

Abstract
Sactipeptides are ribosomally-synthesized peptides that contain a characteristic thioether bridge (sactionine bond) that is installed posttranslationally and is absolutely required for their antibiotic activity. Sactipeptide biosynthesis requires a unique family of radical SAM enzymes, which contain multiple [4Fe-4S] clusters, to form the requisite thioether bridge between a cysteine and the α-carbon of an opposing amino acid through radical-based chemistry. Here we present the structure of the sactionine bond-forming enzyme CteB, from Clostridium thermocellum ATCC 27405, with both SAM and an N-terminal fragment of its peptidyl-substrate at 2.04 Å resolution. CteB has the (β/α)6-TIM barrel fold that is characteristic of radical SAM enzymes, as well as a C-terminal SPASM domain that contains two auxiliary [4Fe-4S] clusters. Importantly, one [4Fe-4S] cluster in the SPASM domain exhibits an open coordination site in absence of peptide substrate, which is coordinated by a peptidyl-cysteine residue in the bound state. The crystal structure of CteB also reveals an accessory N-terminal domain that has high structural similarity to a recently discovered motif present in several enzymes that act on ribosomally-synthesized and post-translationally modified peptides (RiPPs), known as a RiPP precursor peptide recognition element (RRE). This crystal structure is the first of a sactionine bond forming enzyme and sheds light on structures and mechanisms of other members of this class such as AlbA or ThnB.
Keywords: sactipeptides, radical SAM enzymes, SPASM domain, RiPPs, RREs
Graphical abstract
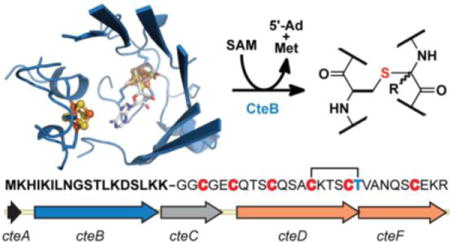
Ribosomally-synthesized and post-translationally modified peptides (RiPPs) are a growing class of natural products that have garnered substantial attention because of their structural diversity and biological activities.1–4 The biosynthesis of RiPPs involves leader peptide-directed enzymatic transformations that are readily exploited for combinatorial biosynthesis and other applications.5–9 Sulfur-to-alpha carbon thioether cross-linked peptides (sactipeptides) are a class of RiPPs characterized by distinctive cysteine-sulfur to α-carbon thioether (sactionine) linkages.10,11 Sactipeptides contain one or more sactionine linkages that form their constrained macrocyclic peptide backbones, making them resistant to heat and proteolysis.12,13 Importantly, sactipeptides exhibit narrow spectrum antibiotic activity against Clostridia and other drug resistant bacteria.13–19 These properties render sactipeptides attractive scaffolds for antibiotic development.
Sactipeptide precursor genes are typically co-localized in operons with members of the radical S-adenosyl-L-methionine (SAM) superfamily of enzymes (termed sactisynthases), which are required for their biosynthesis.10,20 Radical SAM (RS) enzymes generally contain a canonical CX3CX2C sequence, where the conserved Cys residues provide ligands for three iron atoms of a [4Fe-4S] cluster. The fourth iron coordinates a SAM molecule and activates it towards reductive cleavage upon electron transfer to an antibonding orbital of the SAM sulfonium group.21 The low-potential electron can be supplied to the [4Fe-4S] cluster by a chemical reductant such as dithionite, or, in some cases, by the enzymatic NADPH/flavodoxin-flavodoxin reductase system.22–26 Reductive cleavage generates methionine and a potent 5’-deoxyadenosyl radical (5’-dA•) that, in most cases, abstracts a hydrogen atom from substrate, which initiates the requisite chemical transformation.27 Recent work has shown that sactisynthases similarly cleave SAM to generate 5’-dA•, which is used to catalyze thioether bond formation by an incompletely understood mechanism (Figure 1a).1–4,28–34 For example, AlbA, a sactisynthase from Bacillus subtilis has been shown to catalyze 5’-dA formation that is coupled to making three sactionine linkages on its cognate peptide SboA as part of Subtilosin A biosynthesis.5–9,29 We recently demonstrated that AlbA can accept substrates with differently positioned cysteines to generate libraries of new sactipeptides with diverse ring sizes.10,11,35 These efforts are limited to substrates that fortuitously undergo modification by the native enzyme, but engineering and rational design of sactipeptide libraries will require a better understanding of sactisynthase structure and mechanism.
Figure 1.
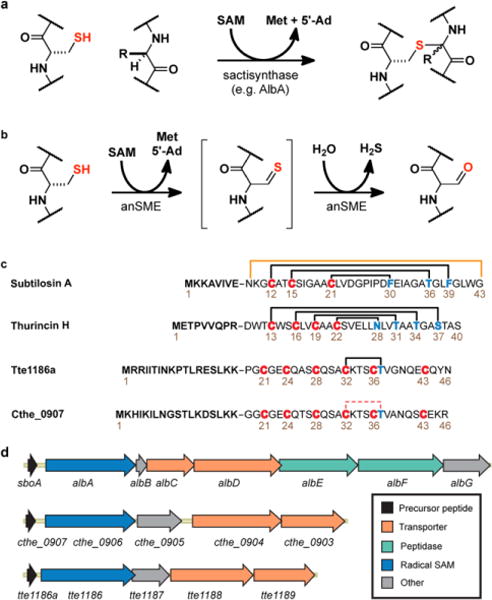
Introduction to sactipeptides. a) Formation of sactionine thioether linkages found in sactipeptides. b) Formation of formyl glycine from cysteine by anSME c) Comparison of known sactipeptides to the bridge formed in CteA. d) Gene clusters of some known sactipeptide producers.
Sequence homology suggests that sactisynthases contain a unique C-terminal extension, termed a SPASM domain in addition to a conserved RS domain.12,13,34,36–40 The defining feature of SPASM domains is their coordination of auxiliary [4Fe-4S] clusters, which are thought to expand and enhance the range of chemistries accessible by the RS domain.13–19,36,38 SPASM-containing enzymes catalyze a variety of chemical transformations: in addition to sactionine carbon-sulfur bonds, SPASM domains have been implicated in C-C bond formation, such as PqqE in PQQ biosynthesis, and the recently characterized StrB from streptide biosynthesis.10,20,41–44 Yet there appears to be significant variation among SPASM domains, in particular in the coordination of the auxiliary iron-sulfur clusters.21,36,38 Only one structure of a full SPASM domain has been reported to date, that of the anaerobic sulfatase maturating enzyme (anSME). anSME diverges significantly from the sactisynthases both in amino acid sequence and in chemistry: anSME cotranslationally catalyzes the formal 2-electron oxidation of a cysteine residue found in the active site of its cognate sulfatase to yield formyl glycine (Figure 1b).39,45–48 To gain further insight into the mechanism of sactionine linkage formation and substrate promiscuity of these enzymes, and to increase their utility, we set out to gain structural information.
In this study, we reconstitute the activity of the sactisynthase, CteB, in the predicted biosynthetic pathway for a sactipeptide, thermocellin (cte) from Clostridium thermocellum ATCC 27405. Through a combination of chemical derivatization and tandem mass spectrometry, we demonstrate that CteB installs a single sactionine thioether linkage between Cys32 and Thr37 of its cognate peptide, CteA, and that the remaining five cysteines in CteA go unmodified. We further report two structures of CteB: a 2.70-Å-resolution structure of CteB with SAM bound and a 2.04-Å-resolution structure of CteB with both SAM and the leader peptide of CteA bound. The structure defines all three [4Fe-4S] clusters predicted by bioinformatics, one of which, auxiliary cluster I (Aux I), displays a novel open coordination site on one of its constituent iron ions. These structures, together with peptide binding assays, provide insight into the mechanism of thioether bond formation for CteA and other members of the sactisynthase family.
Results
In vitro reconstitution of CteB: a sactionine synthase
Bioinformatics analyses predict sactipeptide clusters in a wide array of bacterial genomes, including several from thermophilic bacteria.36,38,49 We anticipated that sactionine synthases from these thermophiles might have the desired stability for efficient heterologous expression and crystallization. In particular, the sactisynthase, Cthe_0906 from Clostridium thermocellum ATCC 27405, here referred to as CteB, is adjacent to a member of a large family of predicted sactipeptides, which Haft and Basu dubbed SCIFF (or six cysteines in forty-five residues) peptides.36 CteB is co-localized with the short peptide Cthe_0907, here referred to as CteA, and is therefore predicted to catalyze thioether bond formation on CteA. We therefore considered CteB a strong candidate for enzymatic reconstitution and structural analysis. Although no native natural products belonging to the SCIFF family have been isolated to date, during the course of our efforts, Bandarian and co-workers reconstituted the activity of Tte1186 from a putative SCIFF pathway in Caldanaerobacter subterraneus subsp. tengcongensis MB4.33
The genes encoding CteB and its putative peptide substrate CteA were codon optimized for expression in E. coli and separately cloned into expression vectors. We had previously observed improved yields when SboA was co-expressed with its modifying enzyme, AlbA, presumably due to protection from proteolysis.35 The precursor peptide, CteA, was therefore co-expressed with CteB in a pETDuet vector. Only CteA was His-tagged in the construct and could be readily purified from inclusion bodies formed during expression at 18° C with generous aeration. No modification of CteA was observed under these aerobic conditions, making recombinant CteA obtained in this manner suitable for enzymatic assay. CteB could be expressed and purified in a manner similar to other radical SAM enzymes (see Supporting Information).50
We first confirmed that reconstituted CteB was able to carry out reductive cleavage of SAM to generate methionine and 5’-dA. When CteB was incubated in the presence of SAM and the strong non-physiological reductant dithionite, we observed the distinctive mass (252.1108) and UV-absorbance of 5’-dA in LC/MS traces of the assay supernatants (Figure S1). This product was not observed in control reactions without CteB or SAM, suggesting that reconstituted CteB carries out this characteristic reaction of radical SAM enzymes.
We next sought to assess whether CteB is in fact a sactisynthase, capable of forming thioethers on its putative substrate CteA. We thus incubated CteB together with CteA and SAM under various conditions and the products were analyzed by HPLC coupled to an Agilent 6520 Accurate-Mass Q-TOF spectrometer. In the presence of stoichiometric amounts of enzyme, we observed complete conversion of CteA to a mass 2.0 atomic mass units (amu) lower than the starting mass, consistent with the loss of two hydrogen atoms and formation of a single thioether bond. Figures 2a and 2b show examples of the mass shift in the envelope corresponding to the +6 charge state of CteA; a 2 amu overall shift in mass corresponds to a decrease of 0.3378 in the +6 charge ion (Figure 2b).
Figure 2.
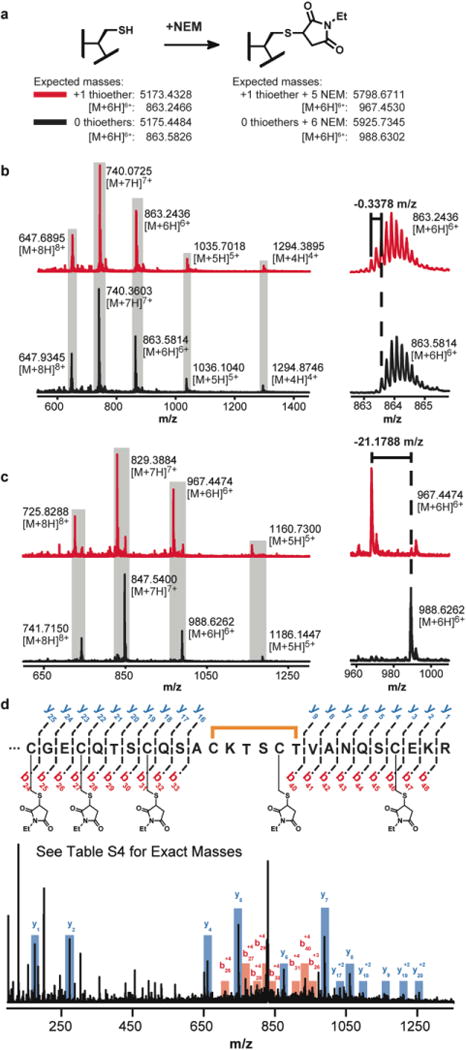
MS analysis of CteA modified by CteB. a) Expected masses of CteA modified by CteB and alkylated with NEM. b) MS of CteA modified by CteB. In red is 1 eq. of CteA treated with 1 eq. of CteB while in black is CteA unmodified. The difference between corresponding charge states is that of one sactionine bridge or two hydrogen atoms. c) MS of CteA product alkylated with NEM after first being modified by CteB. In red is 1 eq. of CteA treated with 1 eq. of CteB then NEM, while in black is CteA treated with just NEM. The difference between corresponding charge states is that of one sactionine bridge and one NEM modification. See Table S3 for expected exact masses. d) MS/MS analysis (+7 charge state) of where the sactionine bridge is forming in modified CteA.
We further sought to confirm that the 2.0 amu change resulted from a thioether and not a disulfide, which would also yield a 2.0 amu change in mass. To rule out disulfides, the reactions were quenched under reducing conditions and reacted with N-ethylmaleimide (NEM), in order to alkylate all free cysteines (Figures 2a and 2c). In a control reaction, where CteA was directly treated with NEM, the m/z values for the various charge states corresponded to the mass of the peptide plus six molecules of NEM (Figure 2c, black trace and Table S3). In contrast, CteA that was treated with CteB and SAM before being quenched with NEM exhibited masses corresponding to m/z for peptide with five alkylated cysteine residues minus two hydrogens (Figure 2c, red trace and Table S3), confirming that a single thioether had been installed by CteB under these conditions.
In order to identify the location of the sactionine crosslink we compared tandem MS/MS spectra of unmodified CteA and CteB-modified CteA. Both samples were treated with reductant and NEM to alkylate free cysteines, and the peptides fragmented by CID. Based on the pattern of b- and y- ions, the newly formed thioether likely resides between Cys32 and Thr37 of CteA. The MS/MS spectrum of CteB and NEM-modified CteA is presented in Figure 2d. This spectrum indicates that all three N-terminal cysteines (Cys21, 24, and 28) as well as the two C-terminal cysteines, Cys36 and 43, can be alkylated with NEM after modification with CteB. Ions from fragments of CteA that contain these residues exhibit increased m/z according to the relative number of NEMs present. For example, y8, which contains an NEM-modified Cys43, appears as an [M+H]1+ mass of 1060.4894, corresponding to the mass of A39-R46 plus one NEM. In contrast, b- and y- ions for fragments containing Cys32 lack one NEM group and two hydrogens. For example, y17, which contains NEM-modified Cys36 and 43, appears as an [M+2H]2+ mass of 1032.4511, corresponding to the mass of S30-R46 plus only two NEMs minus two hydrogens. A full table of observed masses and the residues to which they correspond is provided in Table S4. To further corroborate the assignment, the Cys32Ala mutant of CteA was prepared via Gibson Assembly mutagenesis, and purified as described for wild-type CteA. Assays with C32A-CteA in presence of CteB and SAM yielded only the unmodified precursor peptide, consistent with thioether formation at this position (Figure S3).
Peptide products with only one thioether bridge were observed regardless of whether CteB was limited or used in large excess. It cannot be completely ruled out that multiple thioether bridges may be formed in the cellular environment of the native producer with the native reductant. Whether this is the active form of CteA in vivo remains to be determined. Interestingly, Bandarian and co-workers observed only a single thioether bridge between Cys32 and Thr37 during their characterization of the related Tte1186 sactipeptide product.33
Crystal Structure of CteB
To obtain structural insights into sactipeptide synthesis, we determined crystal structures of CteB in two different states: 1) the 2.7 Å structure with bound SAM and no substrate present and 2) the 2.04 Å structure with a 21-residue N-terminal fragment of CteA (CteA-M1-C21) and SAM; the portion of CteA that undergoes crosslinking is not present in this structure. We attempted co-crystallization with the full-length CteA precursor peptide, but were unable to obtain diffraction quality crystals. To date, there have been no structures reported for any RiPP enzyme and its cognate full-length precursor peptide bound and fully defined.
The two CteB structures, CteA-bound and unbound, superimpose with a root mean square deviation (RMSD) of 1.3 A2 based on 415 Cα atoms (Figure S11). Owing to this structural similarity, our discussion focuses on the higher resolution CteB+SAM+CteA-M1-C21 complex, with reference to the unbound structure where relevant. Crystals of the enzyme-peptide complex exhibit diffraction consistent with the orthorhombic space group P21212, with a monomer in the asymmetric unit (Figure 3a). The final model consists of residues 1 to 449 (out of 450) of CteB, residues 1 – 9, and 20-21 of the CteA-M1-C21 peptide, 12 iron ions, 12 sulfide ions, 2 calcium ions, 1 SAM molecule and 146 water molecules. Residues 115-121 of CteB are not defined by electron density, and reside in a disordered loop immediately following the radical SAM cluster binding motif. A similar disordered loop was also seen in the structure of anSME.39 In addition, residues 334-336 are not defined in a disordered loop that joins α6 in the RS domain to the SPASM domain. This region lies on a symmetry axis and is difficult to model (Figure S8A). This region contains a conserved cysteine (C336), which may form a disulfide bond with the adjacent Cys336 from a symmetry mate (Figure S8B). We have not modeled this disulfide bond due to the weak electron density in this region (Figure S8B). In addition, this entire region (residues 330-341) is disordered in the CteB+SAM structure. Interestingly, the crystallization solution contained about 500 μM dithiothreitol that carried over with the CteB added to the solution. In addition, all Fe-S clusters are intact in the crystals indicating a lack of oxidative damage. To determine if a disulfide exists in the crystalline state, crystals of CteB+SAM+CteA-M1-C21 were dissolved in buffer and the solution chromatographed on a size exclusion column equilibrated in buffer that did not contain reductants (Figure S5). The majority (~ 65 %) of protein in this sample migrated with an apparent molecular weight of ~ 95 kDa, which is consistent with a dimer, while the remaining protein migrated as a monomer. The monomer fraction of this solution is likely CteB that was present in the crystallization drop and did not form crystals. We next tested the oligomeric state of CteB in solution, both in presence and absence of full-length CteA, under reducing conditions by size exclusion chromatography (SEC). Under theses conditions, CteB migrates with an apparent mass of 42 kDa, consistent with a monomer (Figure S6). In the presence of CteA, the apparent mass of CteB increases by ~ 4 kDa, consistent with a CteA-CteB complex. Thus, in solution, under reducing conditions CteB does not form a dimer. Therefore, we removed reductant from CteB and mixed protein with varying ratios of reduced glutathione (GSH)/oxidized glutathione (GSSG) to survey the redox potential of the mixture from −377 mV to −223 mV. These mixtures were subsequently electrophoresed on a non-reducing SDS-PAGE gel (Figure S7). Importantly, increasing the redox potential by increasing the ratio of GSSG does not lead to intermolecular disulfide bond formation in solution. Thus, this disulfide is most likely a spurious artifact of crystallization.
Figure 3.
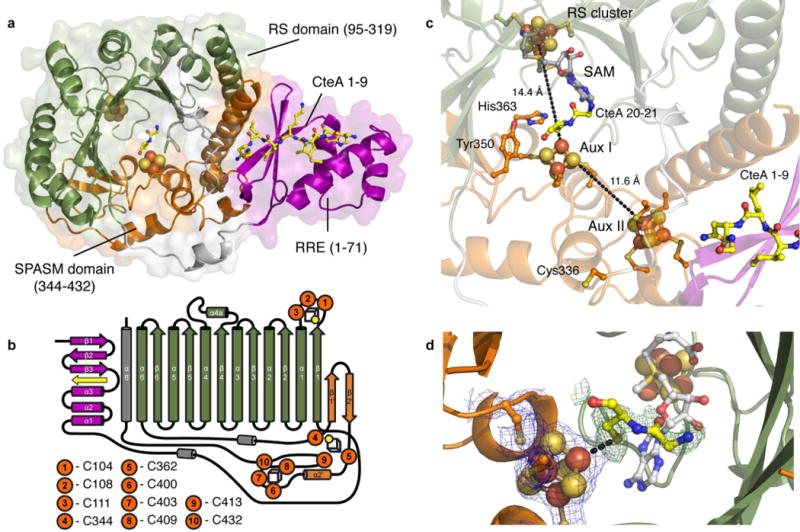
Structure of CteB. a) Overall structure of CteB. The β6/α6 core of the RS domain (green) contains one [4Fe-4S] cluster that coordinates one molecule of SAM. The C-terminal SPASM domain (orange) contains the [4Fe-4S] clusters Aux I and Aux II and comprises residues 344 - 432. The N-terminal RRE domain (magenta) of CteB provides the binding specificity for the peptide substrate leader sequence of CteA (yellow, stick representation). b) Topology figure of CteB. c) Zoom of [4Fe-4S] clusters present in CteB along with their distances from one another. The distance from RS to Aux I is 14.4 Å while the distance from Aux I to Aux II is 11.6Å. RS, radical SAM cluster, Aux I, and, Aux II d) The Fo-Fc omit map contoured to 3.0 σ (green mesh) of Gly20 and Cys21 from CteA-M1-C21 substrate bound to Aux I. The distance between the Fe and Sϒ of Cys21 is 2.7 Å. The 2Fo-Fc map (blue mesh) for the Aux I cluster is contoured to 2.0 σ.
The structure of CteB exhibits three discernable domains (Figure 3): (1) a partial (β/α)6 triose phosphate isomerase (TIM) barrel (residues 95-319) containing one [4Fe-4S] cluster (canonical radical SAM domain), which is flanked by (2) an N-terminal winged helix-turn-helix (wHTH) motif (residues 1-71) and (3) a C-terminal extension (residues 344-432), which contains two additional [4Fe-4S] clusters. These domains are discussed individually below.
The central region of the CteB structure exhibits the characteristic (β/α)6-TIM barrel, common to nearly all other members of the RS superfamily. The SAM activating [4Fe-4S] cluster within the RS domain is bound by the conserved CX3CXφC motif (where φ is an aromatic residue) located in the loop between the α1 helix and β1 loop (residues 100-125). This cluster is ligated by three cysteines (residues 104, 108, and 111), leaving one site open to chelate the α-amino-nitrogen and α-carboxyl oxygen of the SAM co-factor.40,51 The SAM binding pocket is similar to that of anSME and exhibits the four common SAM binding motifs: the GGE motif (residues 153-156), ribose motif (residues S210 and D212), the GXIXGXXE motif (residues 254-262), and the β6 or adenine-binding motif (residues 281-284). In addition, Tyr110 hydrogen bonds to the N6 of adenine and Arg222 stabilizes the ribosyl and carboxyl moieties of AdoMet; these interactions are also present in the anSME SAM binding pocket (Figure S9). Interestingly, two new interactions are present in the SAM binding pocket of CteB: Arg253 and Thr255 in the β5 strand hydrogen bond to N3 of the adenine base. These residues reside in a highly conserved RGT motif found in thermophilic sactisynthases. In sum, a total of eight residues make side chain or backbone polar contacts with SAM (Figure S9). Presumably these numerous interactions stringently position and orient SAM for radical-based hydrogen abstraction from its substrate.
The C-terminal extension of CteB contains the predicted seven-cysteine SPASM domain that binds two additional auxiliary [4Fe-4S] clusters. The CteB SPASM domain exhibits structural homology (Figure S9, R.M.S.D. of 2.3 Å over 113 α-carbons) to the SPASM domain from anSME with some notable differences. The SPASM domain in CteB extends from the C-terminus of the TIM barrel RS domain via a partially ordered loop to coordinate the first auxiliary [4Fe-4S] cluster (Aux I) at Cys344 and Cys362 with β1’ and β2’ interspersed between these residues. In anSME, a short insertion harbors C261, which is the fourth ligand to Aux I. This insertion is absent in CteB. As in anSME, the CX2CX5CX3C motif in the central region of the conserved SPASM domain provides three ligands for the second auxiliary [4Fe-4S] cluster (Aux II) and one additional cysteine ligand for Aux I. Cys400, Cys403, and Cys409 from CteB all coordinate Aux II, while the fourth cysteine of the motif, Cys413, crosses back to provide a third ligand for Aux I. Cys432 provides the fourth and final ligand for Aux II (Figure 3b). The absence of the fourth coordinating ligand to Aux I results in the positioning of the Fe/S cluster closer to the RS cluster. The RS cluster resides 14.4 Å from the open coordination site of Aux I (Figure 3c), which is ~ 2.5 Å closer than the separation between the RS cluster and Aux I in anSME (16.9 Å). The distance between Aux I and Aux II in CteB is 11.6 Å, which is slightly compressed compared to anSME (12.9 Å). These differences indicate that the overall arrangement/separation of Fe/S clusters within the SPASM family of proteins is likely tuned to support the different chemistries that are catalyzed by these enzymes.
In contrast to anSME, Aux I from CteB lacks one protein-derived cysteine residue. In the CteB+SAM+CteA-M1-C21 structure, electron density is observed around the open coordination site of Aux I. We attempted to model this density as a weakly bound DTT molecule as observed in the crystal structure of lipoyl synthase52; however, this exercise did not result in a satisfactory fit of the electron density. Therefore, we postulated that the free thiol of Cys21 from the CteA-M1-C21 peptide could reach into the active site and coordinate the cluster. The modeling the Cys21 and Gly20 of the peptide into this density provided a plausible fit (Figure 3d). The lack of density for the peptide sequence between Gly9 and Gly20 is likely due to inherent flexibility and the lack of contacts with CteB in this region. Because only a fragment of the precursor (residues 1-21) peptide was found to co-crystallize with CteB, it is possible that the Cys21-Fe ligation is not mechanistically relevant, but rather represents a thermodynamically stable state of the peptide in the absence of the native sequence. However, we propose that this Cys21 coordination may be analogous to an enzyme-substrate interaction that occurs during the catalytic cycle involving at least one of the six cysteines from CteA and Aux I.
The N-terminal winged helix-turn-helix (wHTH) motif of CteB is also not present in anSME. This wHTH is structurally homologous to RiPP recognition elements (RREs), which have recently been identified in the structures of other RiPP modifying enzymes, such as LynD, PaaA, and NisB.53–60 CteB represents just the fourth report of a leader-bound RRE structure (Figure 4a).54,55 The structures all exhibit a common pattern in the conserved wHTH domain architecture and β-strand conformation of bound substrate (Figure 4a-c). Despite sharing only 13 % sequence identity to the RRE domains of LynD and NisB, the overall R.M.S.D is strong at 2.16 Å and 3.05 Å, respectively over 71 α-carbons. The RRE domain provides one of the primary structural determinants for leader peptide recognition. The three stranded β-sheet, or wing, of the RRE hydrogen bonds with the backbone of the N-terminus of the CteA fragment in the co-crystal structure in a manner similar to LynD and NisB (Figure 4a–c). An extensive hydrogen bond network is formed by backbone carbonyl and amide interaction of the RRE and CteA (Figure 4d and Table S6). Hydrogen bonds are also seen between side-chain and main-chain atoms of CteA. CteA His3 forms a series of salt bridges with the absolutely conserved CteB residues Asp27, Glu60 and Glu64. CteA also makes advantageous van der Waals interactions with the RRE via Ile4 and Ile6, both of which fit into hydrophobic pockets in the cleft between α3 and β3. The RRE is connected to the N-terminus of β1 by a long, flexible linker, which passes across the face of the SPASM domain to position the RRE next to the α6’ helix (Figure 3b). β1 and β2 of the RRE make hydrophobic contacts with the α6’ helix emanating from the SPASM domain, weakly stabilizing its position relative to the active site pocket. In addition, the RRE domain makes limited crystallographic contacts with symmetry molecules and as results shows higher average β-factors than the core of CteB. This explains why the density for the leader peptide is weaker than the resolution would predict.
Figure 4.
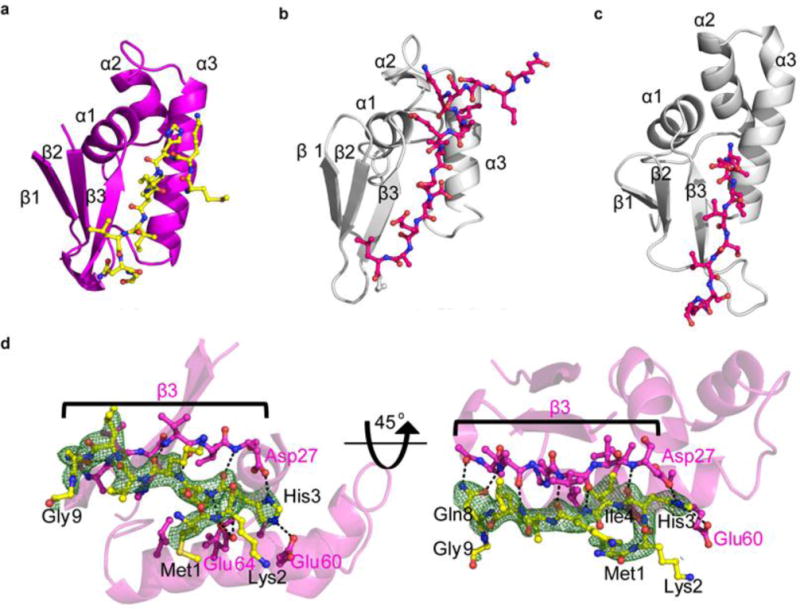
Leader peptide and binding to RRE of CteB. Comparison of RRE domains from CteB (a), LynD (b), NisB (c). d) Simulated annealing omit composite map (2Fo-Fc) contoured to 1.0 σ of residues 1 – 9 of the leader peptide (yellow sticks) of CteA. Residues from CteA involved in binding of the leader peptide are shown in yellow. Hydrogen bond interactions are shown as dashed lines. β3 from the RRE domain is shown in pink sticks. β3 forms extensive hydrogen bonds to the leader peptide of CteA, while α3 forms mostly hydrophobic interactions, with the exception of Glu60 and Glu64. For full list of interactions and distances see supplementary Table S6.
Homology and Comparison to SPASM and Twitch Domain Enzymes anSME, BtrN, and MoaA
CteB is only the second example of a SPASM domain to be structurally characterized, after anSME. In addition, BtrN61 and MoaA62,63 exhibit smaller, single cluster domains, which have been dubbed “Twitch” domains.37 Taken together, the four structures provide four different coordination architectures for Aux I (Figure 5). All four enzymes use the two conserved cysteines on either side of the β1’/β2’ hairpin loop (Cys344 and Cys362 in CteB) and differ in positioning of the remaining coordination residues. MoaA has an open coordination site on Aux I, similar to CteB, however, the open iron sites in these two structures are on alternate sides of Aux I. While Cys413 from the CX2CX5CX3C motif loops back to provide the third ligand in CteB, this feature is not present in the MoaA twitch domain, and Aux I is instead ligated by an additional cysteine, Cys264, upstream of the hairpin loop. Cys264 in MoaA is analogous to Cys261 in anSME, but is absent in CteB. This difference in coordination pattern results in the open coordination site on Aux I of CteB being oriented towards the active site entrance, appropriately positioned for coordination by an incoming peptide substrate. In contrast, the open coordination site on Aux I on MoaA is oriented towards the interior of the active site, to aid in sequestering its smaller substrate.62,64 The specific orientation of these [4Fe-4S] clusters also impacts their distance from the SAM activating cluster: this distance is 14.4 Å in CteB (Figure 3c), whereas it is ~17.0 Å in anSME, BtrN, and MoaA (16.8, 16.9, and 17.3 Å, respectively). The more compact architecture in CteB could facilitate the intermolecular bond-formation reaction between Cys32 and Thr37 of the CteA substrate.
Figure 5.
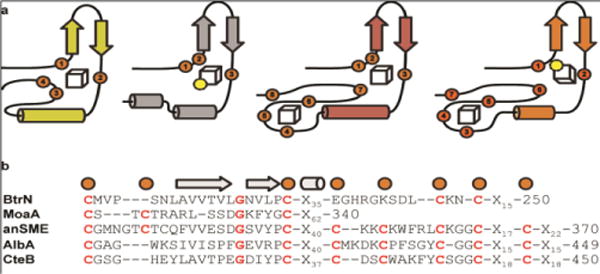
Comparison of Aux I and Aux II clusters. a) Topology diagrams of known crystallized enzymes that hold either one or both Aux I and Aux II clusters. Yellow-BtrN, gray-MoaA, red-anSME, and orange-CteB. b) Sequence alignments of those domains.
Overall, the RS and SPASM domains of CteB and anSME adopt a similar structural organizations (R.M.S.D. of 2.52 Å over 300 Cα atoms) (Figure S9), despite sharing only 20% sequence identify. Interestingly, the conserved Asp277 and Tyr24 active site residues of anSME are absent in CteB. In particular, Asp277 was shown to be absolutely required for anSME activity and was proposed to act as a base to deprotonate a cysteine thiol in the anSME reaction mechanism.39 His363 and Tyr350 in CteB are within 8 Å of Aux I and these residues could similarly take a proton from the crosslinking thiol in the CteB reaction mechanism. However, when we prepared and tested the H363A and Y350A mutants, we observed formation of the thioether bridge on CteA (Figure S1 and S2). These observations suggest that these residues do not act as an essential base during the reaction. Studies are under way to determine which active site residues are critical for the activity of CteB.
Contributions to Binding Affinity of CteA
In order to examine the contributions of leader and core regions of CteA to binding CteB, a fluorophore-labeled probe was prepared. To this end, CteA (M1-C21) was synthesized with a 5,6-TAMRA label on the N-terminus for use in fluorescence polarization binding assays. This leader region alone exhibits a 0.7 μM binding affinity, in good agreement with affinities for similar leader peptide/RRE interactions.53,65 The unlabeled peptides CteA(1-21), CteA(1-21)-H3A, wild-type CteA, and CteA-C32A were used in competition assays. Both wild-type CteA and CteA-C32A exhibit a slightly lower IC50, and therefore higher affinity, than the leader peptide alone (Figure S4). Notably, the C32A mutant did not substantially impact binding, but the H3A variant peptide was unable to compete for binding with the labeled peptide. The fact that the H3A variant exhibits greatly reduced binding to CteB provides strong evidence that the histidine side chain interactions with the RRE domain of CteB are critical for CteA recognition by CteB. These observations also provide evidence that the peptide substrate sequence is modeled correctly. Taken together, these observations suggest that there is an extended binding interface involving determinants from both the leader and core regions of CteA, which diminishes the impact of a single cysteine mutation. However, key interactions between the leader peptide and RRE (e.g. His3) can significantly impact binding.
Discussion
Perhaps the most salient feature of the CteB structure is the clear presence of an open coordination site on Aux I in the substrate free state, which appears to be satisfied by a cysteine from the substrate peptide in the bound structure. The coordination state of this predicted [4Fe-4S] cluster within sactisynthases has been the subject of debate. Berteau34 and Drennan37,39 both noted the insufficient number of cysteines for complete ligation of two [4Fe-4S] clusters in the sactisynthase AlbA. Berteau conjectured that the ligation state may be satisfied by a pendant serine or arginine as in LipA66 and BioB67, respectively, whereas Drennan and co-workers suggested that an open site on the cluster might be involved in substrate binding as in MoaA.37 The structures of CteB are consistent with a mechanism in which the open coordination of Aux I in CteB is involved in substrate binding. Substrate coordination at this open site also appears to be consistent with spectrophotometric data reported by Marahiel et al. for AlbA, where substrate titration was accompanied by a shift in the UV-spectrum, which is absent in mutants that disrupt Aux I.29 Although the current structure shows a distal cysteine, Cys21, coordinating to Aux I, we hypothesize that in the native substrate, coordination of the reacting cysteine would serve to orient and activate it for thioether bridge formation.
Two mechanisms have been proposed (Figure 6a). The first mechanism involves separate activation of the bridging partner α-carbon and the cysteine sulfur by distinct [4Fe-4S] clusters, followed by attack of the carbon centered radical on the activated sulfur atom (Figure 6, Mechanism A). An alternative mechanism, in which the intermediate α-carbon radical undergoes a one-electron oxidation to the ketoimine (Figure 6, Mechanism B), has also been proposed.33 In the latter mechanism, the thioether is formed by nucleophilic attack of the cysteine sulfur on the ketoimine; either a fully ligated Aux I or the radical SAM cluster itself, after reductive cleavage of SAM, could potentially act as the one-electron oxidant of the intermediate radical (Figure 6a). Mechanism B has previously been promoted on the basis of mixtures of both D- and L- sactionine linkages observed in sactipeptides like Subtilosin A, because a polar mechanism could support nucleophilic addition on either the re- or si-face of the ketoimine.33 However, the putative radical intermediate, which is captodatively stabilized on the α-carbon, may well be more planar than pyramidal and could similarly undergo attack on either face; there fore, Mechanism A cannot be ruled out.
Figure 6.
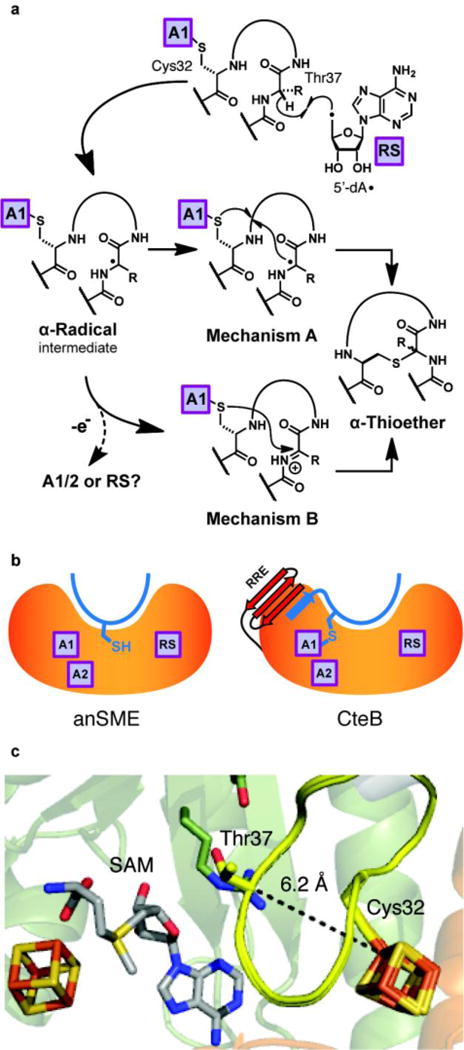
Proposed mechanisms of sactionine bridge formation. a) Mechanisms describing either separate activation of the bridging partner α-carbon and the cysteine sulfur by distinct [4Fe-4S] clusters followed by attack of the carbon centered radical on the coordinated sulfur atom (Mechanism A) or the intermediate α-carbon radical undergoes a one-electron oxidation to the ketoimine which is then subject to nucleophilic attack of the cysteine sulfur (Mechanism B). b) Proposed binding of substrates in their enzymes. c) Rosetta model of CteA:CteB complex with Cys32 ligated to Aux I. c) Computational model generated with Rosetta showing possible interactions between CteA (yellow) and CteB. In the model Cys32 from CteA ligates the free coordination site on Aux I and Thr37 is placed in close proximity to where the 5’-dA radical is formed from SAM (gray)
This newly revealed coordination site has potential ramifications for the mechanism of thioether bond formation. Based on coordination of Cys21—a site that does not make a thioether bridge in CteA—it seems possible that the observed coordinating cysteine could be spurious. An alternative is that the bound Cys21 in the current structure is a mimic of the physiologically relevant cysteine, Cys32, which would be highly activated for crosslinking by coordinating to this [4Fe-4S] cluster. By analogy to anSME, CteA would bind the RRE with its N-terminus and likely project down into the bowl-like active site, where the reactive cysteine sulfur may coordinate to Aux I (Figure 6b). We generated a computational model with Rosetta (Figure 6c and see Supporting Information, Figure S17 and S18) to examine whether placement of Thr37 in proximity to the 5’-carbon of SAM is spatially compatible with coordination of Cys32 to Aux I. Although only a model, this suggests that the two residues are in appropriately close proximity for coupling (Figure 6c). Moreover, both mechanisms can reasonably be drawn with an Aux I-ligated Cys being incorporated into the thioether linkage, as in Figure 6b. The open coordination site on Aux I could provide either an electron sink for the radical mechanism or an oxidant and intermediate Lewis acid for the polar mechanism. The CteB structure demonstrates that Aux I and Aux II of CteB are in sufficient proximity to act as electron transfer partners as previously proposed for anSME.47,68 Patches of highly conserved surface residues border the RS and Aux II clusters of CteB (Figure 7), delineating possible recognition surfaces for single electron donors and acceptors, such as ferrodoxins or the flavodoxin/flavodoxin reductase system.27
Figure 7.
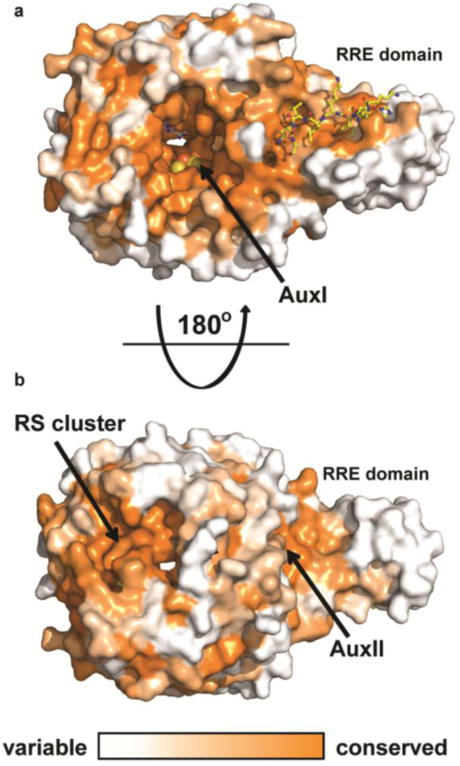
Conservation of CteB homologs. Surface map (ConSURF server) of sequence conservation based on 150 sequences with homology ranging from 35% to 90% identity. Conservation scores are based on Bayesian method. a) The highest sequence conservation can be found around the active site and peptide binding surface of the RRE domain. b) 180 ° rotation showing the bottom of CteB. A patch of highly conserved residues is found around the RS and Aux II clusters. These sites may have a role in the recognition of redox partners.
Based on our sequence analyses, the open site on Aux I appears to be conserved in several sactisynthases. For example, multiple sequence alignments (Figure S12-S15) indicate that sactisynthases from thurincin H (ThnB - 4 bridges) and subtilosin A (AlbA - 3 bridges) biosynthesis should have very similar architectures to CteB, despite being capable of making multiple sactionine linkages. Moreover, significant variations on this SPASM architecture are predicted in sactisynthases from thuricin CD (TrnC and TrnD) and sporulation killing factor (SkfB). The relationship of these structural variations to chemistry and mechanism remains to be defined. Two other SPASM-containing enzymes, PqqE and StrB, also align well with the CteB SPASM architectures. Both PqqE and StrB are involved in C-C bond forming reactions that are very different from the chemistry carried out by CteB. It will be interesting to see how the structures of the SPASM domains of these distant relatives relate to CteB.
It remains unclear how CteB-related sactisynthases catalyze the formation of multiple nested thioether linkages. For example, AlbA catalyzes formation of three sactionine linkages in subtilosin A and ThnB makes four in thurincin H biosynthesis. Active site dynamics and substrate control could play roles in the formation of additional thioethers. Initial substrate coordination may well act to “set the register” for thioether positioning in these multiply bridged systems. The long RRE linker would presumably allow greater flexibility of the N-terminus, and enable a more diverse ensemble of approaches to the catalytic site.
In conclusion, we have biochemically characterized a new sactionine synthase, CteB from C. thermocellum ATCC 27405, which installs a single sactionine bridge on the thermocellin precursor peptide. We also determined the X-ray crystal structures of CteB both in presence and in absence of a fragment of the peptide substrate, which represent the first structures of a sactionine synthase. The structures reveal a conserved SAM activating domain, as well as a new SPASM domain motif displaying a single open coordination site on the internal auxiliary iron-sulfur cluster (Aux I). These structures provide insight into the enzymatic mechanism of sacti-bridge formation and suggest that substrate ligation to an open iron coordination site may be involved. We anticipate that this structure will have utility for the continued mechanistic understanding and engineering of sactionine synthases and other related RiPP enzymes.
Supplementary Material
Acknowledgments
The authors would like to thank P. Hanzelmann (University of Wuerzburg for pPH151 and the Tamayo Lab (UNC) for allowing us to use their anaerobic chamber. We would also like to thank B. Li (UNC Chapel Hill) and C. Neumann (Seattle Genetics) for informative discussions and careful reading of the manuscript. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). GM/CA CAT has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104).
Funding Sources
AAB is a Beckman Young Investigator and acknowledges support by Arnold & Mabel Beckman Foundation. This work was supported in part by NIH P01 GM118303-01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Materials, experimental methods, and supplementary figures can be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.” Coordinates and structure factors have been deposited in the Protein Data Bank with accession codes TBD (CteB+SAM+CteA-M1-C21) and TBD (CteB+SAM).
Notes
The authors declare no competing financial interest.
References
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian K-D, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Muller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJT, Rebuffat S, Ross RP, Sahl H-G, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Süssmuth RD, Tagg JR, Tang G-L, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Nat Prod Rep. 2012;30(1):108. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ortega MA, van der Donk WA. Cell Chem Biol. 2016;23(1):31. doi: 10.1016/j.chembiol.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunbar KL, Mitchell DA. ACS Chem Biol. 2013;8(3):473. doi: 10.1021/cb3005325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McIntosh JA, Donia MS, Schmidt EW. Nat Prod Rep. 2009;26(4):537. doi: 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowers AA, Acker MG, Koglin A, Walsh CT. J Am Chem Soc. 2010;132(21):7519. doi: 10.1021/ja102339q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menzella HGH, Reeves CDC. Curr Opin Microbiol. 2007;10(3):8. doi: 10.1016/j.mib.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Sardar D, Schmidt EW. Curr Opin Chem Biol. 2015;31:15. doi: 10.1016/j.cbpa.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim E, Moore BS, Yoon YJ. Nat Chem Biol. 2015;11(9):649. doi: 10.1038/nchembio.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruffner DE, Schmidt EW, Heemstra JR. ACS Synth Biol. 2015;4(4):482. doi: 10.1021/sb500267d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flühe L, Marahiel MA. Curr Opin Chem Biol. 2013;17(4):605. doi: 10.1016/j.cbpa.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 11.Lohans CT, Vederas JC. J Antibiot. 2014;67(1):23. doi: 10.1038/ja.2013.81. [DOI] [PubMed] [Google Scholar]
- 12.Babasaki K, Takao T, Shimonishi Y, Kurahashi K. J Biochem. 1985;98(3):585. doi: 10.1093/oxfordjournals.jbchem.a135315. [DOI] [PubMed] [Google Scholar]
- 13.Shelburne CE, An FY, Dholpe V, Ramamoorthy A, Lopatin DE, Lantz MS. J Antimicrob Chemother. 2007;59(2):297. doi: 10.1093/jac/dkl495. [DOI] [PubMed] [Google Scholar]
- 14.Rea MC, Sit CS, Clayton E, O’Connor PM, Whittal RM, Zheng J, Vederas JC, Ross RP, Hill C. Proc Natl Acad Sci U S A. 2010;107(20):9352. doi: 10.1073/pnas.0913554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sit CS, McKay RT, Hill C, Ross RP, Vederas JC. J Am Chem Soc. 2011;133(20):7680. doi: 10.1021/ja201802f. [DOI] [PubMed] [Google Scholar]
- 16.Wang G, Feng G, Snyder AB, Manns DC, Churey JJ, Worobo RW. FEMS Microbiol Lett. 2014;357(1):69. doi: 10.1111/1574-6968.12486. [DOI] [PubMed] [Google Scholar]
- 17.Sit CS, van Belkum MJ, McKay RT, Worobo RW, Vederas JC. Angew Chem Int Ed. 2011;50(37):8718. doi: 10.1002/anie.201102527. [DOI] [PubMed] [Google Scholar]
- 18.Sutyak KE, Wirawan RE, Aroutcheva AA, Chikindas ML. J Appl Microbiol. 2008;104(4):1067. doi: 10.1111/j.1365-2672.2007.03626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thennarasu S, Lee D-K, Poon A, Kawulka KE, Vederas JC, Ramamoorthy A. Chem Phys Lipids. 2005;137(1–2):38. doi: 10.1016/j.chemphyslip.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Jarrett JT. J Biol Chem. 2015;290(7):3972. doi: 10.1074/jbc.R114.599308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dey A, Peng Y, Broderick WE, Hedman B, Hodgson KO, Broderick JB, Solomon EI. J Am Chem Soc. 2011;133(46):18656. doi: 10.1021/ja203780t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brazeau BJ, Gort SJ, Jessen HJ, Andrew AJ, Liao HH. Appl Environ Microbiol. 2006;72(9):6402. doi: 10.1128/AEM.01143-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruender NA, Young AP, Bandarian V. Biochemistry. 2015;54(18):2903. doi: 10.1021/acs.biochem.5b00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bianchi V, Eliasson R, Fontecave M, Mulliez E, Hoover DM, Matthews RG, Reichard P. Biochemical and Biophysical Research Communications. 1993;197(2):792. doi: 10.1006/bbrc.1993.2548. [DOI] [PubMed] [Google Scholar]
- 25.Ifuku O, Koga N, Haze S, Kishimoto J, Wachi Y. Eur J Biochem. 1994;224(1):173. doi: 10.1111/j.1432-1033.1994.tb20009.x. [DOI] [PubMed] [Google Scholar]
- 26.Eliasson R, Pontis E, Ballinger MD, Reichard P. Journal of Biological. 1992 [PubMed] [Google Scholar]
- 27.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114(8):4229. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frey PA, Booker SJ. Adv Protein Chem. 2001;58:1. doi: 10.1016/s0065-3233(01)58001-8. [DOI] [PubMed] [Google Scholar]
- 29.Flühe L, Knappe TA, Gattner MJ, Schäfer A, Burghaus O, Linne U, Marahiel MA. Nat Chem Biol. 2012;8(4):350. doi: 10.1038/nchembio.798. [DOI] [PubMed] [Google Scholar]
- 30.Flühe L, Burghaus O, Wieckowski BM, Giessen TW, Linne U, Marahiel MA. J Am Chem Soc. 2013;135(3):959. doi: 10.1021/ja310542g. [DOI] [PubMed] [Google Scholar]
- 31.Wieckowski BM, Hegemann JD, Mielcarek A, Boss L, Burghaus O, Marahiel MA. FEBS Lett. 2015;589:1802. doi: 10.1016/j.febslet.2015.05.032. [DOI] [PubMed] [Google Scholar]
- 32.Bruender NA, Bandarian V. Biochemistry. 2016;55(30):4131. doi: 10.1021/acs.biochem.6b00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruender NA, Wilcoxen J, Britt RD, Bandarian V. Biochemistry. 2016;55(14):2122. doi: 10.1021/acs.biochem.6b00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benjdia A, Guillot A, Lefranc B, Vaudry H, Leprince J, Berteau O. Chem Commun. 2016;52(37):6249. doi: 10.1039/c6cc01317a. [DOI] [PubMed] [Google Scholar]
- 35.Himes PM, Allen SE, Hwang S, Bowers AA. ACS Chem Biol. 2016;11(6):1737. doi: 10.1021/acschembio.6b00042. [DOI] [PubMed] [Google Scholar]
- 36.Haft DH, Basu MK. J Bacteriol. 2011;193(11):2745. doi: 10.1128/JB.00040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grell TAJ, Goldman PJ, Drennan CL. J Biol Chem. 2015;290(7):3964. doi: 10.1074/jbc.R114.581249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haft DH. BMC Genomics. 2011;12(21):1. doi: 10.1186/1471-2164-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldman PJ, Grove TL, Sites LA, McLaughlin MI, Booker SJ, Drennan CL. Proc Natl Acad Sci U S A. 2013;110(21):8519. doi: 10.1073/pnas.1302417110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vey JL, Drennan CL. Chem Rev. 2011;111(4):2487. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schramma KR, Bushin LB, Seyedsayamdost MR. Nat Chem. 2015;7(5):431. doi: 10.1038/nchem.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barr I, Latham JA, Iavarone AT, Chantarojsiri T, Hwang JD, Klinman JP. J Biol Chem. 2016;291(17):8877. doi: 10.1074/jbc.C115.699918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puehringer S, Metlitzky M, Schwarzenbacher R. BMC Biochem. 2008;9(1):8. doi: 10.1186/1471-2091-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schramma KR, Seyedsayamdost MR. ACS Chem Biol. 2017;12(4):922. doi: 10.1021/acschembio.6b01069. [DOI] [PubMed] [Google Scholar]
- 45.Benjdia A, Subramanian S, Leprince J, Vaudry H, Johnson MK, Berteau O. FEBS J. 2010;277(8):1906. doi: 10.1111/j.1742-4658.2010.07613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benjdia A, Leprince J, Guillot A, Vaudry H, Rabot S, Berteau O. J Am Chem Soc. 2007;129(12):3462. doi: 10.1021/ja067175e. [DOI] [PubMed] [Google Scholar]
- 47.Grove TL, Ahlum JH, Qin RM, Lanz ND, Radle MI, Krebs C, Booker SJ. Biochemistry. 2013;52(17):2874. doi: 10.1021/bi400136u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grove TL, Lee KH, Clair JS, Krebs C, Booker SJ. Biochemistry. 2008 doi: 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy K, O’Sullivan O, Rea MC, Cotter PD, Ross RP, Hill C. PLoS ONE. 2011;6(7):e20852. doi: 10.1371/journal.pone.0020852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lanz ND, Grove TL, Gogonea CB, Lee K-H, Krebs C, Booker SJ. Meth Enzymol. 2012;516:125. doi: 10.1016/B978-0-12-394291-3.00030-7. [DOI] [PubMed] [Google Scholar]
- 51.Dowling DP, Vey JL, Croft AK, Drennan CL. Biochim Biophys Acta. 2012;1824(11):1178. doi: 10.1016/j.bbapap.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harmer JE, Hiscox MJ, Dinis PC, Fox SJ, Iliopoulos A, Hussey JE, Sandy J, Van Beek FT, Essex JW, Roach PL. Biochem J. 2014;464(1):123. doi: 10.1042/BJ20140895. [DOI] [PubMed] [Google Scholar]
- 53.Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA. Nat Chem Biol. 2015;11(8):564. doi: 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ortega MA, Hao Y, Zhang Q, Walker MC, van der Donk WA, Nair SK. Nature. 2015;517(7535):509. doi: 10.1038/nature13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koehnke J, Mann G, Bent AF, Ludewig H, Shirran S, Botting C, Lebl T, Houssen WE, Jaspars M, Naismith JH. Nat Chem Biol. 2015;11(8):558. doi: 10.1038/nchembio.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ghodge SV, Biernat KA, Bassett SJ, Redinbo MR, Bowers AA. J Am Chem Soc. 2016;138(17):5487. doi: 10.1021/jacs.5b13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ortega MA, Hao Y, Walker MC, Donadio S, Sosio M, Nair SK, van der Donk WA. Cell Chem Biol. 2016;23(3):370. doi: 10.1016/j.chembiol.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsai T-Y, Yang C-Y, Shih H-L, Wang AHJ, Chou SH. Proteins. 2009;76(4):1042. doi: 10.1002/prot.22461. [DOI] [PubMed] [Google Scholar]
- 59.Regni CA, Roush RF, Miller DJ, Nourse A, Walsh CT, Schulman BA. EMBO J. 2009;28(13):1953. doi: 10.1038/emboj.2009.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheung WL, Chen MY, Maksimov MO, Link AJ. ACS Cent Sci. 2016;2(10):702. doi: 10.1021/acscentsci.6b00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goldman PJ, Grove TL, Booker SJ, Drennan CL. Proc Natl Acad Sci U S A. 2013;110(40):15949. doi: 10.1073/pnas.1312228110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hänzelmann P, Schindelin H. Proc Natl Acad Sci U S A. 2006;103(18):6829. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hänzelmann P, Schindelin H. Proc Natl Acad Sci U S A. 2004;101(35):12870. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lees NS, Hänzelmann P, Hernández HL, Subramanian S, Schindelin H, Johnson MK, Hoffman BM. J Am Chem Soc. 2009;131(26):9184. doi: 10.1021/ja903978u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koehnke J, Bent AF, Zollman D, Smith K, Houssen WE, Zhu X, Mann G, Lebl T, Scharff R, Shirran S, Botting CH, Jaspars M, Schwarz-Linek U, Naismith JH. Angew Chem Int Ed. 2013;52(52):13991. doi: 10.1002/anie.201306302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McLaughlin MI, Lanz ND, Goldman PJ, Lee K-H, Booker SJ, Drennan CL. Proc Natl Acad Sci U S A. 2016;113(34):9446. doi: 10.1073/pnas.1602486113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Science. 2004;303(5654):76. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moser CC, Anderson JLR, Dutton PL. Biochim Biophys Acta. 2010;1797(9):1573. doi: 10.1016/j.bbabio.2010.04.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.