

Abstract
T cells infiltrating lymphedematous tissues have a mixed T helper 1 (Th1) and Th2 differentiation profile. Treatment with neutralizing antibodies targeting cytokines that promote Th2 differentiation [interleukin 4 (IL-4) and IL-13] decreases the severity of lymphedema in preclinical models, suggesting that Th2 cells play a key role in the pathology of this disease. However, these previous studies do not address the contribution of Th1 cells and it remains unknown if IL-4 and IL-3 blockade acts primarily on T cells or decreases the pathological changes of lymphedema by other mechanisms. Therefore, this study sought to analyze the effect of lymphatic injury in transgenic mice with mutations that cause defects in Th1 and Th2 cell generation (T-bet knockout or T-betKO and STAT6 knockout or STAT6KO mice, respectively). Using both the mouse tail and popliteal lymph node dissection models of lymphedema, we show that Th2-deficient (STAT6KO) mice are protected from developing lymphedema, have decreased fibrosis, increased collateral vessel formation, and preserved collecting lymphatic vessel pumping function. In contrast, mice with defective Th1 cell generation (T-betKO) develop disease with the same severity as wild-type controls. Taken together, our results suggest that Th2 differentiation is necessary for development of lymphedema following lymphatic injury and that Th1 differentiation does not significantly contribute to the pathology of the disease. Such findings are important as immunotherapy directed at Th2 cells has been found to be effective in well-studied Th2-mediated diseases such as asthma and atopic dermatitis and may therefore be similarly useful for lymphedema management.
Brief Commentary
Background
Lymphedema management is limited due to a poor understanding of its pathophysiology. In this study, we seek to delineate the importance of T helper 1 (Th1) and Th2 cells in this disease by evaluating the effect of lymphatic injury on mice that have impaired Th1 or Th2 cell generation. We found that Th1 differentiation does not significantly contribute to the disease, whereas the inability to develop Th2 cells is protective.
Translational Significance
Our findings suggest that Th2 cells play a critical role in lymphedema and that biologics directed against Th2 cells may be valuable.
Introduction
Secondary lymphedema is a common complication of cancer management that occurs as a consequence of lymph node dissection for tumor staging or treatment [1]. As many as 1 in 3 patients with breast cancer and 1 in 6 patients with other solid tumors go on to develop this dreaded disease [2]. The incidence of lower extremity lymphedema is even higher in some gynecological cancers, affecting as many as 75% of patients [3]. Although recent advancements in lymph node staging such as sentinel lymph node biopsy have been helpful in decreasing the incidence of lymphedema, these gains have been offset by increasing rates of patient and treatment factors such as obesity, advanced age, and radiation therapy that favor lymphedema development. As a result, it is estimated that nearly 10 million Americans suffer from lymphedema.
Recent studies from our laboratory and others have suggested that CD4+ T cells play a key role in direct or indirect regulation of fibroadipose deposition and lymphatic dysfunction, both of which are clinical hallmarks of lymphedema [4–8]. Using antibody depletion techniques, we previously demonstrated that loss of CD4+ T cells, but not CD8+ T cells or macrophages, markedly decreases the pathological changes in a mouse tail model of lymphedema [4, 9]. Furthermore, we have found that nude mice lacking T cells entirely or mice lacking CD4+ T cells specifically (CD4 knockout, CD4KO) do not develop lymphedema following lymphatic injury [5]. Similarly, Ogata et al. noted that CD4+ T cells promote the excessive generation of immature lymphatic vessels, which is necessary for initial edema development in the early stages of the disease [7]. Importantly, we have also shown that the degree of CD4+ T cell inflammation is positively correlated with disease severity in patients with upper extremity lymphedema [5].
Further characterization of the CD4+ T cell infiltrate in lymphedema has revealed that it is comprised of a heterogeneous population of subtypes, including T helper 1 (Th1) and T helper 2 (Th2) cells [4, 5, 10, 11]. Th2 cells in particular appear to be necessary for fibrosis and lymphatic dysfunction, as monoclonal antibody-based inhibition of cytokines that regulate Th2 differentiation [interleukin-4 (IL-4) and IL-13] is an effective means of both prevention and treatment of these features in the mouse tail model of lymphedema [5]. This is consistent with in vitro and in vivo studies demonstrating that physiologic doses of these cytokines potently inhibit lymphatic endothelial cell survival, proliferation, migration, and tubule formation [6]. However, while these findings are interesting, these studies do not elucidate the role of Th1 cells in lymphedema, nor do they specifically implicate Th2 cells since it remains unknown if IL-4 and IL-3 blockade acts primarily on T cells or decreases the pathological changes of lymphedema by other mechanisms. Further understanding of the role of these T helper cells is critical, especially because immunotherapy against specific cellular subtypes have been shown to be effective in other inflammatory diseases and may be similarly useful in lymphedema [12].
To more specifically study the role of Th1 and Th2 cells in the regulation of lymphedema development, we evaluated T-bet knockout (T-betKO) and signal transducer and activator of transcription 6 knockout (STAT6KO) mice because these animals have impaired Th1 and Th2 differentiation capacity, respectively. T-bet is a T-box transcription factor encoded by Tbx21 and is considered to be the master regulator of Th1 differentiation [13, 14]. Early commitment to the Th1 lineage is induced by the activation of STAT1, which then promotes T-bet expression, which in turn results in the production of IL-12Rβ2 and interferon gamma (IFN-γ). As such, the absence of T-bet results in a deficiency of Th1 cells and, ultimately, the failure to activate antimicrobial mechanisms against a variety of pathogens [13, 15]. This has been demonstrated by Finotto et al., who showed that mice possessing a targeted mutation for T-bet have markedly reduced IFN-γ levels and dysfunctional immune responses [14]. In contrast, activated STAT6 is required for the differentiation of activated CD4+ T cells to the Th2 phenotype [16, 17]. Latent cytoplasmic STAT6 becomes phosphorylated by Janus kinases after interaction with the cytokines IL-4 and IL-13, which then allows for translocation into the nucleus to induce the expression of the Th2 differentiation genes GATA3 and Crth2 [17, 18]. Further research has also shown that STAT6 is also important in the regulation of alternative signaling pathways involved in Th2 differentiation [19]. Consistent with such data, Kaplan et al. provided evidence that mice lacking STAT6 have nearly completely abrogated Th2 differentiation even in the setting of IL-4 or IL-13 incubation. Such a deficiency in Th2 cells consequently results in defective antibody production and elimination of extracellular organisms [20].
In this study, we found that T-betKO mice have a similar phenotype as wild-type (WT) mice following lymphatic injury with resultant edema, fibrosis, inflammation, and lymphatic dysfunction following lymphatic injury. Meanwhile, STAT6KO mice have a phenotype similar to CD4KO mice with features of lymphedema. Taken together, our findings suggest that Th2 but not Th1 cells are necessary for the development of lymphedema and that Th2 differentiation coordinates the pathologic changes resulting from lymphatic injury. Such results indicate that immunotherapy directed at Th2 cells may prove to be an useful preventative or therapeutic treatment option for patients with lymphedema.
Materials and Methods
Mouse models
All experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee at Memorial Sloan Kettering Cancer Center, which adheres to the National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals and operates under the Animal Welfare Act and Animal Scientific Procedures Act of 1986. Female WT (C57BL/6J; #000664), CD4KO [CBY.129S2 (B6)-Cd4tm1mak/J; #002663], T-betKO (B6.129S6-Tbx21tm1Glm/J, #004648), and STAT6KO (B6.129S2(C)-Stat6tm1Gru/J; #005977) mice were purchased from The Jackson Laboratory (Bar Harbor, Maine) and maintained in a pathogen-free, temperature- and light-controlled environment and fed ad libitum. CD4KO, T-betKO, and STAT6KO mice have previously been established as models for CD4+ T cell, Th1 cell, and Th2 cell deficiency, respectively, and were therefore chosen for this study to determine the effect of these particular cell types on the development of lymphedema following lymphatic injury [15, 17, 21]. A minimum of 5 mice were used for each experimental group and assays were performed in triplicate. All counts were performed by 2 reviewers blinded to the intervention.
For all survival procedures, anesthesia was induced with isoflurane (Henry Schein Animal Health; Dublin, OH) and depth of anesthesia was monitored through observation of respiratory rate and reaction to tail pinching. Terminal tissue harvest was performed following euthanasia by carbon dioxide asphyxiation as recommended by the American Veterinary Medical Association (AVMA).
Surgical models of lymphedema
We used both the mouse tail and the popliteal lymph node dissection (PLND) models of lymphedema in this study, as each model has its own advantages and disadvantages [22]. In the tail model, the superficial and deep lymphatic vessels are carefully ligated after circumferential excision of a 3-mm portion of skin 2 cm from the base of the tail and identification using Evans’ blue dye (Sigma-Aldrich; St. Louis, MO) injection into the distal tail. This results in complete or nearly complete dissociation of the lymphatic vasculature in the distal tail from the proximal tail. We and others have shown that this results in sustained tail edema, fibroadipose deposition, and lymphatic dysfunction for as long as 10–12 weeks after surgery [23–26]. When indicated, animals that underwent tail skin incision without lymphatic vessel injury (sham surgery) served as controls. In other experiments, we utilized the PLND model, which is particularly valuable for the evaluation of collateral lymphatic formation and collecting lymphatic vessel pumping capacity [27–29]. In this procedure, the popliteal lymph node is excised along with the surrounding fat, which includes the afferent and efferent lymphatic vessels, after Evans’ blue injection into the distal dorsal hindlimb. The incision is closed with 3–0 non-absorbable sutures in a continuous fashion. Any animal noted to have wound infection or skin ulceration following either surgical procedure was excluded and sacrificed per AVMA guidelines.
Tail volume measurements
The tail volumes of mice that endured tail surgery were calculated weekly using the truncated cone formula: V = 1/4π (C1C2 + C2C3 + C3C4) [24]. Tail diameters were measured in a standard fashion using digital calipers every 1 cm starting from the surgical site going distally toward the tip of the tail.
Flow cytometry
Flow cytometry was performed using single-cell suspensions obtained from tail skin and subcutaneous tissues after tail surgery. Harvested tissues were mechanically dissociated and enzymatically digested using a combination of DNase I, Dispase II, and collagenase D (all from Roche Diagnostics; Indianapolis, IN). After endogenous Fc receptors were blocked using rat monoclonal anti-CD16/CD32 (#14-0161-85; eBioscience; San Diego, CA), cells were stained with varying combinations of the following fluorophore-conjugated monoclonal antibodies: rat anti-CD4 (GK1.5; #11-0041-82), Armenian hamster anti-CXCR3 (CXCR3-173; #45-1831-80), and Armenian hamster anti-CCR5 (7A4; #12-1951-82) from eBioscience; and Armenian hamster anti-CCR4 (2G12; #131211) and rat anti-CCR8 (SA214G2; #150311) from BioLegend (San Diego, CA). All cells were also incubated with 4,6-diamidino-2-phenylindole (DAPI; #D1306, Molecular Probes/Invitrogen; Eugene, OR) to allow for exclusion of dead cells. Single-stain compensation samples were created using UltraComp eBeads™ (#01-2222-42, Affymetrix, Inc.; San Diego, CA). Flow cytometry was performed using a Fortessa flow cytometer (BD Biosciences; San Jose, CA) with BD FACS Diva. Resultant data were analyzed with FlowJo software (Tree Star; Ashland, OR).
Histology and immunohistochemistry
Tail and hindlimb tissues were harvested, fixed in 4% paraformaldehyde (Sigma-Aldrich) at 4°C overnight, decalcified using 5% ethylenediaminetetraacetic acid (Santa Cruz Biotechnology; Santa Cruz, CA), embedded in paraffin, sectioned at 5 μm, and rehydrated for histological and immunohistochemical staining. Unless otherwise specified, cross-sections were harvested at a point 1 cm distal to the zone of injury.
Hematoxylin and eosin staining was performed per a standard protocol using Mayer’s hematoxylin (Lillie’s Modification; Dako North America; Carpinteria, CA) and eosin Y solution (Thermo Fisher Scientific; Waltham, MA). Sections were dehydrated with increasing concentrations of alcohol, which was extracted with xylene (Sigma-Aldrich) prior to mounting with VectaMount Permanent Mounting Medium (Vector Laboratories, Inc.; Burlingame, CA).
For immunofluorescent staining, antigen unmasking was achieved using sodium citrate (Sigma-Aldrich) in a 90°C water bath. After non-specific binding was blocked with 20% donkey serum (Sigma-Aldrich) in phosphate-buffered saline for 1 hour in room temperature, sections were incubated at 4°C with the appropriate combinations of primary antibodies overnight. The following primary antibodies were used: rat monoclonal anti-CD4 (1:100; GK1.5; #BAM554), rat monoclonal anti-CD45 (1:100; 30-F11; #MAB114), goat polyclonal anti-LYVE-1 (1:400; #2125-LY), and goat polyclonal anti-podoplanin (1:100; #AF3244) from R&D Systems (Minneapolis, MN); rabbit polyclonal anti-collagen I (1:100; #ab34710), rat monoclonal anti-F4/80 (1:100; #ab16911), and rabbit polyclonal anti-inducible nitric oxide synthase (iNOS) (1:100; #ab3523) from Abcam (Cambridge, MA); and mouse monoclonal anti-alpha smooth muscle actin (α-SMA) (1:100; #A2547) from Sigma-Aldrich. After washing, the sections were then incubated with corresponding fluorescent-labeled secondary antibody conjugates (1:1000; Life Technologies; Thermo Fisher; Waltham, MA) at room temperature for 5 hours and DAPI for 10 minutes before being mounted with Mowiol (Sigma-Aldrich).
All mounted sections were scanned with Zeiss Mirax slide scanner and analyzed with Pannoramic Viewer (3D Histech; Budapest, Hungary). Analysis was performed by 2 blinded reviewers using 4–5 high-powered sections per animal and with a minimum of 5 animals per group. Cell counts and calculation of capillary lymphatic vessel area are quantified in standardized areas measuring 0.25 mm2. Fibroadipose thickness was measured as the width of tissues bounded by the reticular dermis and deep fascia in 4 standardized regions of tail cross-sections. Type I collagen deposition was quantified as a ratio of positively stained dermis and subcutaneous tissues within a fixed threshold to total tissue area using Metamorph Offline Software (Molecular Devices; Sunnyvale, CA). Alpha-smooth muscle actin accumulation around collecting vessels following PLND was evaluated at the points of greatest thickness in each cross-section.
Analysis of lymphatic function
Near-infrared lymphangiography using indocyanine green (ICG) was utilized for assessment of ipsilateral hindlimb collecting lymphatic vessel function after PLND per a previously validated protocol [30–33]. Briefly, 15 μL of 0.15 mg/mL (Sigma-Aldrich) was injected intradermally into the first webspace of the hindpaws of anesthetized mice. The mice were allowed to awaken and move freely for 30 minutes to promote uptake of the ICG into the lymphatic vasculature. They were then placed under anesthesia again for imaging, which was performed using a custom-made EVOS EMCCD camera (Life Technologies; Carlsbad, CA) and LED light source (CoolLED; Andover, UK) mounted on a SteREO Lumar.v12 microscope (Zeiss; Jena, Germany). Images were obtained every 8 seconds for 30 minutes for each mouse hindlimb using the same machine and settings. Lymphatic pumping was analyzed with Fiji software (National Institutes of Health; Bethesda, MD). A region-of-interest was selected over the dominant collecting vessel of each hindlimb and noise-subtracted fluorescence intensity was plotted over time as arbitrary units, which are representative of the brightness of the near-infrared signal. Fluctuations in intensity or brightness correspond to lymphatic contractions and can therefore be utilized to assess lymphatic vessel pumping frequency and strength. The initial 10 minutes of each image set were excluded to avoid inaccuracies from inadvertent lymphatic stimulation due to positioning.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software; San Diego, CA). Differences between two groups were compared with unpaired student’s t test, while comparisons between four groups or over time were conducted using one-way or two-way analysis of variance, respectively, with Tukey’s multiple comparisons test. Data are presented as mean ± standard deviation unless otherwise noted and P < 0.05 was considered significant.
Results
Mice with impaired Th2 differentiation do not develop lymphedema
Using multiple mouse models of lymphedema, including the tail surgery model, the axillary lymph node dissection (ALND) model, and the PLND model, we have previously shown that lymphatic injury reliably results in a distinct inflammatory infiltrate largely comprised of CD4+ T cells [4, 11, 33]. Importantly, this finding was also demonstrated in human lymphedematous tissues [5]. Subsequent studies that sought to further characterize these inflammatory responses noted that both Th1 and Th2 cells were increased in lymphedema. These studies relied largely on histological staining [5]. In the current study, however, we chose to utilize flow cytometric evaluation of single-cell suspensions derived from lymphedematous tissues to improve cellular analysis and quantification. The tail tissues of WT mice that underwent tail sham surgery (skin incision without lymphatic excision) or skin and lymphatic excision were harvested, digested, and analyzed 6 weeks post-operatively (Fig. 1a). The tail model was specifically chosen in this case because we and others have found that inflammation is better analyzed in this model compared to the lymphadenectomy models [22]. Consistent with prior findings, we noted that there was a significant increase in CD4+ T cell infiltration in lymphedematous tissues as compared with sham controls (Fig. 1b; P = 0.0015). Further characterization revealed that this overall increase in the percentage of CD4+ T cells corresponded to a 4.3-fold increase in Th1 cells (CD45+CD4+CXCR3+CCR5+) and a 2.6-fold increase in Th2 cells (CD45+CD4+CCR4+CCR8+; Fig. 1c, d; P < 0.0001 for both).
Fig. 1. Lymphedema is associated with a mixed CD4+ T cell infiltrate.

(a) Schematic diagram of experimental protocol. Flow cytometric analysis performed 6 weeks after tail skin incision without lymphatic excision (sham surgery) or tail skin and lymphatic excision (lymphedema). (b-d) Representative flow cytometric plots and quantification of CD45+CD4+ T cells (b), CD45+CD4+CXCR3+CCR5+ Th1 cells (c), and CD45+CD4+CCR4+CCR8+ Th2 cells (d) in distal tail skin and subcutaneous tissues (n = 5–6 per group). Mean ± s.d.; *P<0.05, **P<0.01, and ***P<0.001 by unpaired student’s t test.
We next used the same mouse tail model of lymphedema to compare pathologic changes following lymphatic injury in WT, global CD4KO, Th1-deficient (T-betKO), and Th2-deficient (STAT6KO) mice (Fig. 2a). We chose to use this model because it is particularly useful for the evaluation of the gross development of edema and quantification of fibroadipose tissue deposition.
Fig. 2. STAT6 deficiency mitigates lymphedema development after tail skin and lymphatic excision.
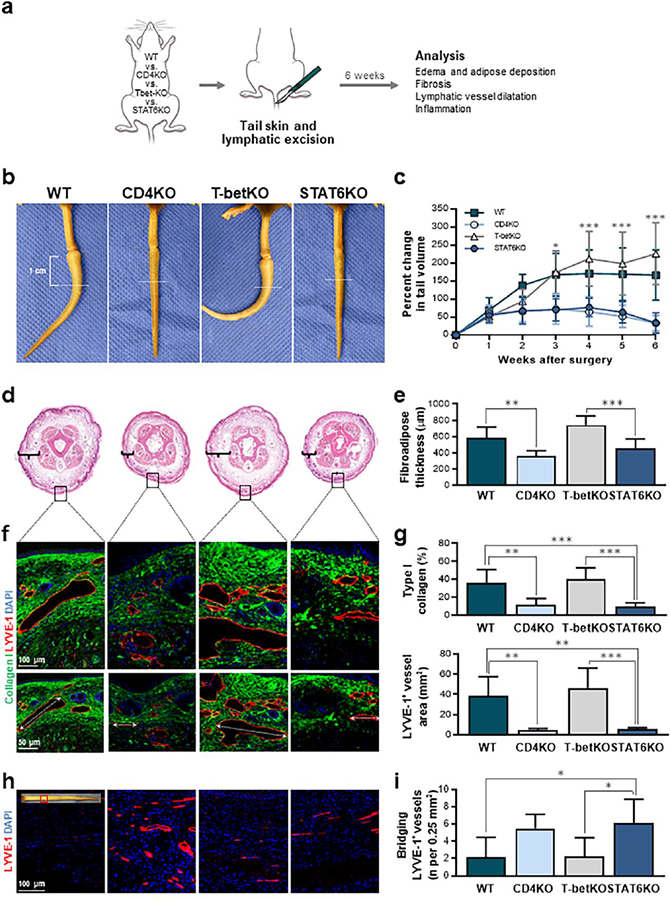
(a) Schematic diagram of experimental protocol. Analysis performed 6 weeks after tail skin and lymphatic excision. (b) Representative photographs of mouse tails with white dotted lines indicating 1 cm distal to incision, the point at which histological features were analyzed. (c) Quantification of changes in tail volumes over time after surgery (n = 7 per group). (d) Representative H&E staining of tail cross-sections obtained at the location indicated by the white dotted lines in part b with brackets indicating fibroadipose thickness. Black boxes indicate areas at which subsequent collagen analysis was performed. (e) Quantification of fibroadipose thickness (n = 7 per group). (f) Representative immunofluorescent images localizing type I collagen with LYVE-1+ lymphatic vessels with higher magnification above and lower magnification below; dotted arrows indicate diameters of the largest lymphatic vessel in the representative images for each group. (g) Quantification of dermal type I collagen deposition (upper) and LYVE-1+ lymphatic vessel area (lower) (n = 6–7 per group). (h) Representative immunofluorescent images localizing LVYE-1+ lymphatic vessels at the surgical site. (i) Quantification of LYVE-1+ lymphatic vessels bridging the surgical site (n = 6–8 per group). Mean ± s.d., unless otherwise specified; *P<0.05, **P<0.01, and ***P<0.001 by one-way ANOVA with Tukey’s multiple comparisons test. H&E, hematoxylin and eosin.
Consistent with previous studies, we found that CD4KO mice had significantly decreased tail edema (Fig. 2b, c; P < 0.01 starting at 3 weeks post-operatively) and fibroadipose tissue deposition (Fig 2d, e; P = 0.0043) as compared to WT mice that also underwent tail skin and lymphatic excision. T-betKO mice had a similar phenotype to WT mice, displaying significant increases in tail volume and fibroadipose deposition. In fact, these Th1-deficient mice had a nearly identical gross appearance of their tails as WT mice with the characteristic J-shaped configuration of the distal lymphedematous tail that occurs as a result of tissue fibrosis. Interestingly, quantification of histological sections comparing WT and T-betKO mice showed that T-betKO mice had a nearly 30% increase in tail fibroadipose deposition, a difference that approached statistical significance (Fig. 2e; P = 0.055), suggesting that the loss of Th1 responses may worsen the lymphedema phenotype. In contrast, the tails of STAT6KO mice displayed a gross appearance that was identical to CD4KO mice with little evidence of swelling and demonstrated significantly decreased tail volumes compared to WT or T-betKO mice starting 3 weeks post-operatively (Fig. 2c; P < 0.001). Furthermore, the Th2 cell-deficient mice displayed minimal increases in fibroadipose thickness 6 weeks after surgery (Fig. 2e; P = 0.0003 for T-betKO versus STAT6KO).
Fibrosis in lymphedema is related to dermal type I collagen deposition [5, 34, 35]. To determine whether the defect in Th2 cell generation in STAT6KO mice corresponded to changes in this characteristic feature, we used immunofluorescent staining to quantify the percentage of type I collagen in the dermis of distal tail cross-sections obtained 6 weeks after skin and lymphatic excision in all experimental groups (Fig. 2f). We found that the accumulation of type I collagen in STAT6KO mice was less than one-third of that found in both WT and T-betKO mice (Fig. 2g upper; P = 0.0005 and P < 0.0001, respectively). In contrast, it was not significantly different from that in CD4KO mice (P = 0.99).
We also noted that the lymphedema phenotype in the WT and T-betKO mice corresponded to dilated LYVE-1+ lymphatic capillary vessels in the distal portions of the tail at least 1 cm from the surgical site (Fig. 2f). These vessels in particular were examined because lymphedema is known to be associated with gradual enlargement of lymphatic capillary vessels, a phenomenon that is especially evident in the tail surgery model of lymphedema, in which there are no alternate drainage pathways to quickly compensate for the obstruction caused by lymphatic injury [8]. In contrast, CD4KO and STAT6KO mice had lymphatic capillary vessels with significantly smaller cross-sectional areas, suggesting that these animals had decreased tissue edema and resistance to lymphatic flow distal to the lymphatic injury (Fig. 2g lower; P = 0.0016 and P = 0.0019, respectively, compared to WT mice). In addition, analysis of the surgical wound in the various groups showed that STAT6KO mice had nearly 3 times as many lymphatic vessels that bridged the wound as compared to WT and T-betKO controls (Fig. 2h, i; P = 0.019 and P = 0.024, respectively). These findings are supportive of prior reports showing that the Th2 cell-derived cytokines IL-4 and IL-13 have potent anti-lymphangiogenic effects [6] and that mice with impaired Th2 differentiation capacity have increased lymphatic regeneration following lymphatic injury resulting in decreased edema and fibroadipose deposition [5].
Impaired Th2 differentiation results in decreased inflammation after lymphatic injury
We then sought to determine whether the absence of the lymphedema phenotype in CD4KO and STAT6KO mice is associated with decreased inflammation, another hallmark of the disease. To test this, we evaluated the number of peri-lymphatic CD45+ leukocytes, CD4+ cells, and F4/80+ cells in lymphedematous tail tissues 6 weeks after surgery. We found no overall changes in peri-lymphatic CD45+ or CD4+ cell infiltration when comparing WT with T-betKO mice (Fig. 3a–c; P = 0.26 and P = 0.17, respectively). However, T-betKO mice did display a decrease in the number of peri-lymphatic F4/80+ cells as compared with WT controls (Fig. 3a, d; P = 0.0034). In contrast, CD4KO mice as well as STAT6KO mice had significantly decreased peri-lymphatic accumulation of CD45+ leukocytes, CD4+ cells, and F4/80+ cells as compared with WT or T-betKO mice. As expected, that inflammatory cell accumulation was lowest in CD4KO mice. Nevertheless, these findings show that impaired Th2 differentiation decreases peri-lymphatic inflammatory responses while a defect in Th1 differentiation results in minimal changes in inflammation following lymphatic injury.
Fig. 3. STAT6 deficiency mice have decreased inflammation after lymphatic injury.

Analysis performed 6 weeks after tail skin and lymphatic excision. (a) Representative immunofluorescent images localizing CD45 (upper), CD4 (middle), and F4/80 (lower) with LVYE-1+ lymphatic vessels. (b-d) Quantification of CD45+ (b), CD4+ (c), and F4/80+ (d) cells within 50 μm of lymphatic vessels in an area measuring 0.25 mm2. Mean ± s.d.; *P<0.05, **P<0.01, and ***P<0.001 by one-way ANOVA with Tukey’s multiple comparisons test.
Impaired Th2 differentiation improves lymphatic collecting vessel pumping and collateral lymphatic vessel development after lymphatic injury
The maintenance of unidirectional flow through spontaneous active contraction of smooth muscle cells around collecting lymphatic vessels together with one-way valves is a major component of lymphatic transport. In fact, it is estimated that collecting lymphatic vessel pumping is responsible for nearly two-thirds of lymphatic volume movement at rest [36]. This mechanism is particularly important in limbs, in which lymphatic fluid is transported against pressure gradients due to gravity. Based on this knowledge and the understanding that lymphedema is associated with lymphatic dysfunction, we sought to study the effect of impaired Th2 differentiation capacity on collecting lymphatic vessel pumping and the cellular mechanisms that regulate this phenomenon using the PLND model of lymphedema (Fig. 4). This model, in contrast to the tail model, does not cause significant edema but is useful for analyzing collecting vessel pumping mechanics [22, 27, 37]. Both lymphatic vessel pumping and collateral lymphatic vessel regeneration were assessed with ICG lymphangiography 4 weeks after mice from each group underwent PLND [38].
Fig. 4. STAT6 deficiency mice have improved collecting vessel pumping and increased collateral vessel formation after lymphatic injury.

(a) Schematic diagram of experimental protocol. Analysis performed 4 weeks after PLND. (b) Representative near-infrared lymphangiography images of the ipsilateral hindlimb following ICG injection into the dorsal hindpaw. (c) Representative graphs depicting lymphatic pumping as indicated by changes in ICG intensity over time. (d) Quantification of ipsilateral hindlimb collecting lymphatic vessel contractions per minute (n = 6–7 mice per group). Mean ± s.d.; *P<0.05, **P<0.01, and ***P<0.001 by one-way ANOVA with Tukey’s multiple comparisons test.
Similar to our findings of increased numbers of bridging lymphatic vessels across wounds following mouse tail surgery (Fig. 2h, i), we observed many collateral lymphatic vessels in the hindlimbs of CD4KO and STAT6KO mice (indicated by arrows in Fig. 4b). These collateral lymphatic vessels appeared to branch from the main hindlimb collecting vessels and drain diagonally across the limb in the direction of the inguinal lymph nodes, the drainage basin immediately proximal to the excised popliteal lymph nodes. Meanwhile, WT and T-betKO mice consistently demonstrated very few collateral vessels in this region. Such finding suggest that CD4KO and STAT6KO have improved capacity for lymphangiogenesis following lymphatic injury, a hypothesis that is supported by prior evidence that Th2 cytokines are potent anti-lymphangiogenic factors [6].
We next analyzed videos of the hindlimbs following ICG injection to quantify differences in hindlimb collecting vessel contraction after PLND. This analysis demonstrated that CD4KO and STAT6KO mice had significantly more frequent lymphatic contractions as compared with WT (Fig. 4c, d; P = 0.0051 and P = 0.0016, respectively). In addition, we once again found that T-betKO mice were similar to WT mice with fewer contractions over time. Notably, lymphatic contractions in WT and T-betKO mice were not only less frequent but also of lower amplitude and less regular as compared with CD4KO or STAT6KO mice.
Studies have shown that inflammation has detrimental effects on lymphatic contractility by promoting the expression of iNOS in inflammatory cells [39]. Increased accumulation of iNOS subsequently results in increased global production of nitric oxide (NO) and is thought to disrupt endogenous NO gradients produced by lymphatic endothelial nitric oxide synthase (eNOS). This, in turn, induces lymphatic vessel dilatation and interferes with the rhythmic sequence of lymphatic collecting vessel contraction and dilatation necessary for sequential propulsion of lymph [40]. Indeed, we have previously found that mice with a global loss of iNOS do not develop impaired lymphatic pumping following PLND [33]. Consistent with this, we noted significant decreases in the number of peri-lymphatic iNOS+ cells in CD4KO and STAT6KO mice as compared with WT or T-betKO mice (Fig 5a, b). We found that STAT6KO mice had 30% fewer iNOS+ cells within 50 μm of dermal lymphatic vessels in the ipsilateral hindlimb than T-betKO mice (Fig. 5b; P = 0.0003). These results indicate that one mechanism by which Th2 cells negatively regulate lymphatic pumping is by mediating peri-lymphatic inflammation and expression of iNOS.
Fig. 5. STAT6 deficiency decreases peri-lymphatic iNOS accumulation and collecting vessel fibrosis.
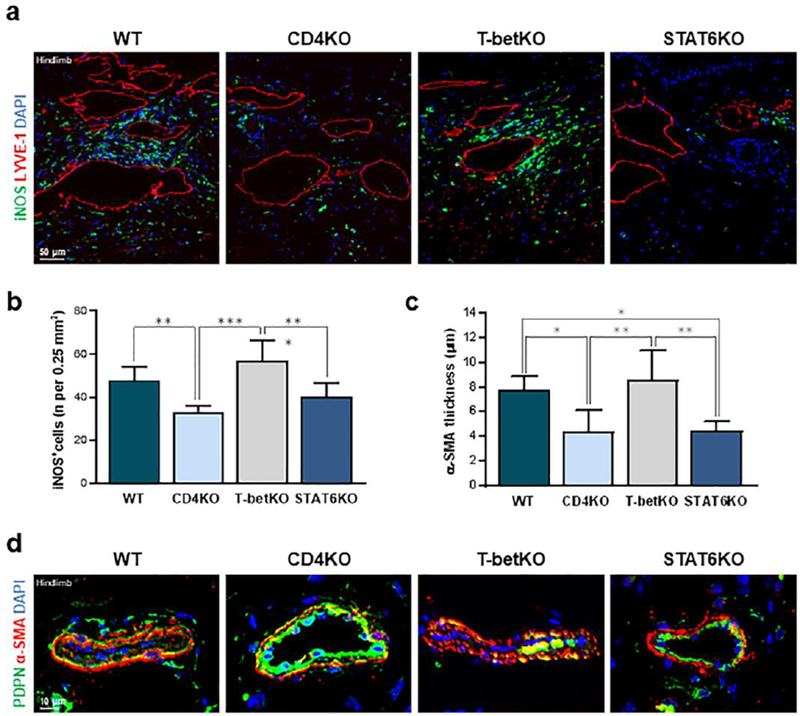
(a) Representative immunofluorescent images localizing iNOS with LYVE-1+ lymphatic vessels 4 weeks following PLND. (b) Quantification of iNOS+ cells within 50 μm of lymphatic vessels in an area measuring 0.25 mm2. (c) Quantification of α-SMA thickness around hindlimb collecting lymphatic vessels (n = 5–7 per group). (d) Representative immunofluorescent images localizing PDPN with α-SMA around hindlimb collecting lymphatic vessels 4 weeks following PLND. Mean ± s.d.; *P<0.05, **P<0.01, and ***P<0.001 by one-way ANOVA with Tukey’s multiple comparisons test. α-SMA, alpha-smooth muscle acting; PDPN, podoplanin; PLND, popliteal lymph node dissection.
Patients with lymphedema have progressive changes in their collecting lymphatic vessels characterized by increased accumulation of lymphatic α-SMA cells and resultant vessel obliteration [35]. We therefore analyzed the distribution and quantity of α-SMA+ cells in the hindlimb collecting vessels of the different mouse groups 4 weeks following PLND (Fig. 5c, d). Co-localization of α-SMA and podoplanin around the vessels distal to the lymphadenectomy demonstrated that CD4KO and STAT6KO mice had significantly decreased α-SMA thickness as compared to WT and T-betKO mice (Fig. 5b; P = 0.029 and P = 0.033, respectively, as compared to WT mice). Consistent with prior results, the collapsed appearance of and α-SMA accumulation surrounding the collecting lymphatic vessels harvested from WT and T-betKO mice mirrored clinical findings.
Discussion
Current lymphedema treatment is largely palliative in nature and designed to prevent disease progression by decreasing fluid accumulation with massage and compressive garments. Advances in therapeutic options have been impaired by the fact that the pathology of lymphedema is still unclear. Combined with previous reports, the findings in this study confirm that inflammatory responses play a key role in the pathogenesis of lymphedema in at-risk patients who endure lymphatic injury [4, 7]. These data provide additional evidence that Th2 cell differentiation in particular is necessary for this response in preclinical models.
Previous studies have found that inhibition of Th2 cell-derived cytokines such as IL-4 and IL-13 is effective at mitigating lymphedema development [5, 6]. Such reports have been valuable and have even led to a human immunotherapy trial testing the efficacy of combined IL-4 and IL-13 blockade for the treatment of lymphedema. However, while these cytokines are characteristically associated with Th2 cells, they are also produced by and have effects on other cell types [41, 42]; as such, inhibition of these cytokines does not specifically target the effects of Th2 cells. In contrast, this study’s use of transgenic mice with defects in Th1 and Th2 differentiation, respectively, allows for more direct delineation of the role of Th2 cells in the development of lymphedema. It is important to note that the cellular and molecular events contributing to Th1 and Th2 differentiation are complex and, as is the case with much of immunologic phenomena, cannot be narrowed down to an isolated pathway. However, although studies have shown that the polarization of CD4+ Th2 cells may be influenced by STAT6-independent pathways, [19, 43, 44] the vast majority of studies support an import role for this transcription factor [16, 19, 45]. Elo et al., for example, used genome-wide profiling of IL-4 and STAT6 to demonstrate that STAT6 is critical to the transcription required for the Th2 phenotype [46]. In addition, it is also important to note that both T-bet and STAT6 contribute to processes other than T cell differentiation. For instance, T-bet has been shown to contribute to natural killer (NK) cell developmental pathways [47]. However, we have previously shown that lymphatic injury did not have any significant effects on NK cells and that depletion of NK cells does not reverse lymphedema, suggesting that changes in NK cell populations in our knockout animals would not have a significant effect on the lymphedema phenotype [4, 33]. Despite these limitations, T-bet and STAT6 continue to be regarded as important regulators of commitment to Th1 and Th2 lineage, respectively, and the understanding of the effect of lymphatic injury in the absence of these factors provides valuable information, particularly when taken in the context of our prior findings in which we found that while lymphedematous tissue are infiltrated by both Th1 and Th2 cells [4], Th2 cells appear to play a more important role in the pathology of lymphedema by regulating fibrosis, overall inflammation, collateral lymphatic vessel development, and collecting lymphatic vessel pumping [5, 6].
We found that the absence of Th2 differentiation resulted in a diminished fibrotic response, as well as decreased inflammation. A less robust immune response with reduced expression of Th2 cytokines also appears to correlate with improved lymphangiogenesis following lymphatic injury. This is demonstrated by the presence of increased bridging lymphatic vessels at the site of injury in the tail surgery model (Fig. 2h), in addition to a denser collateral lymphatic vasculature as visualized by near-infrared lymphangiography in the PLND model (Fig. 4b). Although it is possible that the visualized collateral lymphatic vessels in the near-infrared images are reflective of pre-existing alternative drainage pathways, it is more likely that they are reflective of an improved capacity for lymphangiogenesis. Not only have no studies have identified a link between inflammation and the dilatation of pre-existing lymphatic collateral vessels, but prior reports have shown that inadequate or impaired lymphangiogenesis following lymphatic injury plays a significant role in the pathogenesis of lymphedema [48]. In fact, researchers have sought to develop therapies that enhance lymphangiogenesis as a potential treatment for lymphedema [49]. Furthermore, our findings are consistent with our prior study in which we demonstrated that the Th2 cytokines IL-4 and IL-13 are potent anti-lymphangiogenic factors [6]. Taken together, these results suggest that targeting of Th2 cells may result in improved lymphangiogenesis, which may =in turn prevent or treat lymphedema.
Our results also indicate that one of the many mechanisms by which Th2 cells contribute to lymphedema pathology is related to the promotion of peri-lymphatic iNOS expression, which disrupts the sequence of lymphatic contraction and dilatation necessary for effective lymph fluid transport. Lymphatic vessel pumping capacity is also concurrently affected by structural vessel changes resulting from the positive effect of Th2 cells on the accumulation of α-SMA around collecting lymphatic vessels. It is likely that this response is progressive and that with worsening lymphedema, the cellular mechanisms that promote α-SMA accumulation increase over time. These findings provide a mechanistic rationale for previous studies demonstrating that lymphatic pump failure is a key mechanism in lymphedema development clinically [50, 51]. In addition, our results may also provide a rationale for the observation that patients with increased lymphatic pump pressures prior to surgery have an increased risk of developing lymphedema after axillary lymph node dissection [51]. It may be that in this patient population, subclinical alterations in lymphatic clearance induced proliferation of α-SMA+ cells around collecting lymphatic vessels, thus increasing pump pressures. Following surgery, these patients are therefore at increased risk for developing lymphedema since the process of α-SMA accumulation around these vessels had already begun. Therefore, increased lymphatic pump pressure in these patients may simply be a clinical correlate of impaired lymphatic function.
This study is important because it suggests that Th2 cells may be an effective target for the prevention and/or treatment of lymphedema. Additional research should be directed at identifying the specific cytokines or, more likely, the combinations of cytokines that Th2 cells produce to induce lymphedema development, in addition to the factors that initiate Th2 differentiation, to maximize the effectiveness of any biologic options. Importantly, lessons may be learned from research into therapies used for other Th2-mediated diseases for which the pathophysiology is better defined. Asthma, for example, is well-known to be regulated by Th2 cells and has been the area of focus for numerous clinical trials aimed at modifying Th2 immunity [52–54]. Interestingly, studies have shown that targeting of IL-13, either alone or in combination with IL-4, is effective in mild-to-moderate asthma, while selective inhibition of IL-4 does not produce significant changes. Recently, however, more attention has been directed at IL-5, another Th2 effector cytokine that is essential for eosinophil regulation. Clinical trials of anti-IL-5 biologics have been shown to be promising for subset of severe asthma and have also been tested for other Th2-mediated diseases such as chronic rhinosinusitis and atopic dermatitis [12, 54]. Research involving atopic dermatitis is particularly interesting, given that it is similar to lymphedema in that it is also an inflammatory disease of the skin. Much like our preliminary findings with monoclonal antibodies directed toward IL-4 and IL-13 in the mouse model of lymphedema [5], the use of dupilumab, a human antibody that blocks the same cytokines, has shown promise for patients with severe atopic dermatitis [12, 55]. It remains unknown whether blockade of these or other cytokines will play the same role in lymphedema, but this direction of research has the potential to change the standard of care for this morbid disease.
In conclusion, we have found strong evidence that the differentiation of Th2 cells isnecessary for the manifestation of edema, fibroadipose deposition, inflammation, and lymphaticdysfunction in lymphedema. These novel findings contribute mechanistic insights andcorroborate previous reports. In addition, our study provides further rationale for modulation ofthe Th2 immune pathway in patients at risk for or with evidence of lymphedema.
Acknowledgements
The authors are grateful to the staff of the Molecular Cytology Core and the Flow Cytometry Core at Memorial Sloan Kettering Cancer Center for assistance with histology, tissue imaging, and flow cytometry.
This work was supported by the following funding sources: NIH R01 HL111130–01 and R21-CA194882 grants awarded to B.J.M., Sharp Foundation Fund philanthropic gift awarded to B.J.M., NIH T32 CA9501–29 grant to C.L.L., NIH T32 CA009501–27 grant to G.D.G.N., Plastic Surgery Foundation/Musculoskeletal Transplant Foundation Allograft Tissue Research Grant 501832 to C.L.L., Plastic Surgery Foundation Pilot Grant 350627 to G.D.G.N., and the NIH/NCI Cancer Center Support Grant P30 CA008748.
All authors have read the journal’s authorship agreement and policy on disclosure of potential conflicts of interest. The manuscript, as submitted, has been reviewed by and approved by all named authors. The authors declare that the corresponding author is empowered by all of the authors to act on their behalf with respect to the submission of the manuscript; that the article is original; that the article does not infringe upon any copyright or other proprietary right of any third party; that neither the text nor the data reported have been published previously; and that the article or a substantially similar article is not under consideration by another article at this time. In addition, the authors report no potential conflicts of interest.
Abbreviations:
- α-SMA
Alpha smooth muscle actin
- AVMA
American Veterinary Medical Association
- CD4KO
CD4 knockout
- DAPI
4,6-diamidino-2-phenylindole
- eNOS
Endothelial nitric oxide synthase
- H&E
Hematoxylin and eosin
- ICG
Indocyanine green
- IFN-γ
Interferon gamma
- IL-4
Interleukin 4
- IL-13
Interleukin 13
- iNOS
Inducible nitric oxide synthase
- NK
Natural killer
- PDPN
Podoplanin
- PLND
Popliteal lymph node dissection
- STAT6KO
Signal transducer and activator of transcription 6 knockout
- T-betKO
T-bet knockout
- Th1
T helper 1
- Th2
T helper 2
- WT
Wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sakorafas GH, Peros G, Cataliotti L, Vlastos G. Lymphedema following axillary lymph node dissection for breast cancer. Surgical oncology. 2006;15:153–65. [DOI] [PubMed] [Google Scholar]
- [2].DiSipio T, Rye S, Newman B, Hayes S. Incidence of unilateral arm lymphoedema after breast cancer: a systematic review and meta-analysis. The Lancet Oncology. 2013;14:500–15. [DOI] [PubMed] [Google Scholar]
- [3].Kuroda K, Yamamoto Y, Yanagisawa M, Kawata A, Akiba N, Suzuki K, et al. Risk factors and a prediction model for lower limb lymphedema following lymphadenectomy in gynecologic cancer: a hospital-based retrospective cohort study. BMC women’s health. 2017;17:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zampell JC, Yan A, Elhadad S, Avraham T, Weitman E, Mehrara BJ. CD4(+) cells regulate fibrosis and lymphangiogenesis in response to lymphatic fluid stasis. PloS one. 2012;7:e49940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Avraham T, Zampell JC, Yan A, Elhadad S, Weitman ES, Rockson SG, et al. Th2 differentiation is necessary for soft tissue fibrosis and lymphatic dysfunction resulting from lymphedema. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27:1114–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Savetsky IL, Ghanta S, Gardenier JC, Torrisi JS, Garcia Nores GD, Hespe GE, et al. Th2 cytokines inhibit lymphangiogenesis. PloS one. 2015;10:e0126908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ogata F, Fujiu K, Matsumoto S, Nakayama Y, Shibata M, Oike Y, et al. Excess Lymphangiogenesis Cooperatively Induced by Macrophages and CD4(+) T Cells Drives the Pathogenesis of Lymphedema. The Journal of investigative dermatology. 2016;136:706–14. [DOI] [PubMed] [Google Scholar]
- [8].Gousopoulos E, Proulx ST, Scholl J, Uecker M, Detmar M. Prominent Lymphatic Vessel Hyperplasia with Progressive Dysfunction and Distinct Immune Cell Infiltration in Lymphedema. Am J Pathol. 2016;186:2193–203. [DOI] [PubMed] [Google Scholar]
- [9].Torrisi JS, Joseph WJ, Ghanta S, Cuzzone DA, Albano NJ, Savetsky IL, et al. Lymphaticovenous bypass decreases pathologic skin changes in upper extremity breast cancer-related lymphedema. Lymphatic research and biology. 2015;13:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gousopoulos E, Proulx ST, Bachmann SB, Scholl J, Dionyssiou D, Demiri E, et al. Regulatory T cell transfer ameliorates lymphedema and promotes lymphatic vessel function. JCI Insight. 2016;1:e89081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Garcia Nores GD, Ly CL, Savetsky IL, Kataru RP, Ghanta S, Hespe GE, et al. Regulatory T Cells Mediate Local Immunosuppression in Lymphedema. J Invest Dermatol. 2018;138:325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nakayama T, Hirahara K, Onodera A, Endo Y, Hosokawa H, Shinoda K, et al. Th2 Cells in Health and Disease. Annual review of immunology. 2017;35:53–84. [DOI] [PubMed] [Google Scholar]
- [13].Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol. 2013;13:777–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH, et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 2002;295:336–8. [DOI] [PubMed] [Google Scholar]
- [15].Amarnath S, Laurence A, Zhu N, Cunha R, Eckhaus MA, Taylor S, et al. Tbet is a critical modulator of FoxP3 expression in autoimmune graft-versus-host disease. Haematologica. 2017;102:1446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhu J, Guo L, Watson CJ, Hu-Li J, Paul WE. Stat6 is necessary and sufficient for IL-4’s role in Th2 differentiation and cell expansion. J Immunol. 2001;166:7276–81. [DOI] [PubMed] [Google Scholar]
- [17].Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–9. [DOI] [PubMed] [Google Scholar]
- [18].Walford HH, Doherty TA. STAT6 and lung inflammation. Jak-stat. 2013;2:e25301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Maier E, Duschl A, Horejs-Hoeck J. STAT6-dependent and -independent mechanisms in Th2 polarization. European journal of immunology. 2012;42:2827–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol. 2018;18:121–33. [DOI] [PubMed] [Google Scholar]
- [21].Rahemtulla A, Fung-Leung WP, Schilham MW, Kundig TM, Sambhara SR, Narendran A, et al. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature. 1991;353:180–4. [DOI] [PubMed] [Google Scholar]
- [22].Ly CL, Kataru RP, Mehrara BJ. Inflammatory Manifestations of Lymphedema. International journal of molecular sciences. 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Avraham T, Daluvoy S, Zampell J, Yan A, Haviv YS, Rockson SG, et al. Blockade of transforming growth factor-beta1 accelerates lymphatic regeneration during wound repair. Am J Pathol. 2010;177:3202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Clavin NW, Avraham T, Fernandez J, Daluvoy SV, Soares MA, Chaudhry A, et al. TGF-beta1 is a negative regulator of lymphatic regeneration during wound repair. American journal of physiology Heart and circulatory physiology. 2008;295:H2113–27. [DOI] [PubMed] [Google Scholar]
- [25].Rutkowski JM, Boardman KC, Swartz MA. Characterization of lymphangiogenesis in a model of adult skin regeneration. American journal of physiology Heart and circulatory physiology. 2006;291:H1402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tabibiazar R, Cheung L, Han J, Swanson J, Beilhack A, An A, et al. Inflammatory manifestations of experimental lymphatic insufficiency. PLoS medicine. 2006;3:e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Blum KS, Proulx ST, Luciani P, Leroux JC, Detmar M. Dynamics of lymphatic regeneration and flow patterns after lymph node dissection. Breast cancer research and treatment. 2013;139:81–6. [DOI] [PubMed] [Google Scholar]
- [28].Proulx ST, Luciani P, Christiansen A, Karaman S, Blum KS, Rinderknecht M, et al. Use of a PEG-conjugated bright near-infrared dye for functional imaging of rerouting of tumor lymphatic drainage after sentinel lymph node metastasis. Biomaterials. 2013;34:5128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sharma R, Wang W, Rasmussen JC, Joshi A, Houston JP, Adams KE, et al. Quantitative imaging of lymph function. American journal of physiology Heart and circulatory physiology. 2007;292:H3109–18. [DOI] [PubMed] [Google Scholar]
- [30].Huang JJ, Gardenier JC, Hespe GE, Garcia Nores GD, Kataru RP, Ly CL, et al. Lymph Node Transplantation Decreases Swelling and Restores Immune Responses in a Transgenic Model of Lymphedema. PloS one. 2016;11:e0168259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Robinson HA, Kwon S, Hall MA, Rasmussen JC, Aldrich MB, Sevick-Muraca EM. Noninvasive optical imaging of the lymphatic vasculature of a mouse. Journal of visualized experiments : JoVE. 2013:e4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gardenier JC, Hespe GE, Kataru RP, Savetsky IL, Torrisi JS, Garcia Nores GD, et al. Diphtheria toxin–mediated ablation of lymphatic endothelial cells results in progressive lymphedema. JCI Insight. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Garcia Nores GD, Ly CL, Cuzzone DA, Kataru RP, Hespe GE, Torrisi JS, et al. CD4(+) T cells are activated in regional lymph nodes and migrate to skin to initiate lymphedema. Nature communications. 2018;9:1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Avraham T, Clavin NW, Daluvoy SV, Fernandez J, Soares MA, Cordeiro AP, et al. Fibrosis is a key inhibitor of lymphatic regeneration. Plastic and reconstructive surgery. 2009;124:438–50. [DOI] [PubMed] [Google Scholar]
- [35].Mihara M, Hara H, Hayashi Y, Narushima M, Yamamoto T, Todokoro T, et al. Pathological steps of cancer-related lymphedema: histological changes in the collecting lymphatic vessels after lymphadenectomy. PloS one. 2012;7:e41126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Olszewski WL, Engeset A. Intrinsic contractility of prenodal lymph vessels and lymph flow in human leg. The American journal of physiology. 1980;239:H775–83. [DOI] [PubMed] [Google Scholar]
- [37].Liao S, Jones D, Cheng G, Padera TP. Method for the quantitative measurement of collecting lymphatic vessel contraction in mice. Journal of biological methods. 2014;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gardenier JC, Kataru RP, Hespe GE, Savetsky IL, Torrisi JS, Nores GD, et al. Topical tacrolimus for the treatment of secondary lymphedema. Nat Commun. 2017;8:14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Scallan JP, Zawieja SD, Castorena-Gonzalez JA, Davis MJ. Lymphatic pumping: mechanics, mechanisms and malfunction. The Journal of physiology. 2016;594:5749–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liao S, von der Weid PY. Inflammation-induced lymphangiogenesis and lymphatic dysfunction. Angiogenesis. 2014;17:325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gadani SP, Cronk JC, Norris GT, Kipnis J. IL-4 in the brain: a cytokine to remember. J Immunol. 2012;189:4213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rael EL, Lockey RF. Interleukin-13 signaling and its role in asthma. The World Allergy Organization journal. 2011;4:54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ariga H, Shimohakamada Y, Nakada M, Tokunaga T, Kikuchi T, Kariyone A, et al. Instruction of naive CD4+ T-cell fate to T-bet expression and T helper 1 development: roles of T-cell receptor-mediated signals. Immunology. 2007;122:210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Usui T, Nishikomori R, Kitani A, Strober W. GATA-3 suppresses Th1 development by downregulation of Stat4 and not through effects on IL-12Rbeta2 chain or T-bet. Immunity. 2003;18:415–28. [DOI] [PubMed] [Google Scholar]
- [45].Goenka S, Kaplan MH. Transcriptional regulation by STAT6. Immunologic research. 2011;50:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Elo LL, Jarvenpaa H, Tuomela S, Raghav S, Ahlfors H, Laurila K, et al. Genome-wide profiling of interleukin-4 and STAT6 transcription factor regulation of human Th2 cell programming. Immunity. 2010;32:852–62. [DOI] [PubMed] [Google Scholar]
- [47].Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cooke JP. Lymphangiogenesis: a potential new therapy for lymphedema? Circulation. 2012;125:853–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Visuri MT, Honkonen KM, Hartiala P, Tervala TV, Halonen PJ, Junkkari H, et al. VEGF-C and VEGF-C156S in the pro-lymphangiogenic growth factor therapy of lymphedema: a large animal study. Angiogenesis. 2015;18:313–26. [DOI] [PubMed] [Google Scholar]
- [50].Cintolesi V, Stanton AW, Bains SK, Cousins E, Peters AM, Purushotham AD, et al. Constitutively Enhanced Lymphatic Pumping in the Upper Limbs of Women Who Later Develop Breast Cancer-Related Lymphedema. Lymphatic research and biology. 2016;14:50–61. [DOI] [PubMed] [Google Scholar]
- [51].Modi S, Stanton AW, Svensson WE, Peters AM, Mortimer PS, Levick JR. Human lymphatic pumping measured in healthy and lymphoedematous arms by lymphatic congestion lymphoscintigraphy. The Journal of physiology. 2007;583:271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bosnjak B, Stelzmueller B, Erb KJ, Epstein MM. Treatment of allergic asthma: modulation of Th2 cells and their responses. Respiratory research. 2011;12:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Levine SJ, Wenzel SE. Narrative review: the role of Th2 immune pathway modulation in the treatment of severe asthma and its phenotypes. Annals of internal medicine. 2010;152:232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Smith KA, Pulsipher A, Gabrielsen DA, Alt JA. Biologics in Chronic Rhinosinusitis: An Update and Thoughts for Future Directions. American journal of rhinology & allergy. 2018:1945892418787132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hamilton JD, Ungar B, Guttman-Yassky E. Drug evaluation review: dupilumab in atopic dermatitis. Immunotherapy. 2015;7:1043–58. [DOI] [PubMed] [Google Scholar]