our previous identification of CDK2 as a relevant target for hearing-loss protection, as confirmed by a CDK2-knockout mouse study.17 The kinase profiles also identified potential targets, other than CDK2, that contribute to the protection against cisplatin-induced ototoxicity conferred by these compounds.
Figure 8.

Kinase Interaction Maps of kenpaullone, AZD5438, AT7519, and analog 7. aReported data for kinome scan against 234 kinases with kenpaullone.26 bKinome scan against 468 kinases by DiscoverX. All compounds were tested at 1 μM. A high level of selectivity for kinases in the CMGC kinase group was observed for AT7519, analog 7, and AZD5438, and significant overlap for GSK3A, GSK3B, and members of the CDK family was observed for all four compounds.
DISCUSSION
Despite decades of research, no drugs have been approved by the FDA to combat cisplatin-induced ototoxicity, and the debilitating effects of cisplatin-related hearing loss on patients with cancer remain a major drawback of this treatment. We recently reported that cisplatin induced mitochondrial ROS production that correlated with increased CDK2 activity and that CDK2 inhibition by kenpaullone or CDK2 ablation in CDK2-knockout mice significantly reduced cisplatin-induced ototoxicity.17 Therefore, we sought to identify other otoprotective compounds by investigating CDK2 as a therapeutic target. We performed dose-response analyses of 187 selected CDK2 inhibitors in HEI-OC1 cells treated with cisplatin and subsequently evaluated the top hits in cochlear explants treated with cisplatin. We identified AT7519 and AZD5438 as the most potent compounds, and they were selected as lead compounds for further SAR studies.
Although AT7519 was one of the most potent compounds in cochlear explant cultures treated with cisplatin, its efficacy declined sharply at higher doses. Nevertheless, because of the impressive activity of AT7519 in cochlear explants and its amendable scaffold, we sought to improve its efficacy and therapeutic index. From a library of 36 analogs, four compounds, identified as analogs 4, 5, 7, and 14, exhibited nanomolar activity in HEI-OC1 cells treated with cisplatin. Each of the four compounds featured either a 2,6-dichlorophenyl or 2,6-difluorophenyl substituent in the R1 position, suggesting that an electron-poor phenyl group locked into a perpendicular orientation is important for activity. In contrast, more flexibility was observed for the R2 substituent, as a comparison of the R2 group of each of the four analogs suggests that their activity in cells does not depend greatly on the polarity and size of this group. None of the four compounds featured an alkyl substituent at the R3 position.
The most potent analogs of the HEI-OC1 cell assay, analogs 4, 5, 7, and 14, were tested in explants at a concentration of 150 nM; however, we observed protection only with analog 7. A structural feature common to AT7519 and analog 7, but not to the others, is that their R2 substituents are solubilizing groups containing hydrogen-bond donors, as piperidine and cyclohexanol, respectively. This suggests that hydrogen bonding or aqueous solubility could be important contributing factors to the activity exhibited by these two compounds in the explant assay. In comparison to AT7519, analog 7 did not lose efficacy at higher doses in cochlear explant cultures with cisplatin but provided comparable potency (EC50 = 28 nM). Additional SAR studies with more diverse analogs are needed to identify the determinant properties required for efficacy in cochlear explants, as well as rationalize the observed differences in efficacy between AT7519 and analog 7.
In adult FVB mice treated with cisplatin, transtympanic delivery of analog 7 resulted in ABR threshold shifts at 16 kHz and 32 kHz that were significantly smaller than those in mice that received the vehicle alone. However, a significant difference (7 dB) was observed between the threshold shifts in mice that received no cisplatin and those in mice treated with analog 7 and cisplatin. Furthermore, our morphologic analysis showed that not all OHCs were preserved by analog 7, and we found no significant differences in the OHC counts of mice treated with vehicle alone and mice treated with analog 7. Together, these data show that analog 7, under the treatment conditions in vivo, provides some—but not complete—protection against cisplatin-induced ototoxicity at 32 kHz at a functional level, but not at a morphologic level.
In comparison, AZD5438 provided complete protection against cisplatin-induced ototoxicity at all frequencies, and we found no significant difference between the untreated control group and the group that received AZD5438 and cisplatin. The threshold shifts at 32 kHz in mice treated with AZD5438 and cisplatin were significantly different from those in mice treated with cisplatin and vehicle alone (14 dB). In addition, a morphologic study of the cochlear middle basal turn (the 32-kHz region) of the organ of Corti in mice treated with AZD5438 and cisplatin found virtually all the OHCs to be present. Together, these data suggest that AZD5438 provides full protection against cisplatin-induced ototoxicity in vivo at both the functional and morphologic levels.
All three lead compounds, AZD5438, AT7519, and analog 7, were screened against a panel of 468 kinases at a concentration of 1 μM and exhibited a high level of selectivity, primarily for the CMGC group of kinases. CDK and MAPK are the two best-studied kinase families within the CMGC group.27 As expected, all three compounds had significant activity for members of the CDK family. However, the compounds exhibited little to no activity for members of the MAPK family, which regulate processes such as cell-fate determination. A comparison of the kinomes of the three compounds with the previously reported kinome of kenpaullone revealed significant overlaps in the activity for GSK3A, GSK3B, and members of the CDK family for all four compounds. These results provide additional evidence that the inhibition of CDK2 is important for the protective effects of these compounds, as we have previously reported,17 are in agreement with a previous report suggesting the role of GSK in cisplatin-induced ototoxicity,16 and provide insight into other potential target kinases.
A critical challenge to developing new drugs for local delivery to treat diseases of the inner ear is the lack of a clear understanding of the properties required for optimal passage through physical barriers such as the round window membrane (RWM).22 It would be valuable to establish predictive guidelines specific for the inner ear, similar to the well-known Lipinski’s rule of five, by determining correlations between the chemical properties obtained in vitro, such as permeability and solubility, and the desired phenotypic response in vivo. For our study, we determined the aqueous solubility and permeability of most of the prepared analogs, along with kenpaullone (Figure S2), to gain some insight into how these chemical properties might contribute to the success of these compounds as otoprotective agents. Of the top three candidates in vivo, AZD5438 was the best in terms of permeability, as measured on the PAMPA assay, and had the highest aqueous solubility (Pe = 304.7 × 10−6 cm and Saq = 68.8 μM), whereas analog 7 was the least permeable (Pe = 30.9 × 10−6 cm/s) and kenpaullone had the lowest aqueous solubility (5.2 μM) (Tables S2 and S3). We believe that it is necessary to determine the same properties in a uniform setting for many more compounds that have been determined to be protective ex vivo, in vivo, or in clinical trials, so that inner ear–specific guidelines can be derived to aid in the future development of drugs to treat hearing disorders.
CONCLUSIONS
Our studies provide an example of target-based drug development for hearing disorders. We took advantage of CDK2 as the drug target and explored diverse CDK2 inhibitors that have been reported with various scaffolds. Upon identifying two lead scaffolds, we performed a small-scale SAR study by analyzing several analogs and identified analog 7 as a compound with potency superior to that of the lead compound AT5719. Both AZD5438 and kenpaullone provide complete protection against cisplatin-induced ototoxicity in mice, whereas analog 7 provides only partial protection. However, it should be pointed out that the comparison of the compounds is complicated by differences in the study designs and the limited hearing loss observed in FVB mice. Although we have made some progress in this area by measuring the permeability and solubility of the compounds, further analysis is needed of the determinant properties of these compounds for protection against cisplatin ototoxicity in vivo when administered by transtympanic injection.
The local delivery of otoprotectants offers advantages such as providing high local concentrations of the agent, reducing the possibility of interference of the antitumoral activity of cisplatin, and allowing for patients to serve as their own controls by comparing the ear injected with the active compound to the other ear injected with vehicle alone in proof of concept studies. In contrast, non-surgical options, such as intravenous or oral administration would offer advantages in patient compliance and convenience. In this regard, the poor aqueous solubility of kenpaullone and its lack of study in vivo make it unattractive for development as a candidate for oral or intravenous administration. In contrast, AZD5438 is a very promising candidate for development as a systematically administered agent, in addition to being an excellent local delivery agent, as it has been shown to have a good bioavailability and safety profile in clinical trials. Future efforts in our laboratory will be directed towards determining the distribution of AZD5438 in the ear and its efficacy against cisplatin-induced ototoxicity when administered systemically. In conclusion, we have identified two additional CDK2 inhibitors (AZD5438 and analog 7) that provide significant protection against cisplatin-induced toxicity in mice and provide further evidence that CDK2 is an attractive target for treating cisplatin-induced ototoxicity.
EXPERIMENTAL
Cell-Based Dose-Response Study.
Cell-based dose-response studies in HEI-OC1 cells28 were performed in 384 well-plates with initial plating of 3200 cells per well. Cells were drugged after overnight incubation with 10 doses of each compound in triplicates and two minutes later cisplatin was added at a final concentration of 50 μmolar. The cells media used did not contain gamma-interferon to reduce cells’ growth and mimic conditions of nonproliferating inner ear cells. The equipment and reagents used are detailed in Teitz et al.17,18 Caspase-3 cleavage activity was measured 22 hours after co-treatment with tested compound and cisplatin employing Caspase-Glo 3/7 assay (Promega), and CellTiter-Glo assay (Promega) was used to measure cell viability of the cells with compound alone. Cells treated with 50 μM cisplatin were assigned 100% caspase-3/7 activity while cells not treated with cisplatin were assigned 0% caspase-3/7 activity.
Cochlear Explant Cultures.
Using the procedure previously described by Tal et al.17 P3 FVB mouse cochlear explants were dissected and maintained in culture on filters (Millicell, PICM03050; Millipore) in 6-well culture plates with 1 mL medium (DMEM 12430–054; GIB CO Life Technologies, with 1% FBS [16000–044; GIB CO Life Technologies] and 50 μg/mL ampicillin) solution both inside and outside the filter. After incubation at 37 °C in 5% CO2 for 1 d, the media was removed from the wells, and fresh media with or without the test compound was added to each well, and the explants were returned to the incubator. After 1 h, the explants were removed from the incubator and the media of each well was exchanged with fresh media containing cisplatin (150 μM), with or without the test compound (in such a way that only the same wells as before received the test compound). The explants were then returned to the incubator for 1 d. The explants were fixed with 4% PFA, stained for actin with Alexa Fluor 568 phalloidin (A12379 or A12380; Invitrogen) to determine the viability of the HCs, and imaged by confocal microscopy. Confocal imaging was performed using a Zeiss 710 or 700 scanning confocal microscope with a 63× objective. Visualization and projections were performed using ZEN 2009 or ZEN 2012 software (Zeiss). Two 160-μm regions from the middle turns were photographed, and the number of intact HCs in each region were counted and averaged.
Chemistry.
Analogs 1–13 were synthesized according to procedures described by Wyatt et al.21 All synthesized compounds had purities >95% as measured by LC-MS -ELSD/UV-Vis and all characterization (1H NMR and HRMS) data matched those reported in the literature.21 Analogs 14–45 were purchased from commercial sources. All purchased compounds had purities >95% as measured by LC-MS-ELSD/UV-Vis, with the exceptions of 16 (92%), 26, (85%), 34 (93%), and 43 (94%).
Solubility.
Solubility assays were carried out on a Biomek FX Laboratory Automation Workstation (Beckman Coulter, Inc., Fullerton, CA) using SOL Evolution software (pION Inc., Woburn, MA). The detailed method was as follows. A 10 μL aliquot of 10 mM compound stock (in DMSO) was added to 190 μL of 1-propanol to make a reference stock plate. From this plate, a 5-μL aliquot was mixed with 70 μL of 1-propanol and 75 μL of citrate phosphate-buffered saline (PBS; isotonic) to make the reference plate, and the UV spectrum (250–500 nm) of the reference plate was read. Then, 6 μL of 10 mM test compound stock was added to 594 μL of buffer in a 96-well storage plate and mixed. The storage plate was sealed and incubated at room temperature for 18 h. The suspension was then filtered through a 96-well filter plate (pION Inc.), and 75 μL of filtrate were mixed with 75 μL of 1-propanol to make the sample plate. The UV spectrum of the sample plate was then read. Calculations were carried out with μSOL Evolution software, based on the area under the curve (AUC) of the UV spectra of the sample and reference plates. All compounds were tested in triplicate.
Permeability.
Our parallel artificial membrane permeability assay (PAMPA) was conducted with a Biomek FX Laboratory Automation Workstation (Beckman Coulter, Inc., Fullerton, CA) and PAMPA Evolution 96 Command software (pION Inc., Woburn, MA). The detailed method for this assay was as follows. First, 3 μL of 10 μM test compound stock in DMSO was mixed with 597 μL of citrate PBS (isotonic) to make a diluted test compound. Then, 150 μL of diluted test compound was transferred to a UV plate (pION Inc.), and the UV spectrum of this reference plate was read. The membrane, on a pre-loaded PAMPA Sandwich (pION Inc.), was painted with 4 μL of GIT lipid (pION Inc.). The acceptor chamber was then filled with 200 μL of ASB (acceptor solution buffer; pION Inc.), and the donor chamber was filled with 180 μL of diluted test compound. The PAMPA Sandwich was assembled, placed on the Gut-Box stirring device, and stirred for 30 min. The aqueous boundary layer was set to 40 μm for stirring. The UV spectrum (250–500 nm) of the donor and the acceptor were then read. The permeability coefficient was calculated using PAMPA Evolution 96 Command software, based on the AUCs of the reference plate, the donor plate, and the acceptor plate. All compounds were tested in triplicate.
CDK2 Inhibition Assay.
Cdk2/Cyclin A (200 pM) was mixed with histone H1 (40 μM; EMD Millipore) and the respective compounds and incubated overnight at 4 °C. Subsequently, ATP (at a 50 μM total concentration, including 10 μCi [γ−32P]-ATP [PerkinElmer, Inc.]) was added to each reaction mixture and incubation was continued for 30 min at 35 °C. Each reaction mixture had a total volume of 30 μL. The sample buffer contained 20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) pH 7.3, 25 mM sodium β-glycerolphosphate, 15 mM MgCl2, 16 mM EGTA, 0.5 mM Na3VO4, and 10 mM DTT. The reactions were quenched by adding SDS-gel–loading buffer (7 μL) and then analyzed by SDS-PAGE (10 μL). The gels were dried at 70 °C under vacuum, then a phosphorimager (GE Healthcare, Piscataway, NJ) was used to quantify the 32P-histone H1 bands. AT7519 was used as a control, and the results were consistent with those previously reported.21
In Vivo Study General Procedures.
FVB breeding mice were purchased from The Jackson Laboratory and bred in the St. Jude animal facility. FVB mice were used for the in vivo cisplatin treatment experiments and for the cochlear explant experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee of St. Jude Children’s Research Hospital.
Procedure for Transtympanic Injection in Adult Mice.
Mice were anesthetized with isoflurane and placed on a heating pad to maintain their body temperature, with their left ear upwards. Lubricant eye ointment was applied to prevent corneal ulcers developing. The tympanic membrane was visualized with a surgical stereomicroscope, and compounds were administered by transtympanic delivery with a 33-gauge cannula. The left ear of each mouse was injected with 5 μL of the compound in PBS + 0.5% DMSO or with 5 μL of PBS + 0.5% DMSO as a control. The mice were then placed in their cage on a heating pad and kept on their side with the left ear upwards for 30 min to facilitate recovery.
Procedure for Cisplatin Treatment in Mice.
A stock solution of cisplatin was prepared by dissolving 10 mg of cisplatin in 10 mL of saline; dissolution was facilitated by mild heating at 37 °C for 30 min. Cisplatin (30 mg/kg) was administered by i.p. injection 1 h after the mice received a transtympanic injection. Mice were weighed each day to monitor bodyweight loss and were given warm saline (1 mL) by subcutaneous injection to prevent dehydration resulting from the cisplatin treatment until their bodyweight started to recover. In addition, the cages were placed on heating pads and fresh mush food was provided each day until all mice began to recover their bodyweight. The humane endpoint was 25% bodyweight loss from the day on which cisplatin was administered. Those mice treated with cisplatin that did not lose more than 10% of their bodyweight exhibited no hearing loss and were, therefore, excluded from the study (Figure S1).
Procedure for ABR Threshold and Wave 1 Amplitude Measurements.
ABR threshold and wave 1 amplitude measurements were performed in accordance with the procedures outlined by Teitz et al. with minor modifications.17 ABR measurements were performed in closed-field for the left ear only. ABR waveforms were recorded in a sound booth (Industrial Acoustic Company) by using subdermal needles positioned in the skull, below the pinna, and at the base of the tail, and the responses were fed into a low-impedance Medusa digital biological amplifier system (RA4L; TDT; 20-dB gain). At each frequency, the stimulus intensity was reduced from 90 to 0 dB in 5-dB steps to determine the threshold decibel sound pressure level (SPL) when the electrical response was just above the noise floor. ABR waveforms were averaged in response to 500 tone bursts. The recorded signals were filtered by a band-pass filter from 300 Hz to 3 kHz. Individual ABR wave 1 amplitudes were measured as the difference between the positive peak and the following negative trough. PreABR recordings of adult FVB mice (aged 4–8 weeks) were acquired before all treatments.
Procedure for Tissue Preparation, Immunofluorescence, and Quantification Analysis.
All tissue preparation, immunofluorescence studies, and quantification of OHCs were performed according to procedures outlined by Teitz et al.17 The cells were stained with phalloidin (Invitrogen, catalog no. A12379 or A12380), a primary antibody, mouse anti-parvalbumin (P3088; Sigma-Aldrich, 1:500 dilution) a secondary antibody, goat anti-mouse IgG1 (488 green, Invitrogen, 1:1000 dilution), and ProLong gold antifade reagent with DAPI (P36941; Invitrogen) was used to counterstain nuclei.
In Vivo Study of Analog 7 in Cisplatin-Treated Adult FVB Mice.
To determine the highest nontoxic dose of analog 7 when delivered transtympanically, mice received either analog 7 (2 μM, 10 μM, or 50 μM) or vehicle alone in a blind, randomized manner. Three mice were used for each of the 2 μM, 10 μM, and vehicle groups, and four mice were used for the 50 μM group, giving a total of 13 mice. Each mouse received a single transtympanic injection. After 15 days, ABRs were recorded at 8, 16, and 32 kHz. No hearing loss or toxicity was observed in any of the three groups; therefore, a dose concentration of 50 μM was used in the subsequent studies. To test the protective effect of analog 7, a total of 35 mice were allocated among three groups, in which the mice were treated with vehicle alone, vehicle + cisplatin, or analog 7 + cisplatin. Mice received the vehicle alone or analog 7 by transtympanic injection 1 h before cisplatin administration (i.p.). A total of 12 mice reached the humane endpoint or died, and no mice lost less than 10% of their bodyweight. After the day 15 ABRs had been recorded, the mice were euthanized and their cochleae were harvested for morphologic analysis.
In Vivo Study of AZD5438 in Cisplatin-Treated Adult FVB Mice.
To determine the highest nontoxic dose of AZD5438 when delivered transtympanically, mice received either AZD5438 (2 μM, 10 μM, or 50 μM) or vehicle alone in a blind, randomized manner. Three mice were used for each group (giving a total of 12 mice), and each group received two transtympanic injections separated by 24 h. One mouse in the 10 μM group died while under isoflurane anesthesia. After 15 days, ABRs were recorded at 8, 16, and 32 kHz. No hearing loss or toxicity was observed in any of the three groups; therefore, a dose concentration of 50 μM was used in the subsequent studies. To test the protective effect of AZD5438, a total of 47 mice were allocated among three groups, in which the mice were treated with vehicle alone, vehicle + cisplatin, or AZD5438 + cisplatin. Mice received the vehicle alone or AZD5438 by transtympanic injection 1 h before cisplatin administration (i.p.) and again 24 h afterwards. A total of 20 mice reached the humane endpoint or died, and six mice were excluded because they lost little or no bodyweight (<10%) in response to cisplatin treatment. After the day 15 ABRs had been recorded, the mice were euthanized and their cochleae were harvested for morphologic analysis.
Kinome Profiling of AZD5438, AT7519, and Analog 7.
Kinome profiling was performed by DiscoverX KINOMEscan™ Profiling Service. Kinase-tagged T7 phage strains were grown in parallel in 24-well blocks in an E. coli host derived from the BL21 strain. E. coli were grown to log-phase and infected with T7 phage from a frozen stock (multiplicity of infection = 0.4) and incubated with shaking at 32°C until lysis (90–150 minutes). The lysates were centrifuged (6,000 × g) and filtered (0.2μm) to remove cell debris. The remaining kinases were produced in HEK-293 cells and subsequently tagged with DNA for qPCR detection. Streptavidin-coated magnetic beads were treated with biotinylated small molecule ligands for 30 minutes at room temperature to generate affinity resins for kinase assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1 % BSA, 0.05 % Tween 20, 1 mM DTT) to remove unbound ligand and to reduce nonspecific phage binding. Binding reactions were assembled by combining kinases, liganded affinity beads, and test compounds in 1× binding buffer (20 % SeaBlock, 0.17x PBS, 0.05 % Tween 20, 6 mM DTT). Test compounds were prepared as 40x stocks in 100% DMSO and directly diluted into the assay. All reactions were performed in polypropylene 384 well plates in a final volume of 0.02 ml. The assay plates were incubated at room temperature with shaking for 1 hour and the affinity beads were washed with wash buffer (1× PBS, 0.05 % Tween 20). The beads were then resuspended in elution buffer (1× PBS, 0.05 % Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR. Compounds AT7519, analog 7, and AZD5438 were screened against a total of 468 kinases at a concentration of 1 μM.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Keith A. Laycock for editing the manuscript and Sourav Das from lead discovery informatics for calculating PAINS descriptors of all of the compounds. This work was supported by the National Institutes of Health [grant nos. 1R01DC015010 (to J.Z.), 1R01DC015444 (to J.Z.), and P30CA21765 (to St. Jude)], by ALSAC, and by the Office of Naval Research [grant no. N000141612315 (to J.Z.)].
ABBREVIATIONS
- ABR
auditory brainstem response
- ASB
acceptor solution buffer
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco’s modified eagle medium
- ELSD
evaporative light scattering detector
- GSK-3
Glycogen synthase kinase 3
- HEI-OC1
The House Ear Institute – Organ of Corti
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- OHC
outer hair cells
- Pe
permeability
- RWM
round window membrane
- Saq
aqueous solubility
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge.
Biological activity of CDK2 inhibitors screened in HEI-OC1 cells treated with cisplatin, structures, biological activity, solubility, and permeability data of all purchased and synthesized analogs of AT7519 and AZD5438, kinome selectivity scores of AT7519, AZD5438, and analog 7, bodyweight loss correlation with hearing loss at 32 kHz for mice treated with cisplatin only. (PDF)
Kinome data table of AT7519, AZD5438, and analog 7. (CSV)
Molecular formula strings. (CSV)
REFERENCES
- 1.Greene JB; Standring R; Siddiqui F; Ahsan SF Incidence of cisplatin induced ototoxicity in adults with head and neck cancer. Advances in Otolaryngology 2015, 2015, 1–4. [Google Scholar]
- 2.Cardinaal RM; De Groot JC; Huizing EH; Smoorenburg GF; Veldman JE Ultrastructural changes in the albino guinea pig cochlea at different survival times following cessation of 8-day cisplatin administration. Acta. Otolaryngol 2004, 124, 144–154. [DOI] [PubMed] [Google Scholar]
- 3.Sheth S; Mukherjea D; Rybak LP; Ramkumar V Mechanisms of cisplatin-induced ototoxicity and otoprotection. Front. Cell Neurosci 2017, 11, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sooriyaarachchi M; Gailer J; Dolgova NV; Pickering IJ; George GN Chemical basis for the detoxification of cisplatin-derived hydrolysis products by sodium thiosulfate. J. Inorg. Biochem 2016, 162, 96–101. [DOI] [PubMed] [Google Scholar]
- 5.Wu YJ; Muldoon LL; Neuwelt EA The chemoprotective agent N-acetylcysteine blocks cisplatin-induced apoptosis through caspase signaling pathway. J. Pharmacol. Exp. Ther 2005, 312, 424–431. [DOI] [PubMed] [Google Scholar]
- 6.Sooriyaarachchi M; White WM; Narendran A; Gailer J Chemoprotection by D-methionine against cisplatin-induced side-effects: insight from in vitro studies using human plasma. Metallomics 2014, 6, 532–541. [DOI] [PubMed] [Google Scholar]
- 7.Treskes M; Nijtmans LG; Fichtinger-Schepman AM; van der Vijgh WJ Effects of the modulating agent WR2721 and its main metabolites on the formation and stability of cisplatin-DNA adducts in vitro in comparison to the effects of thiosulphate and diethyldithiocarbamate. Biochem. Pharmacol 1992, 43, 1013–1019. [DOI] [PubMed] [Google Scholar]
- 8.Haake SM; Dinh CT; Chen S; Eshraghi AA; Van De Water TR Dexamethasone protects auditory hair cells against TNFalpha-initiated apoptosis via activation of PI3K/Akt and NFkappaB signaling. Hear. Res 2009, 255, 22–32. [DOI] [PubMed] [Google Scholar]
- 9.Hazlitt RA; Min J; Zuo J Progress in the development of preventative drugs for cisplatin-induced hearing loss. J. Med. Chem 2018, 61, 5512–5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sridhar R; Hanson-Painton O; Cooper DR Protein kinases as therapeutic targets. Pharmaceutical Research 2000, 17, 1345–1353. [DOI] [PubMed] [Google Scholar]
- 11.Ryals M; Pak K; Jalota R; Kurabi A; Ryan AF A kinase inhibitor library screen identifies novel enzymes involved in ototoxic damage to the murine organ of Corti. PLoS One 2017, 12, e0186001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciarimboli G; Deuster D; Knief A; Sperling M; Holtkamp M; Edemir B; Pavenstadt H; Lanvers-Kaminsky C; am Zehnhoff-Dinnesen A; Schinkel AH; Koepsell H; Jurgens H; Schlatter E Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am. J. Pathol 2010, 176, 1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sprowl JA; Ong SS; Gibson AA; Hu S; Du G; Lin W; Li L; Bharill S; Ness RA; Stecula A; Offer SM; Diasio RB; Nies AT; Schwab M; Cavaletti G; Schlatter E; Ciarimboli G; Schellens JH; Isacoff EY; Sali A; Chen T; Baker SD; Sparreboom A; Pabla N A phosphotyrosine switch regulates organic cation transporters. Nat. Commun 2016, 7, 10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pabla N; Gibson AA; Buege M; Ong SS; Li L; Hu S; Du G; Sprowl JA; Vasilyeva A; Janke LJ; Schlatter E; Chen T; Ciarimboli G; Sparreboom A Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 5231–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bielefeld EC; Tanaka C; Chen GD; Coling D; Li M; Henderson D; Fetoni AR An Src-protein tyrosine kinase inhibitor to reduce cisplatin ototoxicity while preserving its antitumor effect. Anticancer Drugs 2013, 24, 43–51. [DOI] [PubMed] [Google Scholar]
- 16.Park HJ; Kim HJ; Bae GS; Seo SW; Kim DY; Jung WS; Kim MS; Song MY; Kim EK; Kwon KB; Hwang SY; Song HJ; Park CS; Park RK; Chong MS; Park SJ Selective GSK3beta inhibitors attenuate the cisplatin-induced cytotoxicity of auditory cells. Hear. Res 2009, 257, 53–62. [DOI] [PubMed] [Google Scholar]
- 17.Teitz T; Fang J; Goktug AN; Bonga JD; Diao S; Hazlitt RA; Iconaru L; Morfouace M; Currier D; Zhou Y; Umans RA; Taylor MR; Cheng C; Min J; Freeman B; Peng J; Roussel MF; Kriwacki R; Guy K; Chen T; Zuo J CDK2 inhibitors as candidate therapeutics for cisplatin- and noise-induced hearing loss. J. Exp. Med 2018, 215, 1187–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teitz T; Goktug AN; Chen T; Zuo J Development of cell-based high-throughput chemical screens for protection against cisplatin-induced ototoxicity. Methods Mol. Biol 2016, 1427, 419–430. [DOI] [PubMed] [Google Scholar]
- 19.Chen EX; Hotte S; Hirte H; Siu LL; Lyons J; Squires M; Lovell S; Turner S; McIntosh L; Seymour L A phase I study of cyclin-dependent kinase inhibitor, AT7519, in patients with advanced cancer: NCIC Clinical Trials Group IND 177. Br. J. Cancer 2014, 111, 2262–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seftel MD; Kuruvilla J; Kouroukis T; Banerji V; Fraser G; Crump M; Kumar R; Chalchal HI; Salim M; Laister RC; Crocker S; Gibson SB; Toguchi M; Lyons JF; Xu H; Powers J; Sederias J; Seymour L; Hay AE The CDK inhibitor AT7519M in patients with relapsed or refractory chronic lymphocytic leukemia (CLL) and mantle cell lymphoma. A Phase II study of the Canadian Cancer Trials Group. Leuk. Lymphoma 2017, 58, 1358–1365. [DOI] [PubMed] [Google Scholar]
- 21.Wyatt PG; Woodhead AJ; Berdini V; Boulstridge JA; Carr MG; Cross DM; Davis DJ; Devine LA; Early TR; Feltell RE; Lewis EJ; McMenamin RL; Navarro EF; O’Brien MA; O’Reilly M; Reule M; Saxty G; Seavers LC; Smith DM; Squires MS; Trewartha G; Walker MT; Woolford AJ Identification of N-(4-piperidinyl)-4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a novel cyclin dependent kinase inhibitor using fragment-based X-ray crystallography and structure based drug design. J. Med. Chem 2008, 51, 4986–4999. [DOI] [PubMed] [Google Scholar]
- 22.El Kechai N; Agnely F; Mamelle E; Nguyen Y; Ferrary E; Bochot A Recent advances in local drug delivery to the inner ear. Int. J. Pharm 2015, 494, 83–101. [DOI] [PubMed] [Google Scholar]
- 23.Freyer DR; Chen L; Krailo MD; Knight K; Villaluna D; Bliss B; Pollock BH; Ramdas J; Lange B; Van Hoff D; VanSoelen ML; Wiernikowski J; Neuwelt EA; Sung L Effects of sodium thiosulfate versus observation on development of cisplatin-induced hearing loss in children with cancer (ACCL0431): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Camidge DR; Pemberton M; Growcott J; Amakye D; Wilson D; Swaisland H; Forder C; Wilkinson R; Byth K; Hughes A A phase I pharmacodynamic study of the effects of the cyclin-dependent kinase-inhibitor AZD5438 on cell cycle markers within the buccal mucosa, plucked scalp hairs and peripheral blood mononucleocytes of healthy male volunteers. Cancer Chemother. Pharmacol 2007, 60, 479–488. [DOI] [PubMed] [Google Scholar]
- 25.Boss DS; Schwartz GK; Middleton MR; Amakye DD; Swaisland H; Midgley RS;Ranson M; Danson S; Calvert H; Plummer R; Morris C; Carvajal RD; Chirieac LR; Schellens JH; Shapiro GI Safety, tolerability, pharmacokinetics and pharmacodynamics of the oral cyclin-dependent kinase inhibitor AZD5438 when administered at intermittent and continuous dosing schedules in patients with advanced solid tumours. Ann. Oncol 2010, 21, 884–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Y; Davies SP; Augustin M; Woodward A; Patel UA; Kovelman R; Harvey KJ A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem. J 2013, 451, 313–328. [DOI] [PubMed] [Google Scholar]
- 27.Varjosalo M; Keskitalo S; Van Drogen A; Nurkkala H; Vichalkovski A; Aebersold R; Gstaiger M The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013, 3, 1306–1320. [DOI] [PubMed] [Google Scholar]
- 28.Kalinec GM; Webster P; Lim DJ; Kalinec F A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol. Neurootol 2003, 8, 177–189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.