

Abstract
Background
Activation of autocrine VEGF-VEGFR2 signalling in tumour cells activates cell proliferation, survival, and angiogenesis, all of which are crucial for tumour progression. Ovarian cancer-associated antigen 66 (OVA66) is now known to be overexpressed in multiple tumours and plays a role in tumour development, but the underlying mechanisms has not been fully investigated.
Methods
We employed ovarian and cervical cancer cells and mouse models to detect the role of OVA66 in angiogenesis, growth, and metastasis of cancer cells. Immunofluorescence and western blot were used to determine the function of OVA66 in regulating autocrine VEGF-VEGFR2 signalling. Immunohistochemistry and bioinformatics analysis were used to detect the correlation of OVA66 and VEGF expression.
Findings
OVA66 overexpression in the cancer cell lines promoted VEGF secretion, tumour growth and angiogenesis in vitro and in vivo. Conversely, shRNA-mediated OVA66 knockdown had the opposite effects. Mechanistically, OVA66 overexpression was found to boost an autocrine VEGF–VEGFR2 positive-feedback signalling loop in the tumour cells, leading to amplified effect of VEGF on tumour angiogenesis and proliferation and increased migration in vitro and in vivo, respectively. Finally, we identified a significant positive correlation between the expression levels of OVA66 and VEGF in ovarian and cervical cancer specimens, and found that OVA66 was significantly associated with advanced ovarian cancer.
Interpretation
We identify a novel function for OVA66 in regulating an autocrine VEGF–VEGFR2 feed-forward signalling loop that promotes tumour progression and angiogenesis.
Fund
This work was supported by the National Natural Science Foundation of China (81602262); and Excellent Youth Scholar Program of Tongji University (2015KJ062).
Keywords: OVA66, Tumour, VEGF, Autocrine, VEGFR2 signalling
Graphical abstract
Highlights
-
•
OVA66 promotes VEGF secretion and angiogenesis of ovarian and cervical cancer cells in vitro and in vivo.
-
•
OVA66 enhances activation of autocrine VEGF-VEGFR2 signalling.
-
•
OVA66 amplifies VEGF-induced angiogenesis and proliferation of ovarian and cervical cancer cells.
-
•
OVA66 can promote migratory potential of ovarian and cervical cancer cells by enhancing autocrine VEGF-VEGFR2 signalling.
-
•
Expression level of OVA66 positively correlated with VEGF expression significantly in ovarian and cervical cancer patients.
1. Introduction
Signalling through vascular endothelial growth factor A (VEGF) and its receptors (VEGFR1/2/3) on tumour cells stimulates angiogenesis and thus plays a crucial role in the progression, dissemination, and metastasis of numerous cancers, including ovarian and cervical cancers [1]. Indeed, the human VEGF-specific monoclonal antibody bevacizumab and small molecule inhibitors of the tyrosine kinase activity of VEGFRs (e.g., sunitinib, pazopanib) have effectively improved the survival of patients with advanced cervical and ovarian cancer [[2], [3], [4], [5]]. Nevertheless, there remain a number of gaps in our understanding of the intracellular and intercellular mechanisms that regulate VEGF production and signalling in ovarian and cervical cancers.
Many studies have demonstrated that VEGF and VEGFRs are co-expressed in subsets of tumour cells, and contributes to tumorigenesis and angiogenesis [6]. Classically, VEGF secreted by tumour cells acts in a paracrine manner to stimulate angiogenesis via interaction with VEGFRs on endothelial cells. However, tumour cell-derived VEGF also functions as an autocrine factor to regulate cancer cells. Recent studies have shown that VEGF can promote cell proliferation, migration, invasion and survival through an autocrine activation of VEGFR1, VEGFR2 and NRP1 [[6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17]]. Autocrine VEGF-VEGFR signalling also stimulates VEGF secretion, thus sustaining an autocrine feed-forward loop in the tumour cells [[10], [11], [12]].
Ovarian cancer-associated antigen 66 (OVA66, Hugo Gene Nomenclature Committee: 24306), also known as NUDC Domain Containing 1 (NUDCD1) and Chronic Myelocytic Leukaemia Tumour Antigen 66 (CML66), one of the highly immunogenic proteins known as a cancer/testis antigens, was first identified by serological analysis of recombinant cDNA expression libraries [18]. Since then, OVA66 has been shown to be overexpressed in multiple tumours and cell lines [19,20]. Previous research in our laboratory demonstrated that OVA66 silencing in HeLa cells inhibited cell proliferation, migration, and invasion in vitro and slowed xenograft growth in nude mice [20]. In NIH3T3 fibroblasts, OVA66 overexpression induces oncogenic transformation by hyperactivating the phosphoinositide 3-kinase (PI3K)–AKT and ERK1/2 signalling pathway [21]. In human ovarian and cervical cancer cells, the effects of OVA66 are at least partially dependent on signalling through the insulin-like growth factor 1 receptor [22]. Intriguingly, inhibition of OVA66 expression in HeLa cells causes significant downregulation of VEGF expression [20]; however, whether or how this might occur in tumour cells is unknown.
To address this knowledge gap, we overexpressed or silenced OVA66 expression in human ovarian and cervical cancer cell lines and examined the effects on VEGF secretion and angiogenesis in vitro and in vivo. We found that OVA66 overexpression increased VEGF secretion, enhanced the effect of VEGF on proliferation, migration, and angiogenesis of cancer cells in vitro, as well as enhanced tumour growth and metastasis formation in mouse models. Moreover, we demonstrated that the mechanism of action of OVA66 involves promotion of an autocrine feed-forward loop of VEGF–VEGFR2 signalling, and that these effects are attenuated by treatment with the receptor tyrosine kinase inhibitor sunitinib. Thus, our data identify a novel role for OVA66 in promoting tumour growth and progression via amplification of autocrine VEGF–VEGFR2 signalling.
2. Materials and methods
2.1. Cell culture and construction of stable cell lines
Human ovarian cancer cell lines (SKOV3 and HO8910), human cervical cancer cell lines (HeLa and SiHa), and human umbilical vein endothelial cells (HUVECs) were purchased from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cell identity was confirmed by short tandem repeat analysis, and mycoplasma tests were negative. All cells were maintained in Dulbecco's modified Eagle's medium (DMEM; HyClone, USA) supplemented with 10% foetal bovine serum (FBS; Gibco, USA). Cell lines with stable knockdown or overexpression of OVA66 were established as previously described [23]. Briefly, OVA66-knockdown or control cells were generated by infection with retrovirus encoding OVA66-specific (OVA66-shRNA) or control short hairpin RNAs (NC-shRNA) in the presence of 4 μg/ml polybrene. Cells were selected by culturing for 3 days in medium containing a lethal concentration of puromycin and then for 1 week in 0.5 μg/ml puromycin. Resistant single cell colonies were isolated and expanded for further study. OVA66-overexpressing or control cells were generated by transfection with pIRESpuro3-OVA66 or empty plasmid (Clontech, USA) using Lipofectamine 2000 (Invitrogen, USA), and stable cell lines were selected with puromycin as described above.
2.2. Cell proliferation and VEGF secretion assays
Cell proliferation was measured using a Cell Counting Kit-8 (Dojindo, Japan). Tumour cell production of VEGF was measured using a human VEGF Quantikine ELISA Kit (R&D Systems, MN) according to the manufacturer's instructions. In brief, equal number of cancer cells were seeded in 6-well plates and serum starved (medium lacking FBS) for 24 h. The cells were then treated for 2 h with 30 ng/ml of recombinant human (rh) VEGF165 (PeproTech, UK) in serum-free medium containing 10 μM Sunitinib (Calbiochem, CA, USA) or vehicle (dimethyl sulfoxide, DMSO) for 2 h. The cells were rinsed twice with phosphate-buffered saline (PBS) and incubated with fresh serum-free medium for an additional 24 h. The culture supernatants were collected and analysed for secreted VEGF by ELISA.
2.3. Preparation of conditioned medium and HUVEC tube formation assay
Cancer cells with stable OVA66 knockdown or overexpression were cultured to 80% confluency in complete medium, washed, and serum starved for 24 h. The culture supernatant (conditioned medium) was then collected and filtered through a 0.22-μm filter (Millipore). HUVECs were serum starved for 3–6 h, resuspended in conditioned medium supplemented with 1% FBS, and added (6 × 104 cells/well) to a 96-well plate pre-coated with 60 μl/well of growth factor-reduced Matrigel (# 354230; BD Biosciences, Sweden). The plates were incubated at 37 °C for 6 h, and the capillary network was then imaged under a light microscope and photographed. The number of branch points in three random fields of view per well were counted.
2.4. Cell migration assay
Cell migration assays were performed using 6.5-mm diameter Transwell chambers (8-μm pore size, BD Biosciences) according to the manufacturer's instructions. Cancer cells were resuspended in 200 μl serum-free medium supplemented with or without 30 ng/μl rhVEGF DMSO, and/or sunitinib (10 μM). The lower chambers were filled with 600 μl medium containing 10% FBS. The plates were incubated at 37 °C for 16 h, the membranes were rinsed with PBS, and the cells on the upper membrane side were removed using cotton-tipped swabs. The migrated cells on the lower side were fixed and stained with crystal violet (Beyotime Biotechnology, China). The membranes were visualised under a light microscope (Olympus, Japan) at 200× magnification and photographed. The number of cells in five randomly selected fields per well were counted and averaged.
2.5. Animal experiments and tumour histology
For the mouse xenograft experiments, OVA66-overexpressing HO8910 cells or OVA66-knockdown HeLa cells were injected subcutaneously into the flank of female nude mice obtained from SLAC Laboratory Animal Co. Ltd. (Shanghai, China) (3–4 weeks of age) at 4 × 106 cells per mouse. Tumour size was monitored twice per week and the volume was calculated using the formula: V = width2 × length/2 (mm3). After 4 weeks, the mice were euthanised and the tumours were removed for analysis. For metastasis assays, OVA66-knockdown HeLa cells (106 cells per mouse) were injected via the tail vein into nude mice. The animals were randomised, divided into two groups (n = 10/group), and administered either 100 mg/kg sunitinib (Sutent, Pfizer) resuspended in a vehicle or same amount of vehicle daily by oral gavage. Three weeks later, the mice were euthanised and the lungs were removed and processed for histology. In brief, the harvested organs and primary tumours were fixed, embedded in paraffin, and stained with haematoxylin and eosin or processed for immunohistochemical staining. All animal protocols were reviewed and approved by the Ethics Review Committee for Animal Experimentation at Shanghai Tenth People's Hospital.
2.6. Receptor tyrosine kinase signalling antibody array
To explore the molecular mechanism by which VEGF signals in OVA66 cells, we used a Human Receptor Tyrosine Kinase Phosphorylation Antibody Array G1 (RayBiotech, USA), which measures the relative levels of phosphorylation of 71 human receptor or non-receptor tyrosine kinases. Briefly, SKOV3 cells transfected with OVA66-shRNA or NC-shRNA (2 × 106/sample) were serum starved for 16 h and then incubated with medium containing 10% FBS for 1 h. Cells were then harvested and processed according to the kit instructions. All experiments were performed according to the manufacturer's instructions. The signal intensities were quantified with Photoshop CS2 software (Adobe, USA). Positive and negative controls were included to normalise the results.
2.7. Analysis of VEGFR2 signalling
Cancer cells were seeded in 6-well plates overnight, serum starved for 24 h, and then treated with or without rhVEGF for the indicated times. For some experiments, the cells were treated with DMSO or sunitinib (10 μM) for 1 h before addition of rhVEGF. After incubation, the cells were rinsed twice with PBS and lysed in M-PER buffer (Thermo Scientific, USA) containing protease and phosphatase inhibitor cocktails (Thermo Scientific). Protein in the lysates was quantified using a BCA Protein Assay Reagent Kit (Thermo Scientific). Samples were then analysed by western blotting (see below). The following primary antibodies were purchased from Cell Signaling Technology (USA): VEGFR2 (#9698, RRID: AB_11178792), phosphorylated (p)-VEGFR2 (Tyr1175) (#2478, RRID: AB_331377), ERK1/2 (#4695, RRID: AB_390779), p-ERK1/2 (#4370, RRID: AB_2315112), AKT (#4685, RRID: AB_2225340), p-AKT (#4060, RRID: AB_2341228), p-FAK (#8556, RRID: AB_10891442), p-P38 (#4511, RRID: AB_2139682), p-PLCγ (#8713, RRID: AB_10890863), p-SRC (#6943, RRID: AB_10013641), and β-actin (#3700, RRID: AB_2242334). Primary VEGFR2 antibody from Abcam (ab39256, RRID: AB_883437) was also used. A primary anti-OVA66 antibody was obtained from Santa Cruz Biotechnology (USA; #sc-271650, RRID: AB_10708575).
2.8. Western blot analysis
Proteins in cell lysates (40 μg protein per sample) were resolved by SDS-PAGE analysis and transferred to PVDF membranes (Bio-Rad, USA). The membranes were blocked by incubation in 5% non-fat milk for 1 h at room temperature and then incubated with the primary antibodies at 4 °C overnight. After washing three times in Tris-buffered saline containing 0.1% Tween, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody (KPL, USA) for 1 h at room temperature. Protein bands were detected with Enhanced Chemiluminescence reagent (Millipore, USA).
2.9. Bioinformatic analysis
The Gene Expression Profiling Interactive Analysis online tool (http://gepia.cancer-pku.cn) was used to explore the relationship between OVA66 and VEGF expression in cervical and ovarian cancer specimens [23,24]. Genes A and B were set as “NUDCD1” and “VEGFA”, and the datasets “Cervical squamous cell carcinoma and endocervical adenocarcinoma” and “Ovarian serous cystadenocarcinoma” were selected. Spearman's correlation coefficient was selected for analysis.
2.10. Ovarian cancer tissue microarray (TMA) and immunohistochemistry (IHC)
A tissue microarray (TMA) containing 5 normal ovarian tissues and 94 ovarian cancer tissues was purchased from Shanghai Super Biotek company (Shanghai, China). TMA sections (4 μm thick) were rehydrated in a graded series of alcohol, and heated for 20 min at 99 °C in 10 mM sodium citrate buffer (pH 6.0) for antigen retrieval. After cooling, the sections were incubated overnight at 4 °C with a mouse monoclonal antibody against OVA66 (CML66 C-4, #sc-271650; Santa Cruz Biotechnology). Antibody binding was detected using an IHC kit (Gene Tech, China; #GK600505). Quantification of immunostaining was based on both the percentage of positive cells and the intensity of staining, and was independently evaluated by two clinical pathologists who were blinded to sample identity. The final staining score was obtained by multiplying the intensity and percentage scores. The median score was used to dichotomise the tumour biopsies into low and high OVA66 expression groups.
2.11. Statistical analysis
All analyses were performed with SPSS version 20.0 (SPSS Inc., USA). Between-group differences were evaluated using Student's t-test for continuous variables and Pearson's χ2, Pearson's corrected χ2, or Fisher's exact test for categorical variables, as appropriate. Data are presented as the mean ± standard deviation (SD). Pairwise gene correlation analysis was performed using Spearman's correlation statistics. All P values were two-sided, and P < .05 was considered statistically significant.
3. Results
3.1. OVA66 overexpression in cancer cells enhances VEGF secretion and angiogenesis in vitro
Previous cDNA microarray analyses revealed that OVA66 knockdown markedly reduces the expression of several metastasis-related genes [20], including VEGF. To determine whether OVA66 regulates VEGF expression in ovarian and/or cervical cancer, we knocked down or overexpressed OVA66 in HeLa, SiHa (cervical), HO8910, and SKOV3 (ovarian) cancer cell lines. Western blot analysis confirmed efficient inhibition and overexpression of OVA66 in the appropriate cell lines (Fig. 1A). VEGF protein in the cell supernatants was quantified by ELISA and found to be significantly reduced and enhanced by OVA66 knockdown and overexpression, respectively (Fig. 1B). Because tumour-secreted VEGF can promote endothelial cell angiogenesis in a paracrine manner [11], we also assessed tube formation by HUVECs after treatment with conditioned medium collected from OVA66-knockdown or -overexpressing cells. The results indicate that conditioned medium from OVA66-overexpressing cells significantly promoted tube formation (Fig. 1C). However, addition of recombinant OVA66 protein itself had no effect on either VEGF production by cancer cells or the angiogenic ability of HUVECs (Supplemental Fig. 1). Moreover, OVA66 was not detected in the conditioned medium from cancer cells (data not shown), suggesting that OVA66 was not a secreted protein, and might play its role within cells. Taken together, these data indicate that OVA66 plays a role in promoting VEGF secretion by tumour cells, which as a result promotes angiogenesis.
Fig. 1.

OVA66 promotes VEGF secretion and in vitro angiogenesis of cancer cells.
(A) HeLa, SKOV3 and SiHa cells were transfected with scramble NC shRNA or OVA66 shRNA followed by puromycin selection. HO8910 cells was transfected with empty vector or OVA66-expressing constructs. The knockdown and overexpression efficiency was detected by immunoblotting with β-actin as a loading control. (B) VEGF protein in the cell supernatants was quantified by ELISA. (C) Representative images of HUVEC tube formation. HUVECs were treated with conditioned medium of stable cell lines. Tube formation was quantified in three randomly selected fields. Magnification, ×100. Bars represent the number of branch points (mean ± SD) of three independent experiments. *P < .05, **P < .01.
3.2. OVA66 promotes tumour angiogenesis, growth, and metastasis in vivo
To verify the in vitro data, we next determined whether OVA66 overexpression promotes VEGF expression by tumour cells in vivo. OVA66-knockdown HeLa cells or OVA66-overexpressing HO8910 cells were injected subcutaneously into nude mice, tumour growth was monitored, and excised tumours were analysed. As shown in Fig. 2A, cancer cells with high OVA66 expression tend to generate bigger tumours. Consistently, more Ki67+ cells were detectable in xenografts with higher OVA66 expression. To detect angiogenesis, we examined expression of CD31, an endothelial cell marker, and VEGF in tumour sections by IHC staining. Indeed, tumours derived from OVA66-knockdown HeLa cells exhibited significantly reduced VEGF expression and microvessel density compared with tumours derived from NC-shRNA-expressing cells (Fig. 2B, D). Conversely, VEGF expression and microvessel density were both higher in HO8910 tumours overexpressing OVA66 than in the corresponding control tumours (Fig. 2C, E). These results strongly suggest that OVA66 promotes VEGF secretion and angiogenesis of ovarian and cervical cancer cells in vivo.
Fig. 2.

OVA66 promotes angiogenesis and tumour growth in vivo in xenograft mouse model.
(A) OVA66-knockdown HeLa cells or OVA66-overexpressing HO8910 cells were injected subcutaneously into nude mice (BALB/C nu/nu, n = 4 for each group). Tumour size (mm3) was monitored twice a week and tumour weights (g) were measured. Data were showed as the mean size ± SD. Difference of OVA66, Ki67, VEGFA and CD31 expression was analysed by IHC. Representative staining images of xenografts from OVA66 knockdown HeLa cells (B) and OVA66 overexpressed HO8910 cells (C). Histograms of statistical MVD in tumour sections from OVA66-knockdown HeLa cells (D) and OVA66-overexpressed HO8910 cells (E) were shown. Bars represent the number of CD31-positive microvessels (mean ± SD) in three randomly selected fields.
3.3. OVA66 enhances autocrine VEGF–VEGFR2 signalling in cancer cells
To investigate the mechanism by which OVA66 promotes VEGF expression, we performed a phospho-proteomics-based study using a high-throughput receptor tyrosine kinase (RTK) phospho-antibody microarray. The results showed that phosphorylation of multiple receptor tyrosine kinases was altered in OVA66-knockdown cells compared with the control cells; of these, the most significant change was a decrease in phosphorylation of VEGFR2 on tyrosine (Tyr) 1175. Phosphorylation of VEGFR2 on Tyr-951, Tyr1059 and Tyr1214 were also significantly downregulated, but to a lesser extent than Tyr1175 (Fig. 3A, Supplemental Table 1). Given that VEGFR2 is the principal receptor for VEGF signalling and that autocrine VEGF–VEGFR2 signalling induces VEGF secretion in many cell types [[10], [11], [12], [13], [14]], these findings suggest that OVA66 might promote VEGF expression in ovarian and cervical cancer cells by enhancing autocrine signalling through VEGFR2. To investigate this, we performed immunofluorescence staining of p-VEGFR2 in OVA66-knockdown HeLa cells incubated with or without rhVEGF. Notably, although rhVEGF treatment markedly increased p-VEGFR2 levels in control cells, this enhancement was dramatically attenuated by OVA66 knockdown (Fig. 3B).
Fig. 3.

OVA66 enhances activation of autocrine VEGF-VEGFR2 signalling pathway of cancer cells.
(A) RTKs with shRNA/NC ratio lower than 0.75 in SKOV3 cells from RTK signalling phospho-antibody microarrays were shown. (B) The phosphorylation level of VEGFR2 with or without VEGF stimulation in stable HeLa cells was analysed by fluorescence microscope microscopy. Magnification, ×400. HeLa (C) and SKOV3 (D) cells with stable OVA66 knockdown and HO8910 cells with OVA66 overexpression (E) were stimulated by 30 ng/ml rhVEGF for indicated time. Phosphorylation of VEGFR2 and downstream Src/AKT, MAPK, PLCγ/ERK1/2 and FAK were analysed by western blot. β-actin served as a loading control.
Next, we examined the effects of rhVEGF on signalling pathways downstream of VEGFR2 in cancer cells with different expression of OVA66. As shown in Fig. 3C–E, rhVEGF stimulation increased the phosphorylated (activated) VEGFR2 as well as multiple downstream kinases, including p-ERK, p-AKT, p-FAK, p-P38, p-SRC, and p-PLCγ, compared with unstimulated cells. These effects were markedly reduced by OVA66 knockdown and, conversely, significantly enhanced by OVA66 overexpression (Fig. 3C, D, E). Taken together, these data demonstrate that OVA66 significantly enhances autocrine VEGF–VEGFR2 signalling in both ovarian and cervical cancer cells.
3.4. Sunitinib attenuates OVA66-mediated enhancement of VEGF secretion, proliferation, and angiogenesis in vitro
To confirm that activation of these downstream events was mediated via VEGF–VEGFR2 signalling, we treated HeLa and HO8910 cells with sunitinib, a multi-target tyrosine kinase inhibitor that blocks VEGFR2, but not VEGFR1/3. As expected, OVA66 overexpression in both cell types significantly increased the appearance of p-VEGFR2, p-ERK1/2, and p-AKT levels after rhVEGF stimulation, whereas OVA66 knockdown had the opposite effect. However, pre-treatment with sunitinib dramatically reduced the rhVEGF-stimulated phosphorylation of VEGFR2, ERK1/2 and AKT, to levels comparable to those observed in OVA66-knockdown HeLa cells or HO8910 cells transfected with empty vector (Fig. 4A, B). Taken together, these data indicate that OVA66-induced amplification of autocrine VEGF–VEGFR2 signalling requires VEGFR2 tyrosine kinase activity.
Fig. 4.

OVA66 amplifies autocrine VEGF-VEGFR2 signalling induced VEGF production, angiogenesis and proliferation of cancer cells.
OVA66-knockdown HeLa cells (A) and OVA66-overexpression HO8910 cells (B) were starved and treated with DMSO or Sunitinib followed by rhVEGF stimulation. P-VEGFR2, P-AKT and P-ERK1/2 as well as total VEGFR2, AKT and ERK1/2 were measured by western blot. β-actin was used as the loading control. The results shown are representative of three independent experiments. (C, D) Secreted VEGF concentration in the supernatant was measured by ELISA assay. (E, F) In vitro angiogenesis was assessed by tube formation assay using HUVECs. The result was quantified by number of branch points per field. (G, H) Proliferation of cancer cells was measured by CCK-8 assay. All bar graphs were expressed by fold change relative to corresponding unstimulated group. All samples were analysed in triplicates (unpaired Student's t-test, mean ± SD, *P < .05, **P < .01, ***P < .001).
Next, we asked whether OVA66 plays a role in the enhanced cell proliferation and VEGF production stimulated by autocrine VEGF–VEGFR2 signalling in the cancer cells. As expected, rhVEGF treatment increased VEGF secretion by control NC-shRNA-expressing HeLa cells compared with unstimulated cells; however, this effect was markedly reduced by OVA66 knockdown (Fig. 4C). Conversely, OVA66-overexpressing HO8910 cells produced significantly more VEGF than control cells in response to rhVEGF stimulation (Fig. 4D). Sunitinib treatment abrogated the enhancement of VEGF secretion induced by OVA66 overexpression in both cell lines (Fig. 4C, D; see also Supplemental Fig. 2A, B for quantification of VEGF in these experiments). In accordance with these findings, OVA66 promoted VEGF induced angiogenesis assessed by tube formation by HUVECs in a manner that precisely mirrored the relative VEGF concentrations in each of the supernatants (Fig. 4E, F; see Supplemental Fig. 2C, D for quantification of branch points).
Similarly, we found that OVA66 also played a crucial role in autocrine VEGF–VEGFR2 signalling for cancer cell proliferation. rhVEGF treatment promoted the growth of NC-shRNA HeLa cells and OVA66-overexpressing HO8910 cells; however, the fold change of proliferation rate was significantly reduced or increased by OVA66 knockdown or overexpression, respectively (Fig. 4G, H and Supplemental Fig. 3A, B). Moreover, sunitinib treatment reduced the proliferation rate of both cell lines to the levels of unstimulated cells (Fig. 4G, H and Supplemental Fig. 3A, B). Interestingly, even in the absence of rhVEGF stimulation, the proliferation of OVA66-knockdown HeLa cells and OVA66-overexpressing HO8910 cells was significantly lower and higher, respectively, than the corresponding control cells (Fig. 4 and Supplemental Fig. 3), which strongly suggests that OVA66 may also be involved in promoting the proliferation of cancer cells through non-VEGF–VEGFR2 pathways.
To confirm these results, we inhibited VEGF–VEGFR2 signalling by transfecting HeLa and HO8910 cells with VEGFR2-specific or scrambled siRNA. Indeed, VEGFR2 silencing abolished rhVEGF-stimulated phosphorylation of VEGFR2, ERK1/2, and AKT in OVA66-knockdown and control HeLa cells as well as in OVA66-overexpressing and control HO8910 cells (Supplemental Fig. 5A, B). Consistent with these effects, VEGFR2-specific siRNA also abolished rhVEGF-induced VEGF secretion by the same cells (Supplemental Fig. 5C, D). Thus, the observed effects of OVA66 upregulation or downregulation are mediated through an autocrine VEGF–VEGFR2 positive-feedback signalling loop in ovarian and cervical cancer cells.
3.5. OVA66 increases the migratory and metastatic potential of cancer cells in vitro and in vivo
We previously demonstrated that OVA66 overexpression can promote the migration of ovarian, breast and hepatic cancer cells [22]. Therefore, we next investigated whether it depends on autocrine VEGF–VEGFR2 signalling. Using in vitro Transwell chemotaxis assays, we found that OVA66 overexpression in HO8910 cells and knockdown in HeLa cells significantly increased and decreased, respectively, cell migration under both control and rhVEGF stimulation conditions (Fig. 5A). In addition, rhVEGF-induced stimulation of migration was abolished in the presence of sunitinib treatment (Fig. 5B), confirming that OVA66-mediated regulation of migration required VEGFR2 signalling.
Fig. 5.

OVA66 enhances VEGF induced cell migration capacity and in vivo metastasis.
Cell migration was determined using a Transwell system. (A) Representative results of VEGF-induced migrated cells of stable OVA66 knockdown or overexpression cell lines with or without sunitinib treatment. (B) Histogram were showed by the fold change of migrated cells relative to corresponding unstimulated cells. (C) Representative lung images of nude mice (n = 10) with macroscopic metastasis tumours generated by OVA66-knockdown HeLa cells with or without Sunitinib treatment. Bar graph showed the statistical result of metastasis among different groups. (D) Representative images of histological examination of mouse lungs for metastatic nodules in different groups. Statistical analysis of differences between groups was performed by unpaired Student's t-test, mean ± SD, *P < .05, **P < .01, ***P < .001.
To further explore the effects on migration, we examined tumour metastasis in vivo. OVA66-knockdown or control HeLa cells were injected intravenously into two groups of nude mice, which were administered with sunitinib (100 mg/kg) or vehicle daily for the following 3 weeks. The mice were then sacrificed and the lungs were excised for analysis. We found that the number of lung metastases was significantly lower in mice injected with cells expressing OVA66-shRNA compared with NC-shRNA (Fig. 5C). Sunitinib administration dramatically reduced the metastasis of both control and OVA66-knockdown HeLa cells, although OVA66 knockdown was more effective than control (Fig. 5C, D). These data indicate that OVA66 overexpression promotes cell migration in vitro and in vivo, at least partly through the autocrine VEGF–VEGFR2 signalling loop.
3.6. OVA66 expression is positively correlated with VEGF expression and disease progression in patients with ovarian cancer
OVA66 is expressed at significantly higher levels in ovarian, colon, and gastric tumour tissues than in normal tissues [18]. To determine whether OVA66 expression correlates with the clinicopathologic characteristics of ovarian cancer patients, we performed IHC staining of a TMA composed of 94 ovarian cancer tissues and 5 normal ovarian tissues. Using the median staining score as the cut-off, we then segregated the patients into two groups based on low and high tumour expression of OVA66. As shown in Table 1, we found that high expression of OVA66 was significantly associated with tumour size (P = .039), advanced stage (P = .020), and distant metastasis (P = .034).
Table 1.
Correlation of OVA66 expression with clinicopathologic features in ovarian cancer tissues.
| Clinicopathologic parameters | OVA66 expression |
P value | |
|---|---|---|---|
| Low | High | ||
| Total cases | 46 | 48 | |
| Age (years) | |||
| ≤60 | 37 | 42 | .350 |
| >60 | 9 | 6 | |
| Histological type | |||
| Serous cystadenocarcinoma | 19 | 25 | .258 |
| Mucinous cystadenocarcinoma | 13 | 7 | |
| Endometrioid adenocarcinoma | 14 | 16 | |
| Histologic grade | |||
| I | 13 | 7 | .609† |
| II | 15 | 14 | |
| III | 18 | 27 | |
| T classification | |||
| T1 | 27 | 18 | .039† |
| T2 | 11 | 15 | |
| T3 | 8 | 15 | |
| Lymph node metastasis | |||
| Negative | 42 | 46 | .634⁎ |
| Positive | 4 | 2 | |
| Distant metastasis | |||
| Negative | 40 | 33 | .034 |
| Positive | 6 | 15 | |
| TNM stage | |||
| I–II | 37 | 28 | .020 |
| III–IV | 9 | 20 | |
P value was calculated by Mantel-Haenszel test.
P values were calculated by Pearson's corrected χ2 test.
To determine whether a correlation existed between OVA66 and VEGF expression, we also analysed the TMA for VEGF levels. This analysis identified a significant positive correlation between OVA66 and VEGF expression levels (R = 0.314, P = .002; Fig. 6A, B). To confirm this result, we performed pairwise gene correlation analysis between the OVA66 and VEGF gene sets using the Gene Expression Profiling Interactive Analysis website. Consistent with our IHC analysis, significant positive correlations between OVA66 and VEGF expression were identified in both ovarian cancer (R = 0.36, P < .001) and cervical cancer (R = 0.34, P < .001; Fig. 6C, D) datasets.
Fig. 6.

Expression of OVA66 is positively correlated with VEGF expression in both ovarian and cervical tumour tissues.
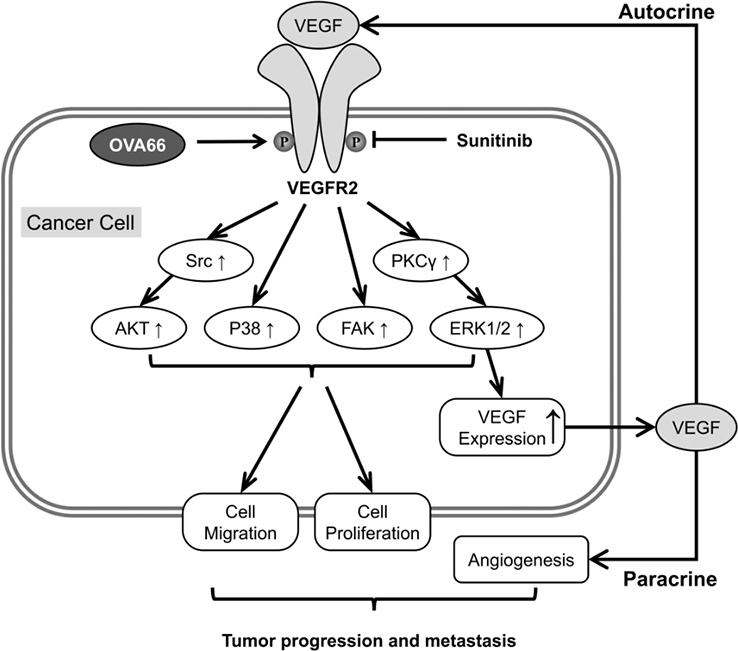
(A) Immunohistochemical staining of ovarian tumour microarray tissues with anti-OVA66 and anti-VEGF antibody. Representative patient samples with low and high pairwise gene expression were shown. (B) Correlation analysis of genes expression between OVA66 and VEGF in ovarian TMA. (C, D) Pairwise gene correlation analysis of OVA66 and VEGF by GEPIA using Spearman statistics in ovarian cancer and cervical cancer. (E) Schematic model of OVA66 promoting autocrine VEGF-VEGFR2 signalling.
4. Discussion
In this study, we identified a new function for OVA66 in tumour angiogenesis and progression. We found that rhVEGF treatment of ovarian and cervical cancer cells activated VEGFR2 signalling and increased VEGF secretion, confirming the existence of an autocrine VEGF–VEGFR2 feed-forward loop in these cells. We also showed that activation of this loop promoted cancer cell proliferation and migration and acted in a paracrine manner to promote tube formation by endothelial cells. Notably, OVA66-overexpression enhanced rhVEGF-stimulated phosphorylation of VEGFR2 and its downstream signalling components AKT and ERK1/2, as well as VEGF secretion, proliferation, and migration in vitro, all of which were also blocked by sunitinib. In addition, sunitinib attenuated the enhancement of lung metastasis caused by OVA66 overexpression in vivo. These data provide strong evidence that OVA66 enhances the autocrine VEGF–VEGFR2 feed-forward loop by increasing VEGFR2 phosphorylation. OVA66 thus synergises with VEGF to play critical roles in angiogenesis and progression of ovarian and cervical cancer.
OVA66 has been shown to function as an oncoprotein in ovarian and cervical cancer [[20], [21], [22]], colorectal cancer [25], and renal cell carcinoma [26,27], and these studies also showed that OVA66 is involved in activating the insulin-like growth factor 1 receptor–ERK1/2 signalling pathway [22,25] and the lissencephaly-1/dynein signalling pathway [26] in these tumours. Here, we confirmed the ability of OVA66 to promote the development of ovarian and cervical cancer, consistent with the observation that OVA66 expression levels are positively associated with metastasis and advanced disease stage in ovarian cancer patients. In addition, in the present study, we uncovered a new mechanism by which OVA66 functions in enhancing autocrine VEGF–VEGFR2 signalling.
In recent years, autocrine VEGF–VEGFR2 signalling has been recognised to contribute to tumourigenesis in angiogenesis-independent and -dependent manners in a wide range of tumour types, including hepatocellular carcinoma, astrocytoma, skin epithelial cancer, ovarian carcinoma, fibrosarcoma, and Barrett's-associated neoplasm [[11], [12], [13], [14], [15], [16], [17]]. PLC–ERK [12,13], PI3K–mTOR [11], FAK, and AKT [16] have been implicated as dominant pathways/components involved in autocrine VEGF–VEGFR2 signalling. Previous studies reported that autocrine VEGF–VEGFR2 signalling can be promoted via NRP1-mediated stabilisation and recycling of cytosolic VEGFR2 to the plasma membrane [14] and via upregulation of NRP1, VEGFR2, and VEGF expression in response to epidermal growth factor receptor signalling [17]. In the present study, we found that OVA66 knockdown in SKOV3 ovarian cancer cells reduced FBS-induced activation of multiple receptor tyrosine kinases, of which VEGFR2 activation was the most affected. OVA66 overexpression increased rhVEGF-induced activation of VEGFR2, P38, FAK, PI3K–AKT, and PLCγ–ERK1/2 pathways, which was abrogated by sunitinib treatment. This regulatory function of OVA66 provides novel insight into autocrine VEGF signalling.
The role of VEGF in inducing angiogenesis is well characterised. In lung cancer, autocrine VEGF–VEGFR2 signalling has been shown to promote VEGF production and trigger angiogenesis [11]. In this study, we showed that OVA66 overexpression increases VEGF production by cancer cells in vitro and in vivo in a VEGFR2 signalling-dependent manner. Using experimental and bioinformatics approaches, we demonstrated that OVA66 and VEGF expression is positively associated in human ovarian cancer specimens, which is consistent with our observation that elevated OVA66 expression increased tumour angiogenesis in vitro and in vivo. Taken together, our results demonstrate that high OVA66 levels promote VEGF production and angiogenesis by enhancing an autocrine VEGF–VEGFR2 feed-forward loop.
Earlier studies demonstrated that autocrine VEGF–VEGFR2 signalling can promote cancer cell proliferation [12,13,17] and protect against apoptosis [15,16] in an angiogenesis-independent manner. Here, we showed that ovarian and cervical cancer cells overexpressing OVA66 exhibited enhanced rhVEGF-stimulated proliferation and migration, which were inhibited by sunitinib. To our knowledge, this is the first report that OVA66 promotes the migration of ovarian and cervical cancer cells depending on autocrine VEGF–VEGFR2 signalling.
In summary, we found that high OVA66 levels increase activation of an autocrine VEGF–VEGFR2 feed-forward loop by enhancing VEGFR2 phosphorylation, and that this promotes tumour progression via an autocrine mechanism and also promotes angiogenesis via a paracrine mechanism (Fig. 6E). Our results identify a potential new strategy for combining OVA66-targeted therapy with anti-angiogenic therapy to achieve synergistic effects on ovarian and cervical cancer.
Acknowledgments
Acknowledgements
We thank Anne M. O'Rourke, PhD, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding sources
This work was supported by grants from the National Natural Science Foundation of China (81602262); and Excellent Youth Scholar Program of Tongji University (2015KJ062). The funders of this work had no role in study design, data collection, data analysis, data interpretation, writing of the report, or decision to submit the article for publication.
Declaration of interests
The authors declare that they have no competing interests.
Authors' contributions
FS designed and performed the experiments, analysed the data, and wrote the manuscript; QC assisted with the design of the experiments and manuscript modification; WR assisted with the experiments relating to signalling pathways; RZ assisted with the animal experiments; YW reviewed the manuscript; HG assisted with the design of the project; and QW designed the project and the experiments, analysed the data, and reviewed the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2019.02.051.
Contributor Information
Ying Wang, Email: ywang@sibs.ac.cn.
Hailiang Ge, Email: gehl@shsmu.edu.cn.
Qing Wei, Email: weiqing1971@tongji.edu.cn.
Appendix A. Supplementary data
Supplementary material
References
- 1.Masoumi Moghaddam S., Amini A., Morris D.L., Pourgholami M.H. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2012 Jun;31(1–2):143–162. doi: 10.1007/s10555-011-9337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tewari K.S., Sill M.W., Penson R.T., Huang H., Ramondetta L.M., Landrum L.M. Bevacizumab for advanced cervical cancer: final overall survival and adverse event analysis of a randomised, controlled, open-label, phase 3 trial (Gynecologic Oncology Group 240) Lancet. 2017 Oct 7;390(10103):1654–1663. doi: 10.1016/S0140-6736(17)31607-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oza A.M., Cook A.D., Pfisterer J., Embleton A., Ledermann J.A., Pujade-Lauraine E. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015 Aug 1;16(8):928–936. doi: 10.1016/S1470-2045(15)00086-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monk B.J., Mas Lopez L., Zarba J.J., Oaknin A., Tarpin C., Termrungruanglert W. Phase II, open-label study of pazopanib or lapatinib monotherapy compared with Pazopanib plus Lapatinib combination therapy in patients with advanced and recurrent cervical cancer. J. Clin. Oncol. 2010 Aug 1;28(22):3562–3569. doi: 10.1200/JCO.2009.26.9571. [DOI] [PubMed] [Google Scholar]
- 5.Ledermann J.A., Embleton A.C., Raja F., Perren T.J., Jayson G.C., Rustin G.J.S. Cediranib in patients with relapsed platinum-sensitive ovarian cancer (ICON6): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2016 Mar 12;387(10023):1066–1074. doi: 10.1016/S0140-6736(15)01167-8. [DOI] [PubMed] [Google Scholar]
- 6.Goel H.L., Mercurio A.M. VEGF targets the tumour cell. Nat. Rev. Cancer. 2013 Dec;13(12):871–882. doi: 10.1038/nrc3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barr M.P., Gray S.G., Gately K., Hams E., Fallon P.G., Davies A.M. Vascular endothelial growth factor is an autocrine growth factor, signaling through neuropilin-1 in non-small cell lung cancer. Mol. Cancer. 2015 Feb 20;14:45. doi: 10.1186/s12943-015-0310-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao Y., Guangqi E., Wang E., Pal K., Dutta S.K., Bar-Sagi D. VEGF exerts an angiogenesis-independent function in cancer cells to promote their malignant progression. Cancer Res. 2012 Aug 15;72(16):3912–3918. doi: 10.1158/0008-5472.CAN-11-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perrot-Applanat M., Benedetto M.D. Autocrine functions of VEGF in breast tumor cells: adhesion, survival, migration and invasion. Cell Adhes. Migr. 2012 Nov 17;6(6):547–553. doi: 10.4161/cam.23332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Golfmann K., Meder L., Koker M., Volz C., Borchmann S., Tharun L. Synergistic anti-angiogenic treatment effects by dual FGFR1 and VEGFR1 inhibition in FGFR1-amplified breast cancer. Oncogene. 2018 Jul;3:1. doi: 10.1038/s41388-018-0380-3. [DOI] [PubMed] [Google Scholar]
- 11.Chatterjee S., Heukamp L.C., Siobal M., Schöttle J., Wieczorek C., Peifer M. Tumor VEGF:VEGFR2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J. Clin. Invest. 2013 Apr 1;123(4):1732–1740. doi: 10.1172/JCI65385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q., Yu C., Peng S., Xu H., Wright E., Zhang X. Autocrine VEGF signaling promotes proliferation of neoplastic Barrett's epithelial cells through a PLC-dependent pathway. Gastroenterology. 2014 Feb 1;146(2):461–472.e6. doi: 10.1053/j.gastro.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng S., Wang Y., Peng H., Chen D., Shen S., Peng B. Autocrine vascular endothelial growth factor signaling promotes cell proliferation and modulates sorafenib treatment efficacy in hepatocellular carcinoma. Hepatology. 2014;60(4):1264–1277. doi: 10.1002/hep.27236. [DOI] [PubMed] [Google Scholar]
- 14.Hamerlik P., Lathia J.D., Rasmussen R., Wu Q., Bartkova J., Lee M. Autocrine VEGF–VEGFR2–Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J. Exp. Med. 2012 Mar 12;209(3):507–520. doi: 10.1084/jem.20111424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sher I., Adham S.A., Petrik J., Coomber B.L. Autocrine VEGF-A/KDR loop protects epithelial ovarian carcinoma cells from anoikis. Int. J. Cancer. 2009 Feb 1;124(3):553–561. doi: 10.1002/ijc.23963. [DOI] [PubMed] [Google Scholar]
- 16.Lee J., Ku T., Yu H., Chong K., Ryu S.-W., Choi K. Blockade of VEGF-A suppresses tumor growth via inhibition of autocrine signaling through FAK and AKT. Cancer Lett. 2012 May 28;318(2):221–225. doi: 10.1016/j.canlet.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 17.Lichtenberger B.M., Tan P.K., Niederleithner H., Ferrara N., Petzelbauer P., Sibilia M. Autocrine VEGF Signaling synergizes with EGFR in tumor cells to promote epithelial Cancer development. Cell. 2010 Jan 22;140(2):268–279. doi: 10.1016/j.cell.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 18.Jin S., Wang Y., Zhang Y., Zhang H.-Z., Wang S.-J., Tang J.-Q. Humoral immune responses against tumor-associated antigen OVA66 originally defined by serological analysis of recombinant cDNA expression libraries and its potentiality in cellular immunity. Cancer Sci. 2008 Aug;99(8):1670–1678. doi: 10.1111/j.1349-7006.2008.00860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang X.-F., Wu C.J., McLaughlin S., Chillemi A., Wang K.S., Canning C. CML66, a broadly immunogenic tumor antigen, elicits a humoral immune response associated with remission of chronic myelogenous leukemia. Proc. Natl. Acad. Sci. 2001 Jun 19;98(13):7492–7497. doi: 10.1073/pnas.131590998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q., Li M., Wang Y., Zhang Y., Jin S., Xie G. RNA interference targeting CML66, a novel tumor antigen, inhibits proliferation, invasion and metastasis of HeLa cells. Cancer Lett. 2008 Sep 28;269(1):127–138. doi: 10.1016/j.canlet.2008.04.035. [DOI] [PubMed] [Google Scholar]
- 21.Rao W., Xie G., Zhang Y., Wang S., Wang Y., Zhang H. OVA66, a tumor associated protein, induces oncogenic transformation of NIH3T3 cells. PLoS ONE. 2014 Mar 14;9(3) doi: 10.1371/journal.pone.0085705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao W., Li H., Song F., Zhang R., Yin Q., Wang Y. OVA66 increases cell growth, invasion and survival via regulation of IGF-1R–MAPK signaling in human cancer cells. Carcinogenesis. 2014 Jul 1;35(7):1573–1581. doi: 10.1093/carcin/bgu070. [DOI] [PubMed] [Google Scholar]
- 23.GEPIA (Gene Expression Profiling Interactive Analysis) http://gepia.cancer-pku.cn/ [Internet]. [cited 2018 Jan 15]. Available from:
- 24.Tang Z., Li C., Kang B., Gao G., Li C., Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017 Jul 3;45(W1):W98–102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han B., Zhang Y.-Y., Xu K., Bai Y., Wan L.-H., Miao S.-K. NUDCD1 promotes metastasis through inducing EMT and inhibiting apoptosis in colorectal cancer. Am. J. Cancer Res. 2018 May 1;8(5):810–823. [PMC free article] [PubMed] [Google Scholar]
- 26.He H., Dai J., Wang X., Qian X., Zhao J., Wang H. NudCD1 affects renal cell carcinoma through regulating LIS1/dynein signaling pathway. Am. J. Transl. Res. 2018 Feb 15;10(2):519–524. [PMC free article] [PubMed] [Google Scholar]
- 27.Wang R.-J., Wang N., Cui G., Chen Y., Zhong H., Tang J. The impact of NudCD1 on renal carcinoma cell proliferation, migration, and invasion. Eur. Rev. Med. Pharmacol. Sci. 2018 Feb;22(3):671–677. doi: 10.26355/eurrev_201802_14292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material