

SUMMARY
Kes1/Osh4 is a member of the conserved, but functionally enigmatic, oxysterol binding protein-related protein (ORP) superfamily that inhibits phosphatidylinositol transfer protein (Sec14)-dependent membrane trafficking through the trans-Golgi (TGN)/endosomal network. We now report that Kes1, and select other ORPs, execute cell-cycle control activities as functionally non-redundant inhibitors of the G1/S transition when cells confront nutrient-poor environments and promote replicative aging. Kes1-dependent cell-cycle regulation requires the Great-wall/MASTL kinase ortholog Rim15, and is opposed by Sec14 activity in a mechanism independent of Kes1/Sec14 bulk membrane-trafficking functions. Moreover, the data identify Kes1 as a non-histone target for NuA4 through which this lysine acetyltransferase co-modulates membrane-trafficking and cell-cycle activities. We propose the Sec14/Kes1 lipid-exchange protein pair constitutes part of the mechanism for integrating TGN/endosomal lipid signaling with cell-cycle progression and hypothesize that ORPs define a family of stage-specific cell-cycle control factors that execute tumor-suppressor-like functions.
Graphical Abstract
In Brief
Huang et al. demonstrate the yeast oxysterol-binding protein (ORP) homolog Kes1, and other ORPs, are inhibitors of the G1/S transition. They show that Kes1 is a non-histone target for the NuA4 lysine acetyltransferase and participates in a phosphatidylinositol-4-phopshate-dependent mechanism for integrating TGN/endosomal lipid signaling with cell-cycle progression.
INTRODUCTION
The trans-Golgi (TGN) network/endosomal system consists of highly dynamic organelles that sit at the confluence of antero-grade and endocytic membrane flow (Glick and Nakano, 2009). Thus, the TGN/endosomal system serves as an important membrane-sorting station, and as a major node for intracellular signaling. TGN/endosomal signaling programs involve multiple branches of lipid metabolism, with a robust and essential interface between lipid metabolism and membrane trafficking through these compartments (Bard and Malhotra, 2006; Graham and Burd, 2011).
Lipid exchange proteins play crucial roles in coordinating lipid metabolism with phosphoinositide signaling and membrane trafficking in TGN/endosomes (Bankaitis et al., 1990, 2010; Graham and Burd, 2011). This circuit is controlled by opposing actions of two lipid-exchange proteins: the PtdIns/PtdCho transfer protein Sec14 and Kes1/Osh4 (Fang et al., 1996; Mousley et al., 2012). Sec14 couples PtdCho biosynthesis with PtdIns-4-P production (Cleves et al., 1991; Schaaf et al., 2008). In turn, PtdIns-4-P signaling drives TGN/endosomal trafficking (Graham and Burd, 2011). Kes1 is one of seven yeast oxysterol binding-related proteins (ORPs) (Fang et al., 1996; Beh et al., 2001), and it antagonizes Sec14 activity by sequestering PtdIns-4-P from its pro-trafficking effectors (Li et al., 2002; de Saint-Jean et al., 2011). Recent studies indicate that a Kes1 sterol/PtdIns-4-P exchange cycle controls amplitude of this trafficking “brake” by tuning the ability of Kes1 to sequester PtdIns-4-P (Mousley et al., 2012).
Although conserved across the Eukaryota, ORPs remain functionally enigmatic, with conflicting data as to whether ORPs are intermembrane lipid transfer proteins or not (Schulz and Prinz, 2009; Georgiev et al., 2011; Stefan et al., 2011; Mousley et al., 2012). This confusion reflects the lack of information regarding the biological activities of these proteins. Kes1 is the exception. It is not only unique among yeast ORP proteins in its role as antagonist to Sec14-dependent PtdIns-4-P signaling in the TGN/endosomal system, but Kes1 is also an antagonist of nitrogen stress responses and mammalian target of rapamycin complex 1(TORC1) signaling (Mousley et al., 2012). Those results forecast complex physiological functions for Kes1, and identify homeostatic regulation of Kes1 activity as a key point of control at the interface of membrane-trafficking, nutrient signaling, and cell-cycle progression. Issues of how cells coordinate membrane growth and trafficking with nutrient signaling and entry into a new mitotic cycle define fundamental, but poorly understood, questions in contemporary cell biology (McCusker and Kellogg, 2012).
In this study, we characterize Kes1 activity as a negative regulator of progression through the G1 stage of the cell cycle when cells confront nutrient-prohibitive environments. We further demonstrate that the antagonistic actions of Kes1 and Sec14 in TGN/endosomal membrane trafficking extend to cell-cycle contexts. Strikingly, these cell-cycle involvements are independent of the roles of these proteins in regulating bulk membrane trafficking. Thus, Kes1 and Sec14 execute dual functions in membrane-trafficking and cell-cycle control, and are well positioned to coordinate cell-cycle progression with membrane flux through the TGN/endosomal system. We also show that Kes1 activity is subject to regulation by the NuA4 lysine acetyltransferase (KAT). Together, the data reveal an interface between TGN/endosomal lipid signaling and cell-cycle control, identify the NuA4 KAT complex as a regulator of that interface, and suggest ORPs as stage-specific inhibitors of cell-cycle progression in eukaryotes.
RESULTS
Kes1-Induced Cell-Cycle Arrest Exhibits Signatures of Quiescence
The first in vivo clues regarding ORP function came from demonstrations that Kes1 is an antagonist of Sec14 and PtdIns-4-P signaling in the context of membrane trafficking through the TGN-endosomal system. That functional readout is complex, however, as it is Kes1 LOF that restores growth and membrane trafficking to cells deficient in normally essential Sec14 activities (Fang et al., 1996; Li et al., 2002). A simpler context was suggested by Mousley et al. (2012), who demonstrated that excessive Kes1 activity evokes an amino acid remedial growth arrest by attenuating TORC1 signaling. Upon elevation of Kes1 activity, most cells arrest in G1, as determined by their terminal pheno-type of large unbudded cells with 1N genome content (Figure 1A). Furthermore, G1 cells with excessive Kes1 activity are unable to initiate a new round of cell division in nutrient-replete media, even though these cells continue to increase their size at a rate similar to that of matched controls, demonstrating that inability to initiate cell division was not due to a general growth deficiency (Figure S1). These collective data forecast a role for Kes1 in the G1 phase when cells evaluate nutrient cues before committing to another round of cell division.
Figure 1. The Quiescence Response Is Activated upon Kes1-Induced Cell-Cycle Arrest.

WT cells harboring the indicated plasmids were cultured in synthetic defined medium lacking uracil ± Dox as indicated. All experiments represent the averages of three independent biological replicates, and error bars indicate standard deviations.
(A) Asynchronous cultures were fixed and stained with propidium iodide and the cell-cycle distribution was determined based on genome content by fluorescence-activated cell sorting analysis. Plotted is the percentage of cells in G1.
(B) Total RNA fractions were isolated and Msn2/4 and Gis1 target gene expression was surveyed by qRT-PCRand normalizedtoACT1 gene expression.
(C) Glycogen and trehalose were extracted and hydrolyzed enzymatically to glucose with amylase and trehalase, respectively. Glucose was quantified and normalized per OD600 of cells.
(D) Cells were washed and resuspended in water to an initial density of OD600 of ~0.9 (starting OD600 indicated as dashed line at top) prior to treatment with zymolyase 20T (150 μg/mL) for 30 min. Cell lysis was assessed by scoring the reduction in the OD600 of the suspended cell cultures. See also Figures S1 and S2.
The MBF/SBF regulon defines a set of genes whose expression is required to negotiate the G1/S transition or START (Spellman et al., 1998; Zaman et al., 2008). Transcription of multiple genes regulated by MBF and SBF was reduced ≥3-fold relative to control in otherwise wild-type (WT) yeast cells programmed for elevated (and doxycycline-repressible) expression of Kes1 or Kes1Y97F (a dominant-active Kes1 defective in sterol-binding but competent for PtdIns-4-P binding; Im et al., 2005; Mousley et al., 2012). The affected genes included those encoding G1 cyclins (CLN1, CLN2, PCL1, PCL2, PCL9), a gene encoding a homeobox transcription factor that promotes early cell-cycle box gene transcription (YOX1), and AXL2 whose product specifies spatial organization of bud emergence from the mother cell during G1 (Figure 1B). That reduced AXL2 expression was of physiological significance was validated by the abnormal bipolar budding phenotype exhibited by Kes1-overex-pressing haploid cells (Figure S2A). Bipolar budding is a signature property of diploid yeast with normal Axl2 levels. Reduced transcription of MBF/SBF-controlled genes were not observed in cells expressing the nonfunctional Kes1K109A defective in PtdIns-4-P binding (Li et al., 2002; de Saint-Jean et al., 2011), nor were these observed in cells where KES1 or kes1Y97F transcription was repressed by Dox (Figure 1B).
Reciprocally, Msn2/4- and Gis1-dependent transcriptional responses were activated in yeast with elevated Kes1 activity. Both Kes1- and Kes1Y97F-arrested cells displayed ≥3-fold elevations in Msn2/4 and Gis1 target gene expression relative to control (HSP12, HSP78, HSP82, and HSP104, and SSA3, GRE1, and CTT1; Figure 1B). Those derangements were not observed in cells expressing Kes1K109A, nor in cells where ectopic KES1 or KES1Y97F expression was repressed by Dox. Activation of the Msn2/4 and Gis1 regulon is a hallmark of cells transitioning into quiescence in response to nutritional stress (Beck and Hall, 1999; Pedruzzi et al., 2000).
Kes1/Kes1Y97F-arrested cells displayed metabolic signatures of quiescent cells as well. The storage carbohydrate glycogen and the stress protectant trehalose accumulate in yeast transitioning into quiescence (Lillie and Pringle, 1980). Both metabolites precociously accumulated in Kes1- and Kes1Y97F-arrested cells incubated under nutrient-replete conditions, but not in cells expressing Kes1K109A (Figure 1C). Phosphorylated long-chain sphingoid base (LCBP) accumulation is repressed by the Pho85 cyclin, and elevated LCBP represents another metabolic signature of quiescent cells (Lester et al., 2013). Intracellular LCBP levels of both the dihydro- and phyto-classes were dramatically increased in Kes1- and Kes1Y97F-arrested yeast relative to control (Figure S2B). Moreover, quiescent yeast exhibit reinforced cell walls. This property was scored by resistance to zymolyase digestion (Krause and Gray, 2002). Either Kes1- or Kes1Y97F-overexpressing induced zymolyase resistance to cells cultured in nutrient-rich media (Figure 1D). Elevated Kes1K109A expression had no such effect.
Kes1 Antagonizes PKA Signaling
Entry into quiescence requires the balanced downregulation of both TORC1 (NH4+-dependent) and protein kinase A ([PKA]; carbohydrate-dependent) pathways for proliferative signaling (Wei et al., 2008). Two readouts confirmed PKA signaling was downregulated in Kes1/Kes1Y97F-arrested yeast. First, PKA-catalyzed phosphorylation of the model substrate choline kinase (Cki1; Ramachandran and Herman, 2011) was reduced in Kes1/Kes1Y97F-arrested cells, but not in cells expressing Kes1K109A (Figure 2A). Second, processing bodies (P-bodies) are sites for storage, degradation and quality control of translationally repressed mRNAs. These structures, marked by Dcp2 and Xrn1, accumulate in cells as cytoplasmic puncta when PKA signaling is depressed (Ramachandran et al., 2011). Elevated Kes1/Kes1Y97F expression (but not of Kes1K109A) induced robust formation and stable maintenance of cytoplasmic Dcp2- and Xrn1-positive P-body puncta (Figure 2B).
Figure 2. Kes1-Dependent Cell-Cycle Arrest Exhibits Reduced PKA Signaling and Is Rim15 Dependent.

(A) Left panel: WT yeast co-transformed with YCp(PCUP1::myc-CKI1), and either YCp(PDOX::KES1), YCp(PDOX :: KES1Y97F), or YCp(PDOX::kes1K109A) were cultured in selective media containing 10 μg/mL Dox. Subsequently, KES, KES1Y97F, or kes1K109A expression was induced for 12 hr (–Dox), or not (+Dox; 10 μg/mL), and cells were challenged with CuSO4 (100 μM for 1 hr) to induce myc-Cki1 expression. Lysates were prepared, normalized for total protein (10 μg loaded), and phosphorylated and non-phosphorylated myc-Cki1species and GAPDH were visualized by immunoblotting with anti-myc and anti-GAPDH antibodies, respectively. Phosphorylated and non-phosphorylated myc-Cki1species (myc-Cki1P and myc-Cki1, respectively) and GAPDH are identified on the left. Fractional proportion of phosphorylated myc-Cki1 as determined by densitometric analysis using image lab software (Bio-Rad) is shown at the bottom. Experiments represent the average of three independent biological replicates.
(B) Yeast cells expressing either Dcp2-GFP or Xrn1-GFP were transformed with the indicated plasmids and cultured in uracil-free medium ± Dox (10 μg/mL), as indicated, at 30°C. GFP profiles are shown with corresponding DIC images. Scale bar, 5 μm
(C) WT (BY4742) and isogenic rim15Δ cells carrying the indicated plasmids were spotted in 10-fold dilution series on uracil-free media ± Dox (10 μg/mL) and incubated at 30°C. Images were taken after 72 hr of incubation.
(D) Glycogen and trehalose were enzymatically hydrolyzed to glucose and quantified, as described in the legend to Figure 1C, in WT or rim15Δ cells harboring YCp(PDOX::KES1), YCp(PDOX :: KES1Y97F), or YCp(PDOX::kes1K109A), and cultured in synthetic defined medium lacking uracil ± Dox (10 μg/mL; black and gray bars, respectively).
(E) WT and rim15D yeast (as indicated), harboring YCp(PDOX::KES1), YCp(PDOX :: KES1Y97F), or YCp(PDOX::kes1K109A) plasmids, were cultured in synthetic defined medium lacking uracil ± Dox (10 μg/mL) as indicated, washed, and resuspended in water to an initial density of OD600 of ~1.0 (starting OD600 depicted as dashed line at top) prior to treatment with zymolyase 20T (150 μg/mL). Cell lysis was assessed by scoring the reduction in the OD600 of the suspended cell cultures after 30 min of incubation. Data represent the average of three independent biological replicates.
Kes1-Chaperoned Entry into Quiescence Is Rim15 Dependent
The Rim15 kinase regulates exit from the cell cycle into quiescence (Cameroni et al., 2004; Swinnen et al., 2006). Ras/PKA, TORC1, and Pho85 cyclin-dependent kinase phosphate- sensing pathways promote cell proliferation, and inhibit entry into quiescence, by attenuating Rim15 activity in response to nutrient availability. If Kes1 operates upstream of Rim15 in the quiescence pathway, Rim15 activity will be required for entry of Kes1/Kes1Y97F-overexpressing cells into quiescence. Indeed, rim15Δ cells were resistant to Kes1/Kes1Y97F-overexpression (Figure 2C). Rim15-deficient cells also failed to present the quiescence signatures typically associated with Kes1/Kes1Y97F-mediated growth arrest. That is, neither glycogen nor trehalose accumulated in rim15Δ mutants induced for Kes1/Kes1Y97F expression upon withdrawal of Dox from the medium (Figure 2D), and rim15Δ mutant cell walls failed to acquire zymolyase resistance (Figure 2E). Steady-state Kes1 and Kes1Y97F levels were appropriately elevated in rim15Δ cells after Dox withdrawal.
Cells Lacking Kes1 Are Less Responsive to Nutrient Deprivation Signals
Kes1 loss-of-function (LOF) mutants did not appropriately exit the cell cycle when confronted by nutrient limitation. Whereas KES1 cells ceased to proliferate upon reaching a density of ca. 1.5 × 108 cells/mL (Figure 3A, 12–48 hr time points), parallel kes1Δ cell cultures consistently reached cell densities of ca. 3 × 108 cells/mL (Figure 3A, 12–48 hr time points). Cells lacking Kes1 ultimately arrested, and the DNA content of kes1Δ cell populations in stationary phase exhibited G1 signatures indistinguishable from those of their isogenic KES1+ counterparts (Figure 3B). However, kes1Δ-arrested cells were only ~75% of the size of arrested WT cells (Figure 3C). Thus, Kes1-deficient mutants were licensed to initiate one supernumerary round of cell division under nutrient-prohibitive conditions of such severity that cell expansion could no longer be supported.
Figure 3. Cells Lacking Kes1 Respond Aberrantly to Nutrient-Restrictive Environments.

Unless otherwise indicated, all experiments represent the averages of three independent biological replicates, and error bars indicate SDs.
(A) Cell densities of the indicated yeast strains (y axis) were quantified scored by light microscope hemacytometer counting at the indicated times as cells ceased to divide (x axis). Averages and standard deviations obtained from at least four independent experiments are shown. **p < 0.01, Student’s t test.
(B) DNA content histograms of the indicated yeast strains collected at the 24 hr time point shown in (A) were generated by flow cytometry analyses. The number of cells analyzed in each case (y axis) is plotted as a function of fluorescence per cell (FL2-A; x axis).
(C) Cell size histograms of the indicated yeast strains collected at the 24 hr time point shown in (A) represent the distributions produced by channelyzer cell volume measurements (>30,000 individual cells measured per biological replicate). Cell number is plotted as a function of cell volume (in fL).
(D) Glycogen and trehalose were extracted from logarithmic (black bars) and stationary phase (gray bars) WT and kes1Δ cells grown in nutrient-rich media (YPD), hydrolyzed enzymatically to glucose, and glucose was quantified and normalized per OD600 cells.
(E) WT and kes1Δ yeast were cultured in YPD to logarithmic (black bars) and stationary phase (gray bars), washed and resuspended in water to a density of OD600 of ~0.9 (indicated as dashed line at top) prior to treatment with zymolyase 20T (150 μg/mL). Cell lysis was assessed by the reduction in the OD600 of suspended cell cultures after 30 min of incubation.
The supernumerary cell division phenotype of kes1Δ cells suggested that Kes1 is part of the quiescence response, which is engaged when yeast are challenged with nutrient deprivation. In support, survey of multiple metabolic signatures of quiescence demonstrated that kes1Δ cells were defective in mounting quiescence responses. Relative to WT cells, Kes1-deficient mutants showed ca. 2-fold reductions in the magnitude of glycogen and trehalose accumulation when starved (Figure 3D). Moreover, nutrient-stressed kes1Δ cell walls failed to acquire zymolyase resistance (Figure 3E). Chronological lifespan assays also showed kes1Δ cells displayed accelerated loss of viability when subjected to chronic nutrient limitation.
Sec14 and Kes1 Function in Cell-Cycle Control in Cycling Cells
Kes1 antagonizes Sec14 and PtdIns-4-P signaling in the context of membrane trafficking through the TGN-endosomal system. Hence, we investigated whether introducing a sec14ts allele into kes1Δ cells restored their ability to arrest properly in the face of nutrient exhaustion. Indeed, the license to undergo the supernumerary round of cell division in nutrient-prohibitive environments was partially suppressed in sec14–1ts kes1Δ cells. Notably, this phenotypic suppression was evident for cells cultured at a permissive temperature (30°C), where the sec14–1ts gene product retains sufficient activity to support WT kinetics of bulk membrane trafficking through the secretory and TGN/endosomal pathways.
We examined whether the antagonistic relationship between Sec14 and Kes1 was manifested in cycling cells in nutrient-replete medium. During exponential growth in rich media (YPD), sec14–1ts cells exhibited doubling times that were ca. 10% longer than those of isogenic SEC14 cells (108.7 ± 2.2 versus 93.4 ± 1.3 min, p < 0.05, Student’s t test). This phenotype was reversed by Kes1 LOF, as sec14–1ts kes1Δ cells exhibited WT population doubling times (92.6 ± 6.4 min). The population doubling time and DNA content of asynchronous and exponentially dividing kes1Δ cells did not differ from the corresponding parameters of isogenic KES1 cells (Figure 4A). However, kes1Δ and sec14–1ts cells had altered size homeostasis. Compared with WT, kes1Δ cells were slightly smaller, while sec14–1ts cells were larger (Figure 4B, left). These differences in the population mean cell size were not due to differences in birth size of these strains (Figure 4B, right), and the large cell size of sec14–1ts cells was corrected in sec14–1ts kes1D cells (Figure 4B, left). “Bypass Sec14” mutations that inactivate PtdCho synthesis via the CDP-choline pathway (pct1Δ; Cleves et al., 1991) failed to reduce the size of sec14–1ts cells (Figure 4B).
Figure 4. Mutual Antagonism of Kes1 and Sec14 in Cell-Cycle Control.

(A) DNA content histograms were determined by flow cytometry. Fluorescence per cell (FL2-A) is plotted on the x axis, while the number of cells analyzed for each condition is plotted on the y axis. The average of the fractional representation of cells with G1 content in the population (% G1), and standard deviations (n = 6), are shown, along with the corresponding generation times (Td) in each case. Genotypes are identified at top.
(B) Boxplots reporting the mean and birth cell size (y axis, in fL) of asynchronously proliferating populations of yeast with the indicated genotypes. Each symbol (open circle) corresponds to an independent biological replicate where 30,000–150,000 individual cells were measured for each replicate.
(C) Boxplots depicting the specific rate of cell size increase, k (in hr−1, left panel), and critical size (in fL, right panel) of synchronous cells of the indicated strains are shown.
(D) The relative growth in size during the G1 phase (kTG1, y axis) for the indicated mutants are plotted as a function of the natural logarithm of normalized birth size values (WT = 1). The line depicts the linear fit (obtained with the regression function of Microsoft Excel) of data from all mutants. Established cell-cycle regulator mutants are shown as controls (in red; whi5, defective in the repressor of passage through G1; cln3, defective in a G1 cyclin; bck2, a transcriptional activator of G1 cyclins; previously reported in Soma et al., 2014). Values for WT (black square), the sec14–1ts mutant (light red), and sec14–1ts kes1Δ cells (gray) are highlighted.
Since the decision to initiate a new round of cell division in response to nutrient signaling is exercised in G1, we examined cell-cycle progression of highly synchronous early-G1 daughter cells obtained by centrifugal elutriation. This approach holds the advantage that cell-cycle synchrony is achieved without applying arrest and release regimens that uncouple cell growth from cell division (e.g., shift of cdcts mutants to restrictive temperatures or pheromone arrest). The elutriated early-G1 cells were allowed to progress in the cell cycle, and evaluated in a time course by scoring the incidence of budded cells, and the volume of cells, with a channelyzer. Since initiation of DNA replication in yeast is coupled to the formation of a bud (Johnston et al., 1977), the critical size is defined as the size at which 50% of the cells in a synchronous population have initiated a new round of cell division. KES1+ and kes1Δ cells increased in size at the same rate (Figure 4C, left), and exhibited the same critical size (Figure 4C, right). However the critical size of sec14–1ts cells was larger than that of isogenic WT cells (47.9 ± 2.1 versus 43.9 ± 1.8 fL, p = 0.003, Student’s t test; Figure 4C, right). Because sec14–1ts mutants increased in size at WT rates at 30°C (Figure 4C, left), but did not commit to divide until cells reached a larger critical size, their G1 phase was extended. This protracted G1 phase, in conjunction with the increase in G2/M DNA content observed for sec14–1ts cells (consistent with a second delay in mitosis; data not shown), accounted for the slower doubling time of sec14ts cells at 30°C.
A proper coupling between cell growth and division was reestablished in sec14–1ts cells by Kes1 LOF, even though kes1Δ mutants by themselves had normal critical size and rate of size increase under nutrient-replete conditions. The corrective effects upon Kes1 LOF were specific because loss of Pct1 (i.e., an independent bypass Sec14 condition) failed to restore a proper critical size to sec14–1ts cells (Figure 4C, right).
sec14ts Mutant Exhibits an Unusual Cell Size Control Phenotype
The critical size data obtained from synchronously cycling cell cultures suggested sec14–1ts cells, despite their normal rates of cell size increase, did not correctly register that they had grown sufficiently to commit to another round of cell division. This defect was visualized by plotting the logarithm of birth size against relative rate of cell size increase in the G1 phase of the cell cycle (Figure 4D). Such plots relate the coherence of cell size control mechanisms, and the relevant values for sec14–1ts mutants were compared with analogous values for a series of mutants analyzed previously (Soma et al., 2014). The increased cell growth requirements of sec14–1ts yeast for the commitment of these cells to a new round of cell division were clear in such representations of the data. The sec14–1ts critical size phenotype was unusual in that mutants in growth control processes (e.g., the rpl20b ribosomal protein gene deletion) attain degrees of overall cell growth from birth to START that are indistinguishable from WT cells (Figure 4D). By contrast, the sec14–1ts phenotype was qualitatively similar to the delay observed in cells lacking the G1 cyclin Cln3, and was of at least the same magnitude as that observed upon loss of the Bck2 cell-cycle regulator which acts in parallel with Cln3 (Di Como et al., 1995). This delay was corrected by Kes1 LOF (Figure 4D).
Cell Size and Cell Division Phenotypes Are Associated with Defects in Other ORP Proteins
In addition to Kes1, yeast express three Kes1-like “short” ORPs (Osh5, Osh6, and Osh7) and three “long” versions (Osh1, Osh2, and Osh3). The functional differences between Kes1 and other members of the yeast ORP family were evident in: (1) phenotypic bypass Sec14 contexts (Figures 5A and S3A), and (2) CPY pulse-chase trafficking assays assessing bypass Sec14 pheno-types (Figures 5B and S3B). To determine whether cell-cycle dysregulation associated with Kes1 LOF was a unique property, we examined cells carrying deficiencies in individual members of the Osh protein family. Again, Kes1 LOF was unique in its ability to restore proper size homeostasis to exponentially growing sec14–1ts cells at 30°C, and in reducing mean cell size of exponentially growing cells (Figure 5C).
Figure 5. Functional Specification of Yeast ORPs.

(A) SEC14 or sec14–1ts yeast (as indicated to the left of vertical line) with either a WT complement of yeast ORPs, or the indicated ORP deletion mutations (indicated to the right of the vertical line), were dilution spotted onto YPD agar and incubated at the indicated temperatures for 48 hr. Growth of sec14–1ts derivative strains at 37°C identifies “bypass Sec14” phenotypes.
(B) Yeast strains of the indicated genotype (at top) were cultured to mid-logarithmic growth phase at 30°C, shifted to 37°C for 2 hr, and pulse-radiolabeled with [35S]amino acids for 30 min. After chase with excess unlabeled methionine and cysteine for the indicated times (at right), CPY species were immunoprecipitated from clarified lysates, resolved by SDS-PAGE, and visualized by autoradiography. p2 CPY and mCPY forms are identified at the left.
(C) Boxplot reporting the mean cell size (in fL) of asynchronously proliferating populations of yeast with the indicated genotypes (at the left) in exponentially growing cultures. Each symbol (open circle) corresponds to an independent biological replicate consisting of 30,000–150,000 individual cells.
(D and E) Survival curves for MATα kes1Δ cells (BY4742 derivative) cells (red), compared with otherwise isogenic and experiment-matched KES1+ cells (black), and cells carrying individual deletions in the indicated genes encoding the short Osh proteins (osh5Δ cells, green; osh6Δ, blue) (D), and survival curves for MATα KES1 cells (black; BY4742) compared with otherwise isogenic and experiment-matched cells (black) carrying deletions in the indicated long Osh protein-encoding genes (osh1Δ cells, green; osh2Δ, blue; osh3Δ, red) (E). Mean lifespans are shown in parentheses, along with the number of cells assayed. See also Figure S3.
In other contexts, however, loss of Osh protein function did evoke phenotypes similar to those associated with kes1Δ. For example, Osh1, Osh3, Osh6, or Osh7 LOF restored WT doubling times to sec14–1ts mutants (Figure S3C). Moreover, as was the case for kes1Δ mutants, nutrient-deprived cells lacking any single Osh protein (other than Osh5) inappropriately passed through G1 into another round of cell division (Figure S3D). Analyses of osh1Δ and osh6Δ cell size profiles during starvation demonstrated that those cells phenocopied kes1Δ cells in their reduced cell size upon arrest. Thus, activities of other Osh proteins in cell proliferation resemble, but are not perfectly redundant with, those of Kes1. That Osh5 showed no functional relatedness to Kes1 was of note given it is the closest Kes1 homolog as defined by primary sequence identity/similarity.
Kes1 and Long ORP Defects Extend Replicative Lifespan
As cells deficient in activities of Kes1 or the other Osh proteins (other than Osh5) showed G1 control defects, we determined the effects of Kes1 LOF and of other Osh protein deficiencies on broader aspects of cell aging and longevity. Those parameters were interrogated in the context of replicative lifespan, i.e., the number of divisions mother cells undergo before reaching senescence. Kes1 LOF extended the mean and maximal replicative lifespan of cells continuously cultured in nutrient-replete media (Figure 5D). In those experiments, the WT strain exhibited a replicative lifespan of 27.7 generations, whereas the kes1D derivative exhibited an extended replicative lifespan (35.4 generations). LOF of any one of the three other “short” Kes1-like yeast ORPs had no such effect (Figure 5D). However, LOF of any one of the three “long” ORPs extended replicative lifespan (Figure 5E). Moreover, the small cell size of kes1 mutants is consistent with those of other long-lived mutants with extended replicative lifespan (He et al., 2014). These findings suggest a trade-off between chronological and replicative lifespan assays for Kes1 activity. That is, Kes1 LOF mutants carry greater replicative potential, but are impaired in their ability to enter a growth-arrested state conducive to long chronological lifespan.
Acetylation of Kes1 Residue K109 Inactivates Lipid Exchange In Vitro
Kes1 involvement as a physiological brake on progression through START raises questions regarding how Kes1 activity is itself regulated. Two independent profiling studies of the yeast acetylome identify Kes1 as a protein modified at as many as nine lysine residues (Figure 6A; Henriksen et al., 2012; Madsden et al., 2015). To assess the significance of each modification, Ala was substituted for each acetylated Kes1 Lys residue, and the activity of each protein was evaluated in a bypass Sec14 complementation assay, where Kes1 LOF permits growth of sec14ts cells at 37°C. K109 was the only site for which an Ala substitution ablated Kes1 activity in cells (Figure 6A). This defect was not for trivial reasons as Kes1K109A is a stable protein in vivo (Li et al., 2002). The K109 acetylation was of special interest because K109: (1) sits within the invariant motif that defines the ORP super-family (Figure 6A; Li et al., 2002), and (2) forms a critical contact with the PtdIns-4-P molecule that occupies the Kes1 lipid-binding pocket (Figure 6B; de Saint-Jean et al., 2011). Thus, K109 acetylation potentially compromises the Kes1::PtdIns-4-P interaction.
Figure 6. Functional Analyses of Kes1 Lysine Acetylation.

(A) Top panel: schematic representation of Kes1 is shown. The ORP signature motif is highlighted (Li et al., 2002), as are the lysine acetylation sites identified by acetylome profiling (Henriksen et al., 2012; Madsden et al., 2015). Bottom panel: SEC14 or sec14–1ts yeast expressing Kes1, or the indicated K→A substitution mutants, were dilution spotted onto YPD agar and incubated at the indicated temperatures for 48 hr. The SEC14 KES1 strain (positive control) and sec14–1ts KES1 (negative control at 37°C) are growth controls (two rows), the sec14–1ts, kes10 mutant defines the bypass Sec14 control (the kes10 frameshift allele produces no Kes1 protein; third row).
(B) K109A helps coordinate binding of the PtdIns-4-P head group. Kes1::PtdIns-4-P interactions are shown (PDB: 3SPW). Intermolecular H bonds between the PtdIns-4-P head group and the labeled Kes1 residues are shown by magenta dotted lines. PtdIns-4-P is rendered as green balls and sticks. Kes1 residues are shown in stick mode. Lys109 makes a critical H bond interaction with the phosphate oxygen of PtdIns-4-P (van der Waals molecular surfaces shown). The Lys amino group modified by acetylation is marked by the arrow.
(C) PtdIns-4-P extraction activities for the indicated mutant/post-translationally modified Kes1 proteins were determined at 25°C in a 30 min endpoint format (see the STAR Methods). Mean ± standard deviations from three independent experiments are shown.
(D) Real-time DHE and PtdIns-4-P transport kinetics measured with Kes1 or versions bearing the indicated substitution for, or acetylation of, Lys109. Top panel: DHE transport. Bottom panel: PtdIns-4-P transfer.
(E) Plot of initial DHE transfer rate versus initial PtdIns-4-P transfer rate for WT Kes1 and indicated mutant/post-translationally modified Kes1 proteins. Mean ± standard deviations from at least three independent experiments are shown. See also Figures S4–S7.
To determine the effect of K109 acetylation on PtdIns-4-P binding, Kes1AcK109 was purified, and the site-specific acetylation was confirmed (Figure S4). Kes1 proteins carrying the inactivating K109A, the acetylation-mimetic K109Q, and non-acetylatable K109R substitutions were also purified. Activities of those proteins were then assessed in a liposome-based PtdIns-4-P extraction assay. Relative to Kes1, the Kes1K109A, Kes1K109Q, Kes1K109R, and Kes1AcK109 proteins all exhibited reduced abilities to extract PtdIns-4-P from liposomes in vitro (Figure 6C).
It is not PtdIns-4-P binding/extraction in isolation, rather a heterotypic ergosterol/PtdIns-4-P exchange cycle, that is central to the biochemical mechanism of Kes1 function in vivo (Mousley et al., 2012; Moser von Filseck et al., 2015). Thus, the consequences of K109 acetylation were also analyzed in dehydroergosterol (DHE)/PtdIns-4-P lipid transfer assays that measure inter-liposomal exchange of those lipids in vitro (de Saint-Jean et al., 2011). Indeed, Kes1 was proficient in transfer of both DHE (~9 molecules/Kes1/min) and PtdIns-4-P (~12 molecules/Kes1/min). Kes1K109A and Kes1AcK109 were each reduced only ca. 30% for DHE transfer activity relative to Kes1 (Figures 6D and 6E). By contrast, both Kes1K109A and Kes1AcK109 exhibited ca. 6-fold reductions in PtdIns-4-P transfer activity. Kes1K109Q was also reduced for PtdIns-4-P transfer (~60% reduction relative to Kes1), although the mutant protein exhibited greater DHE and PtdIns-4-P transfer activity than did Kes1AcK109. Kes1K109R showed a ca. 70% increase in DHE transfer activity, but only modest reduction in PtdIns-4-P transfer activity relative to WT Kes1 (Figures 6D and 6E). Purified Kes1K168Q and Kes1K168R represented controls and were at least as active as Kes1 for DHE and PtdIns-4-P transfer in vitro (Figure 6E).
Identifying Candidate KATs that Regulate the Kes1/Sec14 Signaling Axis
The biochemical data indicated that K109 acetylation is a viable mechanism for downregulating Kes1 activity in vivo, and that KATs and lysine deacetylases are important regulators of the Kes1 DHE/PtdIns-4-P exchange cycle in cells. Two screens identified candidate KATs that regulate Kes1 activity. The first took advantage of the Kes1Y97F PtdIns-4-P clamp whose expression is detrimental to yeast (Mousley et al., 2012). The strategy was to assess the relative toxicity of Kes1Y97F expression to mutants with deficiencies in each of the yeast KATs. Kes1Y97F expression was controlled by a Dox-inducible promoter, and defects in KATs that acetylate K109 (i.e., compromise dampening of Kes1 activity) were expected to exacerbate yeast growth sensitivities to Kes1Y97F expression. Whereas all KAT mutants exhibited growth profiles similar to the corresponding controls cultured under non-inducing conditions, the eaf1Δ, rtt109Δ, and gcn5Δ mutants were sensitized to Kes1Y97F expression relative to WT controls (Figure S5A).
The second screen assessed which KAT deficiencies further sensitized sec14–1ts mutants to elevated temperatures. Because enhanced Kes1 activity is especially detrimental to growth of sec14–1ts mutants (Fang et al., 1996; Li et al., 2002), such a phenotype was expected to be associated with LOF in KAT activities that downregulate Kes1 via K109 acetylation. Again, eaf1Δ and rtt109Δ alleles enhanced the temperature sensitivity of sec14–1ts mutants (Figure S5B). Whereas gcn5Δ showed no synthetic effects in this context, elp3Δ mutants prohibited growth of sec14–1ts cells at 33.5°C. Thus, only eaf1Δ and rtt109Δ satisfied both of the genetic criteria for KATs that target Kes1 in vivo, with eaf1Δ consistently showing the most pronounced effects.
Eaf1 is a scaffolding component of the 13-subunit NuA4 KAT complex (Auger et al., 2008). Deficiencies in other subunits of the NuA4 complex also evoked eaf1Δ-like phenotypes. The eaf7Δ and esa1ts (esa1-L254P; Clarke et al., 1999) mutations satisfied the dual criteria of rendering WT cells less tolerant of Kes1Y97F toxicity, and enhancing temperature sensitivity for growth of sec14–1ts cells (Figure S5C). Moreover, those growth deficits associated with NuA4 deficiencies were alleviated in kes10 and kes1K109A genetic backgrounds (Figure S5D). Bypass Sec14 mechanisms associated with inactivation of the CDP-choline pathway for PtdCho biosynthesis failed to rescue growth of sec14–1ts eaf1D double mutants at restrictive temperatures (Figure S5E). These phenotypic data were reinforced by assays monitoring recycling of the Snc1 t-SNARE through the endosomal system to the plasma membrane (Figure S6A), and CPY trafficking through the secretory pathway to the vacuole (Figure S6B). These results identified Kes1 as a key non-histone target of the NuA4 KAT.
Kes1K109R Is Defective In Vivo
If Kes1 regulation is a case of the NuA4 KAT complex tuning Kes1 activity via K109 acetylation, then the acetylation mimic Kes1K168Q should not be functional in vivo. Moreover, expression of a non-acetylatable Kes1 that retains PtdIns-4-P and sterol-binding activities (i.e., Kes1K109R) should phenocopy the G1-arrest phenotypes that come with enhanced Kes1 or Kes1Y97F expression. The first prediction was fulfilled while the second was not. Whereas Kes1 re-expression reversed the bypass Sec14 phenotype of kes1 null mutants, Kes1K109A, Kes1K109Q, or Kes1K109R re-expression was not able to do so (Figure S7A), even though these K109 mutant proteins were stable in vivo. Similar conclusions were forthcoming from pulse-chase experiments where CPY-trafficking kinetics were examined (Figure S7B). Kes1K109A, Kes1K109Q, or Kes1K109R expression did not reverse the bypass Sec14 status of kes1 null cells. Moreover, in contrast to Kes1 or Kes1Y97F, elevated Kes1K109R did not inhibit growth of WT or sec14–1ts strains. Thus, despite the fact that Kes1K109R was nearly as active as the WT protein in a coupled DHE/PtdIns-4-P transfer assay in vitro, Kes1K109R was nonfunctional in vivo.
DISCUSSION
Understanding how ORPs are integrated into the eukaryotic lipid signaling program requires a description of their individual biological activities. Herein, we identify a role for Kes1, and other ORPs, in regulating cell-cycle progression and replicative lifespan in yeast. Those findings outline four conclusions: (1) Kes1 functions as a key brake on progression through G1 and initiation of a new round of cell division upon nutrient limitation, (2) the antagonistic actions of Kes1 and Sec14 in regulating PtdIns-4-P signaling extend from membrane trafficking to cell-cycle contexts where the Sec14/Kes1 lipid signaling axis plays an important role in coordinating cell growth with initiation of cell division in nutrient-replete environments, (3) Kes1 is a key nonhistone target for acetylation by the NuA4 lysine acetyltransferase, and (4) other members of the yeast ORP family also exhibit activities consistent with stage-specific inhibitors of cell-cycle progression and regulators of replicative lifespan. These data report that Kes1 and Sec14 execute dual membrane-trafficking and cell-cycle functions, and nominate this pair of lipid-exchange proteins as components of a regulatory axis that coordinates TGN/endosomal lipid signaling with cell-cycle progression. Moreover, these results offer fresh perspectives for interpreting ORP function in cell-cycle control, and suggest that ORPs execute unappreciated “tumor-suppressor”-like activities in mammalian cells.
Kes1 Promotes Exit from the Cell Cycle into Quiescence
Kes1 action as an inhibitor of progression through G1 is manifested by excessive Kes1 activity overriding the nutrient-responsive pro-proliferative signals that drive progression through G1 into another round of cell division and, reciprocally, Kes1 LOF resulting in the licensing of nutrient-deprived cells to initiate and complete one extra cell division. Interestingly, accurate recapitulations of this kes1Δ phenotype, and of the premature entry into quiescence associated with excessive Kes1 activity, are apparent in complex developmental settings where exit from the cell cycle must be precisely timed. Drosophila embryonic cells lacking the Cdk inhibitor Dacapo undergo one extra cell division before the midblastula transition, whereas premature Dacapo expression induces G1 arrest (de Nooij et al., 1996; Lane et al., 1996). Moreover, we also note the phenotypes of yeast expressing excessive Cln2 G1 cyclin activity resemble kes1D cells in their ability to pass through G1 in the face of severe nutrient deprivation (Hadwiger et al., 1989).
Sec14 and Kes1 Function at START in Cycling Cells
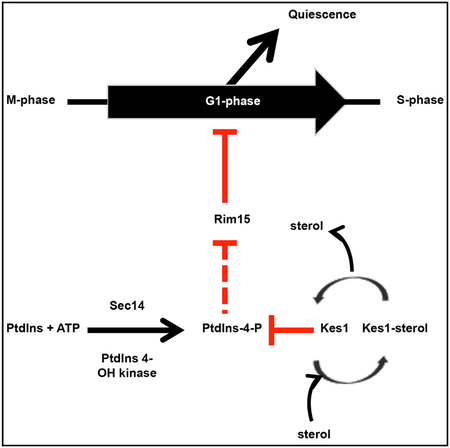
The Sec14/Kes1 signaling axis also operates in cycling cells at the commitment step before initiation of DNA replication. The larger critical size of sec14–1ts cells indicates that these cells were unable to properly interpret that they had reached a sufficient size to commit to a new round of cell division. This defective coupling is qualitatively similar to that observed in cells deficient in the G1 cyclin Cln3 and was corrected by Kes1 LOF. Hence, the Sec14/Kes1 axis not only regulates when cells stop dividing upon nutrient exhaustion, but also when cells initiate a new round of cell division in nutrient-rich conditions. Importantly, those interactions were evident in growing cells and without bulk membrane-trafficking deficits. Kes1 specificity in this level of control deserves emphasis. Other mechanisms for bypass Sec14 that correct the trafficking deficits associated with Sec14 LOF failed to correct the inability of sec14–1ts mutants to properly register critical size. These results point to more specialized roles for Sec14/Kes1-regulated PtdIns-4-P-dependent signaling in cell-cycle control (Figure 7). This specific pool relevant to cell-cycle control is either: (1) functionally distinct from other Sec14/Kes1-regulated PtdIns-4-P pool(s) dedicated to bulk membrane-trafficking functions, or (2) involves the same pool but cell-cycle control exhibits a higher threshold requirement for PtdIns-4-P signaling.
Figure 7. A Sec14/Kes1 PtdIns-4-P Signaling Axis in Cell-Cycle Control.

Heterotypic PtdCho/PtdIns exchange defines the machine by which Sec14 stimulates PtdIns 4-OH kinase-mediated production of a PtdIns-4-P pool that regulates progression through the G1 phases of the cell cycle. This signaling is antagonized by activity of the Sac1 PtdIns-4-P phosphatase and Kes1 binding and sequestering PtdIns-4-P from its effectors. The Kes1 brake is itself tuned by a heterotypic sterol/PtdIns-4-P exchange cycle and the activity of NuA4 KAT in acetylating K109. The Sec14/Kes1 checkpoint requires activity of the Rim15 kinase, and we propose this PtdIns-4-P pool is produced in a TGN/endosomal compartment. Functionally distinct, roles for other Osh proteins (other than Osh5) in regulating G1 progression are also indicated. Pathway brakes are highlighted in red.
Kes1 Is a Key Non-histone Substrate for the NuA4 Lysine Acetyltransferase
The role of Kes1 as inhibitor of progression through the START checkpoint argues for tight regulation of its activity. One such mechanism involves lipid binding/exchange as Kes1 is a brake on TGN/endosomal PtdIns-4-P signaling. The strength of that brake is regulated by a heterotypic sterol and PtdIns-4-P binding/exchange cycle that is itself responsive to the availability of these lipids on TGN/endosomal membranes (Mousley et al., 2012). Our data indicate a second mechanism that involves K109 acetylation (Figure 7). This modification is an inactivating one as determined by both in vivo and in vitro assays. Those defects are reconciled by the unmodified K109 side chain engaging a PtdIns-4-P molecule in the Kes1 lipid-binding pocket (de Saint-Jean et al., 2011).
Our data indicating that impaired activity of the NuA4 KAT complex enhanced Kes1 activity in cells, and that an unmodified K109 was required for those effects, argue that Kes1 is an important non-histone target for this enzyme complex. In that regard, synthetic genetic array studies identified strong interactions of NuA4 deficiencies with cell-cycle and membrane-trafficking mutants (Lin et al., 2008; Mitchell et al., 2008). Those interaction networks: (1) resemble those of Sec14 and Kes1 (Mousley et al., 2008), (2) establish that NuA4 potentiates PKA signaling (Filteau et al., 2015), and (3) show NuA4 depresses both expression of the Msn2/4 regulon and trehalase (Lindstrom et al., 2006; Mitchell et al., 2008). Such pleiotropic NuA4 interactions are interpreted to reflect broad alterations in the transcriptome as a result of deranged histone acetylation landmarks. We posit that these synthetic phenotypes also report NuA4-dependent modulation of Kes1 activity, which contributes to the role of NuA4 as global genetic “buffer” that governs cell fitness (Lin et al., 2008; Mitchell et al., 2008).
Mechanistic Implications for Kes1 Lipid-Exchange Activity
Regardless of whether one favors intermembrane lipid transfer models for Kes1 function (de Saint-Jean et al., 2011), or a lipid-exchange-dependent sensing mechanism that does not involve intermembrane lipid transfer (Mousley et al., 2012), there is general agreement that the Kes1 lipid-exchange cycle is intimately linked to its biological activity. Modulation of Kes1 activity by K109 acetylation has implications for how the Kes1 lipid exchange cycle relates to protein function in the cell.
The data also present a paradox that indicates we do not yet fully understand the details of Kes1 activity in cells. While Kes1AcK109 was defective in the coupled sterol/PtdIns-4-P countercurrent assay in vitro, the non-acetylated Kes1K109R showed robust activity, with only modest reduction in PtdIns-4-P transfer. Yet, Kes1K109R was nonfunctional in vivo. Those data suggest: (1) that Kes1K109R represents a striking case where lipid transfer protein function has been uncoupled from lipid exchange, (2) that even modest defects in the PtdIns-4-P component of the heterotypic DHE/PtdIns-4-P exchange cycle are sufficient to render Kes1K109R biologically inactive, or (3) that K109R interferes with, as yet, unidentified protein:protein or protein:lipid interactions critical for Kes1 biological activity. Either way, those results do not easily fit with models invoking intermembrane Kes1 lipid transfer mechanisms driven solely by differential sterol/PtdIns-4-P gradients.
ORPs and Cell-Cycle Control
The long ORP Osh1 and short ORP Osh6 both exhibited features suggesting that these too operate as nutrient-regulated brakes on G1 progression. Such Osh activities, while resembling those of Kes1, were not precise redundancies, as the corresponding phenotypes were apparent in the face of Kes1 activity. Rather, we interpret the data to report specific roles for these ORPs (Figure 7). Osh1 localization to the nuclear/vacuolar junction (and localization of its mammalian ortholog ORP1L to nuclear/lysosomal junctions) is interesting given the role of lysosomal compartments in nutrient signaling (Kvam and Goldfarb, 2004; Johansson et al., 2007). Osh6 is a PtdIns-4-P- and phosphatidylserine-binding protein (Maeda et al., 2013). We speculate that Osh6 couples its differential lipid-binding activities to other inputs into cell-cycle control.
While the LOF of the short ORPs, Osh5 or Osh6, had no effect on replicative lifespan, Kes1 LOF (short ORP) or LOF of any of the three long ORPs (Osh1, Osh2, and Osh3) extended both mean and maximal replicative lifespan. Mechanisms underlying the long replicative and short chronological lifespan of strains lacking Kes1 remain to be determined. Candidate pathways include TOR signaling and activation of the Gcn4 transcription factor (Mousley et al., 2012), altered trafficking processes related to PtdIns-4-P signaling (Li et al., 2002), and altered cell-cycle dynamics. Evidence to suggest that membrane trafficking integrates cell size homeostasis and cell-cycle progression (Anastasia et al., 2012), and building evidence for the involvement of cyclin-dependent kinase Cdk1 in this process (McCusker and Kellogg, 2012), suggest other possibilities.
Finally, the data hold implications for ORP involvement in mammalian cell-cycle control from the perspective of balancing cell proliferation with differentiation. High expression of Kes1-like short ORPs in mammalian tissues rich in post-mitotic cells is at least consistent with such a view (Johansson et al., 2003). We suggest that ORPs offer flexible mechanisms for coordinating endosomal membrane-trafficking flux and PtdIns-4-P signaling with cell-cycle progression, senescence, and transition of actively dividing cells into programs of differentiation and post-mitotic life.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for resources, reagents or further information should be directed to and will be fulfilled by the Lead Contact, Vytas A. Bank-aitis (vytas@tamhsc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strain genotypes and plasmids are listed in Key Resources. Standard yeast-peptone-dextrose, synthetic complete media and lithium acetate transformation methods were used throughout (Cleves et al., 1991; Schaaf et al., 2008).
METHOD DETAILS
Expression Vectors
Yeast and bacterial expression plasmids are listed in Key Resources. The Ptet-KES1 and sterol-binding deficient mutant Ptet-kes1Y97F vector was generated as follows. KES1 or kes1Y97F and the tetracycline/doxycycline-responsive promoter were amplified by PCR (Q5® High-Fidelity DNA Polymerase kit, NEB #E0555S), using primers that includes homologous ends to each other and to the pRS415 vector (for Ptet-KES1) or pRS413 vector (for Ptet-kes1Y97F). The linearized DNA sequences were cloned together by homologous recombination using SLICE (Zhang et al., 2008). Cloned vectors were isolated and confirmed by PCR. Tetracycline/doxycycline-inducible expression of KES1or kes1Y97F was confirmed by immunoblot.
RT-PCR
Total RNA (1mg) templated reverse transcription to generate cDNA (20ml final volume). Expression of AXL2, CLB1, CLN1, CLN2,PCL1, PCL9, ECM33, SCW10, SRL1, YOX1, CYC7, HSP104, CTT1, GPG1, GAD1, HSP12, HSP30, HSP82, HSP78, GRE1, SSA3 and SSA4 relative to ACT1 was determined using 0.5ml cDNA fraction as template and gene specific oligonucleotides as primers and the SensiFast Sybr Lo-Rox Kit (Bioline). Reactions were performed using a VIIA 7 Real-Time PCR System (Applied Biosystems).
Glycogen and Trehalose Measurements
Glycogen and trehalose were measured as described by Parrou and François (1997). Ten OD600nm of cells were incubated in 250μl of 0.25M Na2CO3 at 95°C for 4 hr. After this the solution was adjusted to pH 5.2 with 150μl 1M acetic acid and 600 μl of 0.2M sodium acetate. For glycogen, samples were incubated overnight with 2 U/ml of amyloglucosidase isolated from Aspergillus niger (Sigma Aldrich) at 57°C with constant agitation. For trehalose, samples were incubated overnight with 0.05 U/ml of trehalase (Sigma Aldrich) at 37°C. The glucose liberated from either glycogen or trehalose was determined by incubating 50μl of sample with 500μl of glucose oxidase reagent (Sigma) for 30 min at 37°C. The reaction was terminated with 500μl of 12M H2SO4 and the absorbance measured at 420 nm.
Zymolyase Resistance Measurements
Cell harboring YCp(PDOX::KES1), YCp(PDOX :: KES1Y97F) or YCp(PDOX::kes1K109A) were cultured to OD600 < 0.05 in synthetic defined medium lacking uracil containing Dox. Cells were isolated, washed and cultured in synthetic defined medium lacking uracil ± Dox for 18 hrs to OD600 < 1.0. Zymolyase resistance assays were performed according to Newman et al. (2005). Cells were grown in synthetic defined medium lacking uracil in the presence or absence of Dox at 30°C for the indicated times. Cells were isolated and resus-pended in water at 1 OD600nm/ml and incubated with zymolyase 20T (150μg/ml) (ICN Biomedicals) at 30°C for 5, 10, 20 and 30 minutes. Cell lysis was monitored by measurement the OD600 nm.
Cell Cycle Methods
Elutriation experiments and cell size measurements were performed as described by Hoose et al. (2012). For elutriation experiments, cells were grown in YPD (1% w/v yeast extract, 2% w/v peptone, 2% w/v dextrose) to a cell density of 1–5 × 107 cells/ml and loaded onto an elutriator chamber at a pump speed of 37 ml/min spinning at 3200 rpm (Beckman J-6M/E centrifuge). During all the centrifugation steps, the temperature in the centrifuge was 25°C. The elutriated small daughter cells were collected at 2400 rpm centrifuge speed and 42 ml/min pump speed, in tubes kept on ice. The elutriated cells were recovered by centrifugation and re-suspended in fresh pre-warmed YPD medium, at a cell density of ca. 1 × 107 cells/ml. The elutriated cultures were then placed in a 30°C incubator and every 20 min we recorded their budding index and measured their cell size with a Beckman Z2 channelyzer. The geometric mean of the cell size of the population was recorded using the Accucomp software package of the instrument. Each cell size measurement represents the average of two technical duplicates taken at two different cell dilutions. The smoothened cell size histograms shown are the splines of the corresponding raw data, generated with the R software package “lattice”.
Replicative lifespan assays were performed on YPD agar at 30°C by serial separations of daughter cells from mother cells using a manual micromanipulator equipped with a fiber-optic needle (Steffen et al., 2009).
Flow Cytometry
Yeast (5 OD600 units) were pelleted and fixed overnight in 70% (v/v) ethanol. Cells were washed with 1 ml of citrate buffer (50 mM sodium citrate, pH 7), briefly sonicated, resuspended in 1 ml of citrate buffer containing 0.25 mg/ml RNase A, and incubated overnight at 37°C. Cells were again washed in 50 mM sodium citrate, pH 7.0, resuspended in 1 ml of 50 mM sodium citrate, pH 7.0 containing 16 mg/ml of propidium iodide, and incubated at room temperature for 30 min. After a last wash in citrate buffer, cells were analyzed on a Beckman MoFlo flow cytometer.
Detection of Lysine Acetylation by Mass Spectrometry
Lysine-acetylated Kes1 were produced using the amber suppression method of Hsu et al. (2016) where E. coli were cultured in Terrific broth supplemented with 0.2% arabinose, 5mM nicotinamide and 5mM Nε-Acetyl-L-lysine, and protein production was induced with IPTG at 16°C. In total, 4mg of purified protein (Kes1 or Kes1AcK109) were digested with trypsin overnight. Resulting samples were analyzed by LC-MS/MS and acetylation sites were identified using a Mascot search engine (Mitchell et al., 2008).
PtdIns-4-P Extraction Assays
Extraction of [14P]-PtdIns-4-P was measured as described (Goto et al., 2016). Liposomes (400 nm) composed of phosphatidylcholine (PtdCho), lactosyl-phosphatidylethanolamine (PtdEtn) and [14P]-PtdIns-4-phosphate (86:10:4 molmol) were prepared by extrusion in liposome buffer (25 mM HEPES [pH 7.4], 150 mM, NaCl and 1 mM EDTA) and pre-cleared by centrifugation at 15,000xg for 5 min at 4°C. Extraction was measured by combining the appropriate Kes1 protein (100 pmol) with liposomes (400 pmol of PtdIns-4-phosphate) and 3 μg BSA in liposome buffer. After incubation at 25°C for 30 min, assays were placed on ice, R. communis agglutinin was added (10 μg) for 10 min and liposomes were sedimented by centrifugation at 15,000xg at 4°C for 5 min. [14P]-PtdIns-4-phosphate was measured in supernatant fractions and PtdIns-4-P extraction was corrected for non-specific extraction (mock).
Lipids and Liposome Preparation for DHE and PtdIns-4-P Transfer Assays
1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), brain L-α-phosphatidylinositol-4-phosphate (PtdIns-4-P), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(5-dimethylamino-1-naphthalenesulfonyl) (DNS-PE), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-lissamine rhodamine B sulfonyl (Rhod-PE) were from Avanti Polar Lipids. Dehydroergosterol (DHE) was from Sigma Aldrich. The concentration of the DHE stock solution in methanol was determined by UV-spectroscopy using an extinction coefficient of 13,000 M−1.cm−1.
Lipids, stored in stock solutions in CHCl3 or methanol, were mixed at the desired molar ratios. Solvent was removed in a rotary evaporator under vacuum. For lipid films containing PtdIns-4-P, the mix was pre-warmed to 33°C for 5 minutes prior to drying. The films were hydrated in 50mM Hepes, pH 7.2, 120mM K-acetate (HK buffer) to obtain a suspension of multilamellar liposomes. After five thawing-freezing cycles with liquid nitrogen, the suspensions were extruded through polycarbonate filters of 0.2mm pore size using a mini-extruder (Avanti Polar Lipids). Liposomes were stored at 4°C in the dark when containing light-sensitive lipids (DHE, DNS-PE, Rhod-PE), and used within 2 days.
DHE and PtdIns-4-P Transfer Assays
DHE and PtdIns-4-P transport assays followed the protocols described (de Saint-Jean et al., 2011; Moser von Filseck et al., 2015) using purified recombinant His6-tagged Kes1 proteins (Li et al., 2002). Experiments were carried out in a Shimadzu RF 5301-PC fluorimeter equipped with a cylindrical quartz cuvette. For measuring DHE transfer, a suspension (570μl) of LB liposomes (200μM lipids, final concentration) made of DOPC and containing 4% (mol/mol) PtdIns-4-P was incubated at 30°C under constant stirring in HKM (HK + 1mM MgCl2) buffer. After 1 min, 30μl of LA liposomes (200μM lipid final concentration) composed of 2.5% of DNS-PE and 5% DHE was added. After 3 min, Kes1 or the appropriate derivative was injected to a final concentration of 375nM. Lipid transport was measured by recording the DNS-PE signal at 525nm (bandwidth 10 nm) upon DHE excitation at 310nm (bandwidth 1.5nm). As the concentration of accessible DHE was equal to 10μM, the amount of DHE (in μM) transported from LA to LB membrane was equal to 10 * ((F-F0)/(Fmax-F0)) where Fmax is the signal before Kes1 injection; F0 is the signal measured upon total DHE extraction by 10mM methyl-β-cyclodextrin (Sigma). Liposomes and proteins were injected from stock solutions with Hamilton syringes through a guide in the cover of the fluorimeter.
To measure PtdIns-4-P transport, a suspension (570μL) of LB liposome (200μM total lipid) composed of 2% Rhod-PE and 4% PtdIns-4-P was incubated with 250nM NBD-PHFAPP at 30°C in HKM buffer under constant stirring. The concentration of accessible PtdIns-4-P (in the outer leaflet) is 4μM. After 1 min, 30μL of LA liposome containing 10% DHE (200μM lipids, final concentration) were injected. After additional 3 min, Kes1 or mutant (375nM final concentration) was injected. PtdIns-4-P transport was followed by measuring the NBD signal at 530 nm (bandwidth 10nm) upon excitation at 460nm (bandwidth 1.5nm). The signal, which mirrors the redistribution of NBD-PHFAPP between LA and LB liposomes, was normalized to determinate the amount of PtdIns-4-P transported by Kes1. For this, the NBD signal (Feq) in a situation where PtdIns-4-P is fully equilibrated between liposomes. NBD-PHFAPP was mixed with LA and LB liposome (200μM total lipid each) that each contains initially 2% PtdIns-4-P. The fraction of PtdIns-4-P on the surface of LB liposome, PtdIns-4-PB/PtdIns-4-PT, is equal to the fraction of PHFAPP on LB liposome and correspond to FNorm = 0.5*(F-F0/Feq-F0) with F0 corresponding to the NBD signal prior to the addition of Kes1 protein. The amount of PtdIns-4-P (in μM) transferred from LB to LA liposomes corresponds to 4*FNorm.
Fluorescence Microscopy
For GFP imaging, cells were grown in synthetic defined medium lacking uracil in the presence or absence of Dox at 30°C. Cells were then fixed in 3.75% formaldehyde and immobilized on concanavalin A coated slides and examined at 25°C. The imaging system employed a CFI plan apochromat lambda 100x oil immersion objective lens NA 1.45 mounted on a Nikon Ti-U microscope base (Nikon, Melville, NY) interfaced to a Photometrics CoolSNAP HQ2 high sensitivity monochrome CCD camera (Roper Scientific, Ottobrunn, Germany) or an Andor Neo sCMOS CCD camera (Andor Technology, Belfast, UK). A Lumen 200 Illumination System (Prior Scientific Inc., Rockland, MA.) was used in conjunction with a B-2E/C (465–495nm/515–555nm;EX/EM) or G-2E/C (528–553nm/590–650nm;EX/EM) filter set (Nikon, Melville, NY). Images were captured using the Nikon NIS Elements software package (Nikon, Melville, NY, version 4.10) and exported as .TIF files. Image analyses were performed using ImageJ (version 1.47t, National Institute of Health) and figures were constructed using Adobe Illustrator and Adobe Photoshop CS6 (version 15.0.0).
Metabolic Radiolabeling and CPY Immunoprecipitation
Yeast strains were grown in minimal media lacking methionine and cysteine to mid logarithmic phase (OD600nm = 0.5) and radiolabeled with [35S]-amino acids (Translabel; New England Nuclear; 100 μCi/ml). Chase was initiated by introduction of unlabeled methionine and cysteine (2 mM each, final concentration), and terminated by addition of trichloroacetic acid (5% w/v – final concentration). CPY was pulled down from clarified cell-free lysates with rabbit polyclonal anti-CPY serum and radiolabeled CPY were resolved by SDS-PAGE and visualized by autoradiography (Bankaitis et al., 1986).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were quantified and analyzed as described in detail in the figure legends, figures, Results, and Method Details in STAR Methods. Statistical methods used included Student’s t-test and one-way ANOVA, values are given as mean ± standard deviation and confidence intervals are identified. Exact definitions of n are identified throughout.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CPY serum | Mousley et al. (2012) | RRID: AB_102312 |
| Bacterial and Virus Strains | ||
| BL21 (DE3) | New England Biolabs | BL21 (DE3) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| His6-Kes1 | Li et al. (2002) | N/A |
| His6-Kes1K109A | This study | N/A |
| His6-Kes1 K109Q | This study | N/A |
| His6-Kes1 K109R | This study | N/A |
| His6-Kes1AcK109 | This study | N/A |
| Critical Commercial Assays | ||
| SensiFast Sybr Lo-Rox Kit | Bioline | N/A |
| VIIA 7 Real-Time PCR System | Applied Biosystems | N/A |
| Q5 High-Fidelity DNA Polymerase kit | New England Biolabs #E0555S | N/A |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae | ||
| MATa, sec14-1, ura3-52, his3-200, lys2-801 | Bankaitis et al. (1989) | CTY1-1A |
| MATa, sec14-1, pct10, ura3-52, his3-200, lys2-801 | Cleves et al. (1991) | CTY102 |
| MATa, sec14-1, kes10, ura3-52, his3-200, lys2-801 | Fang et al. (1996) | CTY159 |
| MATa, sec14-1, cki1-1, ura3-52, his3-200, lys2-801 | Cleves et al. (1991) | CTY160 |
| MATa, ura3-52, his3-200, lys2-801 | Bankaitis et al. (1989) | CTY182 |
| MATa, sec14-1, ura3-52, his3-200, lys2-801, cki1Δ::URA3 | This study | CTY392 |
| MATa, ura3-52, his3-200, lys2-801, cki1Δ::URA3 | This study | CTY393 |
| MATa, ura3-52, his3-200, lys2-801, pct1Δ::URA3 | This study | CTY1784 |
| MATa, ura3-52, his3-200, lys2-801, kes1Δ::URA3 | This study | CTY1683 |
| MATa, ura3-52, his3-200, lys2-801, cki1Δ::URA3 | This study | CTY1784 |
| CTY1683 leu2Δ::KES1-3XFLAG::HIS3 | This study | JHY2 |
| CTY1683 leu2Δ::kes1 K109A-3XFLAG::HIS3 | This study | JHY3 |
| CTY1683 leu2Δ::kes1 K109Q-3XFLAG::HIS3 | This study | JHY4 |
| CTY1683 leu2Δ::kes1 K109R-3XFLAG::HIS3 | This study | JHY5 |
| CTY159 leu2Δ::KES1-3XFLAG::HIS3 | This study | JHY6 |
| CTY159 leu2Δ::kes1 K109A-3XFLAG::HIS3 | This study | JHY7 |
| CTY159 leu2Δ::kes1 K109Q-3XFLAG::HIS3 | This study | JHY8 |
| CTY159 leu2Δ::kes1 K109R-3XFLAG::HIS3 | This study | JHY9 |
| CTY159 leu2Δ::kes1 K168Q-3XFLAG::HIS3 | This study | JHY10 |
| CTY159 leu2Δ::kes1 K168R-3XFLAG::HIS3 | This study | JHY11 |
| CTY182 osh1Δ::URA3 | This study | JHY14 |
| CTY182 osh2Δ::URA3 | This study | JHY15 |
| CTY182 osh3Δ::URA3 | This study | JHY16 |
| CTY182 osh5Δ::URA3 | This study | JHY17 |
| CTY182 osh6Δ::URA3 | This study | JHY18 |
| CTY182 osh7Δ::URA3 | This study | JHY19 |
| CTY1-1A osh1Δ::URA3 | This study | JHY20 |
| CTY1-1A osh2Δ::URA3 | This study | JHY21 |
| CTY1-1A osh3Δ::URA3 | This study | JHY22 |
| CTY1-1A osh5Δ::URA3 | This study | JHY23 |
| CTY1-1A osh6Δ::URA3 | This study | JHY24 |
| CTY1-1A osh7Δ::URA3 | This study | JHY25 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 DCP2-GFP::HIS3MX6 | Invitrogen | CJM1 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 XRN1-GFP::HIS3MX6 | Invitrogen | CJM2 |
| MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0rim15Δ::KanMX4 | Dharmacon | CJM3 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Brachmann et al. (1998) | BY4741 |
| MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Brachmann et al. (1998) | BY4742 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 eaf1 Δ::KANMX | GE Life Sciences | YKB3333 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rtt109Δ::KANMX | GE Life Sciences | YKB3484 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 elp3Δ::KANMX | GE Life Sciences | YKB3482 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 gcn5Δ::KANMX | GE Life Sciences | YKB3489 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 hat1 Δ::KANMX | GE Life Sciences | YKB3485 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sas2Δ::KANMX | GE Life Sciences | YKB3486 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sas3Δ::KANMX | GE Life Sciences | YKB3487 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 hpa2Δ::KANMX | GE Life Sciences | YKB3488 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 hpa3Δ::KANMX | GE Life Sciences | YKB3297 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX eaf1Δ::KANMX | This study | YKB3935 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX rtt109Δ::HIS3 | This study | YKB4074 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX elp3Δ::KANMX | This study | YKB3444 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX gcn5Δ::KANMX | This study | YKB4080 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX hat1Δ::KANMX | This study | YKB3442 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX sas2Δ::KANMX | This study | YKB3447 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX sas3Δ::KANMX | This study | YKB3449 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX hpa2Δ::KANMX | This study | YKB4065 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX hpa3Δ::KANMX | This study | YKB3451 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 eaf7Δ::KANMX | This study | YKB3292 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 esa1 Δ::HIS3 esa1ts-URA3 | GE Life Sciences | YKB4236 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX eaf7Δ::KANMX | This study | YKB4068 |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sec14ts-NATMX esa1Δ::HIS3 esa1ts-URA3 | This study | YKB4242 |
| Recombinant DNA | ||
| Plasmids | ||
| YCp(HIS3) | Sikorski and Hieter (1989) | pRS413 |
| pEVOL (mmAcKRs, araC) | Hsu etal. (2016) | pEVOL-MmAcKRS |
| YCp(LEU2) | Sikorski and Hieter (1989) | pRS415 |
| pRS413 Ptet::KES1Y97F | This study | pKB302 |
| pRS415 Ptet::KES1 | This study | pKB321 |
| YCp(URA3, Pdox::KES1) | Mousley et al. (2012) | pCJM1 |
| YCp(URA3, PDOX:: KES1Y97F) | Mousley et al. (2012) | pCJM2 |
| YCp(URA3, PDOX:: kes1K109A) | Mousley et al. (2012) | pCJM3 |
| pRS313 Cki11−200:: Myc6xHIS3 | Ramachandran and Herman (2011) | pPHY2328 |
| pQE30 (KES1) | Li et al. (2002) | pRE540 |
| pQE30 (KES1Y97F) | This study | pRE1454 |
| pQE30 (kes1K109A) | This study | pJH1 |
| pQE30 (kes1K109Q) | This study | pJH2 |
| pQE30 (kes1K109R) | This study | pJH3 |
| pQE30 (kes1K109amber) | This study | pJH4 |
| pQE30 (KES1K168Q) | This study | pJH5 |
| pQE30 (KES1K168R) | This study | pJH6 |
| pDR196(GFP-SNC1, URA3) | This study | pJH7 |
| Software and Algorithms | ||
| Nikon NIS Elements software package version 4.10 | Nikon, Melville, NY | N/A |
| ImageJ version 1.47t | National Institute of Health | N/A |
| Accucomp Beckman Z2 channelyzer software package | Beckman Instruments | N/A |
| R software package “Lattice” | Trellis Graphics (Cleveland, OH) | Lattice graphics |
Highlights.
Kes1 is a key brake on progression through G1 upon nutrient limitation
Kes1 and Sec14 coordinate cell growth with initiation of cell division
Kes1 is a key non-histone target for the NuA4 lysine acetyltransferase
Yeast ORPs inhibit cell-cycle progression and regulate replicative lifespan
ACKNOWLEDGMENTS
This work was supported by grants GM44530 from the NIH and BE-0017 from the Robert A. Welch Foundation to V.A.B. C.J.M. was supported by start-up funds from Curtin University, Faculty of Health Sciences. K.B. and M.K. were supported by grant MOP-142403 from the Canadian Institutes of Health Research and a Natural Sciences and Engineering Research Council (NSERC) of Canada Discovery Grant (to K.B.). L.D. was supported by an NSERC Canada Graduate Scholarship. N.M. and M.P. were supported by grant GM123139 from the NIH to M.P. and by Texas A&M University Agrilife funds. G.D. was supported by the Center National de la Recherche Scientifique. B.K.K. is an Ellison Medical Foundation Senior Scholar in Aging and was supported by NIH grant R01 AG043080. C.H. was supported by a postdoctoral fellowship from the Glenn Foundation for Medical Research, and N.D.R. was supported by Canadian Institutes of Health Research grant MOP-136809. V.A.B. dedicates this paper to the memory of H. Alex Brown, an outstanding scientist, an exactingly rigorous experimentalist, and a most generous colleague and friend.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.devcel.2017.12.026.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anastasia A, Nguyen DL, Thai V, Meloy M, Doyon Y, MacDonough T, and Kellogg DR (2012). A link between mitotic entry and membrane growth suggests a novel model for cell size control. J. Cell Biol 197, 89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger A, Galarneau L, Altaf M, Nourani A, Doyon Y, Utley RT, Cronier D, Allard S, and Côté J (2008). Eaf1 is the platform for NuA4 molecular assembly that evolutionarily links chromatin acetylation to ATP-dependent exchange of histone H2A variants. Mol. Cell Biol 28, 2257–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis VA, Aitken JR, Cleves AE, and Dowhan W (1990). An essential role for a PL transfer protein in yeast Golgi function. Nature 347, 561–562. [DOI] [PubMed] [Google Scholar]
- Bankaitis VA, Johnson L, and Emr SD (1986). Mutants that fail to properly target protein to the yeast vacuole. Proc. Natl. Acad. Sci. USA 83, 9075–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis VA, Malehorn DE, Emr SD, and Greene R (1989). The Saccharomyces cerevisiae SEC14 gene encodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgi complex. J. Cell Biol 108, 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis VA, Mousley CJ, and Schaaf G (2010). Sec14-superfamily proteins and the crosstalk between lipid signaling and membrane trafficking. Trends Biochem. Sci 35, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, and Malhotra V (2006). The formation of TGN-to-plasma membrane transport carriers. Annu. Rev. Cell Dev. Biol 22, 439–455. [DOI] [PubMed] [Google Scholar]
- Beck T, and Hall MN (1999). The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402, 689–692. [DOI] [PubMed] [Google Scholar]
- Beh CT, Cool L, Phillips J, and Rine J (2001). Overlapping functions of the yeast oxysterol-binding protein homologues. Genetics 157, 1117–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, and Boeke JD (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132. [DOI] [PubMed] [Google Scholar]
- Cameroni E, Hulo N, Roosen J, Winderickx J, and De Virgilio C (2004). The novel yeast PAS kinase Rim 15 orchestrates G0-associated antioxidant defense mechanisms. Cell Cycle 3, 462–468. [PubMed] [Google Scholar]
- Clarke AS, Lowell JE, Jacobson SJ, and Pillus L (1999). Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol. Cell. Biol 19, 2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleves AE, McGee TP, Whitters EA, Champion KM, Aitken JR, Dowhan W, Goebl M, and Bankaitis VA (1991). Mutations in the CDP-choline pathway for phospholipid biosynthesis bypass the requirement for an essential phospholipid transfer protein. Cell 64, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nooij JC, Letendre MA, and Hariharan IK (1996). A cyclin-dependent kinase inhibitor, Dacapo, is necessary for timely exit from the cell cycle during Drosophila embryogenesis. Cell 87, 1237–1247. [DOI] [PubMed] [Google Scholar]
- de Saint-Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B, and Drin G (2011). Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J. Cell Biol 195, 965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Como CJ, Chang H, and Arndt KT (1995). Activation of CLN1 and CLN2 G1 cyclin gene expression by BCK2. Mol. Cell. Biol 15, 1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Kearns BG, Gedvilaite A, Kagiwada S, Kearns M, Fung MKY, and Bankaitis VA (1996). Kes1p shares homology with human oxysterol binding protein and participates in a novel regulatory pathway for yeast Golgi-derived transport vesicle biogenesis. EMBO J 15, 6447–6459. [PMC free article] [PubMed] [Google Scholar]
- Filteau M, Diss G, Torres-Quiroz F, Dubé AK, Schraffl A, Bachmann VA, Gagnon-Arsenault I, Chrétien AÈ, Steunou AL, Dionne U, et al. (2015). Systematic identification of signal integration by protein kinase A. Proc. Natl. Acad. Sci. USA 112, 4501–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev AG, Sullivan DP, Kersting MC, Dittman JS, Beh CT, and Menon AK (2011). Osh Proteins regulate membrane sterol organization but are not required for sterol movement between the ER and PM. Traffic 12, 1341–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BS, and Nakano A (2009). Membrane traffic within the Golgi apparatus. Annu. Rev. Cell Dev. Biol 25, 113–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto A, Charman M, and Ridgway ND (2016). Oxysterol-binding protein activation at endoplasmic reticulum-Golgi contact sites reorganizes phosphatidylinositol 4-phosphate pools. J. Biol. Chem 291, 1336–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TR, and Burd CG (2011). Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol. 21, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadwiger JA, Wittenberg C, Richardson HE, de Barros Lopes M, and Reed SI (1989). A family of cyclin homologs that control the G1 phase in yeast. Proc. Natl. Acad. Sci. USA 86, 6255–6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Tsuchiyama SK, Nguyen QT, Plyusnina EN, Terrill SR, Sahibzada S, Patel B, Faulkner AR, Shaposhnikov MV, Tian R, et al. (2014). Enhanced longevity by ibuprofen, conserved in multiple species, occurs in yeast through inhibition of tryptophan import. PLoS Genet. 10, e1004860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen P, Wagner SA, Weinert BT, Sharma S, Bacinskaja G, Rehman M, Juffer AH, Walther TC, Lisby M, and Choudhary C (2012). Proteome-wide analysis of lysine acetylation suggests its broad regulatory scope in Saccharomyces cerevisiae. Mol. Cell. Proteomics 11, 1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoose SA, Rawlings JA, Kelly MM, Leitch MC, Ababneh QO, Robles JP, Taylor D, Hoover EM, Hailu B, McEnery KA, et al. (2012). A systematic analysis of cell cycle regulators in yeast reveals that most factors act independently of cell size to control initiation of division. PLOS Genet. 8, e1002590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu WW, Wu B, and Liu WR (2016). Sirtuins 1 and 2 are universal histone deacetylases. ACS Chem. Biol 11, 792–799. [DOI] [PubMed] [Google Scholar]
- Im YJ, Raychaudhuri S, Prinz WA, and Hurley JH (2005). Structural mechanism for sterol sensing and transport by OSBP-related proteins. Nature 437, 154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Bocher V, Lehto M, Chinetti G, Kuismanen E, Ehnholm C, Staels B, and Olkkonen VM (2003). The two variants of oxysterol binding protein-related protein-1 display different tissue expression patterns, have different intracellular localization, and are functionally distinct. Mol. Biol. Cell 14, 903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Rocha N, Zwart W, Jordens I, Janssen L, Kuijl C, Olkkonen VM, and Neefjes J (2007). Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor βIII spectrin. J. Cell Biol 176, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston GC, Pringle JR, and Hartwell LH (1977). Coordination of growth with cell division in the yeast Saccharomyces cerevisiae. Exp. Cell Res 105, 79–98. [DOI] [PubMed] [Google Scholar]
- Krause SA, and Gray JV (2002). The protein kinase C pathway is required for viability in quiescence in Saccharomyces cerevisiae. Curr. Biol 12, 588–593. [DOI] [PubMed] [Google Scholar]
- Kvam E, and Goldfarb DS (2004). Nvj1p is the outer-nuclear-membrane receptor for oxysterol-binding protein homolog Osh1p in Saccharomyces cerevisiae. J. Cell Sci 117, 4959–4968. [DOI] [PubMed] [Google Scholar]
- Lane ME, Sauer K, Wallace K, Jan YN, Lehner CF, and Vaessin H (1996). Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell 87, 1225–1235. [DOI] [PubMed] [Google Scholar]
- Lester RL, Withers BR, Schultz MA, and Dickson RC (2013). Iron, glucose and intrinsic factors alter sphingolipid composition as yeast cells enter stationary phase. Biochim. Biophys. Acta 1831, 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Rivas MP, Fang M, Marchena J, Mehotra B, Chaudhary A, Feng L, Prestwich GD, and Bankaitis VA (2002). Analysis of oxysterol binding protein homologue Kes1p function in regulation of Sec14p-dependent protein transport from the yeast Golgi complex. J. Cell Biol 157, 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YY, Qi Y, Lu J, Pan X, Yuan DS, Zhao Y, Bader JS, and Boeke JD (2008). A comprehensive synthetic genetic interaction network governing yeast histone acetylation and deacetylation. Genes Dev. 22, 2062–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom KC, Vary JC Jr., Parthun MR, Delrow J, and Tsukiyama T (2006). Isw1 functions in parallel with the NuA4 and Swr1 complexes in stress-induced gene repression. Mol. Cell. Biol 26, 6117–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie SH, and Pringle JR (1980). Reserve carbohydrate metabolism in Saccharomyces cerevisiae: responses to nutrient limitation. J. Bacteriol 143, 1384–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsden CT, Sylvesteren KB, Young C, Larsen SC, Poulson JW, Andersen MA, Palmqvist EA, Hey-Morgensen M, Jensen PB, Treebak JT, et al. (2015). Biotin starvation causes mitochondrial protein hyperacetylation and partial rescue by the SIRT3-like deacetylase Hst4p. Nat. Commun 6, 7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Anand K, Chiapparino A, Kumar A, Poletto M, Kaksonen M, and Gavin A-C (2013). Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature 501, 257–261. [DOI] [PubMed] [Google Scholar]
- McCusker D, and Kellogg DR (2012). Cdk1 coordinates cell-surface growth with the cell cycle. Curr. Opin. Cell Biol 24, 845–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell L, Lambert J-P, Gerdes M, Al-Madhoun AS, Skerjanc IS, Figeys D, and Baetz K (2008). Functional dissection of the NuA4 histone acetyltransferase reveals its role as a genetic hub and that Eaf1 is essential for complex integrity. Mol. Cell. Biol 28, 2244–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser von Filseck J, Vanni S, Mesmin B, Antonny B, and Drin G (2015). A phosphati-dylinositol-4-phosphate powered exchange mechanism to create a lipid gradient between membranes. Nat. Commun 6, 6671. [DOI] [PubMed] [Google Scholar]
- Mousley CJ, Tyeryar K, Ile KE, Schaaf G, Brost RL, Boone C, Guan X, Wenk MR, and Bankaitis VA (2008). Trans-Golgi network and endo-some dynamics connect ceramide homeostasis with regulation of the unfolded protein response and TOR signaling in yeast. Mol. Biol. Cell 19, 4785–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousley C, Yuan P, Gaur NA, Trettin KD, Nile AH, Deminoff S, Dewar BJ, Wolpert M, Macdonald JM, Herman PK, et al. (2012). A sterol binding protein integrates endosomal lipid metabolism with TOR signaling and nitrogen sensing. Cell 148, 702–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman HA, Romeo MJ, Lewis SE, Yan BC, Orlean P, and Levin DE (2005). Gpi19, the Saccharomyces cerevisiae homologue of mammalian PIG-P, is a subunit of the initial enzyme for glycosylphosphatidylinositol anchor biosynthesis. Eukaryot. Cell 4, 1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrou JL, and François J (1997). A simplified procedure for a rapid and reliable assay of both glycogen and trehalose in whole yeast cells. Anal. Biochem 248, 186–188. [DOI] [PubMed] [Google Scholar]
- Pedruzzi I, Bürckert N, Egger P, and De Virgilio C (2000). Saccharomyces cerevisiae Ras/cAMP pathway controls post-diauxic shift element-dependent transcription through the zinc finger protein Gis1. EMBO J. 9, 2569–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran V, and Herman PK (2011). The cAMP-dependent protein kinase signaling pathway is a key regulator of P body foci formation. Genetics 187, 441–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran V, Shah KH, and Herman PK (2011). The cAMP-dependent protein kinase signaling pathway is a key regulator of P body foci formation. Mol. Cell 43, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf G, Ortlund E, Tyeryar K, Mousley C, Ile K, Woolls M, Garrett T, Raetz CRH, Redinbo M, and Bankaitis VA (2008). The functional anatomy of PL binding and regulation of PIP homeostasis by proteins of the Sec14-superfamily. Mol. Cell 29, 191–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TA, and Prinz WA (2009). Sterol transport in yeast and the oxysterol binding protein homologue (OSH) family. Biochim. Biophys. Acta 1771, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, and Hieter P (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma S, Yang K, Morales MI, and Polymenis M (2014). Multiple metabolic requirements for size homeostasis and initiation of division in Saccharomyces cerevisiae. Microb. Cell 1, 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, and Futcher B (1998). Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9, 3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan CJ, Manford AG, Baird D, Yamada-Hanff J, Mao Y, and Emr SD (2011). Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 144, 389–401. [DOI] [PubMed] [Google Scholar]
- Steffen KK, Kennedy BK, and Kaeberlein M (2009). Measuring replicative life span in the budding yeast. J. Vis. Exp 10.3791/1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen E, Wanke V, Roosen J, Smets B, Dubouloz F, Pedruzzi I, Cameroni E, De Virgilio C, and Winderickx J (2006). Rim15 and the crossroads of nutrient signalling pathways in Saccharomyces cerevisiae. Cell Div. 1, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Fabrizio P, Hu J, Ge H, Cheng C, Li L, and Longo VD (2008). Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 4, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman S, Lippman SI, Zhao X, and Broach JR (2008). How Saccharomyces responds to nutrients. Annu. Rev. Genet 42, 27–81. [DOI] [PubMed] [Google Scholar]
- Zhang F, Gaur NA, Hasek J, Kim SJ, Qiu H, Swanson MJ, and Hinnebusch AG (2008). Disrupting vesicular trafficking at the endosome attenuates transcriptional activation by Gcn4. Mol. Cell. Biol 28, 6796–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.