

Abstract
Phosphate is essential for skeletal mineralization, and its chronic deficiency leads to rickets and osteomalacia. Skeletal mineralization starts in matrix vesicles (MVs) derived from the plasma membrane of osteoblasts and chondrocytes. MVs contain high activity of tissue non-specific alkaline phosphatase (TNSALP), which hydrolyzes phosphoric esters such as pyrophosphates (PPi) to produce inorganic orthophosphates (Pi). Extracellular Pi in the skeleton is taken up by MVs through type III sodium/phosphate (Na+/Pi) cotransporters and forms hydroxyapatite. In addition to its roles in MV-mediated skeletal mineralization, accumulating evidence has revealed that extracellular Pi evokes signal transduction and regulates cellular function. Pi induces apoptosis of hypertrophic chondrocytes, which is a critical step for endochondral ossification. Extracellular Pi also regulates the expression of various genes including those related to proliferation, differentiation, and mineralization. In vitro cell studies have demonstrated that an elevation in extracellular Pi level leads to the activation of fibroblast growth factor receptor (FGFR), Raf/MEK (mitogen-activated protein kinase/ERK kinase)/ERK (extracellular signal-regulated kinase) pathway, where the type III Na+/Pi cotransporter PiT-1 may be involved. Responsiveness of skeletal cells to extracellular Pi suggests their ability to sense and adapt to an alteration in Pi availability in their environment. Involvement of FGFR in the Pi-evoked signal transduction is interesting because enhanced FGFR signaling in osteoblasts/osteocytes might be responsible for the overproduction of FGF23, a key molecule in phosphate homeostasis, in a mouse model for human X-linked hypophosphatemic rickets (XLH). Impaired Pi sensing may be a pathogenesis of XLH, which needs to be clarified in future.
Keywords: phosphate, mineralization, matrix vesicle, osteoblast/osteocyte, rickets, sensing
Introduction
Phosphorus mediates almost all biological processes including composition of cell membrane, maintenance and inheritance of genetic materials as nucleic acids, energy metabolism, and regulation of proteins by phosphorylation/dephosphorylation, as well as skeletal mineralization in vertebrates (1). In human adult body, about 90% of total phosphorus is stored in bone as hydroxyapatite (calcium-phosphate) crystals (2). Most of the remaining phosphorus is distributed in soft tissues, and phosphate is predominantly an intracellular ion. Less than 1% of phosphorus exists in extracellular fluid (2), and phosphorus in serum mostly occurs as inorganic phosphate (Pi) such as and H2, and the former corresponds to 80% at physiological pH (3). Although serum Pi level is influenced by age, diet, and pH (1), its abnormality may lead to undesirable consequences.
Since phosphate is an essential constituent of hydroxyapatite, its chronic deficiency or wasting leads to impaired skeletal mineralization characteristic to rickets, and osteomalacia. In addition to its role in hydroxyapatite formation, Pi also induces apoptosis of hypertrophic chondrocytes as a direct effect on cells (4–8). Moreover, extracellular Pi triggers signal transduction to regulate gene expression (9–13).
In this article, after overviewing the mechanism of phosphate homeostasis and hyperphosphatemic and hypophosphatemic disorders, we will describe the versatile roles of phosphate in the skeleton.
Phosphate Homeostasis in Mammals
Phosphate Balance
In mammals, phosphate homeostasis as a total body is maintained by influx and efflux via the intestines, kidneys, bone, and soft tissue. In growing children, the phosphate balance should be positive to allow accrual of phosphate in the skeletons and soft tissues. To meet the needs of Pi for the growing skeleton and soft tissues, serum Pi levels are maintained higher in children than in adults (14). This is contrast to serum calcium, which is kept constant. Since intracellular concentrations of Pi are higher than its extracellular concentrations, Pi is taken up into cells by active transport through sodium/phosphate (Na+/Pi) co-transporters (15). Although the mechanism for the age-dependent change in serum Pi levels remains unclear, we speculate that the sensing of Pi and the set point of optimal Pi levels might be influenced by skeletal growth and maturation.
Intestinal Absorption of Phosphate
Dietary phosphate is absorbed in the small intestine by a passive, paracellular diffusion, and an active, transcellular transport of Pi (16). The latter is mediated by type IIb Na+/Pi co-transporter (NaPi-IIb) localized in the apical membrane of the small intestine epithelial cells. The intestinal expression of NaPi-IIb is increased by low dietary intake of phosphate and 1,25-dihydroxyvitamin D (1,25(OH)2D), an active metabolite of vitamin D (17). Dietary deficiency of calcium is common because calcium content is relatively low in most foods (18). On the other hand, dietary deficiency of phosphate is rare, because all foods are derived from cells consisting of high amount of phosphate.
Renal Reabsorption of Phosphate
Pi is excreted from the kidneys. After filtered by the glomeruli, majority of Pi is reabsorbed by type IIa and IIc Na+/Pi co-transporters (NaPi-IIa and NaPi-IIc) predominantly expressed in proximal tubules (19). Loss-of function mutations in the SLC34A3 gene encoding NaPi-IIc cause hereditary hypophosphatemic rickets with hypercalciuria, which is characterized by hypophosphatemia due to an increased urinary loss of phosphate (20). The Pi-transport activity of NaPi-IIa and NaPi-IIc in the proximal tubules is determined by their protein amounts in the brush border membrane (BBM).
Parathyroid Hormone (PTH) and Fibroblast Growth Factor 23 (FGF23)
The amount of NaPi-IIa and NaPi-IIc in the BBM is regulated both transcriptionally and post-transcriptionally through protein synthesis, degradation and subcellular localization, and is regulated by several hormones such as parathyroid hormone (PTH) (21, 22) and fibroblast growth factor 23 (FGF23) (23–26) as well as dietary phosphate intake (27). PTH causes a rapid decrease of NaPi-IIa protein on the BBM (21, 22). The amount of NaPi-IIc on the BBM is also reduced by PTH, although it takes longer time (28, 29). FGF23 is mainly produced by the osteoblasts and osteocytes and exerts its effects on distant organs such as the kidneys in an endocrine manner (30). FGF23 requires αKlotho as a co-receptor to evoke signals through FGF receptors (FGFRs) at physiological concentrations (31, 32). In the kidneys, FGF23 decreases the expression of NaPi-IIa and NaPi-IIc to increase the renal Pi excretion (23–26). In addition, FGF23 reduces the production of 1,25(OH)2D by suppressing the expression of vitamin D 1α-hydroxylase and inducing that of 24-hydroxylase, which leads to the decreased Pi absorption in the intestine (24, 33). Considering that FGF23 is unable to normalize serum Pi levels in hyperparathyroidism and that inactivating mutations of FGF23 cause hyperphosphatemia despite the presence of PTH (34), both hormones appear to be necessary to maintain Pi homeostasis.
Type III Na+/Pi Co-transporters
Type III Na+/Pi co-transporters include PiT-1 and PiT-2, which are encoded by SLC20A1 and SLC20A2 genes, respectively, in humans (35). They are expressed in a broad range of tissues with different amounts, and PiT-2 is suggested to have a role in renal Pi reabsorption. Inactivating mutations in SLC20A2 are responsible for familial idiopathic basal ganglia calcification (IBGC), a disease characterized by vascular deposits of calcium/phosphate in the basal ganglia of the brain (36, 37). Thus, PiT-2 is likely to be involved in the maintenance of the physiological Pi level in cerebrospinal fluid. Recently, xenotropic and polytropic retrovirus receptor 1 (XPR1) has been shown to mediate Na+-independent Pi export from cells in mammals, and its inactivating mutations also cause IBGC (38). Regarding the type I Na+/Pi co-transporters, their physiological role has been shown to be the transport of organic anions rather than the Pi transport (39–41).
Clinical Symptoms of Hyperphosphatemia and Hypophosphatemia
Hyperphosphatemia is associated with reduced renal Pi excretion or an excessive phosphate load. It causes ectopic calcification, which may lead to organ failure, gastroenteral bleeding, skin itchiness, keratitis, and tumoral calcinosis (42).
Hypophosphatemia is caused by insufficient intestinal Pi absorption, renal Pi wasting, or shift of Pi into cells (43). Chronic hypophosphatemia is often associated with renal Pi wasting diseases and leads to rickets/osteomalacia (43). Acute hypophosphatemia is associated with respiratory alkalosis, refeeding, diabetic ketoacidosis, malnutrition, and alcoholism (43). Since hypophosphatemia can be caused by transcellular shift of Pi into cells, low serum Pi levels do not always reflect the insufficient storage of Pi within cells. Extra-skeletal symptoms of hypophosphatemia include muscle dysfunction, arrhythmia, low cardiac contractility, hemolysis, white blood cell dysfunction, platelet dysfunction, myopathy, seizures, and fatigue (43). Systemic and extra-skeletal symptoms of hypophosphatemia are not so common as those of hypocalcemia, probably because Pi stored in the skeleton and the cells may prevent acute, severe hypophosphatemia. Hypocalcemia causes an increase in the permeability of plasma membrane of muscle and nerve cells to sodium ions, leading to tetany, cramps, and seizures. Since the intracellular level of calcium is much lower than its extracellular level, intracellular calcium signaling is easily influenced by an alteration in extracellular calcium level. Therefore, serum calcium levels should be more strictly controlled than serum Pi levels (44).
FGF23-related Hyperphosphatemic and Hypophosphatemic Disorders
Hyperphosphatemic Familial Tumoral Calcinosis
Since the FGF23/FGFR/αKlotho signaling is central in maintaining phosphate homeostasis, a disrupted or excessive signaling of this pathway will cause diseases with abnormal phosphate metabolism. Inactivating mutations in FGF23, KLOTHO, and GALNT3 encoding GalNAc-T3, an enzyme required for O-glycosylation of FGF23, are responsible for hyperphosphatemic familial tumoral calcinosis (HFTC) associated with hyperphosphatemia, normal to elevated serum 1,25(OH)2D levels, and massive ectopic calcification (34, 45, 46).
Autosomal Dominant Hypophosphatemic Rickets
An excessive action of FGF23 results in hypophosphatemic diseases with increased renal Pi wasting, an inappropriately low levels of serum 1,25(OH)2D and impaired skeletal mineralization (47). Autosomal dominant hypophosphatemic rickets (ADHR) is caused by missense mutations in the FGF23 gene at Arg176 or Arg179, which make the protein resistant to inactivation by cleavage (48). Iron deficiency triggers the accumulation of uncleaved FGF23 in ADHR patients, leading to the manifestation of the hypophosphatemia and rickets/osteomalacia (49).
X-Linked Hypophosphatemic Rickets and Autosomal Recessive Hypophosphatemic Rickets
Hypophosphatemic rickets/osteomalacia related to an excessive action of FGF23 is also caused by inactivating mutations in the phosphate-regulating gene with homologies to endopeptidases, on the X chromosome (PHEX), dentin matrix protein 1 (DMP1), ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), and family with sequence similarity 20 C (FAM20C) genes (48, 50–55). PHEX is responsible for X-linked hypophosphatemic rickets (XLH), the most common form of hereditary hypophosphatemic rickets (56). Although PHEX protein is suggested to function as a zinc-dependent protease based on its structure, its physiological substrates remain to be identified. DMP1, which is responsible for autosomal recessive hypophosphatemic rickets 1 (ARHR1), is an extracellular matrix protein belonging to the SIBLING (small integrin-binding ligand, N-linked glycoproteins) family. ENPP1 encodes an enzyme which produces pyrophosphates (PPi) and is responsible for ARHR2. FAM20C is a kinase that phosphorylates various secreted proteins which include FGF23 and the SIBLING family such as DMP1. Inactivating mutations of FAM20C have been identified in patients with FGF23-related hypophosphatemia and dental abnormalities (54). Interestingly, PHEX, DMP1, and FAM20C are highly expressed in the osteocytes as well as FGF23 (57), suggesting that these molecules function as local negative regulators of FGF23 production and that osteocytes play a key role in phosphate homeostasis.
Enhanced FGFR Signaling Might be Involved in FGF23 Overproduction in XLH and ARHR1
The mechanisms underlying the FGF23 overproduction are still largely unknown in most of the FGF23-related hypophosphatemic disorders. However, recent studies have suggested that an enhanced FGFR signaling might be a pathogenesis for the overproduction of FGF23 in osteocytes of XLH. In Phex-deficient Hyp mice (a murine model of human XLH), the osteocytic expressions of Fgf1, Fgf2, Fgfr1–3, and Egr-1, which is a target of activated FGFR signaling, were markedly increased compared to in wild-type mice, as well as that of Fgf23 (57). In addition, the conditional deletion of Fgfr1 in osteocytes partially restored the FGF23 overproduction and rescued the hypophosphatemia and mineralization defect in Hyp mice (58). These findings indicate the possible involvement of activated FGFR signaling in the FGF23 overproduction in Hyp osteocytes. Overproduction of FGF23 in Dmp1-knockout mice, a model for ARHR1, has also been attributed to enhancement of FGFR signaling in the bones (59). There is yet no human data on FGFR signaling in the osteoblasts/osteocytes of XLH and ARHR patients. However, a human disease called osteoglophonic dysplasia caused by activating mutations in FGFR1 is often associated with hypophosphatemia due to increased FGF23 levels (60), which also suggests the regulation of FGF23 production by FGFR signaling.
Role of Phosphate in the Skeleton
Matrix-Vesicle Mediated Mineralization and Pi
Although the underlying mechanism for skeletal mineralization is not fully understood yet, involvement of matrix vesicles (MVs) has been suggested. MVs are the extracellular, small membranous structures produced by budding from the plasma membrane of osteoblasts and chondrocytes (61). They serve as the initial site of mineralization by rapidly taking up calcium and Pi ions to form hydroxyapatite crystals. The hydroxyapatite formed in MVs will then propagate on the collagen fibrils to mineralize the extracellular matrix (61, 62). MVs possess high activity of tissue-non-specific alkaline phosphatase (TNSALP), which functions as an ectoenzyme on the outer surface of the vesicles to hydrolyze PPi, adenosine triphosphate (ATP), and the protein-bound form of phosphate to generate orthophosphates (61, 63). PPi acts as an inhibitor against the formation of hydroxyapatite, and TNSALP facilitates the mineralization through the hydrolysis of PPi and the production of Pi. Inactivating mutations in TNSALP cause hypophosphatasia characterized by impaired skeletal mineralization (64). Another phosphatase called PHOSPHO1 has been identified to initiate mineralization by producing Pi from phosphocholine and phosphoethanolamine within MVs (65) (Figure 1).
Figure 1.
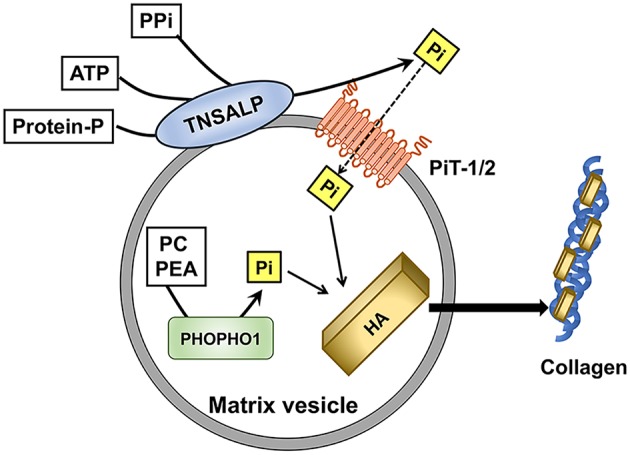
Role of Pi in the initiation of skeletal mineralization in matrix vesicles (MVs). Tissue-non-specific alkaline phosphatase (TNSALP) on the outer membrane of MVs hydrolyzes pyrophosphate(PPi), adenosine triphosphate (ATP), and protein-bound phosphate to produce Pi. Type III Na+/Pi co-transporters PiT-1 and PiT-2 mediate the Pi uptake into MVs. PHOSPHO1 produces Pi from phosphocholine (PC) and phosphoethanolamine (PEA) within MVs. Pi contributes to the formation of hydroxyapatite (HA), which will be deposited on the collagen fibrils in the extravesicular matrix.
Pi is transported into MVs by both Na+-dependent and Na+-independent components (66). The Na+-dependent Pi uptake into MVs appeared to be mediated by PiT-1 and PiT-2, similarly to the Pi uptake by the cells from which the MVs budded (67). Skeletal mineralization was normal in mice with hypomorphic expression of PiT-1 probably because a compensatory increase in the PiT-2 expression accounted for the sufficient Pi uptake (68). Thus, the net influx of Pi into MVs rather than the expression of each transporter seems to be more critical in the mineralization. With regard to the uptake of calcium ions into MVs, annexins which are calcium-binding proteins have been suggested to be involved (69).
Sufficient Pi supply is critical in MV-mediated mineralization. In rickets/osteomalacia, TNSALP is up-regulated compensatorily to supply the needs of Pi. In chondrocytic cells in culture, treatment with high Pi suppressed the expression of TNSALP within 24 h, indicating its responsiveness to Pi availability (9).
Roles of Pi in Chondrocyte Apoptosis
In addition to its profound role in MV-mediated mineralization which occurs extracellularly, Pi also exerts its effects directly on the skeletal cells. In terminally differentiated chondrocytes, an elevated Pi level induces apoptosis (4–8), a process critical in endochondral ossification. Hypophosphatemia caused reduced apoptosis of hypertrophic chondrocytes and led to rickets in the XLH model mouse (Hyp) (70). Reduced apoptosis of hypertrophic chondrocytes was also reported in vitamin D receptor (VDR) knockout mice (71), and hypophosphatemia has been suggested to be a common etiologic factor among all types of rickets (72).
Role of Extracellular Pi as a Regulator of Gene Expression in the Skeleton
Extracellular Pi also regulates gene expression. In 2000, Beck et al. demonstrated that extracellular Pi induced the expression of osteopontin (Opn) gene using a murine osteoblastic cell line MC3T3-E1 (12). Since then, a number of molecules have been identified to be responsive to the alteration in extracellular Pi levels. The Pi-responsive genes identified in osteoblasts include a cell-cycle related gene cyclin D1 (73), and Dmp1 (11). Since Dmp1 is highly expressed in osteocytes, its up-regulation by an elevated extracellular Pi may facilitate the differentiation of osteoblastic cells into osteocytes. Extracellular Pi appears to regulate PPi metabolism as well, since Enpp1 encoding a PPi-generating enzyme and Ank encoding a PPi transporter were also up-regulated by an elevated extracellular Pi (74).
The effects of extracellular Pi on the FGF23 expression in osteoblast-lineage cells has also been extensively investigated, but the results seem inconsistent both in vivo and in vitro (57, 75–79). In a recent human study, plasma FGF23 levels were transiently elevated 4 weeks after high phosphorus intake but returned to the baseline after 8 weeks (80). Treatment of cultured osteoblasts or osteocytes with elevated extracellular Pi increased the FGF23 expression only in a context-dependent manner (57, 78, 79, 81). Considering that an elevation of extracellular Pi induces the expression of Dmp1 (11), Pi may increase FGF23 expression rather indirectly through facilitating the osteoblastic differentiation into osteocytes.
During endochondral skeletogenesis, phosphate content in the extracellular milieu of chondrocytes gradually increases (82). The effects of Pi on chondrocytes differ among the differentiation stages. In early chondrocytes, an increased extracellular Pi induced cyclin D1 expression to facilitate proliferation, and down-regulated Alpl encoding TNSALP (9). In more matured chondrocytes, high extracellular Pi up-regulated Col10a1 encoding type X collagen (83) which is a marker for hypertrophic chondrocytes, and matrix Gla protein (Mgp1) (84) whose product inhibits mineralization.
Signal Transduction Evoked by an Increased Extracellular Pi
Increased Extracellular Pi Induces the Activation of Raf/MEK/ERK Pathway
Accumulating evidence has suggested the involvement of Raf/MEK (mitogen-activated protein kinase/ERK kinase)/ERK (extracellular signal-regulated kinase) pathway in the gene regulation by extracellular Pi. An increased extracellular Pi rapidly activated Raf/MEK/ERK pathway but did not other pathways such as p38MAPK pathway or JNK pathway (13). The activation of Raf/MEK/ERK pathway mediated the Pi-induced regulation of various genes including Opn (13), Dmp1 (11), and Mgp (85) in osteoblastic MC3T3-E1 cells and Mgp (84), Cyclin D1 and Alpl (9) in chondrocytic ATDC5 cells. Pi-induced activation of Raf/MEK/ERK pathway is also shared in the cells derived from extraskeletal tissues such as HEK293 cells (10).
Increased Extracellular Pi Induces the Activation of FGFR
Interestingly, an increased extracellular Pi also induces the activation of FGFR, which is one of the upstream signaling pathways of Raf/MEK/ERK cascade. In MC3T3-E1 cells, both an FGFR inhibitor and an MEK inhibitor abolished the up-regulation of Dmp1 by an increased extracellular Pi (11). In HEK293 cells, knockdown of FGFR1 diminished the phosphorylation of ERK1/2 induced by an increased extracellular Pi (10). These results suggest that FGFR plays a critical role in the transduction of the signaling evoked by an increased extracellular Pi. Knockdown experiments have also implicated that PiT-1 might mediate the Pi-induced signal transduction upstream of FGFR (Figure 2).
Figure 2.
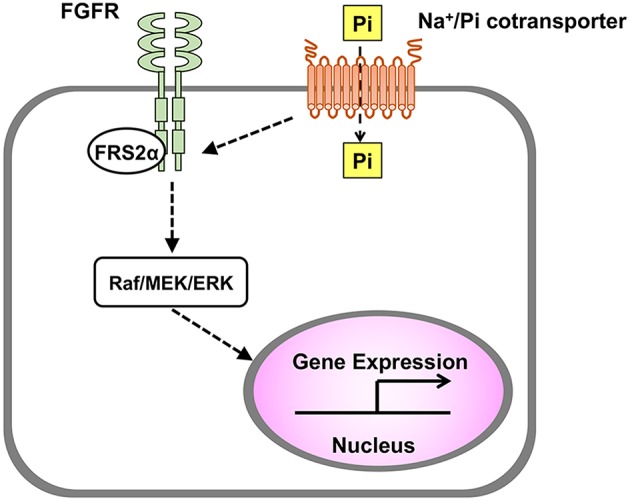
Transduction of signal evoked by extracellular Pi. In various cell types including osteoblasts and chondrocytes, an increased extracellular Pi induces the activation of Raf/MEK/ERK pathway to regulate gene expression, and this process is mediated by Na+/Pi cotransporter and FGFR.
A Possible Relationship Between FGFR and Pi-Sensing
Detection of Pi availability and adaptation are critical to maintain phosphate homeostasis. In unicellular organisms such as bacteria and yeast, the molecular mechanisms for Pi sensing and adaptation are well-defined (1). They use some types of Pi transporters and other molecules such as kinases to sense Pi levels. Although mammalian Pi sensors have not been identified yet, the responsiveness of mammalian cells to an alteration in extracellular Pi suggests that they also might sense and adapt to the Pi availability in their microenvironment. Considering the involvement of FGFR in the transduction of Pi-induced signal and the enhanced FGFR signaling in the osteoblasts/osteocytes in Phex-deficient Hyp mice (57, 58), impaired Pi-sensing in these cells might underlie the overproduction of FGF23 in XLH. Further study is required to clarify the molecular mechanisms by which mammals sense the Pi availability.
Conclusion
Phosphate plays multiple functions in the skeleton. Outside of cells, it plays a pivotal role in the MV-mediated mineralization as a constituent of hydroxyapatite. Moreover, Pi induces apoptosis of hypertrophic chondrocytes. Extracellular Pi also triggers signals within the cell to regulate gene expression, although the role of intracellular Pi remains unclear. Hypophosphatemia causes rickets by reducing hydroxyapatite formation, impairing apoptosis of hypertrophic chondrocytes, and probably altering gense expression in the skeletal cells. In vitro studies have demonstrated the involvement of Na+/Pi cotransporter, FGFR, and Raf/MEK/ERK pathway in the transduction of Pi-evoked signal, and abnormalities in Pi-sensing might be the pathogenesis of some hypophosphatemic diseases such as XLH. Clarification of the mechanisms for Pi-sensing in human will contribute to the development of better strategies to treat the conditions with abnormal phosphate metabolism.
Author Contributions
This manuscript was prepared by a joint effort of TM and KO. TM coordinated the process of manuscript preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. Preparation of the manuscript was supported by a grant from Japan Society for the Promotion of Science (JSPS KAKENHI Grant Number 18K07838) to TM.
References
- 1.Michigami T, Kawai M, Yamazaki M, Ozono K. Phosphate as a signaling molecule and its sensing mechanism. Physiol Rev. (2018) 98:2317–48. 10.1152/physrev.00022.2017 [DOI] [PubMed] [Google Scholar]
- 2.Mitchell H, Hamilton T, Steggerda F, Bean H. The chemical composition of the adult human body and its bearing on the biochemistry of growth. J Biol Chem. (1945) 158:625–37. [Google Scholar]
- 3.Peters JP, Wakeman AM, Lee C. Total acid-base equilibrium of plasma in health and disease: XI. hypochloremia and total salt deficiency in nephritis. J Clin Invest. (1929) 6:551–75. 10.1172/JCI100211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mansfield K, Rajpurohit R, Shapiro IM. Extracellular phosphate ions cause apoptosis of terminally differentiated epiphyseal chondrocytes. J Cell Physiol. (1999) 179:276–86. 10.1002/(SICI)1097-4652(199906)179:3<276::AID-JCP5>3.0.CO;2-# [DOI] [PubMed] [Google Scholar]
- 5.Mansfield K, Pucci B, Adams CS, Shapiro IM. Induction of apoptosis in skeletal tissues: phosphate-mediated chick chondrocyte apoptosis is calcium dependent. Calcif Tissue Int. (2003) 73:161–72. 10.1007/s00223-002-1056-z [DOI] [PubMed] [Google Scholar]
- 6.Mansfield K, Teixeira CC, Adams CS, Shapiro IM. Phosphate ions mediate chondrocyte apoptosis through a plasma membrane transporter mechanism. Bone. (2001) 28:1–8. 10.1016/S8756-3282(00)00409-9 [DOI] [PubMed] [Google Scholar]
- 7.Teixeira CC, Mansfield K, Hertkorn C, Ischiropoulos H, Shapiro IM. Phosphate-induced chondrocyte apoptosis is linked to nitric oxide generation. Am J Physiol Cell Physiol. (2001) 281:C833–9. 10.1152/ajpcell.2001.281.3.C833 [DOI] [PubMed] [Google Scholar]
- 8.Pucci B, Adams CS, Fertala J, Snyder BC, Mansfield KD, Tafani M, et al. Development of the terminally differentiated state sensitizes epiphyseal chondrocytes to apoptosis through caspase-3 activation. J Cell Physiol. (2007) 210:609–15. 10.1002/jcp.20857 [DOI] [PubMed] [Google Scholar]
- 9.Kimata M, Michigami T, Tachikawa K, Okada T, Koshimizu T, Yamazaki M, et al. Signaling of extracellular inorganic phosphate up-regulates cyclin D1 expression in proliferating chondrocytes via the Na+/Pi cotransporter Pit-1 and Raf/MEK/ERK pathway. Bone. (2010) 47:938–47. 10.1016/j.bone.2010.08.006 [DOI] [PubMed] [Google Scholar]
- 10.Yamazaki M, Ozono K, Okada T, Tachikawa K, Kondou H, Ohata Y, et al. Both FGF23 and extracellular phosphate activate Raf/MEK/ERK pathway via FGF receptors in HEK293 cells. J Cell Biochem. (2010) 111:1210–21. 10.1002/jcb.22842 [DOI] [PubMed] [Google Scholar]
- 11.Nishino J, Yamazaki M, Kawai M, Tachikawa K, Yamamoto K, Miyagawa K, et al. Extracellular phosphate induces the expression of dentin matrix protein 1 through the FGF receptor in osteoblasts. J Cell Biochem. (2017) 118:1151–63. 10.1002/jcb.25742 [DOI] [PubMed] [Google Scholar]
- 12.Beck GR, Jr, Zerler B, Moran E. Phosphate is a specific signal for induction of osteopontin gene expression. Proc Natl Acad Sci USA. (2000) 97:8352–7. 10.1073/pnas.140021997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beck GR, Jr, Knecht N. Osteopontin regulation by inorganic phosphate is ERK1/2-, protein kinase C-, and proteasome-dependent. J Biol Chem. (2003) 278:41921–9. 10.1074/jbc.M304470200 [DOI] [PubMed] [Google Scholar]
- 14.Yamazaki Y, Imura A, Urakawa I, Shimada T, Murakami J, Aono Y, et al. Establishment of sandwich ELISA for soluble alpha-Klotho measurement: age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochem Biophys Res Commun. (2010) 398:513–8. 10.1016/j.bbrc.2010.06.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biber J, Hernando N, Forster I. Phosphate transporters and their function. Annu Rev Physiol. (2013) 75:535–50. 10.1146/annurev-physiol-030212-183748 [DOI] [PubMed] [Google Scholar]
- 16.Marks J, Debnam ES, Unwin RJ. The role of the gastrointestinal tract in phosphate homeostasis in health and chronic kidney disease. Curr Opin Nephrol Hypertens. (2013) 22:481–7. 10.1097/MNH.0b013e3283621310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee DB, Walling MW, Brautbar N. Intestinal phosphate absorption: influence of vitamin D and non-vitamin D factors. Am J Physiol. (1986) 250 (3 Pt 1):G369–73. [DOI] [PubMed] [Google Scholar]
- 18.Munns CF, Shaw N, Kiely M, Specker BL, Thacher TD, Ozono K, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. (2016) 101:394–415. 10.1210/jc.2015-2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagner CA, Hernando N, Forster IC, Biber J. The SLC34 family of sodium-dependent phosphate transporters. Pflugers Arch. (2014) 466:139–53. 10.1007/s00424-013-1418-6 [DOI] [PubMed] [Google Scholar]
- 20.Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. (2006) 78:179–92. 10.1086/499409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA. The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int. (2006) 69:495–503. 10.1038/sj.ki.5000148 [DOI] [PubMed] [Google Scholar]
- 22.Bacic D, Schulz N, Biber J, Kaissling B, Murer H, Wagner CA. Involvement of the MAPK-kinase pathway in the PTH-mediated regulation of the proximal tubule type IIa Na+/Pi cotransporter in mouse kidney. Pflugers Arch. (2003) 446:52–60. 10.1007/s00424-002-0969-8 [DOI] [PubMed] [Google Scholar]
- 23.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA. (2001) 98:6500–5. 10.1073/pnas.101545198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun. (2004) 314:409–14. 10.1016/j.bbrc.2003.12.102 [DOI] [PubMed] [Google Scholar]
- 25.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. (2004) 113:561–8. 10.1172/JCI200419081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol. (2009) 297:F282–91. 10.1152/ajprenal.90742.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kido S, Miyamoto K, Mizobuchi H, Taketani Y, Ohkido I, Ogawa N, et al. Identification of regulatory sequences and binding proteins in the type II sodium/phosphate cotransporter NPT2 gene responsive to dietary phosphate. J Biol Chem. (1999) 274:28256–63. 10.1074/jbc.274.40.28256 [DOI] [PubMed] [Google Scholar]
- 28.Segawa H, Yamanaka S, Onitsuka A, Tomoe Y, Kuwahata M, Ito M, et al. Parathyroid hormone-dependent endocytosis of renal type IIc Na-Pi cotransporter. Am J Physiol Renal Physiol. (2007) 292:F395–403. 10.1152/ajprenal.00100.2006 [DOI] [PubMed] [Google Scholar]
- 29.Picard N, Capuano P, Stange G, Mihailova M, Kaissling B, Murer H, et al. Acute parathyroid hormone differentially regulates renal brush border membrane phosphate cotransporters. Pflugers Arch. (2010) 460:677–87. 10.1007/s00424-010-0841-1 [DOI] [PubMed] [Google Scholar]
- 30.Kovesdy CP, Quarles LD. Fibroblast growth factor-23: what we know, what we don't know, and what we need to know. Nephrol Dial Transplant. (2013) 28:2228–36. 10.1093/ndt/gft065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. (2006) 281:6120–3. 10.1074/jbc.C500457200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. (2006) 444:770–4. 10.1038/nature05315 [DOI] [PubMed] [Google Scholar]
- 33.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. (2004) 19:429–35. 10.1359/JBMR.0301264 [DOI] [PubMed] [Google Scholar]
- 34.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. (2005) 14:385–90. 10.1093/hmg/ddi034 [DOI] [PubMed] [Google Scholar]
- 35.Virkki LV, Biber J, Murer H, Forster IC. Phosphate transporters: a tale of two solute carrier families. Am J Physiol Renal Physiol. (2007) 293:F643–54. 10.1152/ajprenal.00228.2007 [DOI] [PubMed] [Google Scholar]
- 36.Wang C, Li Y, Shi L, Ren J, Patti M, Wang T, et al. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat Genet. (2012) 44:254–6. 10.1038/ng.1077 [DOI] [PubMed] [Google Scholar]
- 37.Yamada M, Tanaka M, Takagi M, Kobayashi S, Taguchi Y, Takashima S, et al. Evaluation of SLC20A2 mutations that cause idiopathic basal ganglia calcification in Japan. Neurology. (2014) 82:705–12. 10.1212/WNL.0000000000000143 [DOI] [PubMed] [Google Scholar]
- 38.Legati A, Giovannini D, Nicolas G, Lopez-Sanchez U, Quintans B, Oliveira JR, et al. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet. (2015) 47:579–81. 10.1038/ng.3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Busch AE, Schuster A, Waldegger S, Wagner CA, Zempel G, Broer S, et al. Expression of a renal type I sodium/phosphate transporter (NaPi-1) induces a conductance in Xenopus oocytes permeable for organic and inorganic anions. Proc Natl Acad Sci USA. (1996) 93:5347–51. 10.1073/pnas.93.11.5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iharada M, Miyaji T, Fujimoto T, Hiasa M, Anzai N, Omote H, et al. Type 1 sodium-dependent phosphate transporter (SLC17A1 Protein) is a Cl(-)-dependent urate exporter. J Biol Chem. (2010) 285:26107–13. 10.1074/jbc.M110.122721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiba T, Matsuo H, Kawamura Y, Nagamori S, Nishiyama T, Wei L, et al. NPT1/SLC17A1 is a renal urate exporter in humans and its common gain-of-function variant decreases the risk of renal underexcretion gout. Arth Rheumatol. (2015) 67:281–7. 10.1002/art.38884 [DOI] [PubMed] [Google Scholar]
- 42.Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. (2002) 15:172–86. 10.1046/j.1525-139X.2002.00052.x [DOI] [PubMed] [Google Scholar]
- 43.Felsenfeld AJ, Levine BS. Approach to treatment of hypophosphatemia. Am J. Kidney Dis. (2012) 60:655–61. 10.1053/j.ajkd.2012.03.024 [DOI] [PubMed] [Google Scholar]
- 44.Hannan FM, Babinsky VN, Thakker RV. Disorders of the calcium-sensing receptor and partner proteins: insights into the molecular basis of calcium homeostasis. J Mol Endocrinol. (2016) 57:R127–42. 10.1530/JME-16-0124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. (2007) 117:2684–91. 10.1172/JCI31330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. (2004) 36:579–81. 10.1038/ng1358 [DOI] [PubMed] [Google Scholar]
- 47.Fukumoto S, Ozono K, Michigami T, Minagawa M, Okazaki R, Sugimoto T, et al. Pathogenesis and diagnostic criteria for rickets and osteomalacia-proposal by an expert panel supported by the Ministry of Health, Labour and Welfare, Japan, the Japanese Society for Bone and Mineral Research, and the Japan Endocrine Society. J Bone Miner Metab. (2015) 33:467–73. 10.1007/s00774-015-0698-7 [DOI] [PubMed] [Google Scholar]
- 48.ADHR-CONSORTIUM Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. (2000) 26:345–8. 10.1038/81664 [DOI] [PubMed] [Google Scholar]
- 49.Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab. (2011) 96:3541–9. 10.1210/jc.2011-1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Endo I, Fukumoto S, Ozono K, Namba N, Tanaka H, Inoue D, et al. Clinical usefulness of measurement of fibroblast growth factor 23 (FGF23) in hypophosphatemic patients: proposal of diagnostic criteria using FGF23 measurement. Bone. (2008) 42:1235–9. 10.1016/j.bone.2008.02.014 [DOI] [PubMed] [Google Scholar]
- 51.Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. (2006) 38:1310–5. 10.1038/ng1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. (2006) 38:1248–50. 10.1038/ng1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. (2010) 86:273–8. 10.1016/j.ajhg.2010.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, et al. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res. (2013) 28:1378–85. 10.1002/jbmr.1850 [DOI] [PubMed] [Google Scholar]
- 55.Takeyari S, Yamamoto T, Kinoshita Y, Fukumoto S, Glorieux FH, Michigami T, et al. Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of Raine syndrome. Bone. (2014) 67:56–62. 10.1016/j.bone.2014.06.026 [DOI] [PubMed] [Google Scholar]
- 56.HYP-CONSORTIUM A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. (1995) 11:130–6. 10.1038/ng1095-130 [DOI] [PubMed] [Google Scholar]
- 57.Miyagawa K, Yamazaki M, Kawai M, Nishino J, Koshimizu T, Ohata Y, et al. Dysregulated gene expression in the primary osteoblasts and osteocytes isolated from hypophosphatemic Hyp mice. PLoS ONE. (2014) 9:e93840. 10.1371/journal.pone.0093840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS ONE. (2014) 9:e104154. 10.1371/journal.pone.0104154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin A, Liu S, David V, Li H, Karydis A, Feng JQ, et al. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J. (2011) 25:2551–62. 10.1096/fj.10-177816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. (2005) 76:361–7. 10.1086/427956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Millan JL. The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int. (2013) 93:299–306. 10.1007/s00223-012-9672-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anderson HC, Garimella R, Tague SE. The role of matrix vesicles in growth plate development and biomineralization. Front Biosci. (2005) 10:822–37. 10.2741/1576 [DOI] [PubMed] [Google Scholar]
- 63.Whyte MP, Landt M, Ryan LM, Mulivor RA, Henthorn PS, Fedde KN, et al. Alkaline phosphatase: placental and tissue-nonspecific isoenzymes hydrolyze phosphoethanolamine, inorganic pyrophosphate, and pyridoxal 5'-phosphate. Substrate accumulation in carriers of hypophosphatasia corrects during pregnancy. J Clin Invest. (1995) 95:1440–5. 10.1172/JCI117814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. (2016) 12:233–46. 10.1038/nrendo.2016.14 [DOI] [PubMed] [Google Scholar]
- 65.Yadav MC, Simao AM, Narisawa S, Huesa C, McKee MD, Farquharson C, et al. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification. J Bone Miner Res. (2011) 26:286–97. 10.1002/jbmr.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Solomon DH, Browning JA, Wilkins RJ. Inorganic phosphate transport in matrix vesicles from bovine articular cartilage. Acta Physiol. (2007) 190:119–25. 10.1111/j.1748-1716.2007.01670.x [DOI] [PubMed] [Google Scholar]
- 67.Solomon DH, Wilkins RJ, Meredith D, Browning JA. Characterization of inorganic phosphate transport in bovine articular chondrocytes. Cell Physiol Biochem. (2007) 20:99–108. 10.1159/000104158 [DOI] [PubMed] [Google Scholar]
- 68.Bourgine A, Pilet P, Diouani S, Sourice S, Lesoeur J, Beck-Cormier S, et al. Mice with hypomorphic expression of the sodium-phosphate cotransporter PiT1/Slc20a1 have an unexpected normal bone mineralization. PLoS ONE. (2013) 8:e65979. 10.1371/journal.pone.0065979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blandford NR, Sauer GR, Genge BR, Wu LN, Wuthier RE. Modeling of matrix vesicle biomineralization using large unilamellar vesicles. J Inorg Biochem. (2003) 94:14–27. 10.1016/S0162-0134(02)00629-3 [DOI] [PubMed] [Google Scholar]
- 70.Sabbagh Y, Carpenter TO, Demay MB. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci USA. (2005) 102:9637–42. 10.1073/pnas.0502249102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Donohue MM, Demay MB. Rickets in VDR null mice is secondary to decreased apoptosis of hypertrophic chondrocytes. Endocrinology. (2002) 143:3691–4. 10.1210/en.2002-220454 [DOI] [PubMed] [Google Scholar]
- 72.Demay MB, Sabbagh Y, Carpenter TO. Calcium and vitamin D: what is known about the effects on growing bone. Pediatrics. (2007) 119 (Suppl. 2):S141–4. 10.1542/peds.2006-2023F [DOI] [PubMed] [Google Scholar]
- 73.Conrads KA, Yi M, Simpson KA, Lucas DA, Camalier CE, Yu LR, et al. A combined proteome and microarray investigation of inorganic phosphate-induced pre-osteoblast cells. Mol Cell Proteomics. (2005) 4:1284–96. 10.1074/mcp.M500082-MCP200 [DOI] [PubMed] [Google Scholar]
- 74.Rendenbach C, Yorgan TA, Heckt T, Otto B, Baldauf C, Jeschke A, et al. Effects of extracellular phosphate on gene expression in murine osteoblasts. Calcif Tissue Int. (2014) 94:474–83. 10.1007/s00223-013-9831-6 [DOI] [PubMed] [Google Scholar]
- 75.Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. (2005) 146:5358–64. 10.1210/en.2005-0777 [DOI] [PubMed] [Google Scholar]
- 76.Nishida Y, Taketani Y, Yamanaka-Okumura H, Imamura F, Taniguchi A, Sato T, et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int. (2006) 70:2141–7. 10.1038/sj.ki.5002000 [DOI] [PubMed] [Google Scholar]
- 77.Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. (2005) 90:1519–24. 10.1210/jc.2004-1039 [DOI] [PubMed] [Google Scholar]
- 78.Ito N, Findlay DM, Anderson PH, Bonewald LF, Atkins GJ. Extracellular phosphate modulates the effect of 1alpha,25-dihydroxy vitamin D3 (1,25D) on osteocyte like cells. J Steroid Biochem Mol Biol. (2013) 136:183–6. 10.1016/j.jsbmb.2012.09.029 [DOI] [PubMed] [Google Scholar]
- 79.Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. (2006) 17:1305–15. 10.1681/ASN.2005111185 [DOI] [PubMed] [Google Scholar]
- 80.Trautvetter U, Jahreis G, Kiehntopf M, Glei M. Consequences of a high phosphorus intake on mineral metabolism and bone remodeling in dependence of calcium intake in healthy subjects - a randomized placebo-controlled human intervention study. Nutr J. (2016) 15:7. 10.1186/s12937-016-0125-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hori M, Kinoshita Y, Taguchi M, Fukumoto S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J Bone Miner Metab. (2016) 34:132–9. 10.1007/s00774-015-0651-9 [DOI] [PubMed] [Google Scholar]
- 82.Mwale F, Tchetina E, Wu CW, Poole AR. The assembly and remodeling of the extracellular matrix in the growth plate in relationship to mineral deposition and cellular hypertrophy: an in situ study of collagens II and IX and proteoglycan. J Bone Miner Res. (2002) 17:275–83. 10.1359/jbmr.2002.17.2.275 [DOI] [PubMed] [Google Scholar]
- 83.Magne D, Bluteau G, Faucheux C, Palmer G, Vignes-Colombeix C, Pilet P, et al. Phosphate is a specific signal for ATDC5 chondrocyte maturation and apoptosis-associated mineralization: possible implication of apoptosis in the regulation of endochondral ossification. J Bone Miner Res. (2003) 18:1430–42. 10.1359/jbmr.2003.18.8.1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Julien M, Magne D, Masson M, Rolli-Derkinderen M, Chassande O, Cario-Toumaniantz C, et al. Phosphate stimulates matrix Gla protein expression in chondrocytes through the extracellular signal regulated kinase signaling pathway. Endocrinology. (2007) 148:530–7. 10.1210/en.2006-0763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Julien M, Khoshniat S, Lacreusette A, Gatius M, Bozec A, Wagner EF, et al. Phosphate-dependent regulation of MGP in osteoblasts: role of ERK1/2 and Fra-1. J Bone Miner Res. (2009) 24:1856–68. 10.1359/jbmr.090508 [DOI] [PubMed] [Google Scholar]