

Abstract
Intratumoral infusion of a nonpathogenic replication-competent recombinant polio-rhinovirus chimera for recurrent glioblastoma demonstrates safety and promising preliminary treatment responses.
Glioblastoma (GBM) is the highest-grade glial tumor and most frequent malignant primary brain tumor in adults. GBM prognosis remains dismal with a median overall survival of approximately 15 months, in part due to its widely infiltrative nature, markedly heterogeneous subpopulations, refuge behind the blood–brain barrier, and tenacious capacity for suppressing the immune system both locally and systemically. The immune microenvironment of GBMs is distinctively ‘cold’ with multidimensional immunosuppressive mechanisms that traditionally make GBM a poor candidate for immunotherapy, of which the aim is to activate the host immune system against the tumor1. In order to combat these multifaceted immunosuppressive hallmarks of GBM, several avenues of immune modulation are under active clinical investigation. These include antigen-specific vaccination, immune checkpoint blockade, adoptive cell transfer, chimeric antigen receptor T cell therapy, and virotherapy. The overall goals of these efforts are twofold: to coax lymphocytes into the GBM microenvironment and to unleash their immune effector processes. One such promising novel immunotherapeutic modality was recently highlighted by Desjardins et al. 2 in the New England Journal of Medicine, who report results of a phase 1 clinical trial of a nonpathogenic, replication-competent, recombinant polio-rhinovirus chimera (PVSRIPO) oncolytic virotherapy for recurrent GBM.
In virotherapy, viral biology is exploited in order to preferentially infect tumor cells with viruses, thereby enabling direct lytic killing by the immune system (i.e., by oncolytic viruses) and/or anticancer gene delivery to tumor cells. The study using PVSRIPO represents one in a growing series of promising GBM virotherapy trials that are the culmination of decades of preclinical and clinical development3. These studies indicate that the success of oncolytic virotherapy depends on myriad factors, including pathogenicity of the virus, its ability to stimulate an immune response, its amenability for transgene editing, its tumor tropism—the selective replication of virus in tumor cells, but not neighboring normal cells—and the accessibility of the tumor.
Tumor tropism of the virus can be achieved by modifications that restrict viral attachment and replication to rapidly dividing tumor cells. In the case of PVSRIPO, its nonpathogenic tropism for GBM cells stems from the substitution by Gromeier et al.4 of the cell-type-specific internal ribosome entry site of the live-attenuated, Sabin type 1 poliovirus with that of the human rhinovirus type 2, resulting in neuron-specific functional deficits during PVSRIPO translation3. In addition, PVSRIPO has an innate preference for GBM cells because they frequently expressed poliovirus’s cell surface adhesion receptor CD155 (refs 4,5). Together, these mechanisms restrict PVSRIPO viral translation to mitotically active GBM cells and prevent neurovirulence, including paralytic poliomyelitis.
In this study, 61 consecutive adult patients with a single focus of supratentorial, recurrent GBMs received an intratumoral infusion of PVSRIPO in a single-arm, nonrandomized fashion at one study center. Of these patients, 19% experienced a PVSRIPO-associated adverse event of grade 3 or higher (i.e., severe, life-threatening, or death-related), despite a dose reduction. After a median follow-up of 27.6 months, an estimated 21% of patients formed a ‘long-tail’ plateau of survivors that was observed throughout 2 and 3 years on survival plots (Fig. 1), compared to <4% among historical controls. Within this context, the long-tail survival results of this study lend cautious optimism but also highlight many crucial unanswered questions related to virotherapy: namely, what are the characteristics of the long-tail survivors; what is distinctive about their underlying biology; and do their responses vary based on GBM molecular subtype?
Fig. 1 |. treatment of GBM with PVSriPO results in extended survival.
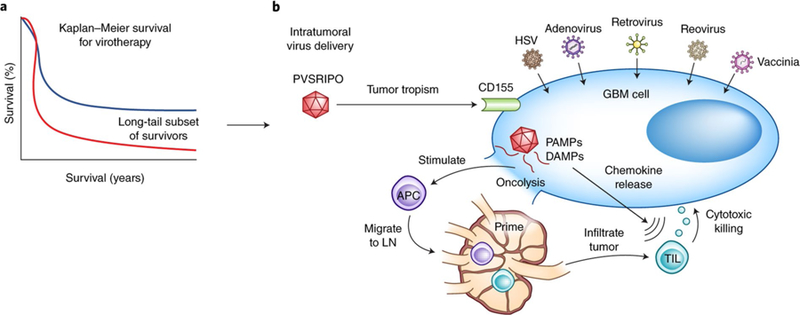
a, Many oncolytic virotherapy trials for GBM, including the one by Desjardins et al.2 using the modified polio virus PVSRIPO, demonstrate a long-tail subset of survivors beyond 2 years (blue line) not seen in historical controls (red line). b, The success of PVSRIPO as seen in the trial by Desjardins et al.2 may, in addition to direct oncolysis, be the result of viral-mediated secondary immune responses. Following intratumoral delivery of virotherapy, viral modifications permit selective GBM cell tropism (e.g., CD155 targeting by PVSRIPO). The subsequent direct viral-mediated oncolysis of tumor cells results in the release of pathogen-associated molecular patterns (PAMPs) and dying cell damage-associated molecular patterns (DAMPs) that are sensed by pattern recognition receptors on antiviral APCs, causing the APCs to mature, release proinflammatory cytokines (for example, interleukin (IL)-12, IL-1β, IL-6, and tumor necrosis factor), and migrate to draining lymph nodes (LN) for the priming of adaptive lymphocytic immune responses. Intracellular PAMP signaling is also thought to lead to upregulation of type 1 interferon pathways and chemokine release (for example, CXCL9 and CXCL10), which promote recruitment of TILs into the GBM microenvironment where they may assist with antitumoral cytotoxicity. In addition to PVSRIPO (NCT02986178*, NCT03043391*), there are many currently open virotherapy clinical trials for high grade gliomas, including several with FDA Breakthrough Therapy/Fast Track status (*) with HSV (NCT02457845, NCT03152318, NCT02062827), adenovirus (NCTs NCT02798406*, NCT03178032*, NCT03072134, NCT01811992, NCT02026271, NCT03576612), retrovirus (NCT02414165*), reovirus (NCT02444546), and vaccinia (NCT03294486) virotherapies.
The PVSRIPO trial, as historically observed among many GBM virotherapy trials, demonstrates a long tail of survivors throughout 2 years, although such responses have rarely persisted beyond this landmark6,7. A granular analysis of the ‘long-tail’ survivors with PVSRIPO is not reported in this study; however, our review of the supplemental patient characteristics suggests that the survival benefit was limited to those with known favorable GBM prognostic variables, such as MGMT promoter methylation or smaller tumors. Additionally, 56% of the patients receiving PVSRIPO also received the angiogenesis inhibitor bevacizumab to treat symptomatic peritumoral edema, which may have confounded radiographic response assessment because bevacizumab can also decrease contrast uptake on brain magnetic resonance imaging (MRI). Follow-up survival, as well as immunologic and biologic correlative results from the PVSRIPO and other virotherapy trials, will be critical in helping to answer these essential questions.
The current study does not provide further mechanistic insights into the therapeutic benefit of PVSRIPO. Evidence from the existing literature gathered in vitro and from preclinical models support the bifunctional potential of virotherapy to selectively target tumor cells for primary lytic killing and to trigger native, viral-induced immunity for antitumoral therapeutic purposes (Fig. 1). The preliminary negative results of the Checkmate 143 (NCT02017717) trial of immune checkpoint PD-1 blockade in relapsed GBM—seemingly due to the activation of immunosuppressed tumor-infiltrating lymphocytes (TILs) without an accompanying increased recruitment of TILs—underscores the need for immunotherapeutic approaches that both recruit and activate TILs8. Toward this end, encouraging results have been observed in the preliminary results of trials in advanced melanoma (NCT01740297, NCT02263508), demonstrating that combination of an oncolytic HSV1 virus (T-VEC, talimogene laherparepvec) with anti-PD-1 therapy can enhance therapeutic benefit because virotherapy upregulates immunosuppressive PD-1 signaling on CD8+ TILs9,10. Also of high interest is an ongoing trail combining adenoviral vectors with immune checkpoint blockade for GBM (NCT03576612). Furthermore, vaccination strategies directed against neoantigens (i.e., new peptides formed as a result of tumor-specific mutations), recently tested in trials for advanced melanoma and GBM11,12, could putatively facilitate the priming of the viral-induced TILs against a spectrum of GBM’s heterogeneous subclones.
Interestingly, CD155 is also expressed on antigen-presenting cells (APCs), leading to chronic and sublethal infection of APCs by PVSRIPO, which may foster priming of T cells against virally infected tumor cells5. Additional important considerations for the design of combination immunotherapies for GBM will mostly likely include: substitute strategies for or minimal dosing of immunosuppressive corticosteroids, which are widely used to alleviate symptomatic peritumoral edema; careful integration with cytotoxic chemotherapies, which can cause severe lymphopenias; delivery methods that bypass the blood–brain barrier; and stratification by GBM molecular subtypes to facilitate extrapolation of trial results to the clinic.
Across the virotherapy clinical trials for GBM, there also have been some, hearteningly, long-tail subsets of patients that demonstrate durable survival for at least 2 years3,6,7. The encouraging PVSRIPO results underscore the need to answer critical questions for GBM and for patients with other cancers, including what determines long-lasting treatment responses for virotherapies and whether oncolytic virotherapy can be combined with other therapies to expand benefit to a wider proportion of patients.
Footnotes
Competing interests
D.A.R. reports receiving honoraria, advisory, and consultant fees from Abbvie; Agenus; Bristol-Myers Squibb; Celldex; EMD Serono; Genentech/Roche; Inovio; Merck; Merck KGaA; Monteris; Novocure; Oncorus; Oxigene; Regeneron; Stemline; and Taiho Oncology, Inc. E.A.C. reports receiving consultant and advisory fees from Advantagene and Dnatrix. C.J.W. is cofounder of Neon Therapeutics and a member of its scientific advisory board.All other authors declare that they have no competing interests.
References
- 1.Lim M, Xia Y, Bettegowda C & Weller M Nat. Rev. Clin. Oncol 15, 422–442 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Desjardins A et al. N. Engl. J. Med 379, 150–161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawler SE, Speranza M-C, Cho C-F & Chiocca EA JAMA Oncol 3, 841–849 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Gromeier M, Alexander L & Wimmer E Proc. Natl Acad. Sci. USA 93, 2370–2375 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown MC et al. Sci. Transl. Med 9, eaan4220 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trask TW et al. Mol. Ther 1, 195–203 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Wheeler LA et al. Neuro-oncol 18, 1137–1145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reardon DA et al. Neuro-oncol 19, abstr. iii21 (2017). [Google Scholar]
- 9.Ribas A et al. Cell 170, 1109–1119.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long GV et al. J Clin. Oncol 34, abstr. TPS9598 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Ott PA et al. Nature 547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keskin DB et al. Cancer Res 78, abstr. 5631 (2018). [Google Scholar]