

Abstract
Previous studies have reported that metformin (MET) has anticancer activity. In combination with chemotherapeutic drugs, MET reduces the dosage of chemotherapeutic drugs required and enhances anticancer efficacy. In the present study, the combination of MET and paclitaxel (PTX) in three human prostate cancer (PCa) cell lines (22RV1, PC-3 and LNCaP) was evaluated to investigate the effects on proliferation and apoptosis of PCa cells. The present study explored whether their effects were associated with reactive oxygen species (ROS). An MTT assay and microscopy were used to study the effect of MET + PTX on cell growth. Half maximal inhibitory concentration (IC50) values were obtained for MET (12.281±1.089 mM for 22RV1, 2.248±0.352 mM for PC-3 cells and 3.610±0.577 mM for LNCaP cells) and PTX (13.170±1.12 nM for PC-3 cells) at 48 h. Since the survival rate of 22RV1 and LNCaP cells did not decrease linearly with increasing PTX concentration, it is difficult to estimate accurate IC50; therefore, only IC50 values for PTX in PC-3 cells were given. When treating the cells with 5 mM MET, the IC50 of PTX decreased to 5.423±0.734 nM for PC-3 cells. Annexin V and propidium iodide staining was used to investigate apoptosis by flow cytometry. The apoptotic mechanisms of MET + PTX in PCa were investigated by detecting the expression of apoptosis-related proteins, activities of caspase-3/7, intracellular ROS accumulation, mitochondrial membrane potential, and intracellular levels of adenosine 5′-triphosphate (ATP). MET + PTX induced PCa apoptosis and ROS accumulation, and decreased mitochondrial membrane potential and intracellular levels of ATP. Taken together, these results indicated that MET + PTX suppressed PCa cell proliferation in a dose- and time-dependent manner. In addition, MET + PTX induced apoptosis by increasing ROS levels, reducing mitochondrial membrane potential, and activating mitochondrial-dependent apoptotic pathways.
Keywords: prostate cancer, metformin, paclitaxel, apoptosis, reactive oxygen species
Introduction
Prostate cancer (PCa) is a malignancy of the urinary system (1). The probability of newly diagnosed PCa is high in European and American men (2). Worldwide it has become the second leading cause of cancer-associated mortality in men (3). Patients with localized PCa are generally treated with hormones, surgery, and chemotherapy or radiation therapy. Hormone therapy is effective at early stages; however, a number of patients slowly develop androgen insensitivity (4,5). When advanced PCa develops into castration-resistant PCa and metastasizes to distant sites, taxanes are often used for treatment (6).
Paclitaxel (PTX) is an alkaloid that has been used as a first-line treatment for PCa in a clinical setting. It exerts its antitumor effects via cell cycle arrest (7). Resistance to PTX limits its therapeutic effect (8). Chemoresistant metastatic PCa is the most lethal form of cancer in adult men (9); therefore, more effective treatments for PCa are required.
Metformin (MET) is an oral anti-diabetic drug, commonly used to treat type 2 diabetes mellitus (10). It is the most commonly used biguanide drug, and has a relatively low incidence of toxicity and side effects (11,12). MET has attracted increased attention in recent years due to its possible anticancer activity; it inhibits several tumor types, including PCa (13–15). A number of in vivo experiments have revealed that MET directly affects cancer cell growth. Its effects have been observed in a wide range of cancer cell lines, including PCa cell lines (16,17). MET induces apoptosis and cell cycle arrest, reducing cancer cell growth (18,19). A previous study reported that MET increases sensitivity to chemotherapy and decreases required chemotherapy drug doses in various cancer cell lines (20).
Given its excellent safety profile, low cost and minimal side effects, MET is an attractive candidate as a potential anticancer agent. Nevertheless, there remains limited knowledge regarding its anticancer molecular mechanisms. Therefore, the present study investigated the effects of MET in combination with PTX on apoptosis of 22RV1, PC-3 and LNCaP cells, as well as the molecular mechanisms underlying these effects. In the present study it was demonstrated that MET augmented the effects of PTX.
Materials and methods
Cell culture
Human PCa cell lines 22RV1, PC-3 and LNCaP were purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China). The three cell lines were cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) for 22RV1 and PC-3 cells, and with 12% FBS for LNCaP cells at 37°C. Finally, a mixture of penicillin and streptomycin (Beyotime Institute of Biotechnology, Shanghai, China) at a final concentration of 1% was added.
Reagents and antibodies
MET and PTX were purchased from Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China). MET was dissolved in 1X PBS to a concentration of 2 M, and PTX was dissolved in 100% dimethyl sulfoxide (DMSO) to create a 10 mM stock solution; these were stored at −20°C. N-acetylcysteine (NAC) and glutathione disulfide (GSSG) were purchased from Beyotime Institute of Biotechnology. NAC (100 mM) and GSSG (10 mM) in PBS stock solutions were stored at −20°C. Antibodies against poly (ADP-ribose) polymerase (PARP; cat. no. 9542), caspase-3 (cat. no. 9665), caspase-9 (cat. no. 9502), B-cell lymphoma 2 (Bcl-2; cat. no. 2872), Bcl-2-associated X protein (Bax; cat. no. 2772), cytochrome c (Cyto-C; cat. no. 11940) and P53 (cat. no. 9284p) were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). GAPDH (cat. no. ab37168) antibody was purchased from Abcam (Cambridge, UK). Immunoglobulin G-horseradish peroxidase (IgG-HRP; cat. no. 030181) was purchased from EarthOx Life Sciences (Millbrae, CA, USA).
Cell viability assay
An MTT assay was used to measure cell viability. Briefly, PCa cells, PC-3/LNCaP (4×103 cells/well) and 22RV1 (1×104 cells/well), were seeded in 96-well plates overnight, and were then incubated with various concentrations of MET and PTX at 37°C for 6, 12, 24, 48 and 72 h. MTT (0.5 mg/ml) was added to each well. After 4 h of incubation, supernatants were removed and 150 µl DMSO was added to each well as a solvent. Using a Multiskan Ascent microplate photometer (EnSpire 2300 Multilabel Reader; PerkinElmer, Inc., Waltham, MA, USA) absorbance was measured at 492 nm. DMSO-treated cells (control group) were regarded as having 100% viability.
Apoptosis assay
Apoptosis was measured using the Apoptosis Detection kit (BD Pharmingen; BD Biosciences, Franklin Lakes, NJ, USA). Cells (1×105 cells/well) plated in 6-cm dishes were treated with MET (5 mM) and PTX (10 nM for PC-3 cells, and 2 µM for 22RV1 and LNCaP cells). After 24 h of treatment, cells were washed with PBS and harvested. The apoptosis assay was performed according to the manufacturer's protocol using flow cytometry and the results were analyzed using BD FACSDiva 6.1 software (BD Biosciences).
Caspase-Glo 3/7 assays
PC-3 cells, LNCaP cells (4×103 cells/well) and 22RV1 cells (1×104 cells/well) were seeded in 96-well plates and exposed to MET and PTX. Equal volumes (100 µl) RPMI-1640 medium and caspase-Glo 3/7 reagent (Promega Corporation, Madison, WI, USA) were added to each well, and the cells were incubated for 30 min at room temperature in the dark. Luminescence was measured by a luminometer (Berthold Sirius L; Titertek-Berthold, Pforzheim, Germany).
Reactive oxygen species (ROS) detection
Cells were pretreated with an antioxidant, NAC (100 µM), or a pro-oxidant, GSSG (5 µM) for 24 h prior to the addition of MET. Following treatment with MET (5 mM) and PTX (10 nM for PC-3 cells, and 2 µM for 22RV1 and LNCaP cells) for 12 h, the Reactive Oxygen Species Assay kit (Beyotime Institute of Biotechnology) was used. Cells (1×105 cells/well) were collected and resuspended in serum-free medium containing DCFH-DA (10 µM). Then cells were incubated at 37°C for 20 min in the dark. ROS levels were measured by flow cytometry and the results were analyzed using BD FACSDiva 6.1 software.
Mitochondrial membrane potential
Cells (1×105 cells/well) were seeded into 6-well plates overnight, and were subsequently treated with MET (5 mM) and PTX (10 nM for PC-3 cells, and 2 µM for 22RV1 and LNCaP cells) for 12 h. According to the Mitochondrial Membrane Potential Assay kit (Beyotime Institute of Biotechnology) manufacturer's protocol, the cells were dyed with JC-1 staining fluid and analyzed by flow cytometry and the results were analyzed using BD FACSDiva 6.1 software.
ATP levels
The ATP Assay kit (Beyotime Institute of Biotechnology) was used for detection of ATP levels. Cells (1×105 cells/well) were treated with MET (5 mM) and PTX (10 nM for PC-3 cells, and 2 µM for 22RV1 and LNCaP cells) for 12 h. Subsequently, cells were treated with 200 µl lysis buffer (Beyotime Institute of Biotechnology) and collected by centrifugation at 12,000 × g for 10 min at 4°C. Subsequently, 50 µl supernatant and 100 µl ATP detection reagent were mixed. Firefly luciferase activity was measured using a luminometer (Berthold Sirius L).
Western blot analysis
Cells were lysed with radioimmunoprecipitation assay buffer (Beyotime Institute of Biotechnology) following MET and PTX treatment for 24 h. Total protein was extracted at 4°C and concentration was determined using a bicinchoninic acid assay. Proteins (30 µg protein) were separated by 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA, USA) at 4°C. The membrane was blocked in 5% milk in Tris-buffered saline with 1% Tween-20 at room temperature for 1 h, and incubated with primary antibodies overnight at 4°C at the following dilutions: PARP, 1:1,000; caspase-3, 1:1,000; caspase-9, 1:1,000; Bcl-2, 1:1,000; Bax, 1:1,000; Cyto-C, 1:1,000; P53, 1:1,000; and GAPDH, 1:100,000. The membranes were then probed with IgG-HRP antibody (dilution, 1:10,000) for 1 h at room temperature. Finally, the proteins were detected using Enhanced Chemiluminescent kit (EMD Millipore).
Statistical analysis
The results are presented as the means ± standard deviation. All experiments were performed in triplicate. Data analysis was performed using a one-way analysis of variance using and the least significant difference post hoc test was used to determine statistical significance. SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA) was used for statistical analysis, and figures were generated using GraphPad Prism 6.0 software (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05, was considered to indicate a statistically significant difference. ImageJ 2.0 software (National Institutes of Health, Bethesda, MD, USA) was used to semi-quantify western blotting images.
Results
Effect of MET on viability of PCa cells
Firstly, the effect of MET on viability of PCa cells was determined using an MTT assay. 22RV1, PC-3 and LNCaP cells were treated with various concentrations of MET (0, 1, 5, 10 and 20 mM) for 48 h. There was a clear reduction in cell viability starting at 5 mM compared with the control group in a dose-dependent manner (P<0.01, Fig. 1). The half maximal inhibitory concentration (IC50) value of MET was 12.281±1.809 mM for 22RV1 cells, 2.248±0.352 mM for PC-3 cells and 3.610±0.557 mM for LNCaP cells at 48 h. These data suggested that MET inhibited the viability of PCa cells in a dose-dependent manner.
Figure 1.
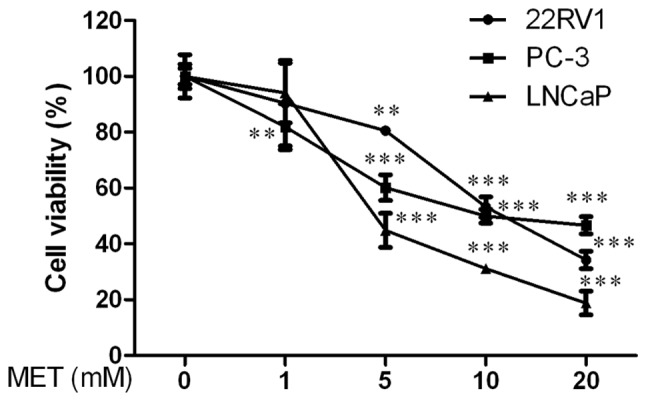
Effects of MET on viability of human prostate cancer cells. Viability of cells following treatment with various concentrations of MET (0, 1, 5, 10 and 20 mM) was detected by MTT assay at 48 h. Cells treated with DMSO were used as the control group with cell viability set at 100%. **P<0.01 and ***P<0.001 compared with the control group (DMSO-treated). DMSO, dimethyl sulfoxide; MET, metformin.
Effects of MET in combination with PTX on viability of PCa cells
To evaluate whether MET improved the chemosensitivity of PTX, MET and PTX were administered together to PCa cells (Fig. 2). The combination of MET (5 mM) and PTX (1, 2, 5, 10 and 20 nM in PC-3 cells, and 0.5, 1, 2, 4 and 8 µM in 22RV1 and LNCaP cells) exhibited a greater inhibitory effect on cell viability (Fig. 2A-C) than MET and PTX did individually. Notably, MET decreased the IC50 of PTX in PC-3 cells (Fig. 2G). These findings suggested that MET inhibited PCa cell proliferation and improved the chemosensitivity of PTX. The concentrations of MET (5 mM) and PTX (10 nM for PC-3 cells, and 2 µM for 22RV1 and LNCaP cells) were selected to verify that MET and PTX suppressed cell proliferation in a time-dependent manner (Fig. 2D-F). Subsequently, MET and PTX-induced growth inhibition in PCa cells was visualized by microscopy; the cells were treated with MET (5 mM) and PTX (10 nM for PC-3 cells, and 2 µM for 22RV1 and LNCaP cells). Cells cultured without these reagents exhibited characteristic normal growth and shape after 24 h. However, confluence was markedly reduced for cells treated with MET in combination with PTX (Fig. 2H). This finding suggested that MET improved the chemosensitivity of PTX. MET in combination with PTX suppressed cell proliferation in a time-dependent manner.
Figure 2.

MET in combination with PTX suppresses cell proliferation. (A-C) Prostate cancer cells were treated with MET (5 mM) and PTX (1, 2, 5, 10, 20 nM for PC-3 cells, and 0.5, 1, 2, 4, 8 µM for 22RV1 and LNCaP cells) for 48 h, and viability was measured by MTT assay. (D-F) Cell viability was measured by MTT assay following treatment with MET (5 mM) and PTX (10 nM for PC-3 cells, and 2 µM for 22RV1 and LNCaP cells) for 6, 12, 24, 48 and 72 h. (G) Changes in IC50 of PTX following MET treatment in PC-3 cells. (H) 22RV1, PC-3 and LNCaP cells were treated with MET (5 mM), PTX (10 nM for PC-3 cells, and 2 µM for 22RV1 and LNCaP cells) and MET + PTX for 24 h. Images of cells were captured using inverted contrast microscopy (magnification, ×100). Cells treated with DMSO were used as the control group with cell viability set at 100%. *P<0.05, **P<0.01, ***P<0.001. DMSO, dimethyl sulfoxide; IC50, half maximal inhibitory concentration; MET, metformin; PTX, paclitaxel.
MET in combination with PTX induces apoptosis of PCa cells
It was also determined whether the augmentation of cell growth inhibition induced by MET in combination with PTX was associated with an increase in apoptosis of PCa cells. The cells were treated with MET (5 mM) and PTX (10 nM for PC-3 cells, and 2 µM for 22RV1 and LNCaP cells). After 24 h of treatment, cells were labeled with Annexin V-fluorescein isothiocyanate/propidium iodide (PI) and analyzed by flow cytometry. The apoptotic effect of MET + PTX was much greater than in the single drug groups (Fig. 3A and B). This suggested that MET in combination with PTX significantly induced early and late apoptosis of PCa cells.
Figure 3.

Analysis of MET + PTX-induced apoptosis by Annexin V/PI double-staining and Caspase-Glo 3/7 assays in prostate cancer cells. Cells were treated with MET (5 mM), PTX (10 nM for PC-3 cells, and 2 µM for 22RV1 and LNCaP cells) and MET + PTX for 24 h. (A) Flow cytometry was employed to analyze apoptotic cells. (B) Quantification of the percentage of apoptotic cells. (C) Caspase-3/7 activity was detected. *P<0.05, **P<0.01, ***P<0.001. MET, metformin; PI, propidium iodide; PTX, paclitaxel.
A caspase-3/7 activity assay was used, as shown in Fig. 3C. MET in combination with PTX markedly increased the activity of caspase-3/7. In addition, the expression levels of apoptosis-associated proteins (PARP, P53, Bcl-2, Bax, Cyto-C, caspase-3 and caspase-9) were measured using western blotting. Caspase family members are key proteins in apoptosis (21). MET in combination with PTX markedly increased the expression levels of cleaved caspase-3/9, Bax, P53, Cyto-C and PARP. However, it was identified that treatment with MET and PTX significantly decreased Bcl-2 expression, compared with levels in the single drug groups (Fig. 4A and B). As PC-3 cells in the control group had undetectable expression of Clv-PARP and Cyto-C, it was not possible to present quantification of the increase in expression in relation to control expression in these cells.
Figure 4.

Western blot analysis of apoptosis-associated proteins in prostate cancer cells. Cells were treated with MET (5 mM), PTX (10 nM for PC-3 cells, and 2 µM for LNCaP cells) and MET + PTX for 24 h. (A) Western blot analysis was used to detect the expression of apoptosis-associated proteins. (B) Semi-quantification of western blotting. **P<0.01, ***P<0.001. Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2; C3, caspase-3; C9, caspase-9; Clv, cleaved; Cyto-C, cytochrome c; MET, metformin; PARP, poly (ADP-ribose) polymerase; PTX, paclitaxel.
MET induces growth suppression and apoptosis of PCa cells via the production of ROS
An MTT assay and a Reactive Oxygen Species Assay kit were used to determine whether MET induced growth suppression and apoptosis via elevation of intracellular ROS levels. Cells were pretreated with an antioxidant, NAC (100 µM), or a prooxidant, GSSG (5 µM), for 24 h prior to the addition of MET. NAC and GSSG in the cells were then removed and MET was added for another 24 h. NAC blocked MET-induced cell growth suppression (Fig. 5A) and increased ROS levels (Fig. 5B). GSSG augmented MET-induced cell growth inhibition and promoted ROS production. This suggested that MET inhibited the growth of PCa cells via the production of ROS.
Figure 5.

MET inhibits cell viability and increases production of ROS. Prostate cancer cells were treated with MET (5 mM) and antioxidant NAC (100 µM) or prooxidant GSSG (5 µM) for 24 h. (A) Viability was detected by MTT assay. (B) ROS levels were detected using a Mitochondrial Membrane Potential Assay kit. *P<0.05, **P<0.01, ***P<0.001. GSSG, oxidized glutathione; MET, metformin; NAC, N-acetylcysteine; ROS, reactive oxygen species.
MET in combination with PTX suppresses cell growth and induces apoptosis by increasing ROS production, decreasing mitochondrial membrane potential and decreasing ATP levels in PCa cells
22RV1, PC-3 and LNCaP cells were treated with MET (5 mM) in combination with PTX (10 nM in PC-3 cells, 2 µM in 22RV1 and LNCaP cells) for 12 h, and ROS production was measured. MET + PTX significantly increased the production of ROS in LNCaP cells (Fig. 6A and B). The imbalance of ROS may promote mitochondrial dysfunction and lead to mitochondria-mediated apoptosis. To evaluate the dysfunction in mitochondrial energy production, the mitochondrial membrane potential was measured. It is known that mitochondrial damage during apoptosis alters the mitochondrial membrane potential and intracellular levels of ATP in PCa cells. The present study identified that the mitochondrial membrane potential (Fig. 6C and D) and intracellular levels of ATP (Fig. 6E) were decreased by MET + PTX, and levels were significantly decreased in the MET + PTX group compared with levels in the single drug groups. This suggested that apoptosis of PCa cells mediated by MET + PTX was associated with damage to the mitochondrial membrane.
Figure 6.

MET in combination with PTX increases ROS production, decreases mitochondrial membrane potential and decreases ATP levels in prostate cancer cells. Cells were treated with MET (5 mM) in combination with PTX (10 nM in PC-3 cells, and 2 µM in 22RV1 and LNCaP cells) for 12 h. (A and B) Intracellular level of total ROS was detected. (C and D) Cells were stained with JC-1 dye and analyzed by flow cytometry. (E) ATP production was detected. *P<0.05, **P<0.01, ***P<0.001. MET, metformin; PTX, paclitaxel; ROS, reactive oxygen species.
Discussion
Chemotherapeutic regimens are commonly used to inhibit tumor growth; nevertheless, these often have side effects. Chemotherapy drugs not only have side effects but cancer cells also often develop resistance to chemotherapeutic agents, limiting their efficacy. Previously, MET was identified as an attractive anticancer adjuvant drug combined with chemotherapeutic drugs, which may improve treatment efficacy and lower the dose of chemotherapeutic agents required.
In the present study, the antitumor activity of MET + PTX was evaluated in PCa cells. MET exhibited potential growth inhibitory activity against PCa cells, as determined using the MTT assay. MET and PTX exhibited enhanced ability to reduce tumor proliferation and growth. It was demonstrated that PTX and MET, on their own or in combination, exhibited anti-proliferative effects against cultured PCa cell lines in a time- and dose-dependent manner. However, there were differential sensitivities, in terms of effectiveness of the treatment dosages, among the cell lines. LNCaP is an early stage androgen-dependent PCa cell line. whereas 22RV1 and PC-3 are androgen-independent PCa cell lines. PC-3 cells have no androgen receptor and exhibit moderate metastatic potential. Therefore, the cell lines exhibit different sensitivity to drugs.
A number of reports have suggested that chemotherapeutic agents exert anti-proliferative effects by inducing apoptosis. It was observed that MET-treated cells exhibit reduced levels of ROS-mediated matrix membrane potential (22). Mitochondria serve a key role in ROS production (23); the present data suggested that MET + PTX induced apoptosis via increasing intracellular ROS levels, and reducing mitochondrial membrane potential and ATP. Notably, an increase in ROS serves a role in the effect of MET + PTX on PCa cells. In the future, we aim to explore whether antioxidant pretreatment can inhibit the effect of MET + PTX on cell proliferation and apoptosis. In the present study, antioxidant NAC and prooxidant GSSG were used to verify that MET increased intracellular ROS levels in PCa cells. NAC pretreatment led to a decrease in MET-mediated production of ROS in 22RV1 and PC-3 compared to MET treatment alone, and had no effect in LNCaP cells. GSSG had no effect on ROS levels in PC-3 and LNCaP cells compared with MET group. In the present study, antioxidant NAC attenuated and prooxidant GSSG increased the effect of MET on ROS production in PCa cells. As is commonly known, the androgen receptor serves an important role in the development of PCa. LNCaP is an early stage androgen-dependent growth PCa cell line, PC-3 cells have no androgen receptor, and 22RV1 is an androgen-independent growth PCa cell line. A previous study demonstrated that physiological stimulation of the androgen receptor increases ROS production (24). The androgen receptor may be an important target to investigate differences among the three studied cell lines concerning ROS levels in response to NAC and GSSG. Therefore, we aim to further investigate whether the androgen receptor is involved in the effect of antioxidant NAC and prooxidant GSSG on ROS levels in PC-3, 22RV1 and LNCaP cells.
Increasing intracellular ROS, and reducing mitochondrial membrane potential and ATP induces mitochondrial damage via Cyto-C release from the mitochondria, which in turn activates downstream caspase activity. Bcl-2 family proteins (Bcl-2, Bax and Bcl-2 homologous antagonist killer) participate in the apoptotic pathway leading to cell death. PTX sensitivity is determined by the anti-apoptotic protein Bcl-extra large (25,26). PTX downregulates Bcl-2 and activates caspases and PARP (27,28), resulting in induction of apoptosis. Annexin V and PI staining and analysis by flow cytometry demonstrated that MET + PTX induced PCa cell apoptosis. Finally, the effects of MET + PTX on the expression levels of various proteins, including caspase-3/9, Bax, Bcl2, PARP, Cyto-C and P53, were studied by western blot analysis. MET + PTX-treated cells exhibited decreased expression of Bcl-2 protein, and increased expression of caspase-3/9, Bax, PARP, Cyto-C and P53 proteins. Taken together, these data suggested that MET + PTX suppressed proliferation and induced apoptosis of human PCa cells via ROS, promoting expression of the pro-apoptotic protein P53, and inducing mitochondrial damage. P53 promoted expression of Bax, inhibited expression of Bcl-2 and mitochondrial damage, and Bcl-2 promoted Cyto-C release from mitochondria. This resulted in the activation of caspase-dependent apoptotic pathways (Fig. 7). A limitation of the present study is that it was limited to in vitro data; therefore, an in vivo study will be seriously considered in the future.
Figure 7.

Cellular pathway of the effects of MET + PTX-induced growth inhibition and apoptosis of PCa cells. MET + PTX increased oxidative stress, and decreased MMP and ATP levels in PCa cells. MET + PTX upregulated the production of P53, PARP, capase3/9, Bax, Cyto-C, and downregulated the production of Bcl-2, promoting the release of Cyto-C, increasing caspase-3/7 activities, and potentiating apoptosis in PCa cells. Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2; CytoC, cytochrome c; MET, metformin; MMP, mitochondrial membrane potential; PARP, poly (ADP-ribose) polymerase; PCa, prostate cancer; PTX, paclitaxel.
In conclusion, this study demonstrated that MET combined with PTX suppressed cell growth and induced apoptosis of PCa cells via mitochondria-mediated apoptotic pathways. These findings provide promising insights into novel, potential therapeutic strategies for PCa.
Acknowledgements
Not applicable.
Glossary
Abbreviations
- MET
metformin
- PTX
paclitaxel
- NAC
N-acetylcysteine
- GSSG
glutathione disulfide
- ROS
reactive oxygen species
Funding
This study was funded by The National Natural Science Funds of China (grant no. 81272833).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
YZ and JL designed the experiments. YZ, XZ, HT and DY performed the experiments. YZ and XZ participated in data and statistical analyses. YZ and JL wrote the article and prepared figures. JL provided the financial support. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Yang Y, Wu XH. Study on the influence of metformin on castration-resistant prostate cancer PC-3 cell line biological behavior by its inhibition on PLCε gene-mediated Notch1/Hes and androgen receptor signaling pathway. Eur Rev Med Pharmacol Sci. 2017;21:1918–1923. [PubMed] [Google Scholar]
- 2.Crawford ED. Epidemiology of prostate cancer. Urology. 2003;62:3–12. doi: 10.1016/S0090-4295(03)00776-3. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 4.Sandblom G, Varenhorst E. Incidence rate and management of prostate carcinoma. Biomed Pharmacother. 2001;55:135–143. doi: 10.1016/S0753-3322(01)00038-5. [DOI] [PubMed] [Google Scholar]
- 5.Zitzmann S, Mier W, Schad A, Kinscherf R, Askoxylakis V, Kramer S, Altmann A, Eisenhut M, Haberkorn U. A new prostate carcinoma binding peptide (DUP-1) for tumor imaging and therapy. Clin Cancer Res. 2005;11:139–146. [PubMed] [Google Scholar]
- 6.Chi K, Hotte SJ, Joshua AM, North S, Wyatt AW, Collins LL, Saad F. Treatment of mCRPC in the AR-axis-targeted therapy-resistant state. Ann Oncol. 2015;26:2044–2056. doi: 10.1093/annonc/mdv267. [DOI] [PubMed] [Google Scholar]
- 7.Weaver BA. How Taxol/paclitacel kills cancer cells. Mol Biol Cell. 2014;25:2677–2681. doi: 10.1091/mbc.e14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobue S, Mizutani N, Aoyama Y, Kawamoto Y, Suzuki M, Nozawa Y, Ichihara M, Murate T. Mechanism of paclitaxel resistance in a human prostate cancer cell line, PC3-PR, and its sensitization by cabazitaxel. Biochem Biophys Res Commun. 2016;479:808–813. doi: 10.1016/j.bbrc.2016.09.128. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, Ma Y, Sheng J, Huang Y, Zhao Y, Fang W, Hong S, Tian Y, Xue C, Zhang L. A multicenter, retrospective epidemiologic survey of the clinical features and management of bone metastatic disease in China. Chin J Cancer. 2016;35:40. doi: 10.1186/s40880-016-0102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatoum D, McGowan EM. Recent advances in the use of metformin: Can treating diabetes prevent breast cancer? Biomed Res Int. 2015;2015:548436. doi: 10.1155/2015/548436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011;9:33. doi: 10.1186/1741-7015-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gou S, Cui P, Li X, Shi P, Liu T, Wang C. Low concentrations of metformin selectively inhibit CD133+ cell proliferation in pancreatic cancer and have anticancer action. PLoS One. 2013;8:e63969. doi: 10.1371/journal.pone.0063969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS One. 2012;7:e33411. doi: 10.1371/journal.pone.0033411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollak MN. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012;2:778–790. doi: 10.1158/2159-8290.CD-12-0263. [DOI] [PubMed] [Google Scholar]
- 15.Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, Martin-Castillo B, Menendez JA. Metformin: Multi-faceted protection against cancer. Oncotarget. 2011;2:896–917. doi: 10.18632/oncotarget.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. metformin Is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 17.Goodwin PJ, Ligibel JA, Stambolic V. Metformin in breast cancer: Time for action. J Clin Oncol. 2009;27:3271–3273. doi: 10.1200/JCO.2009.22.1630. [DOI] [PubMed] [Google Scholar]
- 18.Wang LW, Li ZS, Zou DW, Jin ZD, Gao J, Xu GM. metformin induces apoptosis of pancreatic cancer cells. World J Gastroenterol. 2008;14:7192–7198. doi: 10.3748/wjg.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shank JJ, Yang K, Ghannam J, Cabrera L, Johnston CJ, Reynolds RK, Buckanovich RJ. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol Oncol. 2012;127:390–397. doi: 10.1016/j.ygyno.2012.07.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011;71:3196–3201. doi: 10.1158/0008-5472.CAN-10-3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abedi H, Aghaei M, Panjehpour M, Hajiahmadi S. Mitochondrial and caspase pathways are involved in the induction of apoptosis by IB-MECA in ovarian cancer cell lines. Tumor Biol. 2014;35:11027–11039. doi: 10.1007/s13277-014-2396-9. [DOI] [PubMed] [Google Scholar]
- 22.Cheng G, Lanza-Jacoby S. Metformin decreases growth of pancreatic cancer cells by decreasing reactive oxygen species: Role of NOX4. Biochem Biophys Res Commun. 2015;465:41–46. doi: 10.1016/j.bbrc.2015.07.118. [DOI] [PubMed] [Google Scholar]
- 23.Ding H, Han C, Guo D, Chin YW, Ding Y, Kinghorn AD, D'Ambrosio SM. Selective induction of apoptosis of human oral cancer cell lines by avocado extracts via a ROS-mediated mechanism. Nutr Cancer. 2009;61:348–356. doi: 10.1080/01635580802567158. [DOI] [PubMed] [Google Scholar]
- 24.Khandrika L, Kumar B, Koul S, Maroni P, Koul HK. Role of Oxidative Stress in Prostate Cancer. Cancer Lett. 2009;282:125–136. doi: 10.1016/j.canlet.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29:2407–2420. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castilla C, Flores ML, Medina R, Perez-Valderrama B, Romero F, Tortolero M, Japón MA, Sáez C. Prostate cancer cell response to paclitaxel is affected by abnormally expressed securin PTTG1. Mol Cancer Ther. 2014;13:2372–2383. doi: 10.1158/1535-7163.MCT-13-0405. [DOI] [PubMed] [Google Scholar]
- 27.Tudor G, Aguilera A, Halverson DO, Laing ND, Sausville EA. Susceptibility to drug-induced apoptosis correlates with differential modulation of Bad, Bcl-2 and Bcl-xL protein levels. Cell Death Differ. 2000;7:574–586. doi: 10.1038/sj.cdd.4400688. [DOI] [PubMed] [Google Scholar]
- 28.Lim SJ, Choi MK, Kim MJ, Kim JK. Alpha-tocopheryl succinate potentiates the paclitaxel-induced apoptosis through enforced caspase 8 activation in human H460 lung cancer cells. Exp Mol Med. 2009;41:737–745. doi: 10.3858/emm.2009.41.10.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.