

Abstract
Background
There is increasing evidence that folate, an important component of one-carbon metabolism, modulates the epigenome. Alcohol, which can disrupt folate absorption, is also known to affect the epigenome. We investigated the association of dietary folate and alcohol intake on leukocyte DNA methylation levels in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Leukocyte genome-wide DNA methylation profiles on approximately 450,000 CpG sites were acquired with Illumina HumanMethylation 450K BeadChip measured among 450 women control participants of a case-control study on breast cancer nested within the EPIC cohort. After data preprocessing using surrogate variable analysis to reduce systematic variation, associations of DNA methylation with dietary folate and alcohol intake, assessed with dietary questionnaires, were investigated using CpG site-specific linear models. Specific regions of the methylome were explored using differentially methylated region (DMR) analysis and fused lasso (FL) regressions. The DMR analysis combined results from the feature-specific analysis for a specific chromosome and using distances between features as weights whereas FL regression combined two penalties to encourage sparsity of single features and the difference between two consecutive features.
Results
After correction for multiple testing, intake of dietary folate was not associated with methylation level at any DNA methylation site, while weak associations were observed between alcohol intake and methylation level at CpG sites cg03199996 and cg07382687, with qval = 0.029 and qval = 0.048, respectively. Interestingly, the DMR analysis revealed a total of 24 and 90 regions associated with dietary folate and alcohol, respectively. For alcohol intake, 6 of the 15 most significant DMRs were identified through FL.
Conclusions
Alcohol intake was associated with methylation levels at two CpG sites. Evidence from DMR and FL analyses indicated that dietary folate and alcohol intake may be associated with genomic regions with tumor suppressor activity such as the GSDMD and HOXA5 genes. These results were in line with the hypothesis that epigenetic mechanisms play a role in the association between folate and alcohol, although further studies are warranted to clarify the importance of these mechanisms in cancer.
Electronic supplementary material
The online version of this article (10.1186/s13148-019-0637-x) contains supplementary material, which is available to authorized users.
Keywords: DNA methylation, Dietary folate, Alcohol intake, DMR, Fused lasso, EPIC cohort
Introduction
DNA methylation is a crucial epigenetic mechanism involved in regulating important cellular processes, including gene expression, cell differentiation, genomic imprinting, and preservation of chromosome stability. DNA methylation refers to the addition of methyl groups (–CH3) to the carbon-5 position of cytosine residues in a cytosine-guanine DNA sequence (CpG) by DNA methyltransferases. DNA methylation changes can be influenced by many factors including aging [17, 19] and environmental exposure such as smoking [1, 24] or specific dietary factors [35]. Experimental evidence suggests a link between B vitamins, including folate (vitamin B9), and epigenetic modifications [3]. B vitamins, especially folate, are essential components of one-carbon metabolism (OCM), the network of interrelated biochemical reaction in which a one-carbon unit is received from methyl donor nutrients and transferred into biochemical and molecular pathways essential for DNA replication and repair. Modifications in OCM can significantly impact gene expression and thereby cellular function [53].
Absorbed folate, circulating in the bloodstream, enters the OCM cycle in the liver where it is metabolized to 5-methyltetrahydrofolate (5-methylTHF) and converted into S-adenosylmethionine (SAM) after several successive transformation steps (Fig. 1). SAM is the methyl donor for numerous methylation reactions including the methylation of DNA, RNA, and proteins. The potential role of specific dietary factors including micronutrients such as folate, alcohol, and soya intake, in modifying breast cancer risk via epigenetic mechanisms, has been proposed [54], although evidence is still scarce and inconsistent.
Fig. 1.
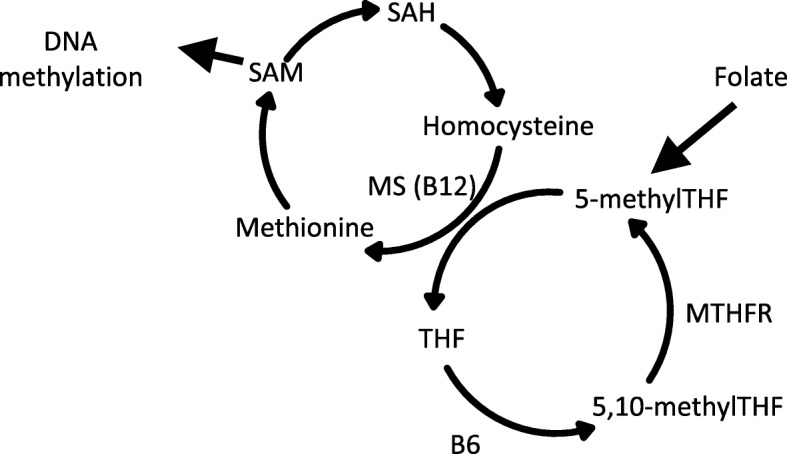
Diagram of the one-carbon metabolism pathway. MS methionine synthase, MTHFR methylenetetrahydrofolate reductase, THF tetrahydrofolate, SAH S-adenosylhomocysteine, SAM S-adenosylmethionine
Alcohol intake affects epigenetic profiles [32]. Ethanol metabolism generates toxins that may directly lead to OCM dysfunction by reducing folate absorption, increasing renal excretion of folate, and inhibiting methionine synthase, the key enzyme in the generation of the methyl donor in the OCM [32, 33]. This antagonistic effect of alcohol on folate could plausibly increase the need of folate intake. Inadequate folate levels may result in abnormal DNA synthesis due to a reduced availability of SAM [27] and disrupted DNA repair and may, hence, influence cancer risk, including breast cancer [4, 60].
The epidemiological evidence linking dietary folate, alcohol intake, and epigenome modifications is, however, not well documented. Therefore, we investigated the relationships between dietary folate and alcohol intake with leukocyte DNA methylation patterns in the controls from the European Prospective Investigation into Cancer and Nutrition (EPIC) study on breast cancer. We complemented standard regression analysis with techniques for the identification of relevant methylated regions.
Methods
Study population
EPIC is a multicenter study that recruited over 521,000 participants, between 1992 and 2000 in 23 regional or national centers in 10 European countries (Denmark, France, Germany, Greece, Italy, The Netherlands, Norway, Spain, Sweden, and the UK) [43]. Among the 367,903 women recruited in EPIC, and after exclusion of 19,583 participants with prevalent cancers at recruitment (except non-melanoma skin cancer), first malignant primary BC occurred for 10,713 women during follow-up between 1992 and 2010. Within a nested case-control study that included 2491 invasive BC cases [34], a subsample of 960 women who completed dietary and lifestyle questionnaires and provided blood samples at recruitment (480 cases and 480 matched controls) from Germany, Greece, Italy, The Netherlands, Spain, and the UK was selected for the DNA methylation analyses [2]. The present study included analysis of 450 controls only originally enrolled in this case-control study on breast cancer (BC) nested within the EPIC study.
Methylation acquisition
Genome-wide DNA methylation profiles in buffy coat samples were quantified using the Illumina Infinium HumanMethylation 450K (HM450K) BeadChip assay [5] in 960 biospecimens from women included in the BC nested case-control study. A total of 20 biospecimens with replicates used to compare technical inter- and intra-assay batch effects and then excluded from the main analysis together with 19 matched pairs, i.e., 38 samples, where at least one of the two samples had a low-quality bisulfite conversion efficiency (intensity signal< 4000) or did not pass all of the Illumina GenomeStudio quality control steps, which were based on built-in control probes for staining, hybridization, extension, and specificity [23]. To prevent collider bias [11], as both alcohol intake and folate intake and DNA methylation profiles are all potentially associated with causes of BC, among the 902 remaining samples from the original case-control study on BC nested within EPIC study, only cancer-free women were selected for the present study. For the 451 controls sample, probes with detection p values higher than 0.05 were assigned “missing” value. After the exclusion of 14,548 cross-reactive probes [10], 47,963 probes overlapping known SNPs with minor allele frequency (MAF) greater than 5% in the overall population (European ancestry) [10] and 1483 low-quality probes (i.e., missing in more than 5% of the samples), 421,583 probes were left for the statistical analyses [2].
For each probe, β values were calculated as the ratio of methylated intensity over the overall intensity, defined as the sum of methylated and unmethylated intensities. The following preliminary adjustment steps were applied to β values: (i) color bias normalization using smooth quantile normalization [13], (ii) quantile normalization [6], and (iii) type I and type II bias correction using the beta-mixture quantile normalization (BMIQ) [56]. Then, M values, defined as , were computed [14]. Surrogate variable analysis (SVA) [30, 31] was used to remove systematic variation due to the processing of the biospecimens during methylation acquisition such as batch, indicating groups of samples processed at the same time, and the position of the samples within the chip [40]. Then M values were standardized to have an identical variance of 1.
The percentage of white blood cell counts, i.e., T cells (CD8+T and CD4+T), natural killer (NK) cells, B cells, monocytes, and granulocytes, was quantified using Houseman’s estimation method [20] and included as covariates in the analysis.
Lifestyle and dietary exposures
Data on dietary habits were collected at recruitment through validated center- or country-specific dietary questionnaires (DQ) [43]. Northern Italy (Florence, Turin, and Varese), UK, Germany, and The Netherlands used self-administered extensive quantitative food-frequency questionnaires (FFQs), whereas Southern Italy (Naples and Ragusa), Spain, and Greece’s centers used interview methods. Usual consumption of alcoholic beverages (number of glasses per day or week) per type of alcoholic beverage (wine, beer, spirits, and liquors) during the 12 months before the administration of dietary questionnaires was collected at recruitment. In addition, 24-h dietary recall (R) harmonized across EPIC countries was collected from a random sample (n = 36,900) in each center to be used as reference measurements [50]. R measurements were used to improve estimation of alcohol content per specific alcoholic beverages using a country-specific estimation of average of glass volume [48]. Dietary folate intake (μg/day) was estimated using the updated EPIC Nutrient Data Base (ENDB) [49], obtained after harmonization from country-specific food composition tables [7]. No specific information on the use of folate supplements was available.
Statistical analyses
After exclusion of one outlier value of dietary folate (value larger than the third quartile plus 10 times the inter-quartile range of the distribution), a total of 450 observations from controls only were retained for statistical analyses.
The association between dietary folate, alcohol intake, and methylation levels was evaluated via (i) CpG site-specific analysis, (ii) identification of differentially methylated regions (DMRs) [41], and (iii) fused lasso (FL) regression [57].
CpG site-specific models
M values expressing methylation levels at each CpG were linearly regressed on dietary folate (log-transformed to reduce skewness) and alcohol intake. Models were adjusted for recruitment center, age at recruitment (year), menopausal status (pre- or post-menopause), and white blood cell counts (proportions of T cells, natural killer cells, B cells, and monocytes in blood). False discovery rate (FDR) was used to control statistical tests for multiple testing.
For the two CpG sites that were associated with alcohol intake, based on q values, the percentage of methylation change for 1 standard deviation (SD) increase of alcohol intake was calculated as follows:
Methylation values in site j were log-transformed and regressed on alcohol intake (Ai), for each site j, and for i = 1, … , n, as:
where α1j estimate the regression coefficient, Zi is a vector of confounding factors related to methylation levels through a vector of regression coefficients γj. The ratio of any two log-transformed methylation values log(Mij1) and log(Mij0) with a difference of alcohol intake of 1 SD () was predicted as . Therefore, the average percentage of methylation change for an increase of 1-SD in alcohol intake was estimated as:
DMR models
Differentially methylated region (DMRs) analyses were identified with the DMRcate package [41]. The rationale of this method is to use kernel smoothing to replace the t test statistics at a given CpG site by a weighted average of t test statistics across its neighboring sites on the same chromosome. More precisely, let pc express the number of sites located on a given chromosome c with c ∈ {1, … , 23} (the 23rd chromosome is chromosome X). For any site k on this chromosome, with k = 1, … , pc, the term tk2 indicates the square of the t test statistics obtained in site-specific analyses. For each site j on chromosome c, tj2 is replaced by the term , defined as
where the terms Kjk express weights, with larger values for sites k closer to j. Let xk express the position of site k on the chromosome, i.e., its chromosomal coordinate in base pairs, these weights are defined using a Gaussian kernel, as
where parameters λ and C represent the bandwidth and the scaling factor, respectively. Here, we used λ = 1000 and C = 2, respectively, as recommended in [41].
Under the null hypothesis of no association between site j and alcohol (or folate), the distribution of can be approximated by a χ2 distribution [41] with degrees of freedom [45]. Accordingly, p values were obtained for each site separately in each chromosome and q values were computed using FDR correction on all the p values to control for multiple testing. Then, DMRs were defined as regions with at least two significant sites separated by a maximal distance λ of 1000 base pairs. In line with [41], t statistics tk were obtained from regression models using an empirical Bayes method to shrink the CpG site variance [51], as implemented in the limma package [52]. For each DMR, the minimum q value, the minimum and maximum coefficients (in absolute value) of the sites included in the region were presented as qDMR, βmin, DMR, and βmax, DMR.
Fused lasso regression
Multivariate penalized regression provides an alternative to DMRs. We implemented a fused lasso (FL) regression [57], which is better suited than the standard lasso when covariates (CpGs) are naturally ordered and the objective is to identify regions on the chromosome of differentially methylated CpG sites. FL is particularly useful when the number of features (p) is way larger than the sample size (n), a situation classically known as p ≫ n.
FL is a multivariable regression method combining two penalties: (i) the lasso penalty, which introduces sparsity of the parameter vector, i.e., many elements of the estimated vector are encouraged to be set to zero, and (ii) the fused penalty, which encourages sparsity of the difference between two consecutive components in the parameter vector, thus introducing smoothness of parameter estimates in adjacent CpG sites [57].
To mimic the DMR analysis, a FL analysis was implemented where dietary folate and alcohol were, in turn, regressed on CpG methylation levels within each chromosome. The vector of methylation coefficient estimates obtained by fused lasso regression was defined as
where yi indicates, in turn, alcohol and dietary folate values for sample i = 1, … , n, Mij is the methylation levels at CpG site j, βj is the associated regression coefficient, Zi is a vector of confounding factors, consistently with linear regression and DMR analyses described above, γ is the corresponding non-penalized vector of coefficients, and ωj and νj are the weights associated with lasso penalty and fused penalty, respectively.
Following the rationale of the adaptive lasso [61] and the iterated lasso [8], the FL procedure was run for the first time with weights ωj and νj set to 1, which returned , an initial estimate of . The final estimates were obtained after running a second FL procedure with weights defined as and , with ε = 10−4.
The FL procedure was implemented on a predefined grid of 50 × 50 = 2500 values for the pair of parameters (λ1, λ2). More precisely, the grid for λ1 consisted of 50 equally spaced values (on a log scale) between and λ1, max, where λ1, max was the lowest λ1 value for which FL returned a null vector for λ2=0, a situation where FL reduces to a standard lasso. For each value λ1on this grid, the grid for λ2consisted of 50 equally spaced values (on a log scale) between and λ2, max(λ1), where λ2, max(λ1) was the lowest λ2 value for which FL returned a vector with all components equal. The optimal pair of tuning parameters (λ1, λ2) was selected as the one minimizing the prediction error estimated by 5-fold cross-validation [16], whose principle can be summarized as follows. The original sample is first partitioned into 5 equally sized subsamples. One subsample is held as the test set while the other 4 are used as a training set, on which FL estimates are computed for the 2500 values for (λ1, λ2). The prediction error is computed on the test set, and the process is repeated 5 times, and for each of the 2500 values of (λ1, λ2). The prediction error is defined as the averaged prediction error on the 5 test sets. FL analysis was implemented using the FusedLasso package.
Preprocessing steps and statistical analyses were carried out using the R software (https://www.r-project.org/) and the Bioconductor packages [21], including lumi, wateRmelon, and sva [29] for the preprocessing steps. The nominal level of statistical significance was set to 5%.
Results
Study population characteristics
Detailed characteristics of the 450 women included in the study are shown in Table 1. The average age at blood collection was 52 years (range 26–73). Participants had an average body mass index (BMI) of 26 kg/m2 (range 16–43) and were mostly post-menopausal (59%), never-smokers (56%), and moderately physically inactive (42%). The average daily intake of dietary folate was 270 μg/day (range 91–1012), and alcohol daily intake was 8 g/day (range 0–72). Non-alcohol consumers, defined as participants consuming less than 0.1 g/day of alcohol at recruitment, represented 15% of the population. Most participants were from the Italian and the German EPIC centers (Additional file 1: Figure S1).
Table 1.
Characteristics of the study population (n = 450)
| Mean (SD) | Min-Max | |
| Age at blood collection (years) | 52 (9) | 26–73 |
| Weight (kg) | 66 (11) | 40–103 |
| Height (cm) | 161 (7) | 143–196 |
| BMI (kg/m2) | 26 (4) | 16–43 |
| Alcohol intake (g/day) | 8 (12) | 0–72 |
| Blood folate level (nmol/L) | 15 (10) | 1–89 |
| Dietary folate (μg/day) | 270 (106) | 91–1012 |
| Cd8t (%) | 7.5 (4) | 0–23 |
| Cd4t (%) | 13.5 (5) | 0–34 |
| Natural killer (%) | 6.7 (5) | 0–27 |
| B cells (%) | 6.1 (2) | 0–17 |
| Monocytes (%) | 5.7 (3) | 0–17 |
| Granulocytes (%) | 60.8 (9) | 27–85 |
| N | % | |
| Menopausal status | ||
| Pre-menopause | 186 | 41.3 |
| Post-menopause | 264 | 58.7 |
| Smoking status | ||
| Never | 250 | 55.6 |
| Former | 93 | 20.7 |
| Smoker | 104 | 23.1 |
| Missing | 3 | 0.7 |
| Physical activity index [58] | ||
| Inactive | 99 | 22.0 |
| Moderate inactive | 187 | 41.5 |
| Moderate active | 75 | 16.7 |
| Active | 78 | 10.7 |
| Missing | 11 | 2.4 |
SD standard deviation, reported for continuous variables only
CpG site-specific models
After FDR correction, dietary folate intake was not significantly associated with methylation levels at any CpG sites (data not shown). Alcohol intake was inversely associated with the cg07382687 CpG site (qval = 0.048) and positively associated with the cg03199996 site (qval = 0.029) (Table 2). Both sites were located in an open sea region, i.e., a genomic region of isolated CpGs. cg07382687 was within the body region of gene CREB3L2, and cg03199996 was within the body region of gene FAM65C.
Table 2.
CpG site-specific model results for the significant CpG sites for alcohol intake (adjusted for recruitment center, age at recruitment, menopausal status, and level of different lymphocyte subtypes)
| CpG names | Alcohol intake | CpG characteristics | ||||||
|---|---|---|---|---|---|---|---|---|
| β (1SD) 1 | q val 2 | % change3 | Associated genes | Gene region4 | Island5 | Chr | ||
| 1 | cg03199996 | 0.263 | 0.029 | 9.7 | FAM65C | Body | Open sea | 20 |
| 2 | cg07382687 | − 0.257 | 0.048 | − 10.3 | CREB3L2 | Body | Open sea | 7 |
1Coefficients for 1 standard deviation alcohol intake (SD = 11.8)
2False discovery rate (FDR) adjusted p values
3Percentage of methylation change for an increase of 1 SD increase of alcohol intake
4Gene region feature category describing the CpG position, from UCSC. TSS200 200 bases upstream of the transcriptional start site (TSS); TSS1500 1500 bases upstream of the TSS; 5′UTR within the 5′ untranslated region, between the TSS and the ATG start site; body between the ATG and stop codon irrespective of the presence of introns, exons, TSS, or promoters; 3′UTR between the stop codon and poly A signal
5The location of the CpG relative to the CpG island. Shore 0–2 kb from island, Shelf 2–4 kb from island, N upstream (5′) of CpG island, S downstream (3′) of CpG island, open sea isolated CpGs in the genome
DMR analysis
A total of 24 regions associated with dietary folate were identified, which included 190 CpG sites over-represented in the TSS1500 and 1st exon regions and under-represented in the body regions and regions outside any gene regions (Fig. 2a). The 15 most significant regions are described in Table 3 and the whole list provided in Additional file 2: Table S1. Among the 24 DMRs, 54% showed an inverse association with dietary folate, i.e., had a βmax, DMR < 0. The DMR most significantly associated with dietary folate (qDMR = 1.3E−13, βmax, DMR = 0.019) was DMR.F1 in chromosome 7, including 49 CpG sites, related to HOXA5 and HOXA6 genes. DMR.F5 was associated with HOXA4, another gene of the homeobox family, (qDMR = 5.8E−4, βmax, DMR = − 0.016).
Fig. 2.

Repartition of gene regions (gene region feature category describing the CpG position, from UCSC. TSS200, 200 bases upstream of the transcriptional start site (TSS); TSS1500, 1500 bases upstream of the TSS; 5′UTR, within the 5′ untranslated region, between the TSS and the ATG start site; body, between the ATG and stop codon; irrespective of the presence of introns, exons, TSS, or promoters; 3′UTR, between the stop codon and poly A signal) among DMRs compare to their repartition within the Illumina 450K (the repartition of CpG sites was done among the 421,583 sites included in this study). a DMRs significant for folate. b DMRs significant for alcohol. c Illumina 450K
Table 3.
The 15 most significant DMRs associated with dietary folate out of 24 significant DMRs (adjusted for recruitment center, age at recruitment, menopausal status, and level of different lymphocyte subtypes)
| DMR characteristics | CpG characteristics | Fused lasso | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Associated genes | Gene regions | hg19coord | Sites1 | q DMR 2 | β min, DMR 3 | β max, DMR 3 | Overlap4 | β FL 5 | |
| F1 | HOXA5,HOXA6 | 1stExon, 5′UTR, TSS200, TSS1500, 3′UTR, body | chr7:27183133-27185512 | 49 | 1.3E−13 | 0.0002 | 0.019 | ||
| F2 | GDF7 | Body | chr2:20869434-20871401 | 8 | 1.4E−08 | − 0.016 | − 0.033 | 7/8 | − 0.0029 |
| F3 | CYP1A1 | TSS1500 | chr15:75018731-75019376 | 13 | 2.4E−05 | 0.0004 | 0.014 | ||
| F4 | PRSS50 | Body, 1stExon, 5′UTR, TSS200, TSS1500 | chr3:46759096-46759698 | 9 | 2.4E−04 | − 0.002 | − 0.020 | 4/4 | − 0.0069 |
| F5 | HOXA4 | 1stExon, 5′UTR, TSS200, TSS1500 | chr7:27170241-27171154 | 14 | 5.8E−04 | − 0.005 | − 0.016 | ||
| F6 | SYNGAP1 | Body | chr6:33401192-33401542 | 6 | 1.0E−03 | 0.004 | 0.008 | ||
| F7 | ZNF833 | TSS1500, TSS200, body | chr19:11784514-11785337 | 13 | 1.1E−03 | − 0.002 | − 0.012 | ||
| F8 | LAMB2 | 1stExon, 5′UTR, TSS200, TSS1500 | chr3:49170496-49170849 | 6 | 3.1E−03 | − 0.008 | − 0.012 | ||
| F9 | GPR19 | 5′UTR, 1stExon, TSS200, TSS1500 | chr12:12848977-12849588 | 9 | 3.7E−03 | 0.001 | 0.023 | 6/7 | 0.0076 |
| F10 | MTMR15 | TSS1500, TSS200, 5′UTR, 1stExon | chr15:31195612-31196075 | 7 | 4.0E−03 | − 0.003 | − 0.017 | ||
| F11 | KCNE1 | 5′UTR, 1stExon, TSS200, TSS1500 | chr21:35831871-35832364 | 8 | 4.2E−03 | 0.007 | 0.019 | ||
| F12 | TNXB | Body | chr6:32054659-32055474 | 20 | 7.2E−03 | 0.0002 | − 0.013 | ||
| F13 | TERT | Body | chr5:1269992-1270152 | 3 | 7.2E−03 | 0.008 | 0.011 | ||
| F14 | C2orf27A | 5′UTR | chr2:132481613-132481826 | 2 | 1.7E−02 | 0.010 | 0.031 | ||
| F15 | ANKRD44 | Body | chr2:198029141-198029332 | 3 | 2.1E−02 | − 0.005 | − 0.018 | ||
1Number of sites located in DMRs significant for dietary folate
2Minimum dietary folate q values of sites located in the DMRs (FDR correction)
3Absolute minimum and maximum of dietary folate coefficient of sites located in the DMRs, for 1 standard deviation of log-transformed diet folate (SD = 0.346)
4Number of sites from the FL region overlapping the DMR/number of sites in the FL region
5Dietary folate changes for an increase of 1 standard deviation of methylation levels of sites located in the FL region
Alcohol intake was associated with methylation levels in 90 DMRs, including 550 CpG sites over-represented in TSS200, 1st exon, and 5′ untranslated regions (5′UTR) and under-represented in the body regions and the regions outside any gene regions (Fig. 2b). The 15 most significant DMRs are detailed in Table 4, and the full list is described in Additional file 3: Table S2. Alcohol intake was positively associated with methylation levels in 66% of the 90 DMRs. The two sites associated with alcohol intake in the CpG site-specific analyses were not included in any DMRs. The most significant DMR associated with alcohol consumption was DMR.A1, 9 sites within the GSDMD gene, (qDMR = 4.7E−14, βmax, DMR = 0.020).
Table 4.
The 15 most significant DMRs associated with alcohol out of 90 significant DMRs (adjusted for recruitment center, age at recruitment, menopausal status, and level of different lymphocyte subtypes)
| DMRs characteristics | CpG characteristics | Fused lasso | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Associated genes | Gene regions | hg19coord | Sites1 | q DMR 2 | β min, DMR 3 | β max, DMR 3 | Overlap4 | β FL 5 | |
| A1 | GSDMD | TSS1500, TSS200, 5′UTR, 1stExon | chr8:144635260-144636462 | 9 | 4.7E−14 | 0.0060 | 0.020 | ||
| A2 | chr6:31650735-31651362 | 21 | 1.8E−13 | 0.0049 | 0.018 | 2/2, 2/2 | 0.390 | ||
| A3 | TRIM4 | Body, 1stExon, 5′UTR, TSS200, TSS1500 | chr7:99516603-99517509 | 14 | 3.0E−06 | − 0.0007 | 0.018 | ||
| A4 | RGL3 | Body | chr19:11517079-11517436 | 5 | 3.3E−06 | 0.0041 | 0.020 | ||
| A5 | COL9A3 | TSS1500 | chr20:61446962-61447992 | 32 | 4.8E−06 | − 0.0004 | − 0.012 | 4/4 | − 1.027 |
| A6 | ADAM32 | TSS1500, TSS200, 1stExon, 5′UTR, Body | chr8:38964500-38965492 | 10 | 1.3E−04 | 0.0019 | 0.014 | ||
| A7 | C21orf56 | 5′UTR, 1stExon, TSS1500 | chr21:47604052-47605174 | 8 | 1.5E−04 | 0.0191 | 0.032 | ||
| A8 | chr2:118616155-118616576 | 5 | 1.9E−04 | 0.0143 | 0.019 | 5/7 | 0.514 | ||
| A9 | LTB4R2, LTB4R, CIDEB | Body, 1stExon, TSS1500, 5′UTR, TSS200 | chr14:24780404-24780926 | 10 | 2.3E−04 | − 0.0031 | − 0.012 | 9/9 | − 0.474 |
| A10 | PTDSS2 | Body | chr11:457256-457304 | 3 | 3.0E−04 | 0.0044 | 0.011 | ||
| A11 | SMC1B, RIBC2 | Body, TSS1500, 1stExon, TSS200, 5′UTR | chr22:45808669-45810043 | 16 | 3.0E−04 | 0.0009 | 0.019 | 4/4, 3/3 | 0.332 |
| A12 | chr10:72013286-72013397 | 2 | 8.4E−04 | − 0.0087 | − 0.014 | ||||
| A13 | TRAF3 | Body | chr14:103366987-103367858 | 5 | 1.4E−03 | − 0.0044 | 0.013 | ||
| A14 | C22orf27 | TSS1500, TSS200, body | chr22:31317764-31318546 | 12 | 1.4E−03 | 0.0016 | 0.015 | 4/4, 2/2 | 0.641 |
| A15 | S100A13, S100A1 | 5′UTR, 1stExon, TSS1500, TSS200 | chr1:153599479-153600156 | 8 | 3.0E−03 | 0.0076 | 0.019 | ||
1Number of sites located in DMRs significant for alcohol
2Minimum alcohol q values of sites located in the DMRs (FDR correction)
3Absolute minimum and maximum of alcohol coefficient of sites located in the DMRs, for 1 standard deviation of alcohol intake (SD = 11.8)
4Number of sites from the FL region overlapping the DMR/number of sites in the FL region, appears twice if two FL regions are included in the DMR
5Alcohol changes for an increase of 1 standard deviation of methylation levels of sites located in the FL region or average of alcohol change if two FL regions are included in a DMR
Methylation levels of each CpG site located in the two most significant DMRs for folate and alcohol, i.e.DMR.F1, DMR.F2, DMR.A1 and DMR.A2, are presented in Additional file 4: Figure S2 by tertiles of dietary folate and alcohol intake, respectively. Correlation heatmaps of CpG sites in DMR.A1, DMR.A2, DMR.F1, and DMR.F2 are displayed in Additional file 5: Figure S3, showing high levels of correlation among methylation levels within the DMR.F2 of dietary folate and the DMR.A2 of alcohol. Other regions showed less correlation, including the DMR.A1 of alcohol intake.
Fused lasso regression
For dietary folate, we identified 71 FL regions, 50 presenting a positive association and 21 an inverse association. Three FL regions were overlapping the 15 most significant DMRs (Table 3). Seven out of 8 sites from a FL region within the GDF7 gene were included in the DMR.F2 (βFL = − 0.0029). All sites from a FL region associated with the PRSS50 gene were part of the DMR.F4 (βFL = − 0.0069). Six out of 7 sites from the FL region within the GPR19 gene were within the DMR.F9 (βFL = 0.0076). None of the 68 other FL regions were overlapping any folate-related DMRs.
For alcohol consumption, we identified 133 FL regions, 71 regions presenting a positive association and 62 an inverse association. Twenty-one regions were included in alcohol-related DMRs. Among them, 9 were overlapping 6 of the 15 most significant DMRs (Table 4). The situation where two close FL regions were part of the same DMR was observed 3 times in the 15 most significant alcohol-related DMRs. In particular, four and three sites from two FL regions located in chromosome 22 were included in DMR.A11, associated with genes SMC1B and RIBC2. All the 9 sites from a FL region were included in DMR.A9 (βFL = − 0.474).
Graphical representations of the DMRs, the FL regions, and their overlap are illustrated for each chromosome in Additional file 6: Figure S4 for dietary folate and Additional file 7: Figure S5 for alcohol intake. For dietary folate, most of FL regions were located in chromosome 3, chromosome 22, and chromosome X. A maximum of four DMRs located in the same chromosome was observed for chromosomes 2 and 3. As for alcohol intake, DMR and FL showed overlap mostly in chromosomes 6 and 22, with, respectively, 4 and 3 DMRs overlapping FL regions.
Discussion
In this study of women from a large prospective cohort, we investigated the association of dietary folate and alcohol intake with leukocyte DNA methylation via three different approaches. The site-specific analysis aimed at identifying single CpG sites independently from each other, whereas DMR and FL analyses aimed at identifying regions of CpG sites using the inter-correlation between methylation levels in close sites, thus exploiting the potential of specific regions of the epigenome to show methylation activity related to lifestyle factors.
While site-specific analysis showed a lack of association between dietary folate, alcohol intake, and individual CpG sites, DMR and FL analyses identified regions of the epigenome associated with dietary folate or alcohol intake. These two sites are located within the body region of the genes FAMB65C and CREB3L2. The FAMB65C gene, also named RIPOR3, is a non-annotated gene. The CREB3L2 gene encodes a transcriptional activator protein and plays a critical role in cartilage development by activating the transcription of SEC23A [18]. Translocation of CREB3L2 gene, located on chromosome 7, and the FUS gene (fused in sarcoma) located on the chromosome 16 has been found in some tumors, including skin cancer and soft tissue sarcoma [37, 38].
Alcohol is known to alter DNA methylation, mostly because it contributes to deregulation of folate absorption, which can lead to a dysfunction of OCM [27]. In our study, alcohol intake was associated with 90 DMRs, some of which may have a role in specific carcinogenesis processes. For example, alcohol intake was inversely associated with methylation levels in DMR.A64 related to the MLH1 gene, which is frequently mutated in hereditary nonpolyposis colon cancer (HNPCC) [39]. A positive association between alcohol intake and methylation in the DMR.A79 was related to the TSPAN32 (tetraspanin 32) gene, also known as the TSSC6 gene, which is one of the several tumor suppressor genes located at locus 11p15.5 in the imprinted gene domain of chromosome 11 [28]. This locus has been associated with adrenocortical carcinoma, lung, ovarian, and breast cancers. Methylations within DMR.A1 were positively associated with alcohol intake, and the related GSDMD gene has also been suggested to act as a tumor suppressor [44]. Alcohol intake was also positively associated with DMR.A6 related to the gene ADAM32, which encodes a protein involved in diverse biological processes, such as brain development, fertilization, tumor development, and inflammation [36].
Several genes, associated with the 24 DMRs identified in our study for dietary folate, were possibly involved in biological processes leading to carcinogenesis. For example, dietary folate was positively associated with methylation in DMR.F16 related to the RTKN (rhotekin) gene, which interacts with GTP-bound Rho proteins. Rho proteins regulate many important cellular processes, including cell growth and transformation, cytokinesis, transcription, and smooth muscle contraction. Dysregulation of the Rho signal transduction pathway has been implicated in many forms of cancer such as bladder cancer, gastric cancer, and breast cancer [9, 15]. Dietary folate was also associated with methylation levels in DMR.F1 and DMR.F5 within the HOXA4, HOXA5, and HOXA6 genes, members of the HOX family, known to be associated with cellular differentiation [46]. Perturbed HOX gene expression has been implicated in multiple cancer types [47]. In addition, HOXA5 may also regulate gene expression and morphogenesis. Methylation of this gene may result in the loss of its expression and, since the encoded protein upregulates the tumor suppressor p53, may play an important role in tumorigenesis [55].
Results from site-specific and DMR analyses were generated with different analytical strategies: methylation levels in different sites were assumed independent in the former, with linear regression models fitted separately in each CpG site, while in the latter, the physical proximity of CpGs was exploited to identify specific regions of the epigenome with similar methylation activity, under the assumption that neighboring CpG sites may share relevant epigenetic information. FL analysis revealed some overlaps with DMRs, particularly for alcohol intake, where 9 FL regions were observed within the 15 most significant DMRs. Yet, the overlap between DMR and FL analyses is relatively low and their results deserve cautious interpretations as they have differences in analytical strategies. Unlike DMRs, FL does not take into account the physical distance between consecutive sites, but rather introduce smoothness of parameters estimated in adjacent mutually adjusted CpG sites. Methylation levels within a chromosome were mutually adjusted in FL regression, while in DMR analysis t test statistics were based on independent associations of methylation levels with folate and alcohol.
The association between folate and DNA methylation has been investigated at different stages of human life, in particular during fetal development and elderly, where folate is especially needed. A meta-analysis of mother-offspring pairs estimated the association between maternal plasma folate during pregnancy and DNA methylation in cord blood [25]. After FDR correction, maternal plasma folate was positively associated with methylation level at 27 CpG sites and inversely associated with methylation level at 416 CpG sites. None of these sites was observed in any of the 24 DMRs related to dietary folate in the present study. This might be explained by the lack of power to identify specific sites due to the sample size: over 2000 samples were included in Joubert’s meta-analysis against 450 in our study. Then, different methods were used to assess folate intake, i.e., plasma folate against dietary folate.
An intervention study was conducted to evaluate the effects of long-term supplementation with folic acid and vitamin B12 on white blood cell DNA methylation in elderly subjects [26]. After the intervention of 2 years, 162 sites were significantly differentially methylated compared to baseline, versus 6 sites only for the placebo group. Folate and vitamin B12 were not significantly associated with methylation level in any CpG sites. Within the same study, 173 and 425 DMRs were identified for folate and vitamin B12, respectively. The gene HOX4, which was inversely associated with dietary folate in our study in DMR.F5, was the only region overlapping with the first 10 DMRs found in the intervention study [26]. However, a higher level of folic acid was observed in the intervention study: averages blood folate of 52 and 23 nmol/L in the intervention and placebo groups, respectively, compared to an average blood folate of 15 nmol/L in our study which might partly explain the different findings.
Within a recent meta-analysis including 9643 participants of European ancestry, aged 42 to 76 years with 54% women [32], 363 CpG sites were significantly associated with alcohol consumption, with 87% of these sites showing inverse associations. In our study, site cg02711608 was part of the 363 identified sites and was also included in DMR.A25 associated with gene SLC1A5. SLC1A5 gene encodes a protein which is a sodium-dependent amino acid transporter [42]. The important difference in the number of significant sites between the meta-analysis and the present study might mostly be explained by the larger study population size and the larger levels of alcohol intake observed in the meta-analysis [32]. Indeed, in the meta-analysis, composed of 46% of men, the medians of alcohol intake ranged from 0 to 14 g/day in the 10 European cohorts, while with a median of 3.5 g/day, alcohol intake was quite low in our study, which included only women. Lastly, cohort-specific approaches were used in the meta-analysis to remove technical variability, while the SVA approach was used in our study, which was shown to produce conservative findings compared to other normalizing techniques [40].
In our study, the sample size was relatively low (n = 450), and women only were included. With a median value of 3.5 g/day, a 95th percentiles equal to 31 g/day, and a percentage of non-consumers equal to 15%, alcohol intake displayed limited variability which potentially constrained the power of the study. In addition, questionnaire measurements used to assess dietary folate and alcohol intake are prone to exposure misclassification, which likely attenuated associations between lifestyle exposures and methylation levels. These elements may alone explain the lack of significant associations in our study. Further studies including men and women, possibly with larger sample size, are needed to further investigate the relationship between dietary folate, alcohol intake, and DNA methylation.
A major strength of this study was the use of ad hoc methodology for normalization of methylation data. Technical management of samples likely introduces systematic technical variability in methylation measurements that might compromise the accuracy of the acquisition process and, if not properly taken into account, could introduce bias in the estimation of the association of interest. The population used in this study included European women from the UK, Germany, Italy, Greece, The Netherlands, and Spain, implying a diversity of diet and lifestyle habits. Three approaches were used to evaluate the relationship between dietary folate, alcohol intake, and DNA methylation. The comparison between DMR and FL analyses was particularly relevant to identify regions of the genome associated with dietary folate and alcohol intake.
Alcohol was classified as group 1 carcinogen in 2012 by the IARC Monograph [22] and was associated with cancer of the upper aero-digestive tract, female breast, liver, and colorectum. Dietary folate has been recently inversely associated with the risk of breast cancer in EPIC [12], although the evidence is not conclusive [59]. Among the DMRs identified in this study for dietary folate or alcohol intake, several regions were associated with genes potentially implicated in cancer development, such as RTKN, the HOX family of genes, and the two tumor suppressor genes GSDMD and TSPAN32. Our study provides some evidence that dietary folate and alcohol intakes may be associated with carcinogenesis through a deregulation of epigenetic mechanisms, although our findings need to be replicated in future evaluations.
In this study, site-specific analyses served as a basis to explore more complex evaluations. By addressing the high dimensionality and complexity of DNA methylation, statistical techniques used in this work may prove useful for future epigenetic studies focusing on the relationship between lifestyle exposures, DNA methylation, and the occurrence of disease outcomes. These tools presented may be adapted to suit specific features of other -omics data.
Conclusion
Weak associations between alcohol intake and methylation levels at two CpG sites were observed. DMR and FL analyses provided evidence that specific regions of CpG sites were associated with dietary folate and alcohol intake, assuming that neighboring features share relevant epigenetic information. Folate and alcohol are known not only to be associated with breast cancer but also to have a mutually antagonistic role in the one-carbon metabolism. In some regions identified by DMRs or FL analysis, mapped genes are known to act as tumor suppressors such as the GSDMD and HOXA5 genes. These results were in line with the hypothesis that folate- and alcohol-deregulated epigenetic mechanisms might have a role in the pathogenesis of cancer.
Additional files
Figure S1. Sample size by recruitment centers. (PDF 10 kb)
Table S1. DMRs associated with dietary folate (log). (DOCX 19 kb)
Table S2. DMRs associated with alcohol intake. (DOCX 34 kb)
Figure S2. Graphical representation of the most 2 significant DMR of dietary folate and alcohol intake. The x-axis represents the position (hg 19 coordinates) of the CpGs included in the plotted DMR. Each tertile of dietary folate, alcohol intake, or their interaction is represented by different colors: green for T1, blue for T2, and red for T3. For all the CpGs included in the plotted DMR, the dashed lines are their 1st and 3rd quartiles of methylation levels and the points represent their median values. (PDF 33 kb)
Figure S3. Correlation heatmap of methylation levels inside the two most significant DMR of folate and alcohol. (PDF 43 kb)
Figure S4. DMRs and FL regions of folate in each chromosome. Dark blue rectangles represent DMRs and light blue FL regions. Overlaps between the two methods are represented by red points. Positive coefficients of the two methods are represented on the top part of each graphic, and negative coefficients are on the bottom part. Positive (negative) coefficients of DMRs were set to 0.5 (− 0.5) and positive (negative) coefficients of FL regions were set to 1 (− 1) to clearly differentiate DMRs from FL regions. The x-axis represents the rank of CpG sites according to their position on the chromosome. (PDF 12 kb)
Figure S5. DMRs and FL regions of alcohol in each chromosome. Dark blue rectangles represent DMRs and light blue FL regions. Overlaps between the two methods are represented by red points. Positive coefficients of the two methods are represented on the top part of each graphic and negative coefficients are on the bottom part. Positive (negative) coefficients of DMRs were set to 0.5 (− 0.5), and positive (negative) coefficients of FL regions were set to 1 (− 1) to clearly differentiate DMRs from FL regions. The x-axis represents the rank of CpG sites according to their position on the chromosome. (PDF 58 kb)
Acknowledgements
The authors would like to thank the financial support provided by La Fondation de France for a doctoral fellowship. They are also grateful for all the women who participated in the EPIC cohort and without whom this work would not have been possible.
Funding
This work was supported by a doctoral fellowship from ‘Fondation de France’ (grant number 2015 00060737) to FP and the grants from the Institut National du Cancer (INCa, France, 2012-070 to IR and ZH), la Ligue nationale contre le cancer (to Z. Herceg). ZH was supported by the European Commission (EC) Seventh Framework Programme (FP7) Translational Cancer Research (TRANSCAN) Framework, the Fondation Association pour la Recherche contre le Cancer (ARC, France). In addition, this study was supported by postdoctoral fellowship to SA from the International Agency for Research on Cancer, partially supported by the EC FP7 Marie Curie Actions – People – Co-funding of regional, national and international programmes (COFUND). SA’s work is supported by Cancer Research UK (grant number: C18281/A19169). SA work in the Medical Research Council Integrative Epidemiology Unit at the University of Bristol which is supported by the Medical Research Council and the University of Bristol (grant number: MC_UU_00011/1, MC_UU_00011/4 and MC_UU_00011/5).
The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF), Deutsche Krebshilfe, Deutsches Krebsforschungszentrum and Federal Ministry of Education and Research (Germany); the Hellenic Health Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, PI13/02633 to EPIC-Navarra), Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (UK).
The funders of the study had no role in study design, data collection, data analysis, data interpretation or writing of the manuscript.
Availability of data and materials
For information on how to submit an application for gaining access to EPIC data and/or biospecimens, please follow the instructions at http://epic.iarc.fr/access/index.php
Abbreviations
- BC
Breast cancer
- BMI
Body mass index
- DMR
Differentially methylated region
- EPIC
European Prospective Investigation into Cancer and Nutrition
- FDR
False discovery rate
- FL
Fused lasso
- HM450K
Illumina Infinium HumanMethylation 450K
- MAF
Minor allele frequency
- NK
Natural killer
- OCM
One-carbon metabolite
- SAM
S-Adenosylmethionine
- SVA
Surrogate variable analysis
Authors’ contributions
FP performed the statistical data analysis and drafted the manuscript. IR and PF developed the concept of the study with FP and contributed to draft the manuscript. SA and CC were responsible for the technical aspects of DNA methylation acquisition. IR and ZH conceived the epigenetics study in the nested case-control study on breast cancer and critically reviewed the manuscript. SA, AG, and HHV contributed to the interpretation of the results. LB, CV, MM, and MJG were involved in the data interpretation. All authors contributed to draft the final versions of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by the Ethical Review Board of the International Agency for Research on Cancer, and by the local Ethics Committees in the participating centres. This study was also conducted in accordance with the IARC Ethic Committee (Project No 10-22).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ambatipudi S, Cuenin C, Hernandez-Vargas H, Ghantous A, Le Calvez-Kelm F, Kaaks R, Herceg Z. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8(5):599–618. 10.2217/epi-2016-0001. [DOI] [PubMed]
- 2.Ambatipudi S, Horvath S, Perrier F, Cuenin C, Hernandez-Vargas H, Le Calvez-Kelm F, Herceg Z. DNA methylome analysis identifies accelerated epigenetic ageing associated with postmenopausal breast cancer susceptibility. Eur J Cancer. 2017;75:299–307. 10.1016/j.ejca.2017.01.014 [DOI] [PMC free article] [PubMed]
- 3.Ba Y, Yu H, Liu F, Geng X, Zhu C, Zhu Q, Zhang Y. Relationship of folate, vitamin B12 and methylation of insulin-like growth factor-II in maternal and cord blood. Eur J Clin Nutr. 2011;65(4):480–85. 10.1038/ejcn.2010.294. [DOI] [PMC free article] [PubMed]
- 4.Baglietto L, English DR, Gertig DM, Hopper JL, Giles GG. Does dietary folate intake modify effect of alcohol consumption on breast cancer risk? Prospective cohort study. Bmj. 2005;331(7520):807. doi: 10.1136/bmj.38551.446470.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98. 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed]
- 6.Bolstad B. M. Probe level quantile normalization of high density oligonucleotide array data. 2001. Retrieved from http://bmbolstad.com/stuff/qnorm.pdf
- 7.Bouckaert KP, Slimani N, Nicolas G, Vignat J, Wright AJ, Roe M, Finglas PM. Critical evaluation of folate data in European and international databases: recommendations for standardization in international nutritional studies. Mol Nutr Food Res. 2011;55(1):166–180. doi: 10.1002/mnfr.201000391. [DOI] [PubMed] [Google Scholar]
- 8.Candès EJ, Wakin MB, Boyd SP. Enhancing sparsity by reweighted ℓ 1 minimization. J Fourier Anal Appl. 2008;14(5):877–905. 10.1007/s00041-008-9045-x.
- 9.Chen M, Bresnick AR, O'Connor KL. Coupling S100A4 to Rhotekin alters Rho signaling output in breast cancer cells. Oncogene. 2012;32:3754. 10.1038/onc.2012.383https://www.nature.com/articles/onc2012383#supplementary-information. [DOI] [PMC free article] [PubMed]
- 10.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–09. 10.4161/epi.23470. [DOI] [PMC free article] [PubMed]
- 11.Cole SR, Platt RW, Schisterman EF, Chu H, Westreich D, Richardson D, Poole C. Illustrating bias due to conditioning on a collider. Int J Epidemiol. 2010;39(2):417–420. doi: 10.1093/ije/dyp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Batlle J, Ferrari P, Chajes V, Park J. Y, Slimani N, McKenzie F, Romieu I. Dietary folate intake and breast cancer risk: European prospective investigation into cancer and nutrition. J Natl Cancer Inst. 2015;107(1):367. 10.1093/jnci/dju367. [DOI] [PubMed]
- 13.Du P, Kibbe WA, Lin SM. Lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24(13):1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 14.Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan J, Ma LJ, Xia SJ, Yu L, Fu Q, Wu CQ, Tang XD. Association between clinical characteristics and expression abundance of RTKN gene in human bladder carcinoma tissues from Chinese patients. J Cancer Res Clin Oncol. 2005;131(3):157–62. 10.1007/s00432-004-0638-8. [DOI] [PubMed]
- 16.Hastie T. The elements of statistical learning: data mining, inference, and prediction. 2009. [Google Scholar]
- 17.Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A. 2012;109. 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed]
- 18.Hino K, Saito A, Kido M, Kanemoto S, Asada R, Takai T, Imaizumi K. Master regulator for chondrogenesis, Sox9, regulates transcriptional activation of the endoplasmic reticulum stress transducer BBF2H7/CREB3L2 in chondrocytes. J Biol Chem. 2014;289(20):13810–13820. doi: 10.1074/jbc.M113.543322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19(6):371–384. doi: 10.1038/s41576-018-0004-3. [DOI] [PubMed] [Google Scholar]
- 20.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13(1):1–16. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Morgan M. Orchestrating high-throughput genomic analysis with bioconductor. Nat Methods. (2015);12(2):115–21. 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed]
- 22.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. (2012). Personal habits and indoor combustions. Volume 100 E. A review of human carcinogens (1017–1606). Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/23193840, https://www.ncbi.nlm.nih.gov/pmc/PMC4781577/. [PMC free article] [PubMed]
- 23.Illumina (Producer) GenomeStudio/BeadStudio Software Methylation Module. 2011. [Google Scholar]
- 24.Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, London SJ. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9(5):436–447. doi: 10.1161/circgenetics.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joubert BR, den Dekker HT, Felix JF, Bohlin J, Ligthart S, Beckett E, London SJ. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat Commun. 2016;7:10577. doi: 10.1038/ncomms10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kok DE, Dhonukshe-Rutten RA, Lute C, Heil SG, Uitterlinden AG, van der Velde N, Steegenga WT. The effects of long-term daily folic acid and vitamin B12 supplementation on genome-wide DNA methylation in elderly subjects. Clin Epigenetics. 2015;7;121. 10.1186/s13148-015-0154-5. [DOI] [PMC free article] [PubMed]
- 27.Kruman II, Fowler AK. Impaired one carbon metabolism and DNA methylation in alcohol toxicity. J Neurochem. 2014;129(5):770–780. doi: 10.1111/jnc.12677. [DOI] [PubMed] [Google Scholar]
- 28.Lee MP, Brandenburg S, Landes GM, Adams M, Miller G, Feinberg AP. Two novel genes in the center of the 11p15 imprinted domain escape genomic imprinting. Hum Mol Genet. 1999;8(4):683–690. doi: 10.1093/hmg/8.4.683. [DOI] [PubMed] [Google Scholar]
- 29.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3. 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed]
- 31.Leek JT, Storey JD. A general framework for multiple testing dependence. Proc Natl Acad Sci U S A. 2008;105(48):18718–18723. doi: 10.1073/pnas.0808709105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu C, Marioni RE, Hedman AK, Pfeiffer L, Tsai PC, Reynolds LM, Levy D. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry. 2016. 10.1038/mp.2016.192. [DOI] [PMC free article] [PubMed]
- 33.Mason JB, Choi S-W. Effects of alcohol on folate metabolism: implications for carcinogenesis. Alcohol. 2005;35(3):235–241. doi: 10.1016/j.alcohol.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 34.Matejcic M, de Batlle J, Ricci C, Biessy C, Perrier F, Huybrechts I, Chajes V. Biomarkers of folate and vitamin B12 and breast cancer risk: report from the EPIC cohort. Int J Cancer. 2017;140(6):1246–1259. doi: 10.1002/ijc.30536. [DOI] [PubMed] [Google Scholar]
- 35.Niculescu MD, Haggarty P. Nutrition in epigenetics: Wiley; 2011.
- 36.O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Pruitt KD. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panagopoulos I, Storlazzi CT, Fletcher CD, Fletcher JA, Nascimento A, Domanski HA, Mertens F. The chimeric FUS/CREB3l2 gene is specific for low-grade fibromyxoid sarcoma. Genes Chromosomes Cancer. 2004;40(3):218–228. doi: 10.1002/gcc.20037. [DOI] [PubMed] [Google Scholar]
- 38.Patel RM, Downs-Kelly E, Dandekar MN, Fanburg-Smith JC, Billings SD, Tubbs RR, Goldblum JR. FUS (16p11) gene rearrangement as detected by fluorescence in-situ hybridization in cutaneous low-grade fibromyxoid sarcoma: a potential diagnostic tool. Am J Dermatopathol. 2011;33(2):140–143. doi: 10.1097/IAE.0b013e318176de80. [DOI] [PubMed] [Google Scholar]
- 39.Peltomaki P, de la Chapelle A. Mutations predisposing to hereditary nonpolyposis colorectal cancer. Adv Cancer Res. 1997;71:93–119. doi: 10.1016/S0065-230X(08)60097-4. [DOI] [PubMed] [Google Scholar]
- 40.Perrier F, Novoloaca A, Ambatipudi S, Baglietto L, Ghantous A, Perduca V, Ferrari P. Identifying and correcting epigenetics measurements for systematic sources of variation. Clin Epigenetics. 2018;10(1):38. doi: 10.1186/s13148-018-0471-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, V Lord R, Molloy PL. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8(1):1–16. doi: 10.1186/1756-8935-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pochini L, Scalise M, Galluccio M, Indiveri C. Membrane transporters for the special amino acid glutamine: structure/function relationships and relevance to human health. Frontiers Chem. 2014;2(61). 10.3389/fchem.2014.00061. [DOI] [PMC free article] [PubMed]
- 43.Riboli E, Hunt KJ, Slimani N, Ferrari P, Norat T, Fahey M, Saracci R. European Prospective Investigation into Cancer and Nutrition (EPIC): study populations and data collection. Public Health Nutr. 2002;5(6B):1113–1124. doi: 10.1079/phn2002394. [DOI] [PubMed] [Google Scholar]
- 44.Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, Sasaki H. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer. 2009;48(3):261–271. doi: 10.1002/gcc.20636. [DOI] [PubMed] [Google Scholar]
- 45.Satterthwaite FE. An approximate distribution of estimates of variance components. Biometrics. 1946;2. 10.2307/3002019. [PubMed]
- 46.Seifert A, Werheid DF, Knapp SM, Tobiasch E. Role of Hox genes in stem cell differentiation. World J Stem Cells. 2015;7(3):583–595. doi: 10.4252/wjsc.v7.i3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010;10(5):361–371. doi: 10.1038/nrc2826. [DOI] [PubMed] [Google Scholar]
- 48.Sieri S, Agudo A, Kesse E, Klipstein-Grobusch K, San-Jose B, Welch AA, Slimani N. Patterns of alcohol consumption in 10 European countries participating in the European Prospective Investigation into Cancer and Nutrition (EPIC) project. Public Health Nutr. 2002;5(6b):1287–1296. doi: 10.1079/phn2002405. [DOI] [PubMed] [Google Scholar]
- 49.Slimani N, Deharveng G, Unwin I, Southgate DA, Vignat J, Skeie G, Riboli E. The EPIC nutrient database project (ENDB): a first attempt to standardize nutrient databases across the 10 European countries participating in the EPIC study. Eur J Clin Nutr. 2007;61(9):1037–1056. doi: 10.1038/sj.ejcn.1602679. [DOI] [PubMed] [Google Scholar]
- 50.Slimani N, Kaaks R, Ferrari P, Casagrande C, Clavel-Chapelon F, Lotze G, Riboli E. European Prospective Investigation into Cancer and nutrition (EPIC) calibration study: rationale, design and population characteristics. Public Health Nutr. 2007;5(6b):1125–1145. doi: 10.1079/PHN2002395. [DOI] [PubMed] [Google Scholar]
- 51.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet and Mol Biol. 2004;3. [DOI] [PubMed]
- 52.Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and computational biology solutions using R. and bioconductor. New York: Springer; 2005. [Google Scholar]
- 53.Szyf M. The implications of DNA methylation for toxicology: toward toxicomethylomics, the toxicology of DNA methylation. Toxicol Sci. 2011;120(2):235–255. doi: 10.1093/toxsci/kfr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teegarden D, Romieu I, Lelievre SA. Redefining the impact of nutrition on breast cancer incidence: is epigenetics involved? Nutr Res Rev. 2012;25(1):68–95. doi: 10.1017/s0954422411000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Teo WW, Merino VF, Cho S, Korangath P, Liang X, Wu Rc, Sukumar S. HOXA5 determines cell fate transition and impedes tumor initiation and progression in breast cancer through regulation of E-cadherin and CD24. Oncogene. 2016;35(42):5539–51. 10.1038/onc.2016.95. [DOI] [PMC free article] [PubMed]
- 56.Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tibshirani R, Saunders M, Rosset S, Zhu J, Knight K. Sparsity and smoothness via the fused lasso. J. R. Stat. Soc. Ser. B Stat Methodol. 2005;67(1):91–108. doi: 10.1111/j.1467-9868.2005.00490.x. [DOI] [Google Scholar]
- 58.Wareham NJ, Jakes RW, Rennie KL, Schuit J, Mitchell J, Hennings S, Day NE. Validity and repeatability of a simple index derived from the short physical activity questionnaire used in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Public Health Nutr. 2003;6(4):407–413. doi: 10.1079/phn2002439. [DOI] [PubMed] [Google Scholar]
- 59.World Cancer Research Fund International, & American Institue for Cancer Research. (2017). Continuous update project report: diet, nutrition, physical activity and breast cancer. Retrieved from https://www.wcrf.org/sites/default/files/Breast-Cancer-2017-Report.pdf
- 60.Zhang S, Hunter DJ, Hankinson SE, Giovannucci EL, Rosner BA, Colditz GA, Willett WC. A prospective study of folate intake and the risk of breast cancer. JAMA. 1999;281(17):1632–1637. doi: 10.1001/jama.281.17.1632. [DOI] [PubMed] [Google Scholar]
- 61.Zou H. The adaptive lasso and its oracle properties. J Am Stat Assoc. 2006;101(476):1418–1429. doi: 10.1198/016214506000000735. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sample size by recruitment centers. (PDF 10 kb)
Table S1. DMRs associated with dietary folate (log). (DOCX 19 kb)
Table S2. DMRs associated with alcohol intake. (DOCX 34 kb)
Figure S2. Graphical representation of the most 2 significant DMR of dietary folate and alcohol intake. The x-axis represents the position (hg 19 coordinates) of the CpGs included in the plotted DMR. Each tertile of dietary folate, alcohol intake, or their interaction is represented by different colors: green for T1, blue for T2, and red for T3. For all the CpGs included in the plotted DMR, the dashed lines are their 1st and 3rd quartiles of methylation levels and the points represent their median values. (PDF 33 kb)
Figure S3. Correlation heatmap of methylation levels inside the two most significant DMR of folate and alcohol. (PDF 43 kb)
Figure S4. DMRs and FL regions of folate in each chromosome. Dark blue rectangles represent DMRs and light blue FL regions. Overlaps between the two methods are represented by red points. Positive coefficients of the two methods are represented on the top part of each graphic, and negative coefficients are on the bottom part. Positive (negative) coefficients of DMRs were set to 0.5 (− 0.5) and positive (negative) coefficients of FL regions were set to 1 (− 1) to clearly differentiate DMRs from FL regions. The x-axis represents the rank of CpG sites according to their position on the chromosome. (PDF 12 kb)
Figure S5. DMRs and FL regions of alcohol in each chromosome. Dark blue rectangles represent DMRs and light blue FL regions. Overlaps between the two methods are represented by red points. Positive coefficients of the two methods are represented on the top part of each graphic and negative coefficients are on the bottom part. Positive (negative) coefficients of DMRs were set to 0.5 (− 0.5), and positive (negative) coefficients of FL regions were set to 1 (− 1) to clearly differentiate DMRs from FL regions. The x-axis represents the rank of CpG sites according to their position on the chromosome. (PDF 58 kb)
Data Availability Statement
For information on how to submit an application for gaining access to EPIC data and/or biospecimens, please follow the instructions at http://epic.iarc.fr/access/index.php