

Summary
The regulatory role of the ribosome in gene expression has come into sharper focus. It has been proposed that ribosomes are dynamic complexes capable of changing their protein composition in response to enviromental stimuli. We applied mass spectrometry to identify quantitative changes in the protein composition of S. cerevisiae 80S ribosomes in response to different environmental stimuli. Using quantitative mass spectrometry, we found that the paralog yeast ribosomal proteins RPL8A (eL8A) and RPL8B (eL8B) change their relative proportions in the 80S ribosome when yeast is switched from growth in glucose to glycerol. Using yeast genetics and polysome profiling, we show that yeast ribosomes containing either RPL8A or RPL8B are not functionally interchangeable. Our quantitative proteomic data support the hypothesis that ribosomes are dynamic complexes that alter their composition and functional activity in response to changes in growth or environmental conditions.
Keywords: Ribosome heterogeneity, carbon source switch, ribosome filter, RPL8A/B
Introduction
Cells growing under one growth condition require a specific proteome, and cells growing in a second condition require a different proteome. A number of cellular mechanisms including transcription, RNA processing, translation, and protein degradation are known to regulate gene expression and ultimately the cell’s proteome when growth conditions change. Translation is the process by which the information encoded in an mRNA is used to synthesize polypeptides. Composed of a small 40S and a large 60S subunit, the eukaryotic 80S ribosome catalyzes the decoding of mRNAs and formation of peptide bonds. Translational control is a major mechanism modulating eukaryotic gene expression [1–5]. Numerous studies have shown that a large number of translation factors, posttranslational protein modifications, and RNA accessory factors play active roles in translational control [3, 6–8]. Initially, the ribosomes themselves were considered only passive players in this process [9].
In recent years, the idea that the ribosomes can function as transcript-specific posttranscriptional regulatory elements has been formulated [10–21]. In the depot model, ribosomes act as reservoirs of regulatory molecules, which are released in response to specific cellular cues [22–24]. An example of this mode of action is the role of the human RPL13A ribosomal protein (r-protein) in translational control during the inflammatory response [22, 25]. A second model the ribosome filter hypothesis proposes a more direct role of ribosomes in translational control [9, 26]. This model derives from studies that reveal ribosomes are not the static, uniform structures described in textbooks. The comparison of ribosomes from rat skeletal muscle and liver using 2-D gel electrophoresis revealed differences in ribosomal composition between the two tissues [27]. In the amoeba, Dictyostelium discoideum, ribosomes from spores and vegetative cells differ both in protein composition and posttranslational modifications [28]. Studies in the Barna lab have shown that the mouse RPL38 gene is needed for efficient translation of specific Hox mRNAs [16]. Another study has shown that human RPL40 was required for translation initiation of vesicular stomatitis virus (VSV) mRNAs in human cell lines [29]. A study in A. thaliana has shown that the protein composition of ribosomes changes upon sucrose feeding [17]. Taken together, these studies point towards an evolutionarily conserved mode gene expression regulation mediated by the ribosome filter.
In the S. cerevisiae genome, the 79 r-proteins in the 80S ribosome are encoded by 138 genes [30] (Table S1). These include 57 duplicate, paralogous ribosomal gene pairs [13, 31] (Table S1), raising the possibility that individual paralogs have distinct finctions. Deletion analysis of S. cerevisiae paralogous pairs showed that the genes are not functionally equivalent [12, 32, 33]. Using mass spectrometry, differences were found in the stoichiometry of core r-proteins in yeast grown under different growth conditions [34]. In higher eukaryotic organisms, previous studies showed specific r-proteins are required for the translation of selected mRNA transcripts [16, 29]. Post-translational modifications, especially phosphorylation, and variations in rRNA composition add further levels of diversity to the different ribosome complexes in cells and tissues [35, 36].
The evidence of heterogeneity in the ribosome’s protein and rRNA composition has generated speculation as to its functional consequences. Mauro and Edelman proposed that ribosomal subunits differing in protein or rRNA composition bind translation factors or specific mRNAs with different affinities, thereby selectively changing the rates of polypeptide synthesis of particular mRNAs [9, 26]. In their model, different or specialized ribosomes in a heterogeneous ribosomal population in the cell function as regulatory elements or ‘filters’ by selectively binding and translating specific mRNAs. In support of this model, Bauer et al. showed that “specialized” ribosomes with specific r-proteins encoded by paralogous genes preferentially translate specific reporter mRNAs [37].
In this study, we hypothesized that the cell’s ribosomes change their protein composition in response to changing growth conditions as a mechanism to regulate expression. To test this hypothesis, we used quantitative mass spectrometry-based proteomics to detect and identify specific r-proteins that change their stoichiometry in the 80S ribosome upon a change in growth condition. Quantitative proteomics using tandem mass spectrometry analysis of protease-digested ribosomes coupled with isotopic labeling was used to both identify and quantify the relative abundances of specific proteins in cellular or subcellular protein fractions [38, 39]. Thus, quantitative proteomics enabled us to identify potential changes in 80S ribosomal protein compositions.
Using our mass spectrometry strategy, we profiled the 80S ribosome composition in yeast cells growing in glucose or glycerol as a carbon source. Abruptly shifting from fermentable glucose to the non-fermentable glycerol carbon source induces a diverse array of changes in S. cerevisiae gene expression [40, 41]. Using proteomic analysis, we identified 75 of the expected 79 r-proteins in the 80S complexes. This set included 55 of the 57 paralogous r-protein gene pairs. Unexpectedly, proteomic analysis revealed that the r-protein paralogs RPL8A and RPL8B change their relative stoichiometry in the population of ribosomes when yeast cells are shifted from glucose to glycerol as a carbon source. Phenotypic analysis and polysome profiling using rpl8aΔ and rpl8bΔ null homozygotes show that the functions of RPS8A and RPS8B are not interchangeable.
Results
We hypothesized that specific r-proteins important for the specialization of ribosome activity will show differential abundances in response to environmental stimuli. In pilot studies, we used quantitative proteomics to identify and quantify changes in individual r-proteins abundances from yeast whole-cell protein extracts after shifts in both carbon source and temperature [41]. The analysis showed most of the 40S and 60S r-proteins were down-regulated in response to the two environmental stimuli (Fig 1). However, selected r-proteins showed significantly larger decreases in abundance [41]. This result suggested that these r-proteins were being differentially expressed compared to the majority of the r-proteins, consistent with the ribosome filter hypothesis [9].
Figure 1. The log2 fold-changes in the distributions of 40S and 60S subunit protein abundances caused by changes in growth conditions.
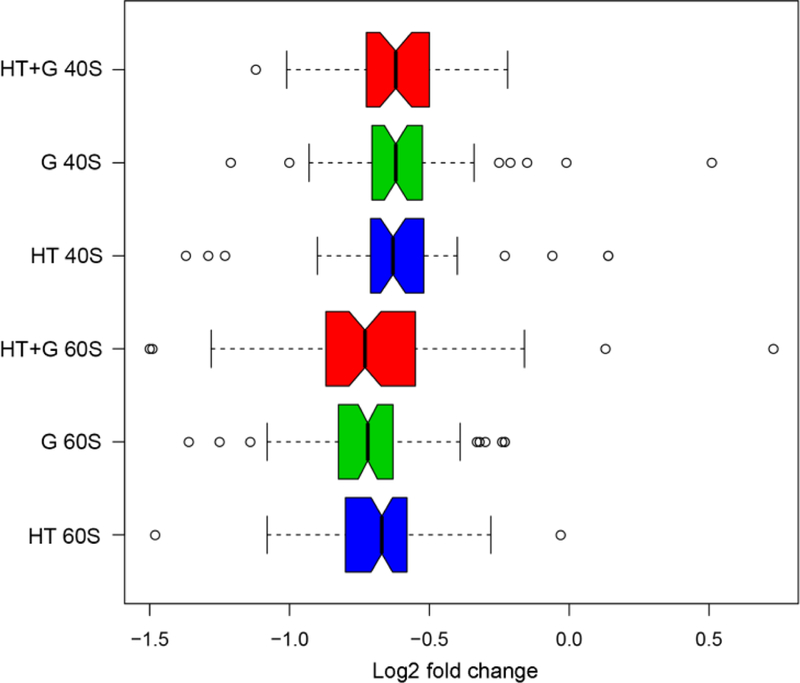
Quantitative proteomics was used to identify and quantify changes in individual r-proteins abundances from S. cerevisiae whole-cell protein extracts after shifts in carbon source and temperature [41]. Yeast was grown at standard conditions (glucose, 30° C) and changed to three different environmental growth conditions or stimuli: high temperature 39° C (HT, blue); glycerol (G, green), and high temperature and glycerol combined (HT+G, red). A custom Python script CompZilla.py was used to analyze changes in the r-proteins in the quantitative proteomic data. Open circles represent r-protein outliers in the log2 fold-changes in their protein abundance.
To identify and quantitate the changes in the protein composition of purified 80S yeast ribosomes, we focused on comparing yeast growing in rich medium with two different carbon sources, glucose and glycerol [41]. From yeast cells cultured in glucose or glycerol, we isolated ribosomes using discontinuous sucrose gradient centrifugation and quantitative proteomic analysis to identify and precisely quantify the relative abundances of ribosomal proteins.
Quantitative proteomic analysis of r-proteins in purified ribosomes
To identify and measure the changes in the protein composition of ribosome populations purified from yeast growing in glucose or glycerol, we analyzed ribosomal protein composition using iTRAQ-labeling and quantitative tandem mass spectrometry [38, 42]. Whole-cell extracts and purified ribosomes from yeast cells grown in glucose (T=0) and the 24h glycerol time point (T=24h) were used. After proteolytic digestion of the proteins with trypsin, the N-termini and amino acid side chain amines of the tryptic fragments were covalently labeled with isotoptic 4-plex mass tags [38, 43]. Replicate ribosome samples from the glucose (T=0) and the 24h glycerol (T=24h) time points were labeled with unique iTRAQ tags and pooled. The labeled ribosomal peptide mixtures were fractionanated using multidimensional microcapillary HPLC [42], and the eluting peptides were analyzed using nanoESI-tandem mass spectrometry (MS/MS) [41]. The peptide fragmentation data were computationally compared to predicted values from a yeast protein database to identify the labeled peptides and hence the corresponding proteins [44]. The fragmentation of the iTRAQ tag generated low molecular mass reporter ions that were used to quantify the identified r-protein peptides and proteins [38, 43].
From three independent biological replicates, we identified 135 of the 139 r-proteins from the purified ribosome complexes using this mass spectrometry-based proteomic approach (Table S2). With a total of 79 yeast r-proteins in the 80S ribosomal complexes, we successfully identified 20 of the 22 r-proteins from single-copy genes and 55 of the 57 r-protein pairs from paralogous genes. The four proteins not identified were from r-protein genes encoding small proteins < 60 amino acids in size. The mass spectrometry analysis identified 21 of the 22 100% identical paralogous r-proteins (Table S2). For 16 of the 35 paralogous gene pairs that are not 100% identical, we successfully identified both unique peptides. Of the remaining 19 non-identical paralogous gene pairs, our mass spectrometry analysis was able to uniquely identify 1 of the 2 paralogous r-proteins in 8 cases (Table S2).
For each of the three replicates, we next calculated the relative fold-change in abundance for 131 r-proteins in glycerol (T=24) compared to glucose (T=0) using the log2 ratio of the iTRAQ reporter ion signal intensities (Table S3). Because ribosomes were previously purified and analyzed from identical whole-cell extracts [41], we were able to directly compare the fold changes of 126 r-proteins quantitated in both the whole-cell extracts and purified ribosomes. A correlation analysis between the fold change of r-proteins in the solubilized whole cell extracts and the purified ribosome extracts shows poor correlation (Fig 2A and B). However, there was strong correlation in the fold-changes of r-proteins between the experimental replicates (Fig 3A and B). The lack of correlation between the fold changes in cell extracts and purified ribosomes suggest that a fraction of r-proteins in the cell are not incorporated into ribosomal complexes. We reasoned that either these r-proteins have not yet been successfully incorporated into ribosomal complexes during ribosome biogenesis or ribosomes were disassembling. The mechanism and significance behind the lack of correlation needs to be investigated in future studies.
Figure 2. Correlation analysis between the fold changes of r-proteins in whole cell extracts and purified ribosomes from cells grown at standard conditions (glucose, 30°C) and glycerol (glycerol, 30°C). (See Tables S2 and S3).
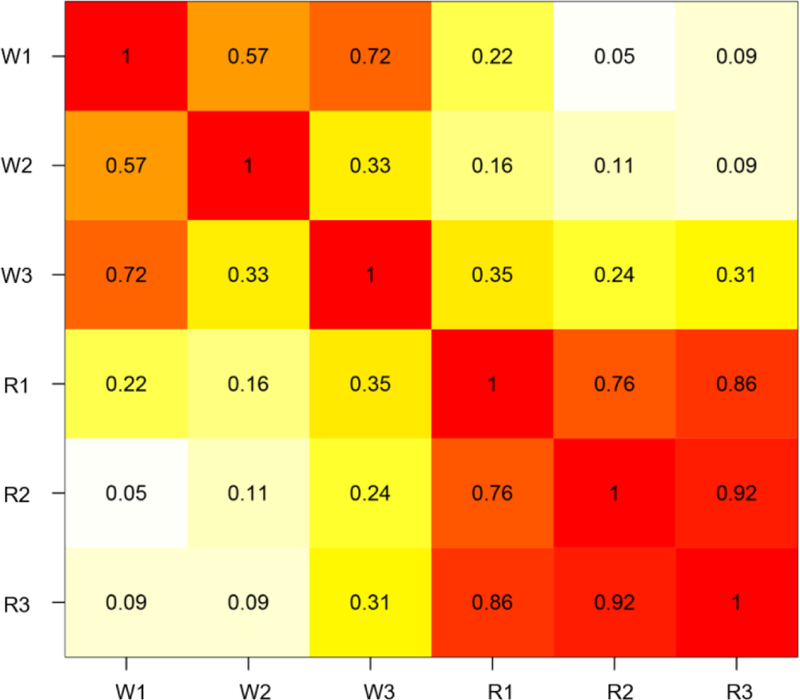
The summary of three replicates is shown. Cross-correlation matrix. Numbers represent Pearson’s R. W# represent whole extract replicates. R# represent purified ribosome replicates.
Figure 3. Scatterplots showing reproducibility among replicates of whole cell extracts and purified ribosomes.
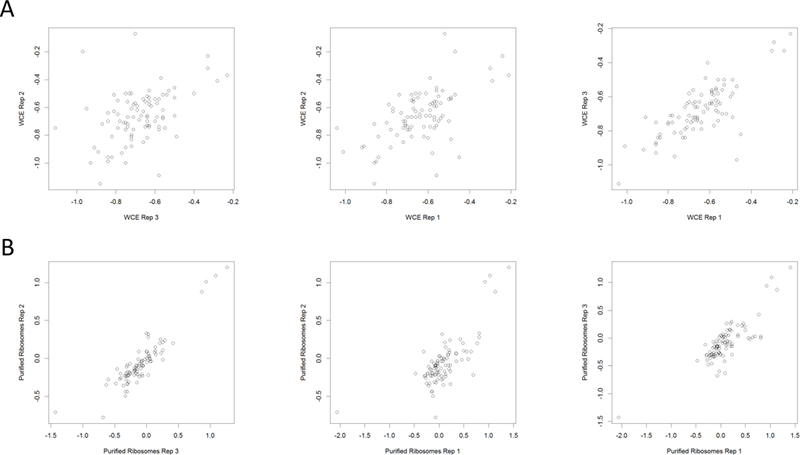
(A) Whole-cell extract replicates. (B) Purified ribosome replicates. Plots were generated in RStudio.
To identify differentially abundant r-proteins in purified yeast ribosomes grown in glucose and glycerol, we used a t-test of independence with an alpha level of 0.05. Since there are only minimal sequence differences between the r-protein paralogs to reliably quantify the paralog-specific changes, we reanalyzed the mass spectrometry data using only the unique peptides for quantitation. We quantitated 45 r-proteins with at least one unique peptide (Fig 4A) and identified 11 r-proteins that were differentially present in the purified ribosome complexes from yeast cells grown in glucose or glycerol (Fig 4B). The list of significantly changing r-proteins in the purified ribosomes contained the paralog pair RPL8A (eL8A) and RPL8B (eL8B) (97% protein sequence identity) whose fold changes were opposite to each other. Since the ribosome filter hypothesis predicts that the ribosomes containing specific paralogs translate different mRNAs with differing efficiencies, and eL8A and eL8B abundances are altered based on carbon source, we chose eL8A and eL8B as candidate ribosome filters for further characterization.To validate the iTRAQ data, the relative difference in abundance of RPL8A (eL8A) and RPL8B (eL8B) proteins in the purified ribosomes was measured using multiple reaction-monitoring mass spectrometry (Fig 4C). The data suggest yeast cells have a differential requirement for RPL8A and RPL8B under the two growth conditions.
Figure 4. Quantitation of r-proteins in ribosomes purified from yeast cells grown in glucose and glycerol.
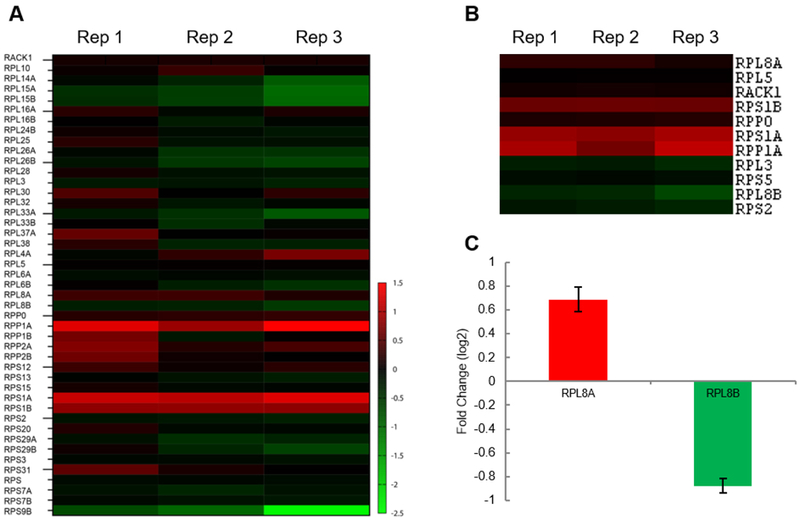
(A) Forty-five ribosomal proteins identified and quantitated using at least one unique peptide in iTRAQ-mass spectrometry experiments. The log2 transformed fold change in the r-protein’s relative abundance 24 h after changing growth in glucose to glycerol was used to generate the heatmap. (B) Eleven statistically significant differentially abundant r-proteins in the purified ribosomes from cells grown in glucose and glycerol identified using a t-test’s p-value less than 0.05. (C) The protein abundance of RPL8A and RPL8B validated using multiple reaction monitoring in three independently purified ribosome replicates. Color Legend: red increased; green decreased abundance of the yeast r-protein in cells growing in glycerol compared to glucose media.
Change of polysomal fractions in response to switch of carbon source
Since we found that with the shift to glycerol, the 80S ribosome population’s r-protein composition changed, we speculated that global translational activities might be affected. We performed polysome profiling experiments using the same time course. These experiments indeed revealed a re-distribution of polysomes (actively translating) into the 80S peak (inactive) 30 minutes following the shift from glucose to glycerol (Fig 5). After 450 min, the polysomes partially recovered (Fig 5). A similar re-distribution of polysomal fractions to the 80S fraction was also observed after cells were shifted from glucose to glycerol for 10 min [40].
Figure 5. The switch of carbon source from glucose to glycerol inhibits translation.
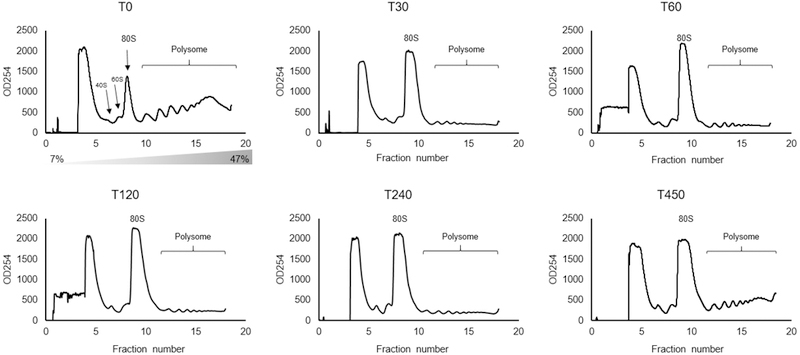
Polyribosome traces from the wild-type S. cerevisiae strain. Yeast was grown in complete medium containing glucose, then re-suspended at T=0 in medium containing glycerol for the indicated times (min). Polyribosomes were analyzed as described in ‘STAR Methods’. The peaks that contain the small 40S ribosomal subunit, the large 60S ribosomal subunit, and complete 80S ribosomes are indicated by arrows. The polysome peaks are bracketed.
Different functional Roles of RPL8A and RPL8B
An increase in eL8a protein in the ribosomes from the cells grown in glycerol suggests that eL8a is important for the translation of proteins required for growth with glycerol as a carbon source. Using growth assays, we tested the paralog-specific roles of RPL8A and RPL8B using yeast genetics. When we compared yeast growth rates in glucose versus glycerol, we observed a functional difference in the activity or specificity of the yeast ribosome depending on the presence of eL8A and eL8B (Fig 6). Under optimal growth conditions in rich media with glucose as the carbon source, the function of either rpl8aΔ or rpl8bΔ null allele can be compensated for by the paralogous gene (Fig 6A-C). However, when the cells encounter non-optimal growth in glycerol, the differential requirement for each paralog becomes apparent (Fig 6B, C, D). Inability of RPL8B to complement loss of RPL8A under the non-optimal growth condition is consistent with the original ribosome filter hypothesis in which the ribosome filter was proposed to fine-tune gene expression [9, 26].
Figure 6. Doubling times comparisons of rpl8AΔ and rpl8BΔ strains.
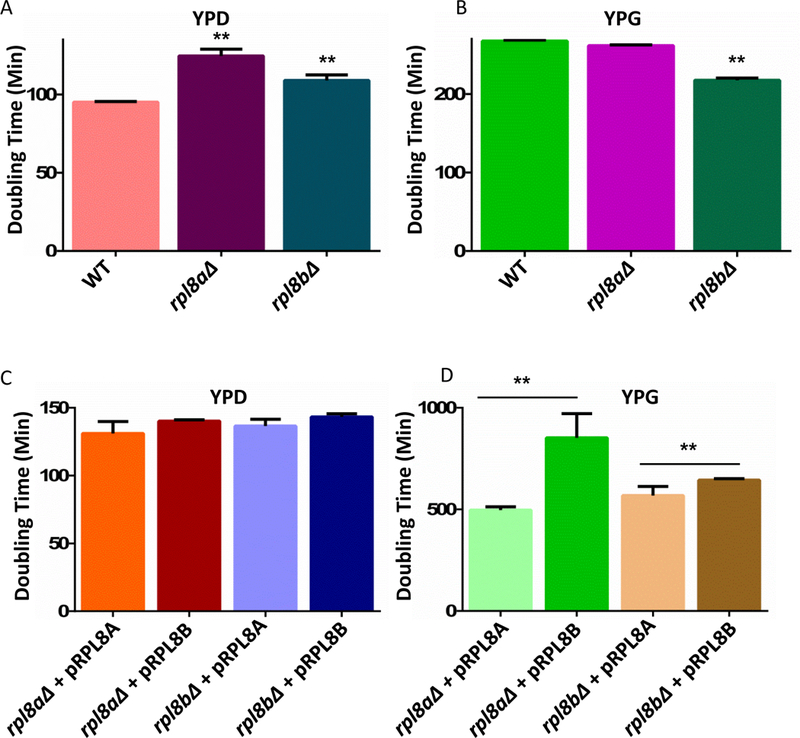
A & B) Stars denote the statistical significance of comparison with the WT strain (p-values < 0.05). A) Doubling times of WT, rpl8aΔ, and rpl8bΔ yeast strains with glucose as a carbon source. B) Doubling times of WT, rpl8aΔ, and rpl8bΔ strains with glycerol as a carbon source. C) Doubling times of rpl8aΔ strain with RPL8A on a plasmid, rpl8aΔ with RPL8B on a plasmid, rpl8bΔ with RPL8B on a plasmid, and rpl8bΔ with RPL8A on a plasmid with glucose as carbon source. D) Doubling times of rpl8aΔ strain with RPL8A on a plasmid, rpl8aΔ with RPL8B on a plasmid, rpl8bΔ with RPL8B on a plasmid, and rpl8bΔ with RPL8A on a plasmid with glycerol as carbon source. Stars denote the statistical significance of comparison denoted by the lines above the bar graph (p-values < 0.05).
Discussion
In this study we have employed quantitative mass spectrometry along with polysome profiling and genetic analysis to follow changes in the composition of the yeast ribosome as a function of time after a shift from growth in glucose to glycerol media. The quantitative proteomic experiments enabled us to identify and quantify changing r-proteins, including paralogs, incorporated into yeast ribosome populations. Isotopic labeling enabled us to perform precise quantitative measurements of individual r-proteins in the purified ribosomal complexes. The quantitative proteomic experiments analyzed the population of purified ribosomal complexes isolated from sucrose gradients. The r-proteins’ low molecular weight and high abundance of lysine and arginine residues limited the number of tryptic peptides that could be surveyed by our mass spectrometry approach. Therefore, the number of r-proteins that could be confidently identified and quantified by the quantitative mass spectrometry was limited. Targeted mass spectrometry approaches would be one solution to this limitation.
Dosage insufficiency has recently been proposed to explain the phenotypes associated with the loss of different ribosomal proteins [45]. However, our growth rate data strongly supports the paralog-specific role for ribosomal proteins when the cells were forced to utilize the non-optimal carbon glycerol. With glycerol as the carbon source overexpression of RPL8A led to a decrease in the doubling times (eg. faster growth) compared to overexpression of RPL8B (Fig 6D). It is interesting to note that under the optimal growth condition (rich media with glucose as the carbon source), the function of either paralogs can be compensated by each other. It is when the cells encounter a non-optimal growth condition that the differential requirement of the paralogs becomes apparent. This contradicts the dosage insufficiency model and is in agreement with the ribosome filter hypothesis where the ribosome filter was proposed to fine tune the gene expression [9, 26].
The heterogeneity in the ribosome composition has been demonstrated for a diverse group of model organisms. One of the earliest reports of differences in composition of ribosomes comes from studies in rat where investigators found that there were differences in the protein composition of ribosomes between different tissues. Studies with the slime mold D. discoideum had revealed differences in the composition of ribosomes based upon the life cycle stage [28]. These studies had led to speculation about the role of ribosome as a translational control element. However, the functional significance of the heterogeneity has only recently been fully appreciated with studies in yeast, plants, mice, and human cell lines providing evidence for ribosome-mediated translational control. The evidences argue for an evolutionarily conserved role of ribosome filter-mediated translational control.
In this study, we have identified another candidate ribosome filter based on the paralog pair RPL8A and RPL8B using iTRAQ labeling and mass spectrometry-based precise quantitative proteomics. The population level changes detected by this approach also identified differences in the levels of singly copy ribosomal proteins. It raises the possibility of super-stoichiometry or sub-stoichiometry in the ribosome composition. Sub-stoichiometric ribosome lacking RPL38 has been shown to be defective in translation of Hox mRNA in mice but are otherwise translation competent [16]. It would be interesting to look at the individual ribosome particles to detect super-stoichiometry or sub-stoichiometry. Another interesting question that still needs to be addressed is the identity of transcripts whose expression is differentially regulated by eL8a or eL8b containing ribosomes. The exact structural mechanism of regulation also remains an open question. Future studies will address these questions.
Materials and Methods
Strains and Media
All experiments used the diploid S. cerevisiae strain BY4743, which has been previously described [46]. The homozygous diploid rpl8aΔ and rpl8bΔ null strains were obtained from the yeast deletion collection. Cells were grown using standard techniques [47]. The genomic region of RPL8A and RPL8B, including ~1kb upstream and ~200b downstream, were cloned into the pFA6a-His3MX6 plasmid with Gibson cloning (New England Biolabs) to generate plasmids pALL8a and pALL8b, respectively [48]. Strains were transformed using the standard yeast transformation protocol [47].
For calculating doubling times in glucose (YPD) and glycerol (YPG) media, 5 mL starter cultures from single colonies of BY4743 (wild-type), and the null strains, rpl8aΔ, and rpl8bΔ, were grown for 24 h as previously described [41]. For calculating doubling times of rpl8aΔ and rpl8bΔ strains complemented with plasmids pALL8a or pALL8b, yeast were grown in SC-His glucose (ScD) or SC-His glycerol (ScG) as previously described [41, 46, 49].
For plasmid rescue experiments, yeast strains were grown in SC–His supplemented with G418. One hundred microliters of selected media was added to wells in a 96-well plate. Next, each well was inoculated with 10 μL of a starter culture. Cells were grown at 30o C in a Synergy Biotek plate reader for 720 min with constant shaking. OD 660 readings were taken every 3 min. A text file containing the OD 660 readings was exported and parsed using a custom Python script. Doubling times were calculated using the lm function in R. A Student’s t-test of independence was used to calculate the statistical significances at alpha level of 0.05. All p-values were adjusted for multiple hypotheses testing using the Bonferroni correction [50, 51]. The source code used in this project is found in supplementary data.
Ribosome isolation and purification
S. cerevisiae cells were grown in glucose medium (YPD) (1% yeast extract, 2% peptone, 2 % glucose) to mid-log phase as determined by OD600 measurement and shifted to glycerol medium (YPG) (1% yeast extract, 2% peptone, 3% glycerol) as previously described [41]. The ribosomes were purified for mass spectrometry analysis at the 0 and 1440 min time points. Ribosomes were purified from yeast in the absence translation inhibitors to avoid potential artefacts. Yeast cells were centrifuged at 2000 rpm for 5 min at 4°C using a Sorvall HLR6/H600A/HBB6 rotor in a Sorvall RC-3B centrifuge and washed with ice-cold deionized H2O. The cell pellets were resuspended in 1 mL ice-cold wash buffer (10 mM Tris pH 8, 5 mM β-mercaptoethanol, 500 mM ammonium chloride, 100 mM magnesium acetate) and lysed at 4°C for 10 min using glass beads and a Bead Beater (BioSpec, Inc) as previously described [52]. The cell suspensions were clarified by centrifugation at 20,000g for 15 min at 4°C. The pellets were discarded and the supernatants were overlaid onto 5/20% discontinuous sucrose gradients (sucrose cushion) prepared in wash buffer. The gradients were centrifuged at 28,000 rpm using a SW-41 swinging bucket rotor for 18 h at 4°C. The ribosome-enriched pellets were resuspended in 1 mL ice cold standard buffer (10 mM Tris pH 8, 5 mM β-mercaptoethanol, 50 mM ammonium chloride, 5 mM magnesium acetate) and centrifuged for 10 min at 10,000g at 4° C. The pellets were discarded, and the ribosome suspensions were stored at −80° C.
iTRAQ labeling, liquid chromatography mass spectrometry, and data analysis
iTRAQ labeling, liquid chromatography tandem mass spectrometry, and database searches for peptide spectrum matching were done as previously described [41, 42, 44]. Proteome Discoverer 1.4 (Thermo Scientific) output files were imported into ProteoIQ (Premier BioSoft) for protein assembly and reporter ion quantitation. Peptide and protein FDR was set to 5%. Reporter ion normalization was based upon their intensities for the single copy r-proteins. This was done for both purified ribosome experiments and the previously published whole cell extract experiment [41]. Briefly, the samples were analyzed using a 12-step MudPIT approach with salt pulses of 0, 25, 50, 75, 100, 150, 200, 250, 300, 500, and 750 mM and 1 M ammonium acetate. Each MudPIT fraction was analyzed using a 120 min reverse phase chromatography. A precursor ion scan in FTMS mode was followed by 4 CID scans and 4 HCD scans. The ion isolation window was set to 2 Da. CID and HCD spectra for a precursor ion were merged using Proteome Discoverer 1.4 (Thermo Fisher Scientific, Inc). Merged spectra were searched for a peptide-spectrum match (PSM) using Sequest running inside Proteome Discoverer 1.4. PSMs were exported and analyzed using ProteoIQ 2.7 (Premier Biosoft, Inc). The purified ribosome experiment was analyzed in two ways using ProteIQ. For comparison with the whole cell extract analysis, all peptides were used for quantitation and protein assembly. To identify differentially present r-proteins, only the peptides uniquely mapping to one r-protein were used for quantitation and protein assembly.
To validate the selected iTRAQ results, proteotypic peptides were selected for targeted quantitation from a database of identified peptides in the iTRAQ experiments. Transitions for unscheduled scout experiments were selected based upon NIST and GPM spectral libraries. Fifty μg of the purified ribosomes was digested with sequencing grade modified trypsin (1:50; Promega Corporation) and desalted essentially as described [41]. Peptides were eluted using an elution buffer composition of 50% acetonitrile, 0.1% trifluoroacetic acid. Peptides were analyzed using a 90 min scheduled SRM analysis. Briefly, peptides were autosampled onto a 200 mm by 0.1 mm (Jupiter 3 micron, 300A), analytical column coupled directly to an TSQ-Vantage (ThermoFisher) using a nanoelectrospray source and resolved using an aqueous to organic gradient (1–45% Buffer B) at a 500 nl/min flow rate. Using a series of unscheduled scout runs to determine retention times and transitions to monitor, a scheduled instrument method encompassing a 10 min window around each retention time along with calculated collision energies was created using Skyline [53]. Q1 peak width resolution was set to 0.7, collision gas pressure was 1 mTorr, and the EZmethod cycle time was 5 s.
The resulting RAW instrument files were imported into Skyline for peak-picking and quantitation [53]. The peak areas of the transitions were exported, and further analysis was done in Microsoft Excel. The sum of the peak areas of all the transitions of a given peptide, the peptide peak area, was used as the quantitative measure of abundance for the peptide. The average of peptide peak areas of all the peptides from a given protein, the protein peak area, was used as the quantitative measure of abundance of the protein. The average protein peak areas of the single copy r-protein RPL5p was used as a control. For differential analysis, in the first step, a ratio of peak area of the test protein to the peak area of the control was calculated across all samples. In the next step, a two-sample t-test with alpha level 0.05 was performed with the ratios to test for statistical significance. Finally, the fold change was calculated by taking the ratio of the average of ratios. The calculated fold change was log2 transformed.
COMPzilla
COMPzilla uses CYC2008 2.0, a manually curated database of biomolecular complexes in yeast to identify complexes that are differentially present [54]. In the first step COMPzilla creates a dictionary with complex names as keys and the proteins that constitute the complex as values. Next, COMPzilla creates a dictionary in which complex names are still the keys, but values are mapped to fold changes of the proteins that constitute the complex. In the third step, COMPzilla creates a list all the fold changes in the population for statistical testing. Finally, COMPzilla compares the fold change distributions associated with protein complexes with population fold change distribution using a two-sample t-test of independence and two-sample Kolomogorv-Smirnov test. COMPzilla exports the results of the two tests in separate tab delimited text files, in which the first column contains the complex names, the second column contains t-statistics or ks-statistics, and the third column contains the corresponding p-value. The CompZilla.py source code is available in the supplementary data.
Polysome profiling
Polysome profiling was modified from previous methods [52]. S. cerevisiae cells were grown in glucose media (YPD) to mid-log phase. Strains were then transferred to glycerol media (YPG) and allowed to continue to grow. At 30 min, 120 min, 240 min and 450 min, cells were collected and lysed as previously described. Protein extracts were layered on top of 7% - 47% sucrose gradients and centrifuged.
Supplementary Material
Highlights.
RPL8A and RPLB change their relative stoichiometry in the ribosome when yeast cells are grown in different carbon sources.
Compositional changes in ribosomal protein paralogs in the ribosomes are reflected in functional changes based on polysome profiles.
Acknowledgements
This study was supported by NIH RO1 grant GM64779 (to AL) and HHMI and NIH R01 GM29169 (to J.F.). CB was supported by NIH training grant T32 AI007611. We would like to thank Marko Jovanovic and Elizabeth M. Link for scientific discussions and editorial assistance in the preparation of this manuscript.
Footnotes
Contact for Reagent and Resource Sharing
Requests for reagents should be directed to Andrew J. Link.
References
- [1].Carpenter S, Ricci EP, Mercier BC, Moore MJ, Fitzgerald KA, Post-transcriptional regulation of gene expression in innate immunity. Nat Rev Immunol 2014, 14, 361–376. [DOI] [PubMed] [Google Scholar]
- [2].Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N, Translational control of long-lasting synaptic plasticity and memory. Neuron 2009, 61, 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hinnebusch AG, Translational control 1995–2015: unveiling molecular underpinnings and roles in human biology. Rna 2015, 21, 636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Holcik M, Sonenberg N, Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 2005, 6, 318–327. [DOI] [PubMed] [Google Scholar]
- [5].Kong J, Lasko P, Translational control in cellular and developmental processes. Nat Rev Genet 2012, 13, 383–394. [DOI] [PubMed] [Google Scholar]
- [6].Dever TE, Green R, The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harbor perspectives in biology 2012, 4, a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kapp LD, Lorsch JR, The molecular mechanics of eukaryotic translation. Annu Rev Biochem 2004, 73, 657–704. [DOI] [PubMed] [Google Scholar]
- [8].Sonenberg N, Hinnebusch AG, Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 2009, 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mauro VP, Edelman GM, The ribosome filter hypothesis. Proc Natl Acad Sci U S A 2002, 99, 12031–12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ramagopal S, Ennis HL, Ribosomal protein synthesis during spore germination and vegetative growth in Dictyostelium discoideum. J Biol Chem 1982, 257, 1025–1031. [PubMed] [Google Scholar]
- [11].Ruggero D, Pandolfi PP, Does the ribosome translate cancer? Nat Rev Cancer 2003, 3, 179–192. [DOI] [PubMed] [Google Scholar]
- [12].Komili S, Farny NG, Roth FP, Silver PA, Functional specificity among ribosomal proteins regulates gene expression. Cell 2007, 131, 557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].McIntosh KB, Warner JR, Yeast ribosomes: variety is the spice of life. Cell 2007, 131, 450–451. [DOI] [PubMed] [Google Scholar]
- [14].Barna M, Pusic A, Zollo O, Costa M, et al. , Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Warner JR, McIntosh KB, How common are extraribosomal functions of ribosomal proteins? Mol Cell 2009, 34, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kondrashov N, Pusic A, Stumpf CR, Shimizu K, et al. , Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 2011, 145, 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hummel M, Cordewener JH, de Groot JC, Smeekens S, et al. , Dynamic protein composition of Arabidopsis thaliana cytosolic ribosomes in response to sucrose feeding as revealed by label free MSE proteomics. Proteomics 2012, 12, 1024–1038. [DOI] [PubMed] [Google Scholar]
- [18].Hummel M, Dobrenel T, Cordewener JJ, Davanture M, et al. , Proteomic LC-MS analysis of Arabidopsis cytosolic ribosomes: Identification of ribosomal protein paralogs and re-annotation of the ribosomal protein genes. J Proteomics 2015, 128, 436–449. [DOI] [PubMed] [Google Scholar]
- [19].Warner JR, Twenty years of ribosome assembly and ribosomopathies. Rna 2015, 21, 758–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xue S, Tian S, Fujii K, Kladwang W, et al. , RNA regulons in Hox 5’ UTRs confer ribosome specificity to gene regulation. Nature 2015, 517, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Smagin DA, Kovalenko IL, Galyamina AG, Orlov YL, et al. , Heterogeneity of Brain Ribosomal Genes Expression Following Positive Fighting Experience in Male Mice as Revealed by RNA-Seq. Mol Neurobiol 2018, 55, 390–401. [DOI] [PubMed] [Google Scholar]
- [22].Mazumder B, Sampath P, Seshadri V, Maitra RK, et al. , Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell 2003, 115, 187–198. [DOI] [PubMed] [Google Scholar]
- [23].Ray PS, Arif A, Fox PL, Macromolecular complexes as depots for releasable regulatory proteins. Trends Biochem Sci 2007, 32, 158–164. [DOI] [PubMed] [Google Scholar]
- [24].Zhou X, Liao WJ, Liao JM, Liao P, Lu H, Ribosomal proteins: functions beyond the ribosome. Journal of molecular cell biology 2015, 7, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kapasi P, Chaudhuri S, Vyas K, Baus D, et al. , L13a blocks 48S assembly: role of a general initiation factor in mRNA-specific translational control. Mol Cell 2007, 25, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mauro VP, Edelman GM, The ribosome filter redux. Cell Cycle 2007, 6, 2246–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sherton CC, Wool IG, A comparison of the proteins of rat skeletal muscle and liver ribosomes by two-dimensional polyacrylamide gel electrophoresis. Observations on the partition of proteins between ribosomal subunits and a description of two acidic proteins in the large subunit. J Biol Chem 1974, 249, 2258–2267. [PubMed] [Google Scholar]
- [28].Ramagopal S, Ennis HL, Regulation of synthesis of cell-specific ribosomal proteins during differentiation of Dictyostelium discoideum. Proc Natl Acad Sci U S A 1981, 78, 3083–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee AS, Burdeinick-Kerr R, Whelan SP, A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAs. Proc Natl Acad Sci U S A 2013, 110, 324–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nakao A, Yoshihama M, Kenmochi N, RPG: the Ribosomal Protein Gene database. Nucleic Acids Res 2004, 32, D168–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Warner JR, The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 1999, 24, 437–440. [DOI] [PubMed] [Google Scholar]
- [32].Breslow DK, Cameron DM, Collins SR, Schuldiner M, et al. , A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods 2008, 5, 711–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Giaever G, Chu AM, Ni L, Connelly C, et al. , Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [DOI] [PubMed] [Google Scholar]
- [34].Slavov N, Semrau S, Airoldi E, Budnik B, van Oudenaarden A, Differential Stoichiometry among Core Ribosomal Proteins. Cell Rep 2015, 13, 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Martin I, Kim JW, Lee BD, Kang HC, et al. , Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell 2014, 157, 472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ramagopal S, Are eukaryotic ribosomes heterogeneous? Affirmations on the horizon. Biochem Cell Biol 1992, 70, 269–272. [DOI] [PubMed] [Google Scholar]
- [37].Bauer JW, Brandl C, Haubenreisser O, Wimmer B, et al. , Specialized yeast ribosomes: a customized tool for selective mRNA translation. PLoS ONE 2013, 8, e67609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ross PL, Huang YN, Marchese JN, Williamson B, et al. , Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 2004, 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- [39].Yates JR, Ruse CI, Nakorchevsky A, Proteomics by mass spectrometry: approaches, advances, and applications. Annual review of biomedical engineering 2009, 11, 49–79. [DOI] [PubMed] [Google Scholar]
- [40].Kuhn KM, DeRisi JL, Brown PO, Sarnow P, Global and specific translational regulation in the genomic response of Saccharomyces cerevisiae to a rapid transfer from a fermentable to a nonfermentable carbon source. Mol Cell Biol 2001, 21, 916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Samir P, Rahul, Slaughter JC, Link AJ, Environmental Interactions and Epistasis Are Revealed in the Proteomic Responses to Complex Stimuli. PLoS ONE 2015, 10, e0134099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Link AJ, Eng J, Schieltz DM, Carmack E, et al. , Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol 1999, 17, 676–682. [DOI] [PubMed] [Google Scholar]
- [43].Unwin RD, Griffiths JR, Whetton AD, Simultaneous analysis of relative protein expression levels across multiple samples using iTRAQ isobaric tags with 2D nano LC-MS/MS. Nat Protoc 2010, 5, 1574–1582. [DOI] [PubMed] [Google Scholar]
- [44].Eng JK, McCormack AL, Yates I, J. R., An approach to correlate tandem mass spectral data of peptides with amino acid sequences. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- [45].Mills EW, Green R, Ribosomopathies: There’s strength in numbers. Science 2017, 358. [DOI] [PubMed] [Google Scholar]
- [46].Brachmann CB, Davies A, Cost GJ, Caputo E, et al. , Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 1998, 14, 115–132. [DOI] [PubMed] [Google Scholar]
- [47].Amberg DC, Burke DJ, Strathern JN, Methods in Yeast Genetics 2005 Edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: 2005. [Google Scholar]
- [48].Gibson DG, Young L, Chuang RY, Venter JC, et al. , Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 2009, 6, 343–345. [DOI] [PubMed] [Google Scholar]
- [49].Winzeler EA, Shoemaker DD, Astromoff A, Liang H, et al. , Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [DOI] [PubMed] [Google Scholar]
- [50].Dunn OJ, Estimation of the median for dependent variables. Ann. Math. Statist. 1959, 30, 192–197. [Google Scholar]
- [51].Dunn OJ, Multiple Comparisons Among Means. Journal of the American Statistical Association 1961, 56, 52–64. [Google Scholar]
- [52].Browne CM, Samir P, Fites JS, Villarreal SA, Link AJ, The yeast eukaryotic translation initiation factor 2B translation initiation complex interacts with the fatty acid synthesis enzyme YBR159W and endoplasmic reticulum membranes. Mol Cell Biol 2013, 33, 1041–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].MacLean B, Tomazela DM, Shulman N, Chambers M, et al. , Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pu S, Vlasblom J, Emili A, Greenblatt J, Wodak SJ, Identifying functional modules in the physical interactome of Saccharomyces cerevisiae. Proteomics 2007, 7, 944–960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.