

Abstract
The 2017 Dagstuhl Seminar on Computational Proteomics provided an opportunity for a broad discussion on the current state and future directions of the generation and use of peptide tandem mass spectrometry spectral libraries. Their use in proteomics is growing slowly, but there are multiple challenges in the field that must be addressed to further increase the adoption of spectral libraries and related techniques. The primary bottlenecks are the paucity of high quality and comprehensive libraries and the general difficulty of adopting spectral library searching into existing workflows. There are several existing spectral library formats, but none capture a satisfactory level of metadata; therefore a logical next improvement is to design a more advanced, Proteomics Standards Initiative-approved spectral library format that can encode all of the desired metadata. The group discussed a series of metadata requirements organized into three designations of completeness or quality, tentatively dubbed bronze, silver, and gold. The metadata can be organized at four different levels of granularity: at the collection (library) level, at the individual entry (peptide ion) level, at the peak (fragment ion) level, and at the peak annotation level. Strategies for encoding mass modifications in a consistent manner and the requirement for encoding high-quality and commonly-seen but as-yet-unidentified spectra were discussed. The group also discussed related topics, including strategies for comparing two spectra, techniques for generating representative spectra for a library, approaches for selection of optimal signature ions for targeted workflows, and issues surrounding the merging of two or more libraries into one. We present here a review of this field and the challenges that the community must address in order to accelerate the adoption of spectral libraries in routine analysis of proteomics datasets.
Keywords: mass spectrometry, spectral libraries, standards, formats, Dagstuhl Seminar, meeting report, Proteomics Standards Initiative
Graphical Abstract
Introduction
Mass spectrometry (MS)-based proteomics has enabled the high-throughput identification of proteins present in biological samples and the measurement of their abundances, post-translational modifications, sequence and splice variants, and interaction partners. Although sample preparation techniques and instrumental setups remain complex and vary greatly, an increasing number of laboratories are applying MS techniques to better understand health and disease and to address basic biological questions. In typical MS-based proteomics experiments, proteins are extracted from samples and enzyme-digested into peptides, which are separated by chromatography and ionized. The mass spectrometer produces digital signatures of these ions at the precursor and fragment ion level. Modern instruments can record the signatures of hundreds of thousands of peptidoforms per experiment.
The translation of these signatures into the desired information about their respective peptides and proteins is crucial for further interpretation. There are many software packages that have been developed over the past 25 years to perform the computational analyses needed to perform this task1. For data dependent acquisition (DDA) workflows, where instruments automatically select which ions to analyze based on simple rules, the most common analysis technique is sequence database searching2. This involves matching observed fragmentation mass spectra to simple simulations of spectra corresponding to peptides that may be present in the sample and selecting the best match for further validation. Once sufficiently confident identifications are made, those peptide-spectrum matches (PSMs) can be stored in a library of previously identified spectra (a spectral library), which could be used for subsequent analyses of other data.
Spectral library searching, as opposed to sequence searching via in silico predicted fragmentation spectra, typically has greater sensitivity for peptide ions included in the library3. Spectral library-based analyses would, therefore, seem like the method of choice for analysis of new datasets, but relatively few DDA datasets are analyzed in this way. A major reason for this is the widespread concern that current libraries are incomplete. Peptide ions for which no corresponding spectrum exists in the reference library will not be identified, and thus some potentially important peptides may be missed. Data independent acquisition (DIA) workflows4,5 have recently undergone rapid growth due to faster and higher mass accuracy instrumentation, affording acquisition methods such as SONAR6, SWATH-MS7, and MSX8. In these techniques, highly multiplexed fragmentation spectra are acquired according to predefined data acquisition patterns, independent of observations within the run, and the analyses of these data have spurred new interest in spectral libraries. Although library-free methods are emerging9–12, the most commonly used analysis techniques for LC-MS DIA data rely on spectral libraries to analyze extracted ion chromatograms to test for the presence of and quantify the abundance of peptide ions in the reference library13–18. Other targeted workflows, such as selected or parallel reaction monitoring (SRM/PRM), increasingly rely on large-scale spectral libraries to determine which proteotypic19,20 peptides and fragment ions to monitor21.
With billions of fragment ion spectra acquired by the research community to date, we argue that it should be possible to leverage these big data for the processing of all new data acquired. However, the current state of spectral libraries, the software that generates them, and software that can use them lag far behind the availability of data. Data from public repositories, such as PeptideAtlas22–24, PRIDE25,26, MassIVE, GPMdb27, ProteomicsDB28 and tools are available29–32; the major hindrances are familiarizing researchers with software tools, rendering them user-friendly, and promoting the use of spectral-based methods to become the norm rather than the exception.
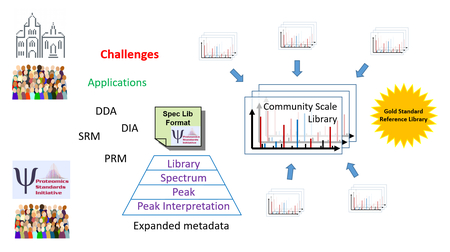
At the 2017 Dagstuhl Seminar on Computational Proteomics (Seminar 17421), hosted October 16–20 at Schloss Dagstuhl in Wadern, Germany, a group of participating researchers (hereafter referred to as “the group”) discussed the current state and future directions of spectral libraries in the field of proteomics. A follow-up meeting at the 2018 Proteomics Standards Initiative33,34 (PSI) Spring Workshop in Heidelberg, Germany (April 18–20) provided an opportunity for further discussion and resulted in a draft of metadata that should be encodable in an eventual PSI spectral library format. In this article, the major topics of discussion and some resulting conclusions are presented, with a special focus on what actions can be taken in the near term to advance the field. The benefits and requirements for a new PSI standard spectral library format are discussed, along with the issues surrounding the development of single-source and community-source spectral libraries and the state of major applications of libraries. The article concludes with a summary of the future opportunities that were discussed by the group.
A New PSI Format
There are several formats for spectral libraries used in proteomics applications. The oldest and most widely used is the simple, text-based MSP format from the National Institute of Standards and Technology (NIST). Highly similar to this is the SpectraST35 splib format, which is essentially a binary indexed version of MSP. SpectraST also writes a companion sptxt format, which is the same as MSP. The Global Proteome Machine27 (GPM) releases libraries in its hlf format for use with its X! Hunter tool36. The bibliospec tool37 began with the original text-based blib format and later moved to a SQLite-based implementation in the blib2 format. The Center for Computational Mass Spectrometry (CCMS) suite of spectral library searching tools18,38,39 and the MassIVE-KB spectral libraries use an extended version of the MGF format originally proposed by MatrixScience. Each of these formats continues to be used, but there is a widespread opinion that none provide the richness of metadata that ought to be available in modern spectral libraries.
To address this, the Human Proteome Organization40 (HUPO) PSI33,34,41 has been gathering participants interested in designing a next-generation standard spectral library format. Funding from the National Institutes of Health has recently been obtained for this development, and initial efforts have begun, with ongoing work accessible in the PSI SpectralLibraryformat GitHub repository (https://github.com/HUPO-PSI/SpectralLibraryFormat). The success of PSI-developed formats largely depends on the breadth of participation in the definition of requirements and design of the format, and the groups gathered at Dagstuhl and Heidelberg offered a great opportunity to gather broad input about the requirements for a community-approved format. Further interactions on GitHub following the meetings allowed additional external inputs. Additional input from the community is welcome via the issue tracker at the above URL.
Note that there is sometimes a distinction drawn between a spectral library and a spectral archive, such that the spectral archive can contain spectra that could not be identified42. Here this distinction is not made and the term “spectral libraries” refers to collections of mass spectra, identified or unidentified, that have been assembled to serve as a reference data after the original data processing.
The greatest identified need for a new format is the introduction of more metadata that can adequately describe the data within the spectral library and the library itself. These metadata can be broadly organized into four levels. Collection-level metadata describe attributes of the library as a whole, such as information about the creation or last update, source of the library, and global false discovery rate (FDR) of the library. Entry-level metadata describe attributes of each spectrum entry in the library, such as its charge, fragmentation type, origin, inferred peptide identification (when known) and retention time. Peak-level metadata describe attributes of each fragmentation ion peak, including its inferred charge, intensity, and fraction of replicates containing the peak. Peak interpretation-level metadata describes attributes of each fragmentation ion peak interpretation (of which there may be multiple per peak), including the probable molecule yielding the peak, the isotope state, and the delta (m/z) between the observed peak and the proposed interpretation. A list of proposed metadata elements at these four levels as drafted by this group and follow-on discussions is provided in the Supplementary Material, which will serve as a design input for the new format. This list does not represent the final specification.
There are several pieces of metadata considered here for the community format that merit further discussion, in part because they are not addressed well in previous formats, or they involve design choices that are not unanimously embraced. Perhaps foremost is the mechanism for specifying residue modifications, of which there are four broad classes: mass delta, chemical formula, English name, and controlled vocabulary term. The mass delta (e.g. “+15.99”) is perhaps the simplest mechanism, but suffers from potential precision or rounding problems that may lead to ambiguity. A chemical formula is precise and specific but will not be easily interpreted to the corresponding molecule by many human readers, and different molecules (e.g. glycans) may have the same formula but be distinct in structure. An English name is typically easily recognized by human readers, but can be context specific and the many synonyms and abbreviations in use make software recognition awkward (e.g., “Ox”, “MetOx”, “oxidation”, “L-methionine sulfoxide”). Finally, the use of controlled vocabulary (CV) terms is usually specific, but accession numbers are not easily recognized by human readers, and implementation of controlled vocabularies in software is often cumbersome, especially with multiple CVs to choose from (e.g., Unimod, PSI-MOD, PTMList). In the end, a design choice will be made to support one or more of these options to the dismay of some in the community.
Current spectral libraries were designed with the notion that each entry would have an associated peptide identification. However, there is good reason to store unidentified spectra as well. There are many spectra that are repeatedly observed in independent experiments but remain unidentified43,44, often because the component mass modifications or sequences are not considered in the search space. Several new searching algorithms, including MSFragger45, support open mass tolerance searches that are able to associate a partial match between a spectrum and a peptide, while leaving part of the identification as an unknown and unspecified mass delta; the new format should also support such matches that are partly identified, but also include an unidentified component. A curated list of commonly observed spectra that are unidentified but known to be often misidentified, leading to erroneous conclusions, would be an especially valuable addition to analysis pipelines. Some library formats support the addition of unidentified spectra, but often as a repurposing of a slot where many software packages already expect to find a parsable peptide sequence. Explicit support for unidentified spectra should be a key feature for the new PSI format. Furthermore, the format should be flexible enough to accommodate predicted spectra46–48 and interconverted spectra49, suitably annotated and differentiable as such, since there is likely to be rapid progress in the field of spectrum prediction and interconversion in the coming years.
It is also important to capture retention times in spectral libraries, as these are often used in downstream analyses. It is easy to capture retention times as acquired, but more useful to report calibrated retention times, along with the associated provenance information and metadata indicating which retention time standard was used and how the calibration was performed.
One reason that the PSI has not yet developed a standard spectral library format is that there is dissent about how the library should be encoded. Most PSI formats are XML-based or tab-separated-value-based, whereas the existing spectral library formats are a mix of plain text and binary formats. Plain text formats are promoted as being universally readable and easy for humans to examine manually and potentially correct when software runs into trouble, but they are inefficient in terms of disk space and computational resources. Custom binary formats are typically the opposite: far more efficient, but hard to restore and fix in case of corrupted or inconsistent data or when suitable supporting software is not easily within reach or no longer available. Broadly supported binary storage systems such as HDF5 or SQLite provide attractive alternatives to some, but are seen as barriers by others in terms of added software complexity or lack of sufficient support in a programming language of choice. In some ways, this conundrum is still being played out with the mzML format50, where every year sees a new publication purveying a format that is better than mzML in demonstrated ways51–53, while downplaying the trade-offs that others will find intolerable. In the end, the best strategy may be to develop a standard archival format where universal readability and carefully defined metadata are the primary design considerations, letting those in the community who demand efficiency transform the primary archival format into a more efficient version locally to suit their needs.
A further important consideration for the development of a library format is the mandatory inclusion of quality metrics at each level. The quality of a library is a crucial parameter that should be considered by all downstream use of that library, as false identifications in the library will potentially lead to false identifications downstream. Therefore, the new format will require a computed posterior error probability or q-value for each spectrum entry, as well the overall estimated FDR for the library as a whole. This will enable tempering probabilities of correctness for downstream identifications with the probabilities in the library. In addition, it may also be necessary to extend the library by including spectra of decoy matches identified in the process of constructing the library, as these may be necessary to properly model false discovery rates in the search process.
Challenges for the creation of libraries
Once a common spectral library format has been established, there will be challenges associated with the creation of libraries to ensure adoption by the community. Indeed, it is important that these challenges should be considered as use cases during the development of the format. One consideration is that the choice of which peaks to retain in library entries is often dictated by the anticipated end use of the library. For example, libraries designed for use by targeted proteomics or DIA methods may contain peaks only within restricted m/z (and/or ion mobility) ranges and only a handful of the most intense yet discriminating peaks, whereas all intense peaks are typically kept for DDA and other applications. Exclusion of reporter ions from isobaric labeling techniques may be advantageous for some applications, but not others. Among the group it was generally felt that the process of filtering libraries for a specific application was undesirable; rather, such filtering should occur at runtime by the analysis software. Yet, the practice may remain common because the precise rules of filtering can be easily controlled by the end user during library transformation, while altering the analysis software may be far more difficult or impossible. Encoding these processing choices in the library metadata is important and must be supported.
Another important consideration is the issue of spectrum variability by instrument. Fragmentation spectra produced by resonant excitation, such as in ion traps, tend to be fairly similar, but for beam-type collisional fragmentation spectra, the variability as a function of collision energy is far more pronounced. While the new spectral library format should easily support differentiation by collision energy, the absolute scales of collision energy numbers varies among instrument manufacturers, or even between instruments from the same vendor. For some applications, the ramping of collision energy from one value to another during acquisition is performed. Even on a single instrument, natural drift in calibration can lead to some differences in the spectra collected at different times54. Adequate metadata fields should be present to capture all cases accurately.
Although spectral libraries can contain multiple spectra from a single peptide ion, most library creation tools will retain only a single representative spectrum for each peptide ion in the final library. There are broadly two categories of approaches to arrive at a single representative spectrum in spectral libraries, the best replicate and the consensus spectrum. In the best replicate approach, the spectrum that is deemed highest quality is retained in the library, although the decision of which is best varies among tools; it might be the spectrum that looks most like the other replicates, the highest SNR (signal-to-noise ratio) spectrum, the spectrum with the highest ratio of explained to unexplained peaks, or some combination of those. The best replicate may be encoded as is or after some noise filtering based on comparison with other replicates. A consensus spectrum approach generally compares the top N replicates to each other, discards outliers, and then only retains peaks that appear in most of the replicates, discarding those that only appear in a few as noise or contamination. Input replicates are generally weighted by an estimate of SNR to compute the final intensities. Such consensus strategies typically filter out nearly all noise. It has been reported that consensus spectra perform better than best replicates55, but incremental addition of new replicates to a consensus library is problematic if the individual spectra are not easily accessible, whereas a new replicate can either be counted as another inferior replicate or supplant the previous best replicate, provided that the metric for best replicate does not require comparison with the other inferior replicates. Clearly the new format must accommodate all of these approaches since a single best approach has not yet emerged. The metadata must encode the choice(s) behind the representative spectrum appropriately.
The ability to merge multiple libraries is an important use case to consider and support. Many current methods for building and merging libraries rely on starting from scratch with each iteration that adds additional data, but as libraries grow substantially in size, this will become far less efficient and eventually infeasible. Therefore, design decisions that enable one library to be subsumed into another will be important. It should be possible to maintain minimum quality thresholds, compute a new overall global FDR at all levels (e.g., spectrum, precursor, peptide and protein), and retain complete pedigree information of the spectra (e.g., provenance from raw data) that remain present in the merged library.
An important new initiative of the PSI that will enable tracking of spectra that comprise a spectral library is the Universal Spectrum Identifier (USI) concept. The design of the USI is not yet complete and implemented, but aims to provide a unique multipart key for every spectrum ever submitted to ProteomeXchange and potentially beyond. This would enable best replicate spectra and even consensus spectra to be traceable to their origins from within the format. More information on the development of the USI concept is provided in a recent summary of PSI activities34 and at http://www.psidev.info/usi. The USI differs from the SPLASH identifier56 (http://splash.fiehnlab.ucdavis.edu/) used by metabolomics reference databases in that is it designed to identify all original experimental spectra via a multipart key rather than an algorithmically generated hash.
Single source and community libraries
Most spectral libraries so far are so-called “single-source” libraries, where a single group processes large numbers of mass spectra made available to them through their own analysis pipeline to produce a library for spectral library searching. A list of major sites providing such libraries is presented in Table 1. These libraries have the advantage that quality filtering is usually uniformly applied and reasonably well understood, either by direct encoding of quality metrics or by reputation. However, the comprehensiveness of these libraries is limited by the data provided to the creator. Most libraries cover a few biological species only and encompass only a subset of commonly used analytical platforms and methodologies.
Table 1.
Major sites for download of peptide spectral libraries
| NIST | http://peptide.nist.gov/ |
| MassIVE | http://massive.ucsd.edu/ProteoSAFe/static/massive-kb-libraries.jsp |
| ProteomeTools | http://www.proteometools.org/index.php?id=53 |
| PRIDE Cluster | https://www.ebi.ac.uk/pride/cluster/#/libraries |
| PeptideAtlas | http://www.peptideatlas.org/speclib/ |
| SWATHAtlas | http://www.swathatlas.org/ |
| SRMAtlas | http://www.srmatlas.org/ |
| GPMDB | ftp://ftp.thegpm.org/projects/xhunter/libs/ |
| BiblioSpec | https://proteome.gs.washington.edu/software/bibliospec/v1.0/documentation/libs.html |
However, it has been shown that new big data approaches could be leveraged to build far more comprehensive community-sourced libraries57. In theory, the application of crowdsourcing efforts throughout the community could lead to a grand library, or set of libraries, that encompass all identifications achieved by the community as a whole thus far. This is in contrast to the previously-described single-source libraries that are generated by a single group, even when the source data are collected from many labs. A core feature of such a community library infrastructure would be how to handle conflicting PSMs from different groups. Such a community library has the potential to transform the field of proteomics, enabling far more sensitive, specific, and comprehensive analyses of all datasets. Yet, in practice, creating such a comprehensive community library will be very challenging to achieve.
One approach towards a comprehensive library would involve setting up a web resource for submitted identified spectra (or commonly seen but as yet unidentified spectra). All submissions will be processed and integrated into a growing community library that can be downloaded and used by everyone. Spectra produced by the same peptide ion by different instrument classes and at different collision energies54 would need to be stored separately and only aggregated when sufficiently similar. Spectra for contaminant PTMs, contaminant peptides (e.g. from a different species than claimed), and different derivatizations (e.g. isobaric labeling) would all need to be tracked appropriately. Spectral library search engines would likely only use a subset of the spectra from the community library as appropriate for the dataset being analyzed.
However, one of the greatest challenges will be maintaining a high degree of quality in the community library. Requiring only the highest quality submissions may dissuade participation. Labeling contributions as either gold, silver, or bronze based on the completeness of metadata, quality of each spectrum (e.g. as measured by SNR) and the quality of each PSM (e.g. as measured by fraction of explainable intensity and number of peaks) is one approach to allow greater inclusiveness. No clear consensus on the precise definitions of gold, silver, and bronze emerged, but in general it was felt that all spectra should have full provenance to the dataset, MS run, and original scan number. A gold spectrum should have or be a corroborating spectrum from a synthetic peptide, have corroborating spectra from a different dataset, and have corroborating spectra from the same peptide sequence but a different ion (peptidoform or charge). Spectra that achieve at least one of these things would be silver, and spectra that achieve none would be bronze. Additional numerical metrics as described above should also apply, but further work is required to set sensible thresholds for the three levels.
Complete automation would likely be required to ensure sustainability. Developing such a community library successfully would be a challenging undertaking. A pioneering example in the field of metabolomics is GNPS57. Single-source libraries have also been very successful in metabolomics and other small molecule analysis; some (such as the NIST/EPA/NIH mass spectral library) started many decades ago and are still actively maintained58. Success of library searching in metabolomics can be attributed to the fact that until recently, there has been no alternative to spectral library searching for metabolite identification59 due to inherent differences between protein and small molecule identification approaches. The number of characterized analytes and the number of biomolecules in reference libraries relevant for metabolomics is, however, usually orders of magnitude smaller than in proteomics; the large number of spectra in, for example, the NIST/EPA/NIH library is mostly due to derivatives60. In contrast to proteomics, reference substances are required to establish small molecule mass spectral libraries with confident identifications, thus generating reference spectra to put into a spectral library is far more difficult and time consuming than in proteomics, as reference substances can be very expensive or impossible to obtain. Due to the inherent differences in applications, past success in metabolomics does not ensure success for proteomics.
There was doubt amongst some participants at the Dagstuhl discussion that such a community library would become widely used. Anecdotes were related of large numbers of researchers preferring to develop their own libraries based on their own samples and instruments, even when an even more comprehensive library fully suitable to their system of study was available. Using a very large library introduces substantial challenges for proper FDR control, and testing many hypotheses that are not relevant for the current sample reduces sensitivity. It remains an unresolved issue under active research in the field whether sample-specific libraries should be preferred to comprehensive libraries, especially as it pertains to DIA analysis.
Application of spectral libraries
Spectral library searching on its own is a powerful technique, demonstrated to be more sensitive and more specific than sequence searching35, but only for peptide ions present in the reference library. In order to identify those ions that are not in the reference library, it seems logical to couple spectral library searching with sequence database searching, where the former assigns those peptide ions that have been previously identified, and the latter identifies peptide species that are not in the library merging the results of the two approaches into a single output for the user. This has been possible for many years in the Trans-Proteomic Pipeline61,62 (TPP) with iProphet63, but still is not commonly performed. Such a workflow has been recently implemented in Mascot Server, which may well increase the adoption of the approach.
The target-decoy approach for estimating the number of false positives at any selected threshold is commonly applied for sequence database searching, either by including the decoy sequences in the searched sequence database or by generating the decoys on-the-fly. There are several approaches to generating decoys, including reversing each protein sequence, generating random sequences based on the relative frequencies of amino acids in the database, reversing tryptic peptide sequences (i.e. holding the positions of lysine and arginine residues fixed and reversing between them), and scrambling the order of amino acids between lysines and arginines. Investigations into the best approach show comparable effectiveness64. An easy metric to assess the usefulness of decoys is to compute the balance between targets and decoys for zero probability identifications; the idea is that the relative ratio of targets to decoys amongst the known incorrect results should be equal to the ratio of targets to decoys in the reference database. The target-decoy approach can also be applied to spectral libraries and spectral library searching, and several ways have been used to produce decoys, for example by adding a fixed value to the precursor and/or fragment m/z, randomly assigning new m/z values to the peaks, and scrambling the letters of the peptide (except for a terminal cleavage residue) while moving the identifiable peaks around to match the scrambled sequence. A comparison of these approaches was performed by Lam et al.65. The results indicate that none of the proposed approaches truly achieve equal probability for target and decoy matches in cases of a zero-probability match. This is likely because these approaches do not produce decoy precursor/spectra that are similar enough to real spectra. The approach of scrambling the peptide sequence and moving the known peaks outperforms the other approaches, however is still somewhat biased and consistently more so than the target-decoy approaches used in sequence database searching. This may in part be due the fact that unannotated peaks are not moved and thus contribute to an incomplete prediction of what the scrambled peptide would be. Similarly, this technique becomes unavailable when libraries contain unassigned spectra since there is no sequence to scramble. Although this bias can be estimated and accounted for in the model to determine FDR, more work in this domain is needed.
Another topic of discussion was the mechanism by which two spectra are compared, typically a library spectrum and a new experimental spectrum. There are two major aspects to this issue, the algorithm used to compare the intensities and m/z values, and how to handle peaks without a match. Several broadly similar algorithms are available for comparison of spectra, most commonly a dot product, a dot product of the Nth root of intensities to reduce the influence of a few intense peaks, a cosine score, and the normalized spectral contrast angle approach66,67. Other approaches have also used probabilistic models of variation in peak intensities39 or proposed machine learning models combining multiple features into a single score38. However, perhaps a greater influence is exactly which peaks are aligned and go into the score. Exclusion of unfragmented precursor peaks, reporter ion peaks, and other non-informative peaks seems logical, but approaches where the absence of a library peak in the acquired data is not penalized could lead to false positives with seemingly high scores if only a few peaks are shared. Some approaches include a training step to calculate characteristic parameter values for each peak39,68. Indeed, when calculating similarity scores between new experimental spectra and reference spectra from synthetic peptides, it is important that all informative peaks are included, even when a peak has no counterpart. In making the decision of which peaks to use, it is important to consider the intent of the comparison38: is spectrum A equivalent to spectrum B? Is spectrum A the primary constituent in spectrum B with minor additional contamination? Is spectrum A one of many constituents in spectrum B?
Comparison of spectra generated from peptides in natural samples with spectra generated from synthetic peptides is a powerful technique for verifying that the spectrum identifications are correct, and is specifically called out in the HPP MS Data Interpretation Guidelines69. SRMAtlas, a large scale effort to develop reference spectra for a few peptides for each human protein has been completed21, and the ProteomeTools project that aims to generate synthetic peptide spectra for nearly all accessible human tryptic peptides is ongoing70. Efforts are underway71 to validate discovery of HPP missing proteins via the comparison with SRMAtlas spectra. This process could be automated such that comparison of newly proposed HPP missing protein detections could easily be checked against available synthetic peptide spectra.
Library Searching for DIA Applications
While initial interest in spectral libraries was driven by spectral library searching of DDA MS datasets, the recent expansion in interest has been driven by applications to DIA workflows. In these workflows, the precursor ion selection window is much wider; thus, the instrument co-fragments many different peptide ion species at once, thereby creating highly multiplexed fragmentation spectra. Although library-free approaches to analyzing such data continue to emerge11,63, the most common methods for analyzing DIA data involve extracting chromatograms for each spectral library fragment ion for a given peptide and determining based on their presence and co-elution whether a given peptide is in the sample. These approaches are asking a fundamentally different question to database searching; i.e. rather than trying to identify a spectrum, they are asking whether there is evidence for a peptide of interest being present in the sample. However, while the original libraries created from the DDA MS input datasets include all peaks from the peptide ion and have been shown to enable peptide identification from DIA data18, the derived libraries destined for use by DIA analysis are typically trimmed such that only the top N (where N is often 5, 6, or 10) most informative peaks are retained. It should be easy to distinguish between the primary archival libraries and the derived, trimmed versions intended for special applications. Some in the group highlighted that while 6 peaks may be sufficient to distinguish most peptides from one another72, 6 peaks may not be enough to confidently distinguish among different post-translational modification (PTM) isomers based on the same peptide sequence. This highlights the need for better encoding of metadata in spectral libraries, since the current formats do not support a uniform mechanism for encoding whether a library has been trimmed to suit a specific application and how that has been done. Also, in the case of PTM isomers, certain peaks in the fragment ion spectrum are highly informative while others are shared between isomers. Current approaches in DIA analysis of peptidoforms include annotation of fragment ions based on their capability to act as “unique ion signature” for a specific peptidoform73,74. The proposed format will need to capture this information on the fragment ion level as well.
The customary workflow for library-based DIA analysis involves the development of the reference library source from DDA input data, where most fragmentation spectra are relatively pure and FDR control is well understood. However, the emergence of library-free DIA analysis techniques with tools such as DIA-Umpire, which use co-elution profiles of precursor and fragment ions to create filtered, simplified MS/MS spectra for searching, enable the possibility of developing spectral libraries from DIA data directly. This has the potential advantage that the reference spectra are created on the same instrument under the same collision energy and selection window conditions as the eventual subsequent analysis. However, most of the Dagstuhl group had serious reservations about such approaches, primarily due to the substantial and insufficiently understood uncertainties in controlling false positives in highly multiplexed spectra when assaying with a limited number of peaks. With DIA data, there is a magnified danger of confusing a peptide ion with another peptide ion that has a similar sequence but with a different mass modification due to the large precursor selection windows employed in DIA data. Also, most approaches to spectral library generation attempt to create high quality libraries from pure compounds to reduce error rates in the library itself and further research is needed on how impurities and low-quality entries in the spectral library affect DIA analysis.
Other complicating factors for DIA analysis include accounting for the use of trimmed spectral peak lists in the initial identification, as the reliability measure attached to the library spectrum should be changed. The use of relative fragment ion intensities, as well as retention and drift (collisional cross section) time are other challenges for reliability estimation. The normalized or indexed retention time of a peptide could be valuable information for improving the confidence of an identification. However, determining retention times for decoy spectra is challenging. Current tools address this issue by ensuring that the overall distribution of retention times is equal between targets and decoys and that peptides of equal AA composition are assigned the same retention time. Retention times for decoy spectra could be estimated with retention time prediction tools, but these are usually less accurate than empirically determined values, so it is not clear how reliability estimates can be calibrated when including retention time as a factor.
Highly Similar Spectra
More broadly, the entire topic of highly similar spectra in a library generated good discussion amongst the group and is an area requiring additional research. Tools such as SpectraST35 include quality control routines that can, at the discretion of the user, prune library entries that have highly similar spectra (and precursor m/z) to another entry that is not simply a sibling peptidoform (e.g. a singly phosphorylated peptide with the phosphorylation at a different site). This can be applied under the assumption that either one of the two similar entries is misidentified, or, if they are both correct, the spectra are so similar that MS cannot effectively distinguish between the two with current technology. A better approach may be to develop more advanced tools that can assess the ambiguity75 and provide the user with probabilistic set of options (e.g., 99.9% confidence that a new spectrum is either peptide ion A or B, but distinguishing amongst those two is only 60%/40%). Clearly, further work is required, and the capture of the metadata on which choices were made for construction of the library will be important.
Gold Standard Test Dataset
A recurring theme of discussion was the need for a gold standard dataset and library that could be used in the uniform testing of various approaches and tools. None of the current set of existing reference datasets summarized at http://compms.org/resources/reference-data were deemed suitable for this purpose. The group decided that a good standard dataset would consist of one spectral library with ~10,000 entries and one mzML file with ~10,000 spectra, in which ~5000 peptide ions (but not exact spectra) were in common. Each of the 10,000 spectra in the two files should be derived from synthetic peptides (e.g. from ProteomeTools74 or SRMAtlas21), and, thus, the corresponding identities are known precisely. There should be a combination of high SNR spectra and low SNR spectra, where the low SNR spectra are derived from fragmentation near the fringes of an elution profile for which conclusive PSM evidence is available from a spectrum obtained near the peak of the same profile. A vetting process conducted by several groups to identify and discard errors in the spectrum identification list will be important to ensure a true gold standard. Efforts are underway to produce such a gold standard dataset.
Conclusion
Spectral libraries remain a substantially underutilized resource in proteomics, with the potential to vastly increase the efficiency of research. Other fields, such as metabolomics, have demonstrated the utility of spectral libraries; however, concepts from metabolomics will not always directly translate to proteomics. Future workflows will likely perform more than one stage of spectral library searching. The first stage will determine the most appropriate libraries to search and suitable parameters, a second stage would search against an extensive collection of the most suitable community libraries including identified and unidentified representative spectra derived from public datasets, and a final stage would perform sequence database searching of only the high quality spectra that remain unmatched after spectral library searching. This complex workflow should be designed to happen with minimal input from the user, and the results from all stages should be presented in a unified manner. Newly identified peptide species should be automatically added to local spectral libraries and optionally contributed to the community libraries, similarly to what is already enabled for metabolomics spectral libraries at GNPS57. Once such workflows become easier, faster, and more effective than current techniques, spectral libraries will be more widely adopted.
However, before that can happen, there are still a substantial number of challenges that must be overcome. Spectral library building, handling, and searching software must become more advanced. The cooperative development and interoperability of spectral library-using software requires a widely adopted community standard format, especially one that could encode extensive metadata about the library and its contents. The PSI is embarking on an effort to create this standard, and wide participation from the community will be a key contributing factor. All contributions are welcome via https://github.com/HUPO-PSI/SpectralLibraryFormat.
Beyond the development of the standard format, there remain many open questions in need of addressing by research in the community as described above, including how to set up community libraries, generate decoys, develop a gold reference standard, and how to compare spectra. By building a standard spectral library format, creating more advanced analysis software that capitalizes on the format, and addressing the remaining open research questions, a broad array of biomedical applications using all MS-based proteomics technologies will be enabled and accelerated.
Supplementary Material
Acknowledgements
This work was funded in part by the National Institutes of Health grants R24GM127667 (EWD), R01GM087221 (EWD), U54EB020406 (EWD), 2P41GM103484-06A1 (NB). This work was in part funded by the German Federal Ministry of Education and Research (BMBF; grant No 031L0008A and 031A535A). YPR, MW and JAV want to acknowledge funding from NIH [R24GM127667], Wellcome Trust [WT101477MA and 208391/Z/17/Z], ELIXIR and BBSRC [BB/P024599/1]. AWD would like to acknowledge BBSRC BB/M024954. HL would like to acknowledge the financial support from the Research Grants Council of the Hong Kong Special Administrative Region Government (Grant No. 16306417). NB is an Alfred P. Sloan Research Fellow.
NB was a co-founder, had an equity interest and received income from Digital Proteomics, LLC through 2017. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. Digital Proteomics was not involved in the research presented here.
Footnotes
Supporting Information
The following supporting information is available free of charge at ACS website http://pubs.acs.org
- Level 1. Metadata at the library level
- Level 2. Metadata specific to spectrum entries
- Level 3. Metadata at Peak Level
- Level 4. Metadata at Peak Interpretation Level
References
- 1.Nesvizhskii AI A Survey of Computational Methods and Error Rate Estimation Procedures for Peptide and Protein Identification in Shotgun Proteomics. J. Proteomics 2010, 73 (11), 2092–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eng JK; Searle BC; Clauser KR; Tabb DL A Face in the Crowd: Recognizing Peptides through Database Search. Mol. Cell. Proteomics MCP 2011, 10 (11), R111.009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang X; Li Y; Shao W; Lam H Understanding the Improved Sensitivity of Spectral Library Searching over Sequence Database Searching in Proteomics Data Analysis. Proteomics 2011, 11 (6), 1075–1085. [DOI] [PubMed] [Google Scholar]
- 4.Venable JD; Dong M-Q; Wohlschlegel J; Dillin A; Yates JR Automated Approach for Quantitative Analysis of Complex Peptide Mixtures from Tandem Mass Spectra. Nat. Methods 2004, 1 (1), 39–45. [DOI] [PubMed] [Google Scholar]
- 5.Silva JC; Denny R; Dorschel CA; Gorenstein M; Kass IJ; Li G-Z; McKenna T; Nold MJ; Richardson K; Young P; et al. Quantitative Proteomic Analysis by Accurate Mass Retention Time Pairs. Anal. Chem 2005, 77 (7), 2187–2200. [DOI] [PubMed] [Google Scholar]
- 6.Moseley MA; Hughes CJ; Juvvadi PR; Soderblom EJ; Lennon S; Perkins SR; Thompson JW; Steinbach WJ; Geromanos SJ; Wildgoose J; et al. Scanning Quadrupole Data Independent Acquisition - Part A. Qualitative and Quantitative Characterization. J. Proteome Res 2017. [DOI] [PubMed] [Google Scholar]
- 7.Gillet LC; Navarro P; Tate S; Röst H; Selevsek N; Reiter L; Bonner R; Aebersold R Targeted Data Extraction of the MS/MS Spectra Generated by Data-Independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics MCP 2012, 11 (6), O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egertson JD; Kuehn A; Merrihew GE; Bateman NW; MacLean BX; Ting YS; Canterbury JD; Marsh DM; Kellmann M; Zabrouskov V; et al. Multiplexed MS/MS for Improved Data-Independent Acquisition. Nat. Methods 2013, 10 (8), 744–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li G-Z; Vissers JPC; Silva JC; Golick D; Gorenstein MV; Geromanos SJ Database Searching and Accounting of Multiplexed Precursor and Product Ion Spectra from the Data Independent Analysis of Simple and Complex Peptide Mixtures. Proteomics 2009, 9 (6), 1696–1719. [DOI] [PubMed] [Google Scholar]
- 10.Tsou C-C; Avtonomov D; Larsen B; Tucholska M; Choi H; Gingras A-C; Nesvizhskii AI DIA-Umpire: Comprehensive Computational Framework for Data-Independent Acquisition Proteomics. Nat. Methods 2015, 12 (3), 258–264, 7 p following 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsou C-C; Tsai C-F; Teo GC; Chen Y-J; Nesvizhskii AI Untargeted, Spectral Library-Free Analysis of Data-Independent Acquisition Proteomics Data Generated Using Orbitrap Mass Spectrometers. Proteomics 2016, 16 (15–16), 2257–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ting YS; Egertson JD; Bollinger JG; Searle BC; Payne SH; Noble WS; MacCoss MJ PECAN: Library-Free Peptide Detection for Data-Independent Acquisition Tandem Mass Spectrometry Data. Nat. Methods 2017, 14 (9), 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Röst HL; Aebersold R; Schubert OT Automated SWATH Data Analysis Using Targeted Extraction of Ion Chromatograms. Methods Mol. Biol. Clifton NJ 2017, 1550, 289–307. [DOI] [PubMed] [Google Scholar]
- 14.MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinforma. Oxf. Engl 2010, 26 (7), 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heaven MR; Funk AJ; Cobbs AL; Haffey WD; Norris JL; McCullumsmith RE; Greis KD Systematic Evaluation of Data-Independent Acquisition for Sensitive and Reproducible Proteomics-a Prototype Design for a Single Injection Assay. J. Mass Spectrom. JMS 2016, 51 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruderer R; Bernhardt OM; Gandhi T; Miladinović SM; Cheng L-Y; Messner S; Ehrenberger T; Zanotelli V; Butscheid Y; Escher C; et al. Extending the Limits of Quantitative Proteome Profiling with Data-Independent Acquisition and Application to Acetaminophen-Treated Three-Dimensional Liver Microtissues. Mol. Cell. Proteomics MCP 2015, 14 (5), 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberger G; Koh CC; Guo T; Röst HL; Kouvonen P; Collins BC; Heusel M; Liu Y; Caron E; Vichalkovski A; et al. A Repository of Assays to Quantify 10,000 Human Proteins by SWATH-MS. Sci. Data 2014, 1, 140031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J; Tucholska M; Knight JDR; Lambert J-P; Tate S; Larsen B; Gingras A-C; Bandeira N MSPLIT-DIA: Sensitive Peptide Identification for Data-Independent Acquisition. Nat. Methods 2015, 12 (12), 1106–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuster B; Schirle M; Mallick P; Aebersold R Scoring Proteomes with Proteotypic Peptide Probes. Nat. Rev. Mol. Cell Biol 2005, 6 (7), 577–583. [DOI] [PubMed] [Google Scholar]
- 20.Mallick P; Schirle M; Chen SS; Flory MR; Lee H; Martin D; Ranish J; Raught B; Schmitt R; Werner T; et al. Computational Prediction of Proteotypic Peptides for Quantitative Proteomics. Nat. Biotechnol 2007, 25 (1), 125–131. [DOI] [PubMed] [Google Scholar]
- 21.Kusebauch U; Campbell DS; Deutsch EW; Chu CS; Spicer DA; Brusniak M-Y; Slagel J; Sun Z; Stevens J; Grimes B; et al. Human SRMAtlas: A Resource of Targeted Assays to Quantify the Complete Human Proteome. Cell 2016, 166 (3), 766–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desiere F; Deutsch EW; Nesvizhskii AI; Mallick P; King NL; Eng JK; Aderem A; Boyle R; Brunner E; Donohoe S; et al. Integration with the Human Genome of Peptide Sequences Obtained by High-Throughput Mass Spectrometry. Genome Biol 2005, 6 (1), R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desiere F; Deutsch EW; King NL; Nesvizhskii AI; Mallick P; Eng J; Chen S; Eddes J; Loevenich SN; Aebersold R The PeptideAtlas Project. Nucleic Acids Res 2006, 34 (Database issue), D655–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deutsch EW; Sun Z; Campbell D; Kusebauch U; Chu CS; Mendoza L; Shteynberg D; Omenn GS; Moritz RL State of the Human Proteome in 2014/2015 As Viewed through PeptideAtlas: Enhancing Accuracy and Coverage through the AtlasProphet. J. Proteome Res 2015, 14 (9), 3461–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martens L; Hermjakob H; Jones P; Adamski M; Taylor C; States D; Gevaert K; Vandekerckhove J; Apweiler R PRIDE: The Proteomics Identifications Database. Proteomics 2005, 5 (13), 3537–3545. [DOI] [PubMed] [Google Scholar]
- 26.Vizcaíno JA; Csordas A; Del-Toro N; Dianes JA; Griss J; Lavidas I; Mayer G; Perez-Riverol Y; Reisinger F; Ternent T; et al. 2016 Update of the PRIDE Database and Its Related Tools. Nucleic Acids Res 2016, 44 (D1), D447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Craig R; Cortens JP; Beavis RC Open Source System for Analyzing, Validating, and Storing Protein Identification Data. J. Proteome Res 2004, 3 (6), 1234–1242. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt T; Samaras P; Frejno M; Gessulat S; Barnert M; Kienegger H; Krcmar H; Schlegl J; Ehrlich H-C; Aiche S; et al. ProteomicsDB. Nucleic Acids Res 2018, 46 (D1), D1271–D1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griss J Spectral Library Searching in Proteomics. Proteomics 2016, 16 (5), 729–740. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z; Burke M; Mirokhin YA; Tchekhovskoi DV; Markey SP; Yu W; Chaerkady R; Hess S; Stein SE Reverse and Random Decoy Methods for False Discovery Rate Estimation in High Mass Accuracy Peptide Spectral Library Searches. J. Proteome Res 2018, 17 (2), 846–857. [DOI] [PubMed] [Google Scholar]
- 31.Burke MC; Mirokhin YA; Tchekhovskoi DV; Markey SP; Heidbrink Thompson J; Larkin C; Stein SE The Hybrid Search: A Mass Spectral Library Search Method for Discovery of Modifications in Proteomics. J. Proteome Res 2017, 16 (5), 1924–1935. [DOI] [PubMed] [Google Scholar]
- 32.Cho J-Y; Lee H-J; Jeong S-K; Paik Y-K Epsilon-Q: An Automated Analyzer Interface for Mass Spectral Library Search and Label-Free Protein Quantification. J. Proteome Res 2017, 16 (12), 4435–4445. [DOI] [PubMed] [Google Scholar]
- 33.Orchard S; Hermjakob H; Apweiler R The Proteomics Standards Initiative. Proteomics 2003, 3 (7), 1374–1376. [DOI] [PubMed] [Google Scholar]
- 34.Deutsch EW; Orchard S; Binz P-A; Bittremieux W; Eisenacher M; Hermjakob H; Kawano S; Lam H; Mayer G; Menschaert G; et al. Proteomics Standards Initiative: Fifteen Years of Progress and Future Work. J. Proteome Res 2017, 16 (12), 4288–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam H; Deutsch EW; Eddes JS; Eng JK; King N; Stein SE; Aebersold R Development and Validation of a Spectral Library Searching Method for Peptide Identification from MS/MS. Proteomics 2007, 7 (5), 655–667. [DOI] [PubMed] [Google Scholar]
- 36.Craig R; Cortens JC; Fenyo D; Beavis RC Using Annotated Peptide Mass Spectrum Libraries for Protein Identification. J. Proteome Res 2006, 5 (8), 1843–1849. [DOI] [PubMed] [Google Scholar]
- 37.Frewen BE; Merrihew GE; Wu CC; Noble WS; MacCoss MJ Analysis of Peptide MS/MS Spectra from Large-Scale Proteomics Experiments Using Spectrum Libraries. Anal. Chem 2006, 78 (16), 5678–5684. [DOI] [PubMed] [Google Scholar]
- 38.Wang J; Pérez-Santiago J; Katz JE; Mallick P; Bandeira N Peptide Identification from Mixture Tandem Mass Spectra. Mol. Cell. Proteomics MCP 2010, 9 (7), 1476–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M; Bandeira N Spectral Library Generating Function for Assessing Spectrum-Spectrum Match Significance. J. Proteome Res 2013, 12 (9), 3944–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanash S; Celis JE The Human Proteome Organization: A Mission to Advance Proteome Knowledge. Mol. Cell. Proteomics MCP 2002, 1 (6), 413–414. [DOI] [PubMed] [Google Scholar]
- 41.Deutsch EW; Albar JP; Binz P-A; Eisenacher M; Jones AR; Mayer G; Omenn GS; Orchard S; Vizcaíno JA; Hermjakob H Development of Data Representation Standards by the Human Proteome Organization Proteomics Standards Initiative. J. Am. Med. Inform. Assoc. JAMIA 2015, 22 (3), 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frank AM; Monroe ME; Shah AR; Carver JJ; Bandeira N; Moore RJ; Anderson GA; Smith RD; Pevzner PA Spectral Archives: Extending Spectral Libraries to Analyze Both Identified and Unidentified Spectra. Nat. Methods 2011, 8 (7), 587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frank AM; Bandeira N; Shen Z; Tanner S; Briggs SP; Smith RD; Pevzner PA Clustering Millions of Tandem Mass Spectra. J. Proteome Res 2008, 7 (1), 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griss J; Perez-Riverol Y; Lewis S; Tabb DL; Dianes JA; Del-Toro N; Rurik M; Walzer MW; Kohlbacher O; Hermjakob H; et al. Recognizing Millions of Consistently Unidentified Spectra across Hundreds of Shotgun Proteomics Datasets. Nat. Methods 2016, 13 (8), 651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry-Based Proteomics. Nat. Methods 2017, 14 (5), 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yen C-Y; Meyer-Arendt K; Eichelberger B; Sun S; Houel S; Old WM; Knight R; Ahn NG; Hunter LE; Resing KA A Simulated MS/MS Library for Spectrum-to-Spectrum Searching in Large Scale Identification of Proteins. Mol. Cell. Proteomics MCP 2009, 8 (4), 857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yen C-Y; Houel S; Ahn NG; Old WM Spectrum-to-Spectrum Searching Using a Proteome-Wide Spectral Library. Mol. Cell. Proteomics MCP 2011, 10 (7), M111.007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Degroeve S; Martens L MS2PIP: A Tool for MS/MS Peak Intensity Prediction. Bioinforma. Oxf. Engl 2013, 29 (24), 3199–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Z; Yang X; Mirokhin YA; Tchekhovskoi DV; Ji W; Markey SP; Roth J; Neta P; Hizal DB; Bowen MA; et al. Interconversion of Peptide Mass Spectral Libraries Derivatized with ITRAQ or TMT Labels. J. Proteome Res 2016, 15 (9), 3180–3187. [DOI] [PubMed] [Google Scholar]
- 50.Martens L; Chambers M; Sturm M; Kessner D; Levander F; Shofstahl J; Tang WH; Römpp A; Neumann S; Pizarro AD; et al. MzML--a Community Standard for Mass Spectrometry Data. Mol. Cell. Proteomics MCP 2011, 10 (1), R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilhelm M; Kirchner M; Steen JAJ; Steen H Mz5: Space- and Time-Efficient Storage of Mass Spectrometry Data Sets. Mol. Cell. Proteomics MCP 2012, 11 (1), O111.011379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bouyssié D; Dubois M; Nasso S; Gonzalez de Peredo A; Burlet-Schiltz O; Aebersold R; Monsarrat B MzDB: A File Format Using Multiple Indexing Strategies for the Efficient Analysis of Large LC-MS/MS and SWATH-MS Data Sets. Mol. Cell. Proteomics MCP 2015, 14 (3), 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Handy K; Rosen J; Gillan A; Smith R Fast, Axis-Agnostic, Dynamically Summarized Storage and Retrieval for Mass Spectrometry Data. PloS One 2017, 12 (11), e0188059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zolg DP; Wilhelm M; Yu P; Knaute T; Zerweck J; Wenschuh H; Reimer U; Schnatbaum K; Kuster B PROCAL: A Set of 40 Peptide Standards for Retention Time Indexing, Column Performance Monitoring, and Collision Energy Calibration. Proteomics 2017, 17 (21). [DOI] [PubMed] [Google Scholar]
- 55.Lam H; Deutsch EW; Eddes JS; Eng JK; Stein SE; Aebersold R Building Consensus Spectral Libraries for Peptide Identification in Proteomics. Nat. Methods 2008, 5 (10), 873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wohlgemuth G; Mehta SS; Mejia RF; Neumann S; Pedrosa D; Pluskal T; Schymanski EL; Willighagen EL; Wilson M; Wishart DS; et al. SPLASH, a Hashed Identifier for Mass Spectra. Nat. Biotechnol 2016, 34 (11), 1099–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang M; Carver JJ; Phelan VV; Sanchez LM; Garg N; Peng Y; Nguyen DD; Watrous J; Kapono CA; Luzzatto-Knaan T; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol 2016, 34 (8), 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stein S Mass Spectral Reference Libraries: An Ever-Expanding Resource for Chemical Identification. Anal. Chem 2012, 84 (17), 7274–7282. [DOI] [PubMed] [Google Scholar]
- 59.Hufsky F; Böcker S Mining Molecular Structure Databases: Identification of Small Molecules Based on Fragmentation Mass Spectrometry Data. Mass Spectrom. Rev 2017, 36 (5), 624–633. [DOI] [PubMed] [Google Scholar]
- 60.Vinaixa M; Schymanski EL; Neumann S; Navarro M; Salek RM; Yanes O Mass Spectral Databases for LC/MS- and GC/MS-Based Metabolomics: State of the Field and Future Prospects. TrAC Trends Anal. Chem 2016, 78, 23–35. [Google Scholar]
- 61.Keller A; Eng J; Zhang N; Li X; Aebersold R A Uniform Proteomics MS/MS Analysis Platform Utilizing Open XML File Formats. Mol. Syst. Biol 2005, 1, 2005.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deutsch EW; Mendoza L; Shteynberg D; Slagel J; Sun Z; Moritz RL Trans-Proteomic Pipeline, a Standardized Data Processing Pipeline for Large-Scale Reproducible Proteomics Informatics. Proteomics Clin. Appl 2015, 9 (7–8), 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shteynberg D; Deutsch EW; Lam H; Eng JK; Sun Z; Tasman N; Mendoza L; Moritz RL; Aebersold R; Nesvizhskii AI IProphet: Multi-Level Integrative Analysis of Shotgun Proteomic Data Improves Peptide and Protein Identification Rates and Error Estimates. Mol. Cell. Proteomics MCP 2011, 10 (12), M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jeong K; Kim S; Bandeira N False Discovery Rates in Spectral Identification. BMC Bioinformatics 2012, 13 Suppl 16, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lam H; Deutsch EW; Aebersold R Artificial Decoy Spectral Libraries for False Discovery Rate Estimation in Spectral Library Searching in Proteomics. J. Proteome Res 2010, 9 (1), 605–610. [DOI] [PubMed] [Google Scholar]
- 66.Stein SE; Scott DR Optimization and Testing of Mass Spectral Library Search Algorithms for Compound Identification. J. Am. Soc. Mass Spectrom 1994, 5 (9), 859–866. [DOI] [PubMed] [Google Scholar]
- 67.Toprak UH; Gillet LC; Maiolica A; Navarro P; Leitner A; Aebersold R Conserved Peptide Fragmentation as a Benchmarking Tool for Mass Spectrometers and a Discriminating Feature for Targeted Proteomics. Mol. Cell. Proteomics MCP 2014, 13 (8), 2056–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mylonas R; Mauron Y; Masselot A; Binz P-A; Budin N; Fathi M; Viette V; Hochstrasser DF; Lisacek F X-Rank: A Robust Algorithm for Small Molecule Identification Using Tandem Mass Spectrometry. Anal. Chem 2009, 81 (18), 7604–7610. [DOI] [PubMed] [Google Scholar]
- 69.Deutsch EW; Overall CM; Van Eyk JE; Baker MS; Paik Y-K; Weintraub ST; Lane L; Martens L; Vandenbrouck Y; Kusebauch U; et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 2.1. J. Proteome Res 2016, 15 (11), 3961–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zolg DP; Wilhelm M; Schnatbaum K; Zerweck J; Knaute T; Delanghe B; Bailey DJ; Gessulat S; Ehrlich H-C; Weininger M; et al. Building ProteomeTools Based on a Complete Synthetic Human Proteome. Nat. Methods 2017, 14 (3), 259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Elguoshy A; Hirao Y; Xu B; Saito S; Quadery AF; Yamamoto K; Mitsui T; Yamamoto T; Chromosome X Project Team of JProS. Identification and Validation of Human Missing Proteins and Peptides in Public Proteome Databases: Data Mining Strategy. J. Proteome Res 2017, 16 (12), 4403–4414. [DOI] [PubMed] [Google Scholar]
- 72.Röst H; Malmström L; Aebersold R A Computational Tool to Detect and Avoid Redundancy in Selected Reaction Monitoring. Mol. Cell. Proteomics MCP 2012, 11 (8), 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sherman J; McKay MJ; Ashman K; Molloy MP Unique Ion Signature Mass Spectrometry, a Deterministic Method to Assign Peptide Identity. Mol. Cell. Proteomics MCP 2009, 8 (9), 2051–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rosenberger G; Liu Y; Röst HL; Ludwig C; Buil A; Bensimon A; Soste M; Spector TD; Dermitzakis ET; Collins BC; et al. Inference and Quantification of Peptidoforms in Large Sample Cohorts by SWATH-MS. Nat. Biotechnol 2017, 35 (8), 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu Y; Orre LM; Johansson HJ; Huss M; Boekel J; Vesterlund M; Fernandez-Woodbridge A; Branca RMM; Lehtiö J Discovery of Coding Regions in the Human Genome by Integrated Proteogenomics Analysis Workflow. Nat. Commun 2018, 9 (1), 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.