

Summary
The Foxp3 transcription factor is a crucial determinant of both regulatory T (TREG) cell development and their functional maintenance. Appropriate modulation of tolerogenic immune responses therefore requires the tight regulation of Foxp3 transcriptional output, and this involves both transcriptional and post-translational regulation. Here, we show that during T cell activation, phosphorylation of Foxp3 in TREG cells can be regulated by a TGF-β activated kinase 1 (TAK1)-Nemo-like kinase (NLK) signaling pathway. NLK interacts and phosphorylates Foxp3 in TREG cells, resulting in the stabilization of protein levels by preventing association with the STUB1 E3-ubiquitin protein ligase. Conditional TREG cell NLK-knockout (NLKΔTREG) results in decreased TREG cell-mediated immunosuppression in vivo, and NLK-deficient TREG cell animals develop more severe experimental autoimmune encephalomyelitis. Our data suggest a molecular mechanism, in which stimulation of TCR-mediated signaling can induce a TAK1-NLK pathway to sustain Foxp3 transcriptional activity through the stabilization of protein levels, thereby maintaining TREG cell suppressive function.
Keywords: Foxp3, phosphorylation, regulatory T cell, NLK, TCR, ubiquitination, immune tolerance
Graphical Abstract
Highlights
-
•
Phosphorylation of Foxp3 is regulated by TAK1-NLK signaling during TREG activation
-
•
Foxp3 phosphorylation decreases its interaction with STUB1 E3-ubiquiting protein ligase
-
•
Foxp3 phorphorylation reduces ubiquitination and proteasome-mediated degradation
-
•
NLK TREG knockout results in reduced immunosuppressive capacity
The maintenance of Foxp3 expression is critical for correct TREG cell function. Fleskens et al. demonstrate a molecular mechanism in which TCR engagement can stabilize Foxp3 protein expression through TAK1-NLK-regulated phosphorylation, thereby maintaining TREG cell suppressive function.
Introduction
Immunological tolerance and homeostasis are established by a specific subset of T lymphocytes called regulatory T (TREG) cells, which are characterized by a highly specialized suppressive phenotype (Fontenot et al., 2003, Khattri et al., 2003). The Forkhead Box transcription factor Foxp3 is a crucial determinant of TREG cell development and function, illustrated by the onset of a fatal complex autoimmune disorder in humans and mice, caused by loss-of-function mutations in the gene encoding Foxp3 (Bennett et al., 2001, Khattri et al., 2003). Foxp3 expression levels are tightly regulated, and the maintenance of Foxp3 expression is crucial for sustained TREG cell suppressive function (Williams and Rudensky, 2007). Foxp3+ TREG cells are frequently found in large numbers at inflammatory loci in diseases, including type 1 diabetes, multiple sclerosis, and rheumatoid arthritis, but they fail to control immune responses at the site of inflammation (Ehrenstein et al., 2004, Lindley et al., 2005, Viglietta et al., 2004). These findings illustrate that physiological cues encountered within the TREG cell microenvironment can modulate their function through a variety of as yet unresolved molecular mechanisms.
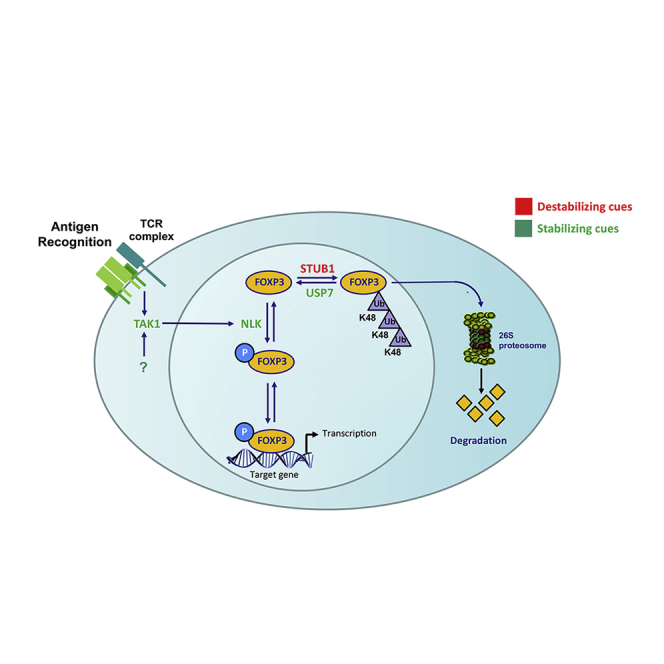
It has been appreciated for some time that there is a requirement for TCR signaling for Foxp3 expression and that T cell receptor (TCR) signaling precedes the induction of FOXP3 transcription (Li and Rudensky, 2016). While it appears that a broad range of high-affinity antigens probably drives TREG cell differentiation, there is also evidence that thymic-derived TREG cell TCRs continually sample (high-affinity) antigens (Moran et al., 2011). TCR expression does not appear to be to be required for the maintenance of resting TREG cells; however, continuous TCR signaling is observed in these cells (Levine et al., 2014, Vahl et al., 2014). Furthermore, TREG cell-specific deletion of the TCR α chain has shown that TCR signaling is critical for the generation and maintenance of activated and suppressive TREG cells (Levine et al., 2014, Vahl et al., 2014). The identification of key signaling pathways induced downstream of TCR engagement is required to fully understand the mechanism driving TREG cell development and maintenance of immune tolerance. While TCR signaling regulates a variety of transcriptional events that are mediated by nuclear factor κB (NF-κB), nuclear factor of activated T cells (NFAT), and Forkhead Box subfamily O (FOXO) transcription factors, what is much less well defined is whether TCR stimulation may also result in the activation of intracellular signaling pathways that directly impinge on Foxp3 function. It is becoming increasingly evident that post-translational modulators can fine-tune Foxp3 transcriptional activity, thereby modulating TREG cell suppressive function. This can include interaction with co-factors that can redirect Foxp3 transcriptional output under distinct environmental conditions (Kwon et al., 2017, Rudra et al., 2012) or through post-translational modifications (Lu et al., 2017, van Loosdregt and Coffer, 2014). The modification of Foxp3 protein by means of acetylation, for example, can modulate several aspects of its transcriptional activity. Depending on the targeted lysine residue, acetylation of Foxp3 can improve its ability to regulate gene transcription by enhancing protein oligomerization, as well as binding to active chromatin sites (Samanta et al., 2008, Song et al., 2012). Furthermore, temporal control of Foxp3 protein stability results from a tight balance between lysine acetylation and poly-ubiquitination, allowing for transient modulation of TREG cell function (van Loosdregt et al., 2010). In response to inflammatory cytokines such as interleukin-6 (IL-6), enhanced proteasomal degradation results from increased poly-ubiquitination by STUB1, as well as decreased ubiquitin-specific peptidase 7 (USP7)-mediated deubiquitination of Foxp3 protein (Chen et al., 2013, van Loosdregt et al., 2013a). In addition, cyclin-dependent kinase 2 (CDK2)-mediated phosphorylation of CDK motifs in the N terminus of Foxp3 was suggested to impede protein stability, whereas tumor necrosis factor α (TNF-α)-induced dephosphorylation of Foxp3 negatively modulated TREG cell function in rheumatoid arthritis (Morawski et al., 2013, Nie et al., 2013). Further understanding of the molecular mechanisms and key players involved in the regulation of Foxp3 function is required to elucidate the mechanisms that can either impede or invigorate TREG function to establish a balanced immune response.
Here, we describe a novel TCR-mediated signaling pathway regulating Foxp3 phosphorylation through the activation of Nemo-like kinase (NLK) in a transforming growth factor β (TGF-β) activated kinase 1 (TAK1)-dependent manner. NLK-mediated phosphorylation of Foxp3 results in the stabilization of protein levels by preventing ubiquitin-mediated proteasomal degradation. Conditional deletion of NLK in TREG cells results in the loss of in vivo suppressive capacity and an age-dependent increase in autoinflammation. The identification of such novel intracellular modulators of Foxp3 that affect TREG cell homeostasis and function provides potential therapeutic targets for controlling immune tolerance and regulating these cells in a variety of pathogenic conditions.
Results
Phosphorylation of Foxp3 Is Regulated by TAK1
Almost 15% of the human Foxp3 protein is composed of amino acids that could be subjected to phosphorylation (∼14% in murine Foxp3), of which the majority is either serine (Ser) or threonine (Thr) (40 and 19 residues, respectively) (Figure 1A). To enable the detection of putatively phosphorylated residues in the Foxp3 protein, we used Phos-tag Acrylamide, followed by SDS-PAGE (Takeda et al., 2003). Human in vitro-induced TREG (iTREG) cells were generated from naive CD4+ T cells isolated from umbilical cord blood (UCB), as previously described (van Loosdregt et al., 2013a, van Loosdregt et al., 2013b), and subsequently treated with increasing concentrations of the Ser/Thr phosphatase inhibitor okadaic acid (OA). A decrease in the electrophoretic mobility of Foxp3 protein, indicative of phosphorylation, was already observed in untreated cells, but markedly increased upon treatment with OA in a dose-dependent manner (Figure 1B). The appearance of multiple bands indicates that phosphorylation likely occurs on several residues. Furthermore, the dependence on OA treatment indicates that this is a dynamic process.
Figure 1.

TAK1 Modulates Foxp3 Phosphorylation in Response to TCR Engagement
(A) Schematic overview of serine and threonine residues in human and mouse Foxp3 protein.
(B) Phos-tag analysis of iTREG cells treated with increasing concentrations of okadaic acid (OA; 0.1–0.2–0.5–1.0–2.0 μM).
(C–G) Phos-tag analysis of lysates from TREG cells deprived from IL-2, TGF-β, and TCR activation for 24 h.
(C) Deprived iTREG cells were restimulated with anti-CD3/CD28 coated beads and OA.
(D) Deprived iTREG cells were restimulated with IL-2, TGF-β, or anti-CD3/CD28 coated beads.
(E) Deprived iTREG cells were restimulated with anti-CD3/CD28 coated beads, PMA, and ionomycin to induce TCR downstream signaling.
(F) Phos-tag analysis of lysates from iTREG cells pre-treated for 2 h with kinase-specific inhibitors, before OA treatment (1 μM, 30 min).
(G) CD4+GFP+ TREG cells were sorted from mouse spleens, pre-incubated with TAK inhibitor (TAKi) (300 nM), and subsequently treated with OA, anti-CD3/CD28 coated beads, PMA, and ionomycin ex vivo for 30 min. Lysates were analyzed by Phos-tag-SDS-PAGE.
Protein phosphorylation is generally regulated upon the activation of signaling pathways by extracellular stimulation. Since we initially detected Foxp3 phosphorylation in a system using iTREG cells generated in the presence of IL-2, TGF-β, and anti-CD3/anti-CD28 antibodies, we postulated that one or all of these signals could be responsible for the observed modification. To investigate this, we deprived differentiated iTREG cells from IL-2, TGF-β, and CD3/CD28 stimulation for 24 h to downregulate intracellular signal transduction pathways. Subsequently, we reactivated signaling with each stimulus or in combination (Figures 1C–1E and S1A). Phos-tag analysis was used to detect phosphorylation, which could be observed only upon reactivation of the TCR by incubation with CD3/CD28 antibodies. A combination of CD3/CD28 antibodies, IL-2, and TGF-β did not further enhance Foxp3 phosphorylation, indicating that TCR activity was sufficient for maximal Foxp3 phosphorylation. To evaluate whether the phosphorylation observed upon OA treatment indeed required prior TCR stimulation, we deprived iTREG cells of differentiation stimuli for 24 h, and then induced TCR activity in the presence or absence of OA (Figure 1C). In deprived iTREG cells, the phosphorylation of Foxp3 was no longer observed upon OA treatment, indicating that additional signals were required. Upon the activation of TCR signaling, Foxp3 phosphorylation was again induced, which was further enhanced when cells were simultaneously treated with OA (Figure 1D). Next, we used PMA (phorbol 12-myristate 13-acetate) and ionomycin as alternative means to activate signaling events downstream of the TCR (Figure 1E). Comparable to CD3/CD28 stimulation, PMA and ionomycin could induce the phosphorylation of Foxp3, supporting the notion that TCR activation is sufficient for Foxp3 phosphorylation.
Next, to investigate which protein kinases downstream of TCR activation may be involved in the regulation of Foxp3 phosphorylation, we took a candidate approach, pretreating iTREG cells with specific inhibitors that targeted a variety of Ser/Thr kinases, followed by OA incubation (Figures 1F, S1B, and S1C). Inhibitors of the phosphatidylinositol 3-kinase-protein kinase B-mammalian target of rapamycin (PI3K-PKB-mTOR) pathway, which is believed to be an important determinant of TREG cell function (Delgoffe et al., 2011, Haxhinasto et al., 2008, Sauer et al., 2008), did not affect Foxp3 phosphorylation in this experimental setup. Neither did the inhibition of glycogen synthase kinase 3 (GSK-3), which we have recently demonstrated can modulate TREG cell function through the regulation of βcatenin/T cell factor 1 (TCF1) activity (van Loosdregt et al., 2013b). A clear reduction in phosphorylation was, however, observed upon the inhibition of TAK1, whereas the inhibition of other mitogen-activated protein kinase (MAPK) family members p38, Jun-N-terminal kinase (JNK), and mitogen activated protein kinase kinase (MEK) had no effect. Since T cell-specific deletion of Map3k7, the gene encoding TAK1, results in the abrogation of TREG cell generation (Chang et al., 2015, Sato et al., 2006, Wan et al., 2006), we hypothesized that the TAK1-mediated phosphorylation of Foxp3 may play a role in the regulation of Foxp3 function.
To establish whether Foxp3 phosphorylation is also regulated by TCR signaling in primary murine TREG cells, freshly isolated TREG cells from Foxp3-GFP mice were incubated with CD3/CD28, PMA, and ionomycin, and phosphorylation was assessed by Phos-tag analysis (Figure 1G). Again, stimulation with CD3/CD28 antibodies induced the phosphorylation of Foxp3. TREG intracellular signaling was apparently already occurring to some extent in these cells, as indicated by the presence of modest phosphorylation in untreated cells (Figure 1G, lane 1), which accumulated upon OA treatment (Figure 1G, lane 3). However, phosphorylation was further enhanced upon the stimulation of TCR signaling by either CD3/CD28 or ionomycin but not PMA treatment (Figure 1G, lanes 2 and 4–7). Ionomycin triggers intracellular Ca2+ release, which was previously demonstrated to induce Ca2+/calmodulin-dependent kinase (CaMKII) to directly phosphorylate TAK1, thereby inducing TAK1 signaling (Ishitani et al., 2003, Wang et al., 2009). To assess the role of TAK1 activity in TCR-mediated Foxp3 phosphorylation, we pretreated cells with TAK inhibitor (TAKi) to inhibit the activity of TAK1 and downstream kinases. As shown in lanes 8–14 in Figure 1G, this inhibition resulted in a marked decrease of all TCR-induced phosphorylation, suggesting that TCR activation may mediate the phosphorylation of Foxp3 through Ca2+-mediated activation of TAK1.
NLK Interacts with and Phosphorylates Foxp3
TAK1 is a member of the MAPK family, in which kinases function as groups of three sequentially acting enzymes that ultimately phosphorylate an effector protein, generally a transcription factor (Figure 2A). TAK1 is an MAPKKK that has been demonstrated to induce the activity of several MAPK family members, including JNK, p38, and NLK (Ishitani et al., 1999, Shim et al., 2005, Wang et al., 1997). To identify a putative TAK1-dependent kinase responsible for Foxp3 phosphorylation, we co-expressed Foxp3 and several MAPKs that were previously described to function downstream of TAK1, and cell lysates were analyzed by Phos-tag SDS-PAGE. Whereas co-expression of p38, JNK, or ERK did not induce phosphorylation, Foxp3 specifically and robustly shifted upon the expression of NLK (Figure 2B). This decreased mobility observed by co-expression with NLK was due to phosphorylation since λphosphatase (λPPase) treatment prevented this shift (Figure 2C).
Figure 2.

NLK Interacts with and Directly Phosphorylates Foxp3
(A) Schematic representation of a conventional MAPK signal transduction pathway.
(B) Phos-tag analysis of Foxp3 co-expressed with MAPKs demonstrated to act downstream of TAK1.
(C) Phos-tag analysis of Foxp3 expressed in HEK293 cells in the presence or absence of ectopic NLK. Foxp3 was immunoprecipitated and immunoprecipitated fractions were incubated with λPPAse for 30 min, as indicated.
(D) Co-immunoprecipitation of FLAG-NLK and HA-Foxp3. HEK293 cells transiently expressing FLAG-NLK and HA-Foxp3 as indicated were lysed, and lysates were incubated with anti-FLAG M2 affinity gel.
(E) Representative confocal microscopy images of sorted CD4+GFP+ TREG cells from the spleen. Endogenous Foxp3 (green) and NLK (red) were visualized using specific antibodies, and DAPI was used to visualize the nucleus.
(F) PLA analysis of Foxp3:NLK association immediately after cell sorting from spleen and MLN.
(G) In vitro kinase assay with ectopic HA-Foxp3 immunoprecipitated from HEK293 cells and purified active GST-NLK protein in the presence or absence of ATP. Samples were analyzed by Phos-tag-SDS-PAGE.
(H) Schematic representation of NLK-dependent phosphorylated residues in Foxp3 identified by mass spectrometry. The asterisk indicates where phosphorylated residues could not be unequivocally determined, and both serine residues conserved in both murine and human Foxp3 were included in further analyses.
(I) In vitro kinase assay with ectopic WT or 7xA mutant HA-Foxp3 immunoprecipitated from HEK293 cells and purified active GST-NLK protein in the presence of ATP. Samples were analyzed by Phos-tag-SDS-PAGE. IVK, in vitro kinase assay.
To determine whether Foxp3 can be a direct substrate of NLK, we first investigated the possible interaction between the two proteins by co-immunoprecipitation. A clear interaction was observed when both proteins were ectopically expressed (Figure 2D). Using the same method, we could not detect an interaction between Foxp3 and TAK1 (data not shown). To confirm that an interaction between Foxp3 and NLK can also occur when both proteins are endogenously expressed in primary TREG cells, we next assessed the localization of NLK and Foxp3 in sorted TREG cells from Foxp3-GFP mice. Co-localization of both proteins was observed in the nucleus (Figure 2E). To further verify that endogenously expressed Foxp3 and NLK can associate in TREG cells, we performed a proximity ligation assay (PLA), enabling the detection of localized protein-protein interactions in cells, as we have previously described (van Loosdregt et al., 2013a, van Loosdregt et al., 2013b). As a control, CD4+GFP− cells that do not express Foxp3 were used. The association of Foxp3 and NLK, appearing as localized dots, was observed and appeared specifically in the nucleus (Figure 2F). The association of NLK and Foxp3 under these experimental conditions could be because either they can interact independently of TCR engagement or isolated CD4+ T cells may already be activated.
To address whether Foxp3 is a direct substrate of NLK, we subsequently performed an in vitro kinase assay using HA-Foxp3 immunoprecipitated from HEK293 cells and subsequently incubated with purified GST-NLK (Figure 2G). In the presence of ATP, GST-NLK induced the phosphorylation of Foxp3, indicating that Foxp3 can indeed be a direct substrate of NLK. The addition of ATP alone was not sufficient to induce the phosphorylation of Foxp3, eliminating the possibility that additional kinases potentially co-precipitating with Foxp3 were able to induce phosphorylation. We next sought to determine which amino acids could be phosphorylated by NLK. Both human and murine Foxp3 were co-expressed with NLK and immunoprecipitated from cell lysates. Using mass spectrometry (MS), as we have previously described (van Loosdregt et al., 2013a), we identified seven distinct residues (S19, S156, S189, S273, S278, S295, and T341), which were detected in both human and murine Foxp3 and phosphorylated exclusively upon the co-expression of NLK (Figures 2H and S2A–S2F). Mutation of all seven phosphorylated residues to alanine (Foxp3 7xA) resulted in decreased phosphorylation as measured by Phos-tag analysis (Figure 2I). These results demonstrate that NLK interacts with Foxp3 and can directly phosphorylate Foxp3 on multiple residues.
NLK Can Modulate TREG Cell Function In Vivo
Given the potential of NLK to phosphorylate Foxp3 on multiple residues, we analyzed the role of NLK in TREG cell-mediated suppression. NLK knockdown in human iTREG cells was performed using two distinct lentiviral small hairpin RNAs (shRNAs), by transducing the cells during in vitro differentiation. This resulted in an approximately 50% reduction in protein levels. Next, iTREG cell suppressive function was again addressed by means of an in vitro suppression assay (Figures S3A and S3B). TREG cell-mediated suppression was significantly abrogated upon the knockdown of NLK compared to the scrambled (Scr) control, supporting the hypothesis that NLK modulates TREG cell function.
To determine whether NLK activity could also modulate TREG cell-mediated suppression in vivo, we used a mouse colitis model. Here, naive CD4+CD25−CD62Lhi T cells were co-transferred with CD4+CD25hi NLK knockdown TREG cells into Rag2−/− mice. In contrast to wild-type (WT) or control knockdown TREG cells, NLK knockdown TREG cells could not suppress the development of disease based on both body weight loss and histological analysis of the colon (Figures 3A–3C, S3C, and S3D). Both interferon γ (IFN-γ and IL-17 production by CD4+ lamina propria lymphocytes was increased in mice that received NLK knockdown TREG cells (Figure 3D). TEFF cell numbers in the spleen, mesenteric lymph node, and lamina propria were increased in mice that received shNLK TREG cells in comparison to controls (Figure 3E). In addition, the numbers of transduced and transferred Thy1.1 TREG cells were similar in both the control and NLK knockdown groups, demonstrating that NLK knockdown did not alter TREG cell survival or homing to lymph nodes (Figure 3F). The percentage of Foxp3+ cells within the Thy1.1 population was decreased in mice that received NLK knockdown TREG cells, indicating that the loss of NLK activity resulted in decreased Foxp3 protein expression. These data demonstrate that NLK activity is required to regulate TREG cell-mediated suppression in vivo.
Figure 3.

NLK Is Required for Optimal TREG Cells’ Suppressive Function In Vivo
Colitis was induced by intravenous co-injection of 1 × 10−6 CD4+CD25−CD62Lhi T cells and 2 × 10−5 CD4+CD25hi TREG cells per Rag2−/− mouse. TREG cells were either untreated or transduced with control or NLK-targeting lentiviral shRNA. Mice were sacrificed 8 weeks post-adoptive transfer, and the level of induced colitis was assessed.
(A) Body weight was analyzed from 0 to 8 weeks post-transfer.
(B) Representative H&E-stained slides of colon sections.
(C) Histological colitis scores of colon sections.
(D–F) Spleen and lymph node (LN) (mesenteric and lamina propria LNs) cells were isolated.
(D) IFN-γ and IL-17 production by CD4+ mesenteric and lamina propria lymphocytes, analyzed by ELISA.
(E) The number of TEFF cells in the spleen and LNs was determined by flow cytometry.
(F) Cell numbers of injected Thy1.1+ TREG cells and Foxp3+ cells were determined by flow cytometry.
TREG Cell-Specific NLK Deficiency Results in Inflammation
To overcome the limitations of using a global NLK knockout mouse to study the role of NLK in TREG cell function in vivo, we generated a TREG-specific NLK-deficient mouse by crossing FoxP3Cre mice onto an NLKf/f background (hereafter referred to as NLKΔTreg). We confirmed that in NLKΔTreg mice, Nlk message was effectively ablated in TREG cells but not in other CD4+ populations (Figures S4A and S4B). By 6 weeks of age, NLKΔTreg mice developed mild splenomegaly (Figure 4A), which is indicative of the development of a spontaneous inflammatory response. Because many autoimmune disorders have an aging component, we also characterized the phenotype of older NLKΔTreg mice. Accordingly, we observed that the spleens of NLKΔTreg mice became substantially larger than those of WT littermate controls when these mice reached 6 months of age (Figure 4B). To further characterize the phenotype of NLKΔTreg mice, we analyzed the cell populations in the spleens of WT and NLKΔTreg littermates of both 6-week-old and 6-month-old mice. While the percentages of dendritic cells, macrophages, B cells, and cytotoxic cells were comparable between both mouse strains (Figures S4C and S4E), we observed an increase in the percentage of CD4+ T cells and a change in the proportions of CD4+ T cell subpopulations (Figures 4C and 4D). Specifically, we observed a reduction in the percentage and number of naive CD4+ T cells and a significant increase in the percentage and number of antigen-experienced CD4+ T cells (Figures 4C, 4D, S4D, and S4F). The differences in T helper cell subpopulation percentages were also pronounced, with age correlating with spleen sizes (Figures 4A–4D). Finally, we observed that young mice had comparable percentages of TREG cells (CD25hiFoxP3+); however, this cell population dramatically decreased with age (Figure 4E). Analysis of the expression of GFP/yellow fluorescent protein (YFP), a reporter of the transcriptional induction of FoxP3, revealed a similar induction of the Foxp3 gene (Figure 4F), suggesting that NLK deletion does not affect the survival or proliferation of TREG cells, but rather it compromises the stability of the Foxp3 protein, as was also observed in the adoptive transfer colitis model (Figures 3F and S4B).
Figure 4.

TREG Cell-Specific NLK-Deficient Mice Naturally Develop Inflammatory Responses
Lymphoid tissues were collected from both 6-week-old and 6-month-old Foxp3Cre NLKf/f mice (NLKΔTreg) and NLKf/f littermate controls for analysis.
(A and B) Inflammation index, calculated as a spleen weight/body weight for NLKΔTreg mice and NLKf/f littermate controls of 6 weeks old (A) or 6 months old (B). The image in (A) and (B) is a representative picture of the spleens to illustrate size.
(C and D) T helper cell subpopulations of the spleens from NLKΔTreg mice and NLKf/f littermate controls were analyzed by flow cytometry at 6 weeks old (C) or 6 months old (D). Total CD4+ T cells, naive CD4+ T cells, antigen-experienced CD4+ T cells, and TREG cells are shown as a percentage of live splenocytes.
(E) Representative fluorescence-activated cell sorting (FACS) plots showing Foxp3 staining in live CD3+CD4+ T cells from 6-week-old and 6-month-old NLKΔTreg mice and NLKf/f littermate controls.
(F) GFP/YFP expression by TREG cells from 6-month-old NLKΔTreg mice and age-matched Foxp3-GFP mice is shown as a percentage of live splenocytes.
(G) Foxp3 expression (MFI) in TREG cells from 6 months old NLKΔTreg mice and age-matched Foxp3-GFP mice. Results are representative from two independent experiments (mean ± SEM) and results for individual mice are shown as dots.
∗p < 0.05, ∗∗p < 0.01, when compared with NLKf/f mice. Non-parametric Mann-Whitney test was used.
NLK Prevents Foxp3 Polyubiquitination and Promotes TREG Cell-Mediated Suppression
To evaluate whether NLK licenses TREG cell-mediated suppression in vivo, we tested the ability of WT and NLKΔTreg TREG cells to suppress the proliferation of naive T cells induced by transfer into lymphopenic hosts. In this experiment, we transferred naive CD45.1/2 T cells alone or mixed 1:1 with CD45.2+ WT or NLKΔTreg TREG cells into lymphopenic RAG-1−/− CD45.1 hosts, allowing for the identification of donor versus host cells at the end of the experiment. Naive T cells were labeled with CellTrace Violet tracker before transfer, and the dilution of the fluorescence of the probe upon cell division was used to quantify proliferation. As shown in Figure 5A, the transfer of naive T cells into lymphopenic hosts induced the division of the CD45.1/2 transferred cells. This proliferation was efficiently suppressed by co-transfer of WT TREG cells; however, the transfer of NLKΔTreg TREG cells did not affect the proliferation of naive T cells, despite being present in the spleens of the hosts in comparable quantities. These results suggest that NLK mediates the phosphorylation of Foxp3, increasing its stability, and in this manner, NLK allows for the optimal suppressive capacity of TREG cells.
Figure 5.

Reduced Levels of Foxp3 Protein from NLK-Deficient TREG Due to Increased Proteasomal Degradation
(A) FACS-sorted CD45.1/2 naive CD4+T cells (CD4+CD25−CD45RBhi) were intravenously (i.v.) injected (1 × 105) alone (Non-Treg [NT]) or mixed 1:1 with CD45.2 NLKf/f or NLKΔTreg TREG cells (CD4+ YFP+ CD25hi) into CD45.1 Rag1−/− recipients. Before transfer, naive T cells were labeled with CellTrace Violet. After 6 days of transfer, spleens were harvested, and single cell suspensions were prepared and stained for CD4, CD45.1, and CD45.2. The dilution of the CellTrace Violet probe was analyzed by FACS in the naive CD4+ population identified as CD4+ CD45.1+ and CD45.2+. Results are representative of two independent experiments (means ± SEMs), and the results for individual mice are shown as dots (except in A). There were three biological replicates per treatment. ∗p < 0.05, ∗∗p < 0.01, ∗p < 0.001, when compared with NLKf/f mice, unless otherwise indicated. Non-parametric Mann-Whitney test was used.
(B) Reduced phosphorylation of Foxp3 in NLKΔTreg TREG cells.
(C) WT and NLKΔTreg TREG cells were FACS sorted from the spleens of Foxp3-reporter mice (Foxp3YFP and Foxp3YFP/Cre NLKf/f) by gating in CD4+ GFP/YFP+ and CD25hi cells. TREG cells were cultured in the presence of 150 μg/mL cycloheximide (CHX), as indicated. Foxp3 expression was determined by flow cytometry.
(D) Analysis of Foxp3 levels in transfected HEK293 cells treated with CHX in the presence or absence of NLK expression.
(E) All seven phosphorylation sites identified by mass spectrometry were mutated to the aspartate phosphomimetic, and samples were analyzed similarly to (D).
(F) Cells were transfected with His-tagged ubiquitin and Myc-tagged Foxp3 in the presence or absence of NLK or STUB1. A ubiquitin pull-down assay was performed as described in Method Details. Foxp3-specific ubiquitination was determined by western blot analysis of Myc-Foxp3.
(G) Lysates of Myc-Foxp3 and HA-STUB1 transfected cells with or without Myc-NLK were immunoprecipitated with anti-Myc coupled beads. Immunoblots were analyzed with anti-Myc, anti-HA, and anti-FLAG.
To evaluate whether NLK-mediated phosphorylation of Foxp3 can indeed regulate Foxp3 stability, we assessed the phosphorylation of Foxp3 in WT compared with NLKΔTreg TREG cells. As shown in Figure 5B, NLKΔTreg TREG cells showed reduced phosphorylation of Foxp3 at steady-state conditions. To determine the impact of the NLK-mediated phosphorylation of Foxp3, we measured Foxp3 expression in WT and NLKΔTreg TREG cells from young mice after CD3/CD28 stimulation in the presence of the protein translation inhibitor cycloheximide (CHX). By inhibiting protein translation, the production of new Foxp3 protein is prevented, and in this way, the stability of Foxp3 protein expression can be qualitatively evaluated. CD3/CD28 stimulation combined with the inhibition of translation led to a rapid decrease in Foxp3 protein in both WT and NLKΔTreg TREG cells (Figure 5C). However, the loss of Foxp3 expression in NLKΔTreg TREG cells occurred significantly faster, even though Foxp3 levels were comparable in both WT and NLKΔTreg TREG cells at the start of the experiment (Figure 5C). These observations suggest that NLK-mediated phosphorylation of Foxp3 enhances its stability.
To determine whether NLK-mediated phosphorylation could result in increased Foxp3 protein levels, we assessed Foxp3 expression in HEK293T cells that were co-transfected with or without NLK in the presence of CHX. As observed in primary TREG cells, the addition of CHX resulted in the loss of Foxp3 expression, while the ectopic expression of NLK noticeably increased Foxp3 protein amounts (Figure 5D). These data further support a model whereby NLK-mediated Foxp3 phosphorylation prevents Foxp3 degradation. Since we had identified several potential NLK phosphorylation sites on Foxp3 (Figures 2H and 2I), we also evaluated the stability of a phosphomimetic mutant (Foxp3 7xD). Here, the Foxp3 7xD mutant demonstrated increased protein expression both in the presence and absence of CHX, suggesting increased protein stability (Figure 5E). We have previously demonstrated that Foxp3 is subject to regulation by K48-linked ubiquitination by the STUB1 E3-ligase and deubiquitination by USP7-regulating levels of Foxp3 protein (Chen et al., 2013, van Loosdregt et al., 2013a). To evaluate whether NLK-mediated Foxp3 phosphorylation could modulate polyubiquitination, HEK293T cells transfected with both His-tagged ubiquitin and hemagglutinin (HA)-tagged Foxp3 were lysed, and ubiquitinated proteins were isolated with nickel-nitrilotriacetic acid (Ni-NTA) beads, as previously described (van Loosdregt et al., 2013a). Foxp3-specific polyubiquitination was determined by immunoblot analysis with anti-HA. A clear polyubiquitination pattern was observed with WT Foxp3 (Figure 5F, lane 2), and this was greatly reduced upon co-transfection of NLK (Figure 5F, lane 3), indicating that Foxp3 polyubiquitination can be inhibited by NLK. Co-transfection of the STUB1 E3-ligase resulted in a robust increase in Foxp3 polyubiquitination, as we have previously reported (Figure 5F, lane 4) (Chen et al., 2013); however, co-transfection of NLK again resulted in a large reduction in Ubi-Foxp3 (Figure 5F, lane 5). Finally, to identify a mechanism by which NLK could interfere in STUB1-mediated Foxp3 polyubiquitination, we evaluated the interaction between STUB1 and Foxp3 in the presence and absence of NLK. Co-transfection of Foxp3 and STUB1 followed by immunoprecipitation of STUB1 revealed a clear interaction between these two proteins (Figure 5G, lane 4). However, the expression of NLK resulted in greatly decreased interactions between Foxp3 and STUB1 (Figure 5G, lane 7), providing a mechanistic explanation of reduced Foxp3 polyubiquitination and increased Foxp3 protein levels under these conditions. No changes in association between Foxp3 and USP7 were observed (Figure S5A).
These results demonstrate that NLK can prevent Foxp3 polyubiquitination, most likely by preventing the association between Foxp3 and STUB1, providing a mechanism for the increased Foxp3 protein turnover in NLK-deficient TREG cells.
NLK-Deficient TREG Cell Animals Develop More Severe Experimental Autoimmune Encephalomyelitis
To test the physiological relevance of our previous observations, we induced experimental autoimmune encephalomyelitis (EAE) in mice with NLK deficiency in Foxp3+ TREG cells. This MOG35–55 mouse model of multiple sclerosis is a CD4+ T helper 1 (TH1)-mediated autoimmune disease in which autoreactive T cells specific for myelin components enter the CNS, initiating a cascade of inflammation and demyelination. CD4+CD25hi TREG cells have been shown to suppress antigen-specific autoreactive immune responses and CNS inflammation during active EAE (Kohm et al., 2002, McGeachy et al., 2005). As also observed under homeostatic conditions (Figure 4), NLKΔTreg animals displayed more advanced, severe disease than WT littermates (Figure 6A). While total cell counts and frequencies of CD4+ infiltrated T cells in CNS were comparable (data not shown), the frequencies of Foxp3+ cells among CD4+ T cells were increased in the brains of NLKΔTreg mice, presumably in a compensatory mechanism, although this was not significant. In keeping with a more robust immune response, NLKΔTreg mice harbored higher proportions of IL-17 and IFN-γ-producing CD4+ cells, with an emphasis on TH1 immune response in their spinal cords during the recovery phase than did NLKfl/fl littermates (p < 0.05) (Figures 6B and S6). These findings support a role for NLK expression in sustaining TREG cell functionality and regulating autoimmune responses in vivo.
Figure 6.

NLK-Deficient TREG Cell Animals Develop More Severe Experimental Autoimmune Encephalmyelitis (EAE)
(A) Both NLKfl/fl and NLKΔTreg mice were injected subcutaneously (s.c.) with 100 μg MOG35–55 in complete Freund’s adjuvant (CFA) and 250 ng pertussis toxin (PT) intraperitoneally (i.p.). Disease severity was scored every 2 days. Scores for each group over time are presented as means ± SEMs (p < 0.05, n = 6–8 per group).
(B) On day 30, CNS (brain and spinal cord)-infiltrating T cells were recovered and stained for CD4 and Foxp3. Secretion of IL-17 and IFN-γ within the CD4+ population was determined after stimulation with PMA and ionomycin. The mean percentages of IL-17+, IFN-γ+, and Foxp3+ cells among CNS-infiltrating CD4+ cells on day 30 were determined.
Results were obtained from three individual experiments and presented as means ± SEMs. ∗p < 0.05.
Discussion
Tight control of TREG cell suppressive function is required to establish a balanced immune response, given the cells’ central role in immune regulation and peripheral tolerance. Human Foxp3+CD4+CD25+ TREG cells may not be stable, encountering the loss of Foxp3 expression or conversion into effector T (TEFF)-like cells (Koenen et al., 2008, Valmori et al., 2010, Voo et al., 2009). Furthermore, it has been observed under inflammatory conditions that the suppressive capacity of TREG cells can be diminished (e.g., through exposure to pro-inflammatory cytokines such as IL-6) (Pasare and Medzhitov, 2003) or enhanced (e.g., by exposure to the epidermal growth factor (EGF)-like growth factor amphiregulin) (Zaiss et al., 2013). Evidence that Foxp3 expression and activity are subjected to post-translational modulation to establish the regulation of TREG cell activity is emerging (Lu et al., 2017, van Loosdregt and Coffer, 2014). Thus, a critical area of TREG cell research remains to understand the mechanisms by which extracellular signals regulate the expression and function of Foxp3.
Here, using a combination of in vitro and in vivo approaches, we have identified and characterized a novel NLK-mediated phosphorylation-ubiquitination “switch” that regulates the Foxp3 protein levels, and we demonstrate that NLK is critical for the maintenance of immune tolerance in vivo. These data may have implications for both natural and peripheral TREG cell stability or plasticity and differentiation, as well as ensure that activation-induced Foxp3 expression in human TEFF cells remains a transient phenomenon and does not lead to the inappropriate generation of peripheral TREG cell populations. Our initial observations identified TAK1 as a regulator of TCR-induced Foxp3 phosphorylation (Figures 1G and 1H). TAK1 functions at the crossroads of multiple signal transduction pathways and regulates the activity of various transcription factors that are important in immune regulation, including activator protein 1 (AP-1) and NF-κB, and TAK1 activation specifically triggered by TCR signaling has been shown to induce both NF-κB and MAPK activity (Shim et al., 2005, Sun et al., 2004, Wan et al., 2006, Wang et al., 1997). In our studies, both pharmacological inhibition and ectopic expression of TAK1 downstream effectors revealed that the regulation of Foxp3 phosphorylation was specifically mediated by NLK. Combined with the finding that the manipulation of Foxp3 expression, and thus transcriptional activity, is sufficient to modulate TREG cell function (Williams and Rudensky, 2007), these findings indicate that TAK1 and NLK regulated TREG cell suppressive capacity at least in part through the modification of Foxp3 protein.
Stimulation of TCR activation is not only a crucial factor for the generation of both thymic and peripheral TREG cells but is furthermore important for TREG cell function (Apostolou and von Boehmer, 2004, Jordan et al., 2001, Kretschmer et al., 2005). The activation of mature TREG cells by TCR ligation induces expansion in vivo, rendering a TREG cell population with enhanced suppressive capacity (Klein et al., 2003, Kretschmer et al., 2005, Walker et al., 2003). TCR ligation is also a requirement for extrathymic differentiation of TREG cells and is dependent on TCR ligand density and affinity (Gottschalk et al., 2010). It has been suggested that the cumulative level of TCR stimulation determines the initial induction of Foxp3 in the periphery, but that TCR ligand density and potency both are important in regulating the persistence of Foxp3+ populations. It could be that NLK activation acts as a sensor of the level of TCR activation by antigen, and in this way can allow the rapid and transient regulation of Foxp3 protein expression levels. A similar role for PKB/AKT and mTOR have been proposed for the regulation of Foxp3 transcription (Haxhinasto et al., 2008, Sauer et al., 2008), and these pathways may complement each other in providing robust and transient regulation of TREG cell functionality. While the activation of TCR signaling was believed to predominantly regulate Foxp3 gene transcription (Josefowicz et al., 2012, Mantel et al., 2006, Walker et al., 2003), we provide novel evidence for the modulation of Foxp3 transcriptional activity by TAK1-NLK-mediated phosphorylation. Mediation of both transcriptional and post-translational regulation of Foxp3 activity by TCR signaling would enable robust temporal control of Foxp3 function. Therefore, manipulation of the TAK1-NLK axis could provide a mechanism to transiently modulate TREG cell function and, ultimately, the intensity and duration of immune responses.
NLK function has been implicated in a variety of developmental processes, including wing and eye development in Drosophila melanogaster, endoderm development in Caenorhabditis elegans, mesoderm as well as anterior head formation in Xenopus laevis, and hematopoiesis in the mouse (Choi and Benzer, 1994, Kortenjann et al., 2001, Ohkawara et al., 2004, Rocheleau et al., 1999, Sato et al., 2006, Zeng et al., 2007). These findings point toward a common role for NLK in embryonic development and cell fate determination. Our current observations demonstrate that specific loss of NLK expression in TREG cells results in an age-dependent reduction in TREG cell numbers, eventually resulting in the development of inflammation. There does not appear to be a problem with TREG cell development per se, since younger animals have normal numbers of CD4+CD25hiFoxp3+ lymphocytes (Figure 4). This suggests that at least thymic-derived TREG cell numbers may not be perturbed by the lack of NLK. However, within 6 months, there is an almost total disappearance of CD4+CD25hiFoxp3+ cells through an as yet unidentified mechanism, which parallels the loss of Foxp3 protein. The observation of a gradual loss of Foxp3 expression and TREG cell numbers suggests that NLK activity is required constitutively after thymic selection to maintain homeostatic TREG cells numbers. Mice deficient in Foxp3 develop fatal autoimmune disease (Fontenot et al., 2003, Hori et al., 2003), while the continuous expression of Foxp3 throughout life prevents autoimmunity (Kim et al., 2007). In accordance with these findings, we found that in contrast to WT mice, NLKΔTreg animals developed signs of colon inflammation at 6 months (data not shown).
Phosphorylation of Foxp3 has been described before, although the physiological relevance of these post-translational modifications remains unresolved. The highly related Pim1 and Pim2 kinases, for instance, have been shown to phosphorylate Foxp3 and negatively regulate transcriptional output, although through distinct mechanisms. Pim1 kinase phosphorylates human, but not murine, Foxp3 at serine-422 and results in reduced DNA binding and activation (Li et al., 2014), while Pim2 kinase was found to phosphorylate the Foxp3 N terminus (Deng et al., 2015). Accordingly, global Pim2 gene deficiency resulted in increased TREG cell lineage stability, and Pim2 knockout animals have increased resistance to dextran sulfate sodium (DSS)-induced colitis. These experiments did not use conditional Pim2 knockout mice, which makes the TREG cell-specific importance of these observations difficult to evaluate. Similarly, the cyclin-dependent kinase CDK2 was reported to directly phosphorylate the N terminus of Foxp3, resulting in increasing protein turnover (Morawski et al., 2013). However, it remains unclear as to when or where such CDK2-mediated regulation occurs under physiological conditions. Lastly, amphiregulin (AREG) signaling through the EGF receptor has been shown to inhibit GSK-3β-mediated phosphorylation of Foxp3 in vitro (Wang et al., 2016). We have previously demonstrated that AREG enhances TREG cell function, and GSK-3β may therefore play a role in this process. While these observations support the idea that Foxp3 phosphorylation may play a role in regulating TREG cell function, none of these studies has explored the relevance of their findings in relevant, conditional knockout in vivo models, making the extrapolation of these data with regard to the regulation of immune tolerance difficult.
Our data reveal a critical role for NLK-mediated phosphorylation of Foxp3. NLK was found to interact with and have the potential to phosphorylate Foxp3 on multiple residues. However, Foxp3 is not the only target of NLK, and while there are few studies that have evaluated NLK substrates in detail, the reported mechanisms by which NLK functions are quite diverse, ranging from regulation of protein stability to DNA binding and recruitment of co-factors, often leading to decreased transcriptional activity (Kurahashi et al., 2005, Sato et al., 2006, Zhang et al., 2014, Zhang et al., 2015). NLK has been both positively and negatively implicated in the stabilization of the protein expression of the transcription factors c-Myb, p53, and activating transcription factor 5 (ATF5) through a variety of mechanisms. Fbxw7, for example, targets c-Myb for ubiquitin-mediated proteosomal degradation in an NLK-dependent manner (Kanei-Ishii et al., 2008), while the association of NLK with ATF5 or p53, for instance, prevented their proteasomal degradation (Zhang et al., 2014, Zhang et al., 2015). Furthermore, NLK was found to promote the acetylation of p53, and Foxp3 expression and activity have also been shown to be regulated through the modulation of acetylation, allowing for rapid and transient control of Foxp3 protein levels (Lu et al., 2017, van Loosdregt and Coffer, 2014). It would be interesting to determine in future experiments whether NLK may also stabilize Foxp3 expression by promoting increased acetylation.
Our data show that NLK positively sustains TREG suppressive function, likely through phosphorylation-dependent deubiquitination of Foxp3. Since the balance between tolerance and autoimmunity can be regulated by Foxp3 stability, our observations concerning the NLK-mediated control of Foxp3 proteasomal degradation can have therapeutic implications. The development of a targeted NLK inhibitor, for example, may provide a way of eliminating TREG cells from the tumor microenvironment with potential beneficial treatment outcome effects.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-Foxp3 | eBiosciences | PCH101 |
| Mouse anti-Foxp3 | eBiosciences | 14-7979; RRID:AB_468499 |
| Mouse anti-FLAG | Sigma-Aldrich | F7425; RRID:AB_439687 |
| Mouse anti-hemaglutanin (HA) | Sigma-Aldrich | 12CA5; RRID:AB_514505 |
| Rabbit anti-NLK | Santa Cruz | H-100; RRID:AB_2112660 |
| Goat anti-actin | Santa Cruz | I-19; RRID:AB_630836 |
| Anti-HSP90 | Cell Signaling | 4875; RRID:AB_2233331 |
| Rat anti mouse CD4 (clone RM4-5) | Biolegend | Cat#100536 |
| Rat anti mouse CD62L (clone MEL-14) | Biolegend | Cat#104424 |
| Rat anti CD44 (clone IM7) | Biolegend | Cat#103032 |
| Rat anti mouse CD45Rb (clone 16A) | BD PharMingen | Cat#553101 |
| Rat anti mouse CD25 (clone PC61) | Biolegend | Cat#102016 |
| Mouse anti mouse CD45.1 (clone A20) | eBiosciences | Cat#17-0453-81 |
| Mouse anti mouse CD45.2 (clone 104) | eBiosciences | Cat#12-0454-81 |
| Rat anti mouse CD8 (clone 53-6.7) | Biolegend | Cat#100706 |
| Rat anti mouse CD19 (clone 6D5) | Biolegend | Cat#115538 |
| Rat anti mouse SiglecF (clone E50-2440) | BD PharMingen | Cat#562681 |
| Rat anti mouse Ly6G (clone 1A8) | Biolegend | Cat#127628 |
| Rat anti mouse CD3 (clone 17A2) | Biolegend | Cat#100228 |
| Armenian Hamster anti mouse CD11c (clone N418) | Biolegend | Cat#117334 |
| Rat anti CD11b (clone M1/70) | Biolegend | Cat#101241 |
| Rat anti mouse F4/80 (clone BM8) | eBiosciences | Cat#25-4801-82 |
| Rat anti mouse MHCII (I-A/I-E) (clone M5/114.15.2) | eBiosciences | Cat#47-5321-82 |
| Mouse anti mouse NK-1.1 (clone PK136) | Biolegend | Cat#108710 |
| Rat anti FoxP3 (clone FJK-16 s) | eBiosciences | Cat#50-5773-82 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phos-tag acrylamide | WAKO Chemicals GmbH | 300-93523 |
| DMEM containing GlutMax | GIBCO Life Technologies | 31966047 |
| RPMI 1640 | GIBCO Life Technologies | 61870044 |
| IMDM | GIBCO Life Technologies | 12440-061 |
| FBS | GIBCO Life Technologies | S1810-500 |
| Penicillin-Streptomycin | GIBCO Life Technologies | 15140-122 |
| Nonidet p40 | US Biologicals | N3500 |
| HALT protease inhibitor cocktail | Thermo Scientific | 78439 |
| Anti-HA conjugated beads | Sigma-Aldrich | A2095 |
| PVDF | Millipore | IPFL00010 |
| iScript cDNA synthesis kit | BioRad | 170-8891 |
| Poly-L-lysine | Sigma-Aldrich | P4707 |
| Anti-CD3/CD28 Dynabeads | ThermoFisher | 11452D |
| TGF-β | R&D systems | 240-B-010 |
| RNasin inhibitor | Promega | N2111 |
| dNTPs | Promega | 28-4065-60 |
| Oligo dT15 | Promega | C1101 |
| PEI | Polysciences | 23966-1 |
| PKB inhibitor VII | Calbiochem | 124018 |
| 5Z-7-oxozeanol | Tocris Biosceince | 3604/1 |
| BIO | Tocris Biosceince | 3194/1 |
| γPPAse | New England Biolabs | P0753s |
| GST-NLK | Sigma-Aldrich | SRP5285 |
| Okadaic acid | Enzo Lifesciences | ALX-350-003-C050 |
| SB203580 | Enzo Lifesciences | BML-EI286-001 |
| SP600125 | Enzo Lifesciences | BML-EI305-010 |
| Rapamycin | Enzo Lifesciences | A275-0005 |
| U0126 | Enzo Lifesciences | BML-EI282-0001 |
| Cycloheximide | Sigma-Aldrich | C4859 |
| Critical Commercial Assays | ||
| Foxp3 staining kit | eBioscience | 00-5521-00 |
| Proximity Ligation Assay kit | Sigma-Aldrich | DUO92101 |
| Live/dead kit | Life Technologies | L3224 |
| CellTrace Violet tracker | ThermoFisher | C34557 |
| Deposited Data | ||
| Raw and analyzed Mass Spectrometry Data | This paper | PRIDE: PXD011164 |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | CRL-11268 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Internal breeding colonies of the University of Edinburgh or of the University of Utrecht | N/A |
| Mouse: BALB/c | Internal breeding colonies of the University of Edinburgh or of the University of Utrecht | N/A |
| Mouse: NLKfl/fl | The Jackson Laboratory | JAX: 024537 B6;129-Nlktm.1.1Ecan/J |
| Mouse: Foxp3CRE | The Jackson Laboratory | JAX: 016959 B6.129(Cg)-Foxp3 tm4(YFP/icre)Ayr/J |
| Mouse: Foxp3-eGFP | The Jackson Laboratory | JAX: 006772 B6.Cg-Foxp3 tm2(EGFP)Tch/J |
| Oligonucleotides | ||
| See Table S1 | N/A | |
| Recombinant DNA | ||
| pMT2-HA_Foxp3 | van Loosdregt et al., 2013b | N/A |
| pMT2-HA_JNK | de Groot et al., 1997 | N/A |
| pMT2-HA_ERK | de Groot et al., 1997 | N/A |
| pMT2-His_Ubi | van Loosdregt et al., 2013a | N/A |
| pMT2-myc_USP7 | van Loosdregt et al., 2013a | N/A |
| pMT2-myc_FoxP3 | van Loosdregt et al., 2013a | N/A |
| pLP/VSVG | van Loosdregt et al., 2013a | N/A |
| psPAX2 | van Loosdregt et al., 2013a | N/A |
| pMT2-FLAG_NLK | Szypowska et al., 2011 | N/A |
| pLKO.1 puro NLK human shRNA | Sigma-Aldrich | TRCN0000002071 |
| pLKO.1 puro NLK human shRNA | Sigma-Aldrich | TRCN0000195090 |
| pRFP-C-Rs Nlk shRNA | OriGene Technologies | TF501483 |
| Stub1 | Harvard Medical School | HsCD00326610 |
| pUC57 Foxp3 7xA and 7x | Genescript Biotech | Custom synthesis |
| Software and Algorithms | ||
| FlowJo 10 | FLOWJO, LLC | https://www.flowjo.com/ |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be addressed to the Lead Contact, Paul Coffer (pcoffer@umcutrecht.nl).
Experimental Model and Subject Details
Cultured Cells
Human embryonic kidney (HEK) 293 cells were maintained in Dulbecco’s modified Eagle’s medium containing Glutamax (GIBCO), supplemented with 8% heat-inactivated fetal calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml streptomycin (GIBCO).
Genetic mouse models
Foxp3-GFP mice were kept in the animal facility of the Utrecht University and experiments were approved by the Animal Experiment Committee of the Faculty of Veterinary Medicine (Utrecht University). C57BL/6J mice (WT, Nlkfl/fl, Foxp3-cre/YFP x Nlkfl/fl) were bred and maintained at the University of Edinburgh in specific-pathogen free conditions. Sex-matched mice were 6-8-weeks or 6 months old at the start of the experiment, and all mice were housed in individually ventilated cages. Experiments were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act of 1986. All researchers were accredited by the UK government Home Office. Dispensation to carry out animal research at The University of Edinburgh was approved by the University of Edinburgh Animal Welfare and Ethical Review Body and granted by the UK government Home Office.
Method Details
Antibodies and reagents
The following antibodies were used: rat anti-Foxp3 clone PCH101 and mouse anti-Foxp3 clone eBio7979 (eBioscience), mouse anti-Flag (Sigma-Aldrich), mouse anti-hemaglutinin (HA) clone 12CAS, rabbit anti-NLK (H-100) and goat anti-Actin (I-19) (Santa Cruz Biotechnology), and anti-HSP90 was purchased from Professor Ineke Braakman (UMC Utrecht, Utrecht, the Netherlands).
The following reagents were used: Phos-tag™ Acrylamide (WAKO Chemicals GmbH), Okadaic acid, SB203580, SP600125, U-0126 and Rapamycin (Enzo Lifesciences), PKB inhibitor VIII (Calbiochem), 5Z-7-Oxozeaenol and BIO (Tocris Bioscience, Bristol, United Kingdom, λPPAse (New England BioLabs) and recombinant GST-NLK active protein (Sigma-Aldrich).
Construct design
pMT2 HA-Foxp3(van Loosdregt et al., 2013b), HA-JNK, HA-P38, HA-ERK (de Groot et al., 1997), and His-Ubi, myc-USP7, myc-Foxp3, pLP/VSVG and psPAX2(van Loosdregt et al., 2013a) were described previously. pMT2-Flag NLK was kindly provided by B. Burgering (Szypowska et al., 2011). MISSION® pLK0.1 puro NLK human shRNA TRCN0000002071 and TRCN0000195090 were purchased from Sigma-Aldrich. pRFP-C-RS Nlk mouse shRNA was purchased from OriGene Technologies. Stub1 was purchased from Harvard Medical school, pUC57 Foxp3 7xA and 7xD were synthesized by Genscript Biotech, and cloned into pMT2 containing an N-terminal HA-tag generating pMT2-HA-Stub1, pMT2-HA-Foxp3 7xA and 7xD respectively.
Immunoprecipitation
For Co-immunoprecipitation, HEK293 cells were transfected with PEI in 60 mm tissue culture dishes, and 48 hours post transfection cells were lysed in NP-40 lysis buffer (50 mM Tris-HCl pH 7.5, 0.5% Nonidet p40 (US Biologicals), 150 mM NaCl, 10 mM EDTA, supplemented with 1% HALT protease inhibitor cocktail (Thermo Scientific). Cell lysates were cleared by high speed centrifugation. Immunoprecipitation was performed utilizing anti-FLAG M2 affinity gel or anti-HA conjugated beads (Sigma-Aldrich). Precipitates were washed 3 times in lysis buffer, boiled, and analyzed by Western Blotting.
Generation of induced regulatory T cells
CD4+CD25- cells were isolated from human umbilical cord blood by magnetic-activated cell sorting and cultured in RPMI 1640 (GIBCO) supplemented 10% FCS, 100 Units/ml penicillin, 100 mg/ml streptomycin, and 5 × 10−5 M 2-mercaptoethanol. Foxp3 expression and TREG cell differentiation was induced by culturing the cells for 5 days in combination with CD3/CD28 Dynabeads (GIBCO), 300 IU IL-2, and 10 ng/ml TGFβ.
Western blotting: conventional and Phos-tag
For conventional western blotting, samples were lysed in NP-40 Lysis buffer, and protein concentrations were determined using a Lowry protein assay. Proteins were separated by SDS-PAGE, electrophoretically transferred to polyvinylidene difluoride (PVDF) membrane (Millipore). Membranes were hybridized with antibodies as indicated. Immunocomplexes were detected using ECL (Amersham Pharmacia Inc.). For Phos-tag SDS-PAGE, 50 μM Phos-tag™ Acrylamide was used and analysis was performed according to the manufacturer’s protocol.
Quantitative RT-PCR
Total RNA was extracted using the RNAeasy kit (QIAGEN) and treated with RNase free DNase (QIAGEN) to eliminate genomic DNA contamination. cDNA synthesis was performed with an iScript cDNA Synthesis Kit (Bio-Rad). cDNA samples were amplified with SYBR Green Supermix (Bio-Rad) in a MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad) according to the manufacturer’s protocol. For quantification the comparative Ct method was used. Relative quantity was defined as 2-ΔΔCt and β2-microglobulin was used as a reference gene.
In vitro suppression assays
HumanCD4+CD25hiCD127lo TREG cells were sorted from human PBMCs and cocultured with PBMCs labeled with 2 mM CFSE (at ratios of 1:10, 1:5, 1:2, and 1:1) in anti-CD3 (clone OKT3)-coated 96-well plates. Cells were cultured for 4 days in RPMI medium supplemented with 10% FCS, 100 U/ml penicillin, 100 mg/ml streptomycin, and 5 × 10−5 M2-β mercaptoethanol. Proliferation of CD4+ cells was determined by measuring CFSE dilution with a FACS CANTOII Analyzer (BD Biosciences). Suppression was determined by a comparison of the percentage of proliferating cells with or without TREG cells.
Confocal imaging
Cells were adhered to poly-L-Lysine (Sigma-Aldrich) coated glass slides (Thermo Scientific), and fixed and permeabilized using the Foxp3 staining kit (eBioscience). Samples were washed in Permeabilization buffer (PB) (eBioscience) and blocked in Block buffer (PB supplemented with 10% normal donkey serum (Jackson Immunoresearch Laboratories)). Subsequently, samples were incubated overnight at 4°C with block buffer containing antibodies recognizing NLK and Foxp3 as indicated. Subsequently, cells were used for localization studies, or Proximity Ligation Assay.
Localization studies
Samples were washed 3x in PB and incubated at 22°C for 1 hour with secondary antibodies anti-mouse-alexa488, and anti-rabbit-alexa546 as indicated. Samples were washed 3 times in PB and mounted in mowiol (3% 1,4-diazabiclo- (2,2,2)-octan followed by a glass cover. Samples were analyzed with a 63X objective on a Zeiss LSM 710 microscope (Carl Zeiss).
Proximity Ligation Assay (PLA):
Cells were washed 3 times in PB and incubated in mouse PLUS and rabbit MINUS probes (Sigma Aldrich) according to the manufacturer’s protocol. Cells were washed in PB before detection using the in situ PLA detection kit (Sigma-Aldrich) as previously described (van Loosdregt et al., 2011). Samples were analyzed with a 63x objective on a Zeiss LSM 710 microscope (Carl Zeiss).
Mass spectrometry and mutational analysis
Human or mouse HA-Foxp3 was ectopically expressed in HEK293 cells with or without ectopic flag-NLK. After immuno-precipitation ofHA-Foxp3, proteins were on-bead digested in 2M Urea, 50 mM Ammonium BiCarbonate (ABC) and finally desalted with homemade C-18 stagetips (3M, St Paul, MN). Peptides were separated on-a 30 cm column (75-μm ID fused silica capillary with emitter tip, New Objective, Woburn, MA) packed with 3-μm aquapur gold C-18 material (Dr Maisch, Ammerbuch-Entringen, Germany) using a 3-hour gradient (0%–80% Acetonitrile), and delivered by an easy nanoflow high-performance liquid chromatography (nHPLC) (Thermo Scientific). Peptides were electrosprayed directly into a LTQ-Verlos-Orbitrap (Thermo Scientific) and analyzed in data-dependent mode with the resolution of the full scan set at 30,000, after which the top 10 peaks were selected for Higher energy Collision-induced Dissociation (HCD) fragmentation (set at a normalized energy of 40%) and detection in the Orbitrap with a target setting of 5000 ions at a resolution of 7500. Raw files were analyzed with Maxquant software, version 1.6.1.0. For identification, the mouse Uniprot database was searched with oxidation of methionine and STY phosphorylation set as variable modifications, and carbamidomethylation of cysteine set as fixed modification, while peptide and protein false discovery rates were set to 1%. ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD011164.
For mutational analysis, Foxp3 phosphorylation was assessed in two independent experiments, each comparing human and mouse Foxp3 with or without overexpression of NLK. Phosphorylated serine and threonine residues that were both identified only upon co-expression of NLK and conserved between human and murine Foxp3 were selected. For the 7 identified conserved NLK-dependent residues, expression plasmids encoding phospho-dead (Foxp3 7xA) or phosho-mimetic (Foxp3 7xD) variants were generated for biochemical and functional analysis (see above).
Colitis induction and histological assessment
Naive CD4+CD25CD62Lhi T cells were isolated from BALB/c and intravenously injected into BALB/c Rag2/ immunodeficient recipients (1x10e6 per mouse). CD4+CD25highTREG cells TREG cells (2x10e5) isolated from Thy1.1 BALB/c were transduced with or shCTR- or shNLK HEK293-derived lentivirus as we have previously described (Chen et al., 2013, van Loosdregt et al., 2013a) and intravenously coinjected where indicated. Mice were monitored weekly for wasting disease. After 8 weeks, mice were sacrificed. Colons were removed from mice 8 weeks after T cells reconstitution and fixed in 10% formalin. Five-micrometer paraffin-embedded sections were cut and stained with hematoxylin and eosin (H&E). Pathology of colon tissue was scored in a blinded fashion, on a scale of 0-5 where a grade of 0 was given when there were no changes observed. Changes associated with other grades were as follows: grade 1, minimal scattered mucosal inflammatory cell infiltrates, with or without minimal epithelial hyperplasia; grade 2, mild scattered to diffuse inflammatory cell infiltrates, sometimes extending into the submusoca and associated with erosions, with mild to moderate epithelial hyperplasia and mild to moderate mucin depletion from goblet cells; grade 3, moderate inflammatory cell infiltrates that were sometimes transmural, with moderate to severe epithelial hyperplasia and mucin depletion; grade 4, marked inflammatory cell infiltrates that were often transmural and associated with crypt abscesses and occasional ulceration, with marked epithelial hyperplasia, mucin depletion; and grade 5, marked transmural inflammation with severe ulceration and loss of intestinal glands.
Isolation of lamina propria leukocytes (LPL)
Mice were euthanized and the colons excised and placed in ice-cold PBS. After extensive washing of the colonic lumen with PBS, colons were minced into 0.3-0.5cm pieces, and repeatedly incubated in Ca- and Mg-free HBSS containing 10% FCS and 5mM EDTA to release intestinal epithelial cells. The remaining tissue was further incubated with a digestion cocktail [Liberase and DNaseI (Roche)] at 37°C for 1h, and the LPL were then layered on discontinuous Percoll gradient (Amersham Biosciences). After centrifugation for 30 min at 2000 rpm at room temperature, the LPL population was recovered at the 40%/75% interphase, washed twice in HBSS/BSA, and counted by trypan blue staining.
Splenocyte preparation and flow cytometry analysis
Single cell suspensions of spleens were obtained by forcing the tissue through a 70 μm cell strainer. Subsequently, cells were treated with red blood cell lysis buffer (Sigma-Aldrich) and counted using an automated cellometer T4 (Peqlab).
Cells were incubated with Fc block (CD16/CD32 and 10% mouse serum) and stained with a combination of the following commercial monoclonal fluorescently conjugated antibodies: CD4, CD62L, CD44, CD45Rb, CD25, CD45.1, CD45.2, CD8, CD19, SiglecF, Ly6G, CD3, CD11c, CD11b, F4/80, I-A/I-E (MHCII), and NK-1.1. For detection of Foxp3, cells were stained for surface markers then fixed and permeabilized using Foxp3 staining buffer set (eBioscience). Cells were then stained with anti-Foxp3 for 30 min at room temperature. Live/Dead (Life Technologies) was used to exclude dead cells from analysis. Samples were analyzed by flow cytometry using Becton Dickinson FACS LSR II and FlowJo software. T cells were defined by the expression of CD45, and CD3. Cytotoxic T cells identified by the expression of CD8 and TH cells by the expression of CD4. Naive and antigen experienced TH cell subpopulations were further characterized by their expression of CD62L, CD44 and CD45RB (naive: CD62L+ CD44lo CD45RBhi and antigen experienced: CD62L- CD44hi CD45RBlo). TREG cells were defined as CD25hi and Foxp3 or YFP positive. In some experiments, TREG cells were sorted using the same gating strategy.
RNA extraction and quantitative real-time PCR
TREG cells were homogenized in TRIzol and RNA was isolated following manufacturer’s instructions. Reverse transcription was performed using 1 mg of total RNA using 200 U of M-MLV reverse transcriptase, 10 mM dNTPs, and 0.5 mg Oligo dT15 and RNasin inhibitor (Promega). Expression of genes of interest was measured by real-time PCR with the Lightcycler 480 II system (Roche) using Taqman Master kit and specific primers for Nlk, as previously described. PCR amplification was analyzed using 2nd derivative maximum algorithm (LightCycler 480 Sw 1.5, Roche) and Nlk expression was normalized to the housekeeping gene Rn18s.
Foxp3 stability assay
TREG cells were cultured in IMDM medium supplemented with 10% FCS, 1% l-glutamine, 1% penicillin/streptomycin and 5x10e-5 M β2-mercaptoethanol at 37°C in a humidified atmosphere at 5% CO2. Sorted TREG cells were treated with 150 μg/ml cycloheximide (CHX) (Sigma) for 0, 0.5, 2 and 4 hr. Next, cells were stained for FoxP3 as described above.
In vivo TREG cell suppression experiments
2.5-5 × 10e5 naive (CD45.1/2+) CD4+ T cells were i.v. injected alone or mixed 1:1 with sorted CD45.2+ NLK-sufficient or NLK-deficient TREG cells into RAG-1−/− CD45.1 hosts. Naive T cells were labeled with the CellTrace Violet tracker (Thermofisher) before transfer per manufacturer’s instructions. 6 days after transfer recipient mice were sacrificed and spleens were collected for quantification of the proliferation of naive T cells.
Lentiviral shRNA knockdown
Virus-containing supernatants were obtained from HEK293T cells transfected using PEI (Polysciences) with pLP/VSV, psPAX2 and pLKO.1 puro lentiviral shRNA vector (1:2:3 ratio). Murine primary CD4+CD25hi TREG cells were transduced with shRNA CTR or shRNA NLK lentivirus as we have previously described (Chen et al., 2013, van Loosdregt et al., 2013a).
Human umbilical cord blood-derived CD4+CD25- T cells were isolated and TREG differentiation was induced by culturing the cells in presence of CD3/CD28 Dynabeads, 300 IU IL-2, and 10 ng/ml TGFβ. After O/N culture, cells were transduced by addition virus supernatant (1:1) supplemented with polybrene (1 mg/ml) and IL-2 and TGFβ added to restore concentrations (300 IU IL-2, and 10 ng/ml resp.), followed by centrifugation (1hr at 2000RPM and 30°C) and O/N Culture. Next, cells were pellet and transduction procedure repeated. After O/N culture media was replaced with RPMI + IL-2 and TGFβ, supplemented with puromycin (1 mg/ml) and cells were cultured for 2 days. Next, Dynabeads were removed by magnet separation, cells were washed 3x and cultured in RPMI + puromycin (1mg/ml) and lL-2 + TGFβ. After 2 days puromycin resistance was used as an indicator of successful transduction (FACS sorting).
EAE induction and scoring
6 to 8 weeks old, sex-matched NLKfl/fl and NLKΔTreg mice littermates were injected subcutaneously in the rear flank with 100 ug MOG35-55 peptide (2HNMEVGWYRSPFSRVVHLYRNGK-COOH) in complete Freund’s adjuvant (CFA) (Sigma) on day 0, and 250ng pertussis toxin (List Biological) was injected intraperitoneally on day 0 and day 2 post-induction. Mice were monitored and disease severity was scored every two days. Clinical signs of EAE were assessed with a 0 to 5-point scoring system: 0, normal; 1, flaccid tail or hind-limb weakness; 2, moderate hind-limb paralysis; 3, full hind-limb paralysis; 4, quadriplegia; 5, death. “In-between” scores (0.5, 1.5, 2.5, 3.5, 4.5) is applied when the clinical picture lies between two defined scores (Taylor and Kitaichi, 2008).
Quantification and Statistical Analysis
If not stated otherwise, data are represented as mean ± SEM p values less than 0.05 were considered statistically significant.
Acknowledgments
The authors thank Boudewijn Burgering for providing constructs. V.F. was supported by a grant from the Dutch Arthritis Foundation (Reumafonds). Work in the D.Z. laboratory is supported by Medical Research Council grant MR/M011755/1 and European Union grant CIG-631413 (“EGF-R for Immunity”). J.M. was supported by the MRC medical fellowship MR/M018911/1. H.-J.V. was supported by the “Proteins at Work” program of the Netherlands Organisation for Scientific Research (NWO) (project no. 184.032.201). Pan lab research was supported by grants from the Bloomberg-Kimmel Institute (BKI), the Melanoma Research Alliance (MRA), the NIH (RO1AI099300, RO1AI089830, and R01AI137046), and the Department of Defense (PC130767). The Li lab was supported by grants from the National Natural Science Committee of China (no. 81725004) and the Shanghai Science and Technology Committee (no. 16410723600).
Author Contributions
D.M.Z. and P.J.C. designed the project; V.F., C.M.M., X.W., P.W., C.E.G.M.P., J.M., S.H., V.G., and H.-J.V. performed the experiments; A.R., F.P., H.L., D.M.Z., and P.J.C. supervised the project and analyzed the data together with contributions from all of the authors; and D.M.Z., P.J.C., V.F., and C.M.M. wrote the manuscript with contributions from all of the authors.
Declaration of Interests
The authors declare no competing interests.
Published: March 26, 2019
Footnotes
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.087.
Contributor Information
Huabin Li, Email: allergyli@163.com.
Dietmar M. Zaiss, Email: dietmar.zaiss@ed.uk.ac.
Paul J. Coffer, Email: pcoffer@umcutrecht.nl.
Supplemental Information
References
- Apostolou I., von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J. Exp. Med. 2004;199:1401–1408. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C.L., Christie J., Ramsdell F., Brunkow M.E., Ferguson P.J., Whitesell L., Kelly T.E., Saulsbury F.T., Chance P.F., Ochs H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- Chang J.H., Hu H., Sun S.C. Survival and maintenance of regulatory T cells require the kinase TAK1. Cell. Mol. Immunol. 2015;12:572–579. doi: 10.1038/cmi.2015.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Barbi J., Bu S., Yang H.Y., Li Z., Gao Y., Jinasena D., Fu J., Lin F., Chen C. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity. 2013;39:272–285. doi: 10.1016/j.immuni.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K.W., Benzer S. Rotation of photoreceptor clusters in the developing Drosophila eye requires the nemo gene. Cell. 1994;78:125–136. doi: 10.1016/0092-8674(94)90579-7. [DOI] [PubMed] [Google Scholar]
- de Groot R.P., van Dijk T.B., Caldenhoven E., Coffer P.J., Raaijmakers J.A., Lammers J.W., Koenderman L. Activation of 12-O-tetradecanoylphorbol-13-acetate response element- and dyad symmetry element-dependent transcription by interleukin-5 is mediated by Jun N-terminal kinase/stress-activated protein kinase kinases. J. Biol. Chem. 1997;272:2319–2325. doi: 10.1074/jbc.272.4.2319. [DOI] [PubMed] [Google Scholar]
- Delgoffe G.M., Pollizzi K.N., Waickman A.T., Heikamp E., Meyers D.J., Horton M.R., Xiao B., Worley P.F., Powell J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng G., Nagai Y., Xiao Y., Li Z., Dai S., Ohtani T., Banham A., Li B., Wu S.L., Hancock W. Pim-2 Kinase Influences Regulatory T Cell Function and Stability by Mediating Foxp3 Protein N-terminal Phosphorylation. J. Biol. Chem. 2015;290:20211–20220. doi: 10.1074/jbc.M115.638221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenstein M.R., Evans J.G., Singh A., Moore S., Warnes G., Isenberg D.A., Mauri C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J. Exp. Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot J.D., Gavin M.A., Rudensky A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Gottschalk R.A., Corse E., Allison J.P. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J. Exp. Med. 2010;207:1701–1711. doi: 10.1084/jem.20091999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haxhinasto S., Mathis D., Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 2008;205:565–574. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S., Nomura T., Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Ishitani T., Ninomiya-Tsuji J., Nagai S., Nishita M., Meneghini M., Barker N., Waterman M., Bowerman B., Clevers H., Shibuya H., Matsumoto K. The TAK1-NLK-MAPK-related pathway antagonizes signalling between beta-catenin and transcription factor TCF. Nature. 1999;399:798–802. doi: 10.1038/21674. [DOI] [PubMed] [Google Scholar]
- Ishitani T., Ninomiya-Tsuji J., Matsumoto K. Regulation of lymphoid enhancer factor 1/T-cell factor by mitogen-activated protein kinase-related Nemo-like kinase-dependent phosphorylation in Wnt/beta-catenin signaling. Mol. Cell. Biol. 2003;23:1379–1389. doi: 10.1128/MCB.23.4.1379-1389.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan M.S., Boesteanu A., Reed A.J., Petrone A.L., Holenbeck A.E., Lerman M.A., Naji A., Caton A.J. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- Josefowicz S.Z., Lu L.F., Rudensky A.Y. Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanei-Ishii C., Nomura T., Takagi T., Watanabe N., Nakayama K.I., Ishii S. Fbxw7 acts as an E3 ubiquitin ligase that targets c-Myb for nemo-like kinase (NLK)-induced degradation. J. Biol. Chem. 2008;283:30540–30548. doi: 10.1074/jbc.M804340200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattri R., Cox T., Yasayko S.A., Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- Kim J.M., Rasmussen J.P., Rudensky A.Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- Klein L., Khazaie K., von Boehmer H. In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc. Natl. Acad. Sci. USA. 2003;100:8886–8891. doi: 10.1073/pnas.1533365100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenen H.J., Smeets R.L., Vink P.M., van Rijssen E., Boots A.M., Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008;112:2340–2352. doi: 10.1182/blood-2008-01-133967. [DOI] [PubMed] [Google Scholar]
- Kohm A.P., Carpentier P.A., Anger H.A., Miller S.D. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- Kortenjann M., Nehls M., Smith A.J., Carsetti R., Schüler J., Köhler G., Boehm T. Abnormal bone marrow stroma in mice deficient for nemo-like kinase, Nlk. Eur. J. Immunol. 2001;31:3580–3587. doi: 10.1002/1521-4141(200112)31:12<3580::aid-immu3580>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Kretschmer K., Apostolou I., Hawiger D., Khazaie K., Nussenzweig M.C., von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 2005;6:1219–1227. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- Kurahashi T., Nomura T., Kanei-Ishii C., Shinkai Y., Ishii S. The Wnt-NLK signaling pathway inhibits A-Myb activity by inhibiting the association with coactivator CBP and methylating histone H3. Mol. Biol. Cell. 2005;16:4705–4713. doi: 10.1091/mbc.E05-05-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.K., Chen H.M., Mathis D., Benoist C. Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nat. Immunol. 2017;18:1238–1248. doi: 10.1038/ni.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine A.G., Arvey A., Jin W., Rudensky A.Y. Continuous requirement for the TCR in regulatory T cell function. Nat. Immunol. 2014;15:1070–1078. doi: 10.1038/ni.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.O., Rudensky A.Y. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat. Rev. Immunol. 2016;16:220–233. doi: 10.1038/nri.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Lin F., Zhuo C., Deng G., Chen Z., Yin S., Gao Z., Piccioni M., Tsun A., Cai S. PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J. Biol. Chem. 2014;289:26872–26881. doi: 10.1074/jbc.M114.586651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindley S., Dayan C.M., Bishop A., Roep B.O., Peakman M., Tree T.I. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes. 2005;54:92–99. doi: 10.2337/diabetes.54.1.92. [DOI] [PubMed] [Google Scholar]
- Lu L., Barbi J., Pan F. The regulation of immune tolerance by FOXP3. Nat. Rev. Immunol. 2017;17:703–717. doi: 10.1038/nri.2017.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel P.Y., Ouaked N., Rückert B., Karagiannidis C., Welz R., Blaser K., Schmidt-Weber C.B. Molecular mechanisms underlying FOXP3 induction in human T cells. J. Immunol. 2006;176:3593–3602. doi: 10.4049/jimmunol.176.6.3593. [DOI] [PubMed] [Google Scholar]
- McGeachy M.J., Stephens L.A., Anderton S.M. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J. Immunol. 2005;175:3025–3032. doi: 10.4049/jimmunol.175.5.3025. [DOI] [PubMed] [Google Scholar]
- Moran A.E., Holzapfel K.L., Xing Y., Cunningham N.R., Maltzman J.S., Punt J., Hogquist K.A. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawski P.A., Mehra P., Chen C., Bhatti T., Wells A.D. Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J. Biol. Chem. 2013;288:24494–24502. doi: 10.1074/jbc.M113.467704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie H., Zheng Y., Li R., Guo T.B., He D., Fang L., Liu X., Xiao L., Chen X., Wan B. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat. Med. 2013;19:322–328. doi: 10.1038/nm.3085. [DOI] [PubMed] [Google Scholar]
- Ohkawara B., Shirakabe K., Hyodo-Miura J., Matsuo R., Ueno N., Matsumoto K., Shibuya H. Role of the TAK1-NLK-STAT3 pathway in TGF-beta-mediated mesoderm induction. Genes Dev. 2004;18:381–386. doi: 10.1101/gad.1166904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasare C., Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- Rocheleau C.E., Yasuda J., Shin T.H., Lin R., Sawa H., Okano H., Priess J.R., Davis R.J., Mello C.C. WRM-1 activates the LIT-1 protein kinase to transduce anterior/posterior polarity signals in C. elegans. Cell. 1999;97:717–726. doi: 10.1016/s0092-8674(00)80784-9. [DOI] [PubMed] [Google Scholar]
- Rudra D., deRoos P., Chaudhry A., Niec R.E., Arvey A., Samstein R.M., Leslie C., Shaffer S.A., Goodlett D.R., Rudensky A.Y. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat. Immunol. 2012;13:1010–1019. doi: 10.1038/ni.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta A., Li B., Song X., Bembas K., Zhang G., Katsumata M., Saouaf S.J., Wang Q., Hancock W.W., Shen Y., Greene M.I. TGF-beta and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc. Natl. Acad. Sci. USA. 2008;105:14023–14027. doi: 10.1073/pnas.0806726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S., Sanjo H., Tsujimura T., Ninomiya-Tsuji J., Yamamoto M., Kawai T., Takeuchi O., Akira S. TAK1 is indispensable for development of T cells and prevention of colitis by the generation of regulatory T cells. Int. Immunol. 2006;18:1405–1411. doi: 10.1093/intimm/dxl082. [DOI] [PubMed] [Google Scholar]
- Sauer S., Bruno L., Hertweck A., Finlay D., Leleu M., Spivakov M., Knight Z.A., Cobb B.S., Cantrell D., O’Connor E. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. USA. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J.H., Xiao C., Paschal A.E., Bailey S.T., Rao P., Hayden M.S., Lee K.Y., Bussey C., Steckel M., Tanaka N. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X., Li B., Xiao Y., Chen C., Wang Q., Liu Y., Berezov A., Xu C., Gao Y., Li Z. Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep. 2012;1:665–675. doi: 10.1016/j.celrep.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Deng L., Ea C.K., Xia Z.P., Chen Z.J. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- Szypowska A.A., de Ruiter H., Meijer L.A., Smits L.M., Burgering B.M. Oxidative stress-dependent regulation of Forkhead box O4 activity by nemo-like kinase. Antioxid. Redox Signal. 2011;14:563–578. doi: 10.1089/ars.2010.3243. [DOI] [PubMed] [Google Scholar]
- Takeda H., Kawasaki A., Takahashi M., Yamada A., Koike T. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of phosphorylated compounds using a novel phosphate capture molecule. Rapid Commun. Mass Spectrom. 2003;17:2075–2081. doi: 10.1002/rcm.1154. [DOI] [PubMed] [Google Scholar]
- Taylor A.W., Kitaichi N. The diminishment of experimental autoimmune encephalomyelitis (EAE) by neuropeptide alpha-melanocyte stimulating hormone (alpha-MSH) therapy. Brain Behav. Immun. 2008;22:639–646. doi: 10.1016/j.bbi.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahl J.C., Drees C., Heger K., Heink S., Fischer J.C., Nedjic J., Ohkura N., Morikawa H., Poeck H., Schallenberg S. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. 2014;41:722–736. doi: 10.1016/j.immuni.2014.10.012. [DOI] [PubMed] [Google Scholar]
- Valmori D., Raffin C., Raimbaud I., Ayyoub M. Human RORγt+ TH17 cells preferentially differentiate from naive FOXP3+Treg in the presence of lineage-specific polarizing factors. Proc. Natl. Acad. Sci. USA. 2010;107:19402–19407. doi: 10.1073/pnas.1008247107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt J., Coffer P.J. Post-translational modification networks regulating FOXP3 function. Trends Immunol. 2014;35:368–378. doi: 10.1016/j.it.2014.06.005. [DOI] [PubMed] [Google Scholar]
- van Loosdregt J., Vercoulen Y., Guichelaar T., Gent Y.Y., Beekman J.M., van Beekum O., Brenkman A.B., Hijnen D.J., Mutis T., Kalkhoven E. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood. 2010;115:965–974. doi: 10.1182/blood-2009-02-207118. [DOI] [PubMed] [Google Scholar]
- van Loosdregt J., Brunen D., Fleskens V., Pals C.E., Lam E.W., Coffer P.J. Rapid temporal control of Foxp3 protein degradation by sirtuin-1. PLoS One. 2011;6:e19047. doi: 10.1371/journal.pone.0019047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt J., Fleskens V., Fu J., Brenkman A.B., Bekker C.P., Pals C.E., Meerding J., Berkers C.R., Barbi J., Gröne A. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. 2013;39:259–271. doi: 10.1016/j.immuni.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt J., Fleskens V., Tiemessen M.M., Mokry M., van Boxtel R., Meerding J., Pals C.E., Kurek D., Baert M.R., Delemarre E.M. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity. 2013;39:298–310. doi: 10.1016/j.immuni.2013.07.019. [DOI] [PubMed] [Google Scholar]
- Viglietta V., Baecher-Allan C., Weiner H.L., Hafler D.A. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J. Exp. Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voo K.S., Wang Y.H., Santori F.R., Boggiano C., Wang Y.H., Arima K., Bover L., Hanabuchi S., Khalili J., Marinova E. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc. Natl. Acad. Sci. USA. 2009;106:4793–4798. doi: 10.1073/pnas.0900408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L.S., Chodos A., Eggena M., Dooms H., Abbas A.K. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J. Exp. Med. 2003;198:249–258. doi: 10.1084/jem.20030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y.Y., Chi H., Xie M., Schneider M.D., Flavell R.A. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat. Immunol. 2006;7:851–858. doi: 10.1038/ni1355. [DOI] [PubMed] [Google Scholar]
- Wang W., Zhou G., Hu M.C., Yao Z., Tan T.H. Activation of the hematopoietic progenitor kinase-1 (HPK1)-dependent, stress-activated c-Jun N-terminal kinase (JNK) pathway by transforming growth factor beta (TGF-beta)-activated kinase (TAK1), a kinase mediator of TGF beta signal transduction. J. Biol. Chem. 1997;272:22771–22775. doi: 10.1074/jbc.272.36.22771. [DOI] [PubMed] [Google Scholar]
- Wang C., Chen T., Zhang N., Yang M., Li B., Lü X., Cao X., Ling C. Melittin, a major component of bee venom, sensitizes human hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating CaMKII-TAK1-JNK/p38 and inhibiting IkappaBalpha kinase-NFkappaB. J. Biol. Chem. 2009;284:3804–3813. doi: 10.1074/jbc.M807191200. [DOI] [PubMed] [Google Scholar]
- Wang S., Zhang Y., Wang Y., Ye P., Li J., Li H., Ding Q., Xia J. Amphiregulin Confers Regulatory T Cell Suppressive Function and Tumor Invasion via the EGFR/GSK-3β/Foxp3 Axis. J. Biol. Chem. 2016;291:21085–21095. doi: 10.1074/jbc.M116.717892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L.M., Rudensky A.Y. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- Zaiss D.M., van Loosdregt J., Gorlani A., Bekker C.P., Gröne A., Sibilia M., van Bergen en Henegouwen P.M., Roovers R.C., Coffer P.J., Sijts A.J. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. 2013;38:275–284. doi: 10.1016/j.immuni.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y.A., Rahnama M., Wang S., Sosu-Sedzorme W., Verheyen E.M. Drosophila Nemo antagonizes BMP signaling by phosphorylation of Mad and inhibition of its nuclear accumulation. Development. 2007;134:2061–2071. doi: 10.1242/dev.02853. [DOI] [PubMed] [Google Scholar]
- Zhang H.H., Li S.Z., Zhang Z.Y., Hu X.M., Hou P.N., Gao L., Du R.L., Zhang X.D. Nemo-like kinase is critical for p53 stabilization and function in response to DNA damage. Cell Death Differ. 2014;21:1656–1663. doi: 10.1038/cdd.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.Y., Li S.Z., Zhang H.H., Wu Q.R., Gong J., Liang T., Gao L., Xing N.N., Liu W.B., Du R.L., Zhang X.D. Stabilization of ATF5 by TAK1-Nemo-like kinase critically regulates the interleukin-1β-stimulated C/EBP signaling pathway. Mol. Cell. Biol. 2015;35:778–788. doi: 10.1128/MCB.01228-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.