

Abstract
Background
Histone methylation mainly occurs on the lysine residues and plays a crucial role during flowering and stress responses of plants, through changing the methylation status or ratio of lysine residues. Histone lysine residues of plants can arise in three forms of methylation (single, double and triple) and the corresponding demethylation can also ensue on certain occasions, by which the plants can accommodate the homeostasis of histone methylation by means of lysine methyltransferase and demethylase. The JmjC domain-containing proteins, an important family of histone lysine demethylases, play a vital role in maintaining homeostasis of histone methylation in vivo.
Results
In this study, we have identified 19 JmjC domain-containing histone demethylase (JHDM) proteins in maize. Based on structural characteristics and a comparison of phylogenetic relationships of JHDM gene families from Arabidopsis, rice and maize, all 19 JHDM proteins in maize were categorized into three different subfamilies. Furthermore, chromosome location and schematic structure revealed an unevenly distribution on chromosomes and structure features of ZmJMJ genes in maize, respectively. Eventually, the 19 ZmJMJ genes displayed different expression patterns at diverse developmental stages of maize based on transcriptome analysis. Further, quantitative real-time PCR analysis showed that all 19 ZmJMJ genes were responsive to heat stress treatment, suggesting their potential roles in heat stress response.
Conclusions
Overall, our study will serve to present an important theoretical basis for future functional verification of JHDM genes to further unravel the mechanisms of epigenetic regulation in plants.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5633-1) contains supplementary material, which is available to authorized users.
Keywords: Epigenetics, Histone demethylation, JHDM gene family, JmjC domain, Heat stress, Zea mays L
Background
In eukaryotic genomes, the histones (H2A, H2B, H3 and H4) wind DNA into nucleosome, and then the DNA-protein complexes are further compressed to form a complex structure known as chromatin. The core nucleosome is composed of histone octamer (containing two copies of each of the four histone proteins H2A, H2B, H3 and H4), and its tail has been elucidated to have posttranslational modifications [1, 2] including methylation, acetylation, phosphorylation, ADP-ribosylation and ubiquitination, which are designated as the histone code. These histone modifications not only affect and change the status of genomic chromatin, but also regulate the transcriptional process of gene expression [3]. Therefore, these histone modifications, such as methylation and demethylation, have become one of the hot topics in epigenetic research in recent years.
Histone methylation and demethylation play crucial roles in regulating gene expression, genome integrity, and epigenetic inheritance [4–7]. Histone methylation can occur at various lysine and arginine residues, including K4, K9, K27, K36, and K79 in histone H3 and K20 in histone H4 [4, 8]. Some protein families including PRMT and SET domains are responsible for histone methylation of specific arginine and lysine residues and have been characterized, respectively. Moreover, two kinds of histone demethylase are known to play important roles in demethylation of lysine residues in organisms [4]. Based on the different ways of their action, these lysine demethylases are sorted into two categories: (1) LSDs, lysine-specific demethylases 1, remove the methyl groups by amine oxidation; (2) JHDMs (JmjC domain-containing histone demethylases), the demethylases containing the JmjC domains, remove the methyl groups through hydroxylation [9, 10]. These two types of enzymes require various cofactors and act on particular substrates. For instance, LSD1 relies on the involvement of Flavin adenine dinucleotide (FAD), which acts only on the mono- and di-methylated lysines. However, JHDM resorts to the participation of Fe and α-ketoglutarate (α-KG), which can act on the methylation of three forms of lysine [6, 11, 12].
The JmjC domain-containing proteins in plants have been demonstrated to be involved in regulating epigenetic processes and in plant growth and development [4, 7, 13]. Many members of the JmjC gene family from various plant species have been identified and characterized in plants. For instance, Arabidopsis contained 21 JmjC proteins, which are involved in the regulation of RNA silencing, DNA methylation, Brassinosteroid (BR) signaling pathway, affecting flowering, and biorhythm and bud regeneration and are subdivided into five groups: JARID1/KDM5, JHDM3/KDM4, JHDM2/KDM3, JHDM6 and a class of proteins just containing the JmjC domain [10]. In the JARID1/KDM5 group, there are a couple of Arabidopsis JmjC genes (AtJMJ14 and AtJMJ15) encoding histone H3K4 demethylases, which are capable of reversing H3K4me3, H3K4me2, and H3K4me1 in vivo and regulate flowering time in Arabidopsis [14]. In the JHDM3/KDM4 group, AtJMJ11/ELF6 (EARLY FLOWERING 6) and its close homologue, AtJMJ12/REF6 (RELATIVE OF EARLY FLOWERING 6) have been shown to play divergent roles in flowering time control. Loss-of-function mutation of AtJMJ11/REF6 leads to increased expression of the flowering repressor FLC (FLOWERING LOCUS C) and hence late flowering [15]. In addition, AtJMJ11/ELF6 and AtJMJ12/REF6 are revealed to interact with BES1, a transcription factor in the brassinosteroid (BR) signaling pathway [16], indicating that histone demethylases can be recruited to target genes by interacting with sequence specific transcription factors. In the JHDM2/KDM3 group, the histone H3K9 demethylase, AtJMJ25/IBM1 (INCREASE IN BONSAI METHYLATION 1) protects genes from CHG (H represents A, T, or G) hypermethylation by CMT3 (CHROMOMETHYLASE 3) [17, 18]. Mutations in AtJMJ25/IBM1 result in increased DNA methylation and a variety of developmental phenotypes that rely on histone H3K9 methylation and can be completely inhibited by mutations in KYP [19, 20]. Furthermore, in the JHDM6 and a class of proteins just containing the JmjC domain groups, AtJMJ30/JMJD5, an evening-expressed gene, is the sole AtJMJ protein to exhibit a robust circadian rhythm of expression [21–24]. By analysis of loss- and gain-of-function mutants, the function of AtJMJ30 has been verified as a genetic regulator of period length in the Arabidopsis circadian clock [21].
Maize (Zea mays L.), as one of the most important crop species in the world, has become one of the important model monocot species for functional genomics analysis. It has been of great significance to the study of molecular biology in plants. However, little is known about the identification and function of histone demethylase gene family in maize. In this study, we performed a comprehensive analysis of ZmJMJ genes, including their phylogenetic relationships, gene structure, domain architecture, chromosome location, duplication patterns, and expression profiles, which will facilitate future studies to elucidate the exact biological functions of the ZmJMJ genes in maize.
Results
Identification of the members of JHDM gene family in maize
To identify all possible homologs of JHDM gene family in maize, the amino acid sequences of Arabidopsis and rice JmjC domain-containing proteins and their JmjC domains (Pfam: PF02373, SMART: SM00558) were adopted as the query sequences to search against the maize genome database with BlastP program. Subsequently, the candidates were surveyed using Pfam and SMART databases to further verify the presence of the JmjC domains, respectively. Consequently, 19 non-redundant JHDM genes were identified and described from the original data after removing the redundant sequences. The total number of JHDM genes identified in maize (19) is similar to that in either Arabidopsis (21) or rice (20) (Table 1). We aligned all the ZmJMJ genes with Arabidopsis and rice JHDM genes to generate a phylogenetic tree for classification of maize JHDM genes. Based on their relationships with Arabidopsis and rice JHDM genes, the 19 ZmJMJ genes were divided into three distinct subfamilies, including the JARID1/KDM5 subfamily (8 genes), the JHDM3/KDM4 subfamily (8 genes) and the JHDM2/KDM3 subfamily (3 genes) (Table 1). A total of 19 JHDM genes are uniformly named as ZmJMJ1-ZmJMJ19 based on the position of their corresponding genes on chromosomes 1–10 (Additional file 1: Table S1). Then, the filtered maize protein sequences were analyzed by Expasy (https://web.expasy.org/compute_pi/) to further confirm some physical parameters including the number of amino acid residues, the isoelectric point and the molecular weight and so on. The length of the ZmJMJ open reading frames (ORFs) varied from 1500 bp for ZmJMJ1 to 4569 bp for ZmJMJ18, with the respective coding potential of 499 and 1522 amino acids (Additional file 1: Table S1). Furthermore, the molecular weight varied from 56,430.79 to 167,759.67, indicating that ZmJMJ proteins share a variety of molecular weight scope.
Table 1.
JHDM gene distribution among species that were used in this study
| Category (Genome size) |
Maize (2300Mbp) |
Arabidopsis
(125Mbp) |
Rice (389Mbp) |
|---|---|---|---|
| JARID1 | 8 | 6 | 4 |
| JHDM3 | 8 | 3 | 5 |
| JHDM2 | 3 | 6 | 4 |
| JHDM6 | 0 | 3 | 2 |
| JmiC-only | 0 | 3 | 5 |
| Total number | 19 | 21 | 20 |
The structures of identified JHDM proteins in maize
To gain more insights into the diversity of the domain architecture, the structure types of 19 maize JHDM proteins were determined by Pfam (http://pfam.xfam.org/) and SMART (http://smart.embl-heidelberg.de/) with default parameters. Based on the phylogenetic analysis of the diverse structural characteristics of Arabidopsis JHDM protein subfamilies [11], the maize JHDM protein family were mainly categorized into three subfamilies: subfamily I(JARID1/KDM5), subfamily II(JHDM3/KDM4) and subfamily III (JHDM2/KDM3), with each subfamily containing 8, 8 and 3 members, respectively (Fig. 1). In JARID1/KDM5 subfamily, the majority of members share five diverse conserved domains including JmjN, JmjC, zf-C5HC2, FYRN and FYRC domains, while ZmJMJ10 contains additional PHD and ARID domains. However, in JHDM2/KDM3 subfamily or JHDM3/KDM4 subfamily, there is only one JmjC domain or three diverse conserved domains, respectively. Among the three subfamilies, all of these proteins all share a conserved JmjC domain, which is characteristic of JHDM proteins and related to the demethylation process of the histone lysine site of action. In addition, the JmjN domain is the second most widespread domain, appearing in the majority of members of two groups, the JARID1/KDM5 and JHDM3/KDM4 subfamilies. The zf-C5HC2 domain was also identified in the JARID1/KDM5 and JHDM3/KDM4 subfamilies. However, there are still some members of the JARID1/KDM5 and JHDM3/KDM4 subfamilies not displaying the zf-C5HC2 domains. It still remains a possibility that the zf-C5HC2 domains in these proteins are lower conserved and hard to be detected under current analysis parameters. Five of members of the JARID1/KDM5 subfamily contain FYRN and FYRC domains, which may hold chromatin-binding activity [11] or contribute to JmjC domain function by collaborating with other proteins.
Fig. 1.

Schematic structure of JHDM proteins in maize. Schematic representation of conserved domains identified among each subfamily of maize JHDM proteins. The protein is represented as a blue box; the N terminus is on the left for each representation. The location and size of domains are shown by different color rectangles as indicated in the key. JmjN, Jumonji N domain; JmjC, Jumonji C domain; zf-C5HC2, Zinc finger of C5HC2-type; FYRN,“FY-rich” domain N-terminal; FYRC, “FY-rich” domain C-terminal; ARID, AT-rich interaction domain; PHD, plant home box domain
Furthermore, two important cofactors, Fe (II) iron and α-KG, have been verified to be necessary to the JmjC domain-containing proteins to perform their demethylase activity [25, 26]. A total of five conserved amino acid residues, namely three conserved residues (His188, Glu/Asp190, and His276) for the Fe (II) cofactor and other two conserved residues (Thr/Phe185 and Lys206) for α-KG are indispensable to bind these cofactors. To further confirm whether these conserved residues bound by cofactors had diverged among the ZmJMJ proteins, we aligned the domain sequences of the JmjC domain-containing proteins from Arabidopsis, rice and maize. On basis of the alignments and conserved amino acid residues, these proteins were sorted into two groups. The first group (JARID1/KDM5, JHDM3/KDM4), contains the three conserved amino acid residues, namely His (H), Glu (E) and His (H) for Fe (II) binding, and other two conserved amino acid ones, namely Phe (F) and Lys (K) for α-KG binding (Additional file 2: Figure S1, and Additional file 3: Figure S2), while the second group (JHDM2/KDM3) comprises the corresponding conserved residues, namely His (H), Asp (D) and His (H) for Fe(II) binding and Thr (T) and Lys (K) for α-KG binding (Additional file 4: Figure S3). Both of groups are compatible with lysine demethylation activity [11]. In short, the majority of members were detected to comprise these conserved amino acid residues bound by cofactors, although there are some special cases. For instance, the third site to interact with Fe (II) iron is missing in ZmJMJ10 protein, while the ZmJMJ19 protein only retains the third conserved His (H) residue for Fe (II) binding and is lack of other conserved sites for interacting with cofactors. Taken together, these conserved residues could play a vital role in involving demethylase activity of the JHDM protein family.
The structures of identified JHDM genes in maize
To illustrate the exon-intron structures of individual maize JHDM genes, comparison of the genomic sequences and their corresponding coding sequences (CDS) was operated using GSDS (Gene Structure Display Server) (http://gsds.cbi.pku.edu.cn/). The unrooted tree categorized the JHDM genes into three major groups with well-supported bootstrap values, indicating that the classification was reliable. The schematic structures clearly revealed that most of the paralogs share a similar exon-intron structure except ZmJMJ8, ZmJMJ10, ZmJMJ12 and ZmJMJ14 has only one exon, respectively (Fig. 2). In JARID1 subfamily, the most coding sequences are disrupted by approximate 7 to 12 exons except ZmJMJ10 and ZmJMJ14. The ZmJMJ2 and ZmJMJ4 in JHDM2 subfamily have the largest number of exon or intron, and share a similar gene structure. Moreover, there was some deference in intron lengths and in the 5’UTR region between ZmJMJ2 and ZmJMJ4, which might be related to the regulation of expression [4].
Fig. 2.

Phylogenetic relationship and gene structure of JHDM genes in maize. a The unrooted tree was constructed with MEGA6.0 program using the full-length sequences of the 19 maize JHDM genes. The bootstrap values are indicated at the branches in black numbers, and the genes were named according to their gene codes. b Exons and introns are indicated by white green boxes and single lines, respectively. Thick blue lines represent the untranslated regions (UTRs). The length of each JHDM gene can be estimated using the scale at the bottom
Phylogenetic analysis of maize JHDM proteins
To better discern the phylogenetic relationship among JHDMs in plants, and assess the evolutionary history of these protein families among maize, rice and Arabidopsis, we performed multiple sequence alignment with MEGA6.0 by using the full-length sequences of each JHDM protein from these plants and constructed an unrooted phylogenetic tree by using Neighbor-Joining method (Fig. 3).
Fig. 3.

Phylogenetic analysis among JHDM proteins of maize, rice and Arabidopsis. Unrooted neighbor-joining (NJ) phylogenetic tree of maize, rice and Arabidopsis JHDM proteins with bootstrap values shown for each clade. The maize ZmJMJs have been highlighted for each group. Five clades are marked as JARID1, JHDM3, JHDM2, JHDM6 and JmjC-only. Protein sequences were downloaded from National Center for Biotechnology Information (NCBI)
The unrooted neighbor-joining phylogenetic tree was generated from aligned full-length protein sequences of 21 AtJMJs, 20 OsJMJs, and 19 ZmJMJs. Based on the comparison and analysis of JHDM diversity and phylogeny, these proteins were categorized into five major subfamilies: JARID1, JHDM3, JHDM2, JHDM6 and a class of proteins just containing the JmjC domain. However, the maize JHDM proteins were only divided into three subfamilies: JARID1, JHDM3, and JHDM2, with each subfamily containing 8, 8 and 3 members, respectively (Fig. 6). In JARID1 subfamily, maize ZmJMJ10 exhibits high homology with rice OsJMJ708 (E = 0.0), OsJMJ718 (E = 0.0) and Arabidopsis AtJMJ17 (E = 3e-161). Maize ZmJMJ14, ZmJMJ13, ZmJMJ7, ZmJMJ6, ZmJMJ5 share great similarity on evolution with Arabidopsis AtJMJ16 (E = 0.0; 0.0; 0.0; 0.0; 0.0) and rice OsJMJ703 (E = 0.0; 0.0; 0.0; 0.0; 0.0), and therefore they belong to orthologous proteins. In JHDM3 subfamily, maize ZmJMJ12, ZmJMJ11and rice OsJMJ707 (E = 0.0; 0.0) belongs to orthologous proteins. ZmJMJ9 and ZmJMJ1 share similarity with rice OsJMJ706 (E = 0.0; 0.0); and maize ZmJMJ19, ZmJMJ17 and ZmJMJ3 have the high similarity with Arabidopsis AtJMJ12 (E = 1e-71; 0.0; 2e-132) and rice OsJMJ705 (E = 9e-76; 0.0; 0.0) and OsJMJ702 (E = 0.0; 0.0; 1e-123). In JHDM2 subfamily, maize ZmJMJ8 and rice OsJMJ719 (E = 0.0) belong to orthologous proteins; and maize ZmJMJ2 and ZmJMJ4 share high similarity with rice OsJMJ715 (E = 0.0; 0.0) and OsJMJ720 (E = 0.0; 0.0).
Fig. 6.

Hierarchical clustering of expression profiles of 19 ZmJMJ genes in 10 tissues. The color scale representing log2 signal values is shown on the left. Green represents a low level and red indicates a high level of transcript abundance (colour figue online). The different tissues and/or organs are noted on the top of each lane. Cluster dendrograms are shown on the left and above, respectively
Chromosomal location and gene duplication of maize JHDM gene family
The physical locations of JHDM genes in maize were investigated by analysis of genomic distribution on chromosomes. The 19 ZmJMJ genes were distributed unevenly across nine of all the ten chromosomes in the maize genome (Fig. 4). A great majority of ZmJMJs are clustered in the bottom of chromosomes and mainly distributed on three (chromosomes 4, 5, 6) out of nine maize chromosomes, which contained the largest number of JHDM gene family members with total12 genes, whereas the least number was detected on chromosomes 3, 7, 8, 9 and 10, containing only one JHDM gene, respectively (Fig. 4). Moreover, gene duplication events were surveyed to explore the evolutionary patterns of the JHDM gene family in maize. Analysis of maize JHDM genes in SyMAP [27] and PLAZA [28] revealed two JHDM genes (ZmJMJ3 and ZmJMJ17) could be assigned to maize segmental duplication and another two JHDM genes (ZmJMJ5 and ZmJMJ6) could be involved in tandem duplication (Fig. 4). These two gene pairs (ZmJMJ5/ZmJMJ6 and ZmJMJ3/ZmJMJ17) were revealed to have very high homology in the sequences by analyses of sequence alignment, indicating that gene duplication events might have occurred during their process of evolution. Moreover, the duplicated genes were inconsistent with those identified in the phylogenetic tree because two ZmJMJ genes (ZmJMJ15 and ZmJMJ16) were not in homologous regions.
Fig. 4.

Chromosomal localization and gene duplication events of maize JHDM genes. a Distribution of ZmJMJ genes in maize chromosomes. Nineteen ZmJMJ genes were mapped on the nine maize chromosomes. The chromosome numbers are indicated at the top of each vertical gray bar. The gene names on the left side of each chromosome correspond to the approximate locations of each ZmJMJ genes. Both segmentally duplicated paralogs and tandemly duplicated homologs are connected and marked by dashed arrows. The scale on the left is in megabases. b Duplicated blocks in maize chromosomes. The circular image retrieved from PLAZA database show inter-chromosome homologous regions connected by bands in different colors. The chromosome numbers and ZmJMJ genes are indicated outside
The cis-acting regulatory elements in the promoter of maize JHDM genes
To further elucidate the possible regulation mechanisms of maize JHDM genes in the abiotic or biotic stress response, the promoter sequences were analyzed using the PlantCARE database to identify cis-regulatory elements in the promoter region. Twelve types of stress- and hormone-related cis-acting regulatory elements were detected in the promoters of maize JHDM genes: eight hormone-related elements (ERE, ABRE, GARE-motif, CGTCA-motif, motif IIb, TCA-element, TGA-element, TGACG-motif) and four stress-related elements (TC-rich repeats, W box, GCC box, HSE) (Fig. 5). All 19 JHDM genes contained 3–8 cis-elements related to stress or hormone response. The ABRE (abscisic acid responsiveness) were detected in 15 and the CGTCA-motif (MeJA-responsiveness) and TGACG-motif (MeJA-responsiveness) were detected in 13 maize JHDM genes. In addition, 11, 10, 9, 9 and 7 JHDM genes contained TGA-element (auxin-responsive element), TC-rich repeats (defense and stress responsiveness), TCA-element (salicylic acid responsiveness), W-box (wounding and pathogen responsiveness), and GARE-motif (gibberellin-responsive element), respectively. Therefore, these results demonstrated that expression of maize JHDM genes would be regulated by various environmental factors.
Fig. 5.

Distribution of major stress-related cis-elements in the promoter sequences of the 19 ZmJMJ genes. Putative ERE, ABRE, GARE-motif, CGTCA-motif, W box, GCC box, motif IIb, TC-rich repeats, TCA-element, TGA-element, HSE and TGACG-motif core sequences are represented by different symbols as indicated in figure key at the bottom. The cis-elements distributed on the sense strand and reverse strand are indicated above and below the black lines, respectively. ERE: ethylene-responsive element; ABRE: cis-acting element involved in the abscisic acid responsiveness; GARE-motif: gibberellin-responsive element; CGTCA-motif: cis-acting regulatory element involved in the MeJA-responsiveness; W box: elicitation; wounding and pathogen responsiveness; GCC box: elicitation; wounding and pathogen responsiveness; motif IIb: abscisic acid responsive element; TC-rich repeats: cis-acting element involved in defense and stress responsiveness; TCA-element: cis-acting element involved in salicylic acid responsiveness; TGA-element:auxin-responsive element; HSE: cis-acting element involved in heat stress responsiveness; TGACG-motif: cis-acting regulatory element involved in the MeJA-responsiveness. The 1.5 kb sequences upstream of the initiation codon (ATG) of the JHDM genes can be estimated using the scale per 250 bp at the above
Analysis of microarray expression profile of maize JHDM genes
To further investigate the possible functions of the ZmJMJs in plant growth and development, a comprehensive expression analysis was firstly accomplished based on whole-genome coverage according to the microarray data released by Sekhon et al. (2011) [29], which provide us with valuable information for gene functional characterization. It can be seen from the heat map that 18 of all 19 ZmJMJ genes (except ZmJMJ15) have distinct expression levels in 10 tissues at different developmental stages as shown in Additional file 1: Table S1, which can be divided into two major groups (Fig. 6). The first group includes all the members of the JADID1 subfamily (ZmJMJ5, ZmJMJ6, ZmJMJ7, ZmJMJ13 and ZmJMJ14), the JHDM3 subfamily (ZmJMJ1, ZmJMJ3, ZmJMJ11 and ZmJMJ18), and the JHDM2 subfamily (ZmJMJ4), which exhibit relatively low expression levels in most of the tissues and/or organs, while the second group, containing all the members of the JADID1 subfamily (ZmJMJ10 and ZmJMJ16), the JHDM3 subfamily (ZmJMJ9, ZmJMJ12, ZmJMJ17 and ZmJMJ19), and the JHDM2 subfamily (ZmJMJ2 and ZmJMJ8), show relatively high expression levels. In addition, some tissue/organ-specific genes were discovered according to the results shown in the heat map. Specifically, ZmJMJ2 and ZmJMJ17 were expressed at high levels in silk and shoot tips respectively, which was remarkably more than any other tissues (Additional file 5: Table S2). Moreover, to further confirm the expression patterns of 19 JHDM genes in maize, we also carried out a comprehensive expression profile analysis of the maize JHDM gene family using GENEVESTIGATOR database (https://genevestigator.com/gv/index.jsp) (Additional file 6: Figure S4). The results further demonstrated that the maize JHDM gene family displayed different expression patterns at diverse developmental stages of maize. Furthermore, to investigate stress-responsiveness of these JHDM genes to environmental stress, we also carried out a comprehensive expression profile analysis of the maize JHDM gene family by using GENEVESTIGATOR database (Additional file 7: Figure S5). The results further demonstrated that the maize JHDM gene family displayed different expression patterns under diverse environmental stress conditions, suggesting that these genes were responsive to stress treatments.
Expression analysis of maize JHDM genes under heat stress treatment
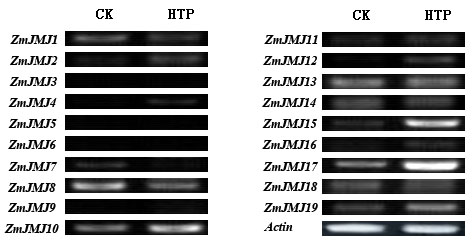
To survey if these predicted genes were expressed in maize and to further confirm their stress-responsiveness to abiotic stress, the transcripts of the 19 genes were firstly detected in the leaves by heat treatment using RT-PCR (Additional file 8: Figure S6). The results revealed that all 19 ZmJMJ genes were responsive to heat stress treatment, suggesting the authenticity of these genes and their potential roles in heat stress response. Secondly, to further confirm their stress-responsiveness to abiotic stress, quantitative real-time PCR was accomplished for 19 ZmJMJ genes in the leaves of maize exposed to heat stress (Fig. 7). Three biological replications were performed in all reactions. The results revealed that these genes are differentially expressed in the leaves under either normal condition (CK) or heat stress condition (HTP). Interestingly, most of members of ZmJMJ gene family exhibited similar up-regulated expressional patterns under heat stress treatment. Among them, there were seven of the expressed genes (ZmJMJ3, ZmJMJ5, ZmJMJ6, ZmJMJ8, ZmJMJ9, ZmJMJ10 and ZmJMJ19), which had lower expression level in the leaves of maize under normal condition, however, they showed significantly up-regulated expression levels (> 2-fold) during heat stress treatment. What’s more, six expressed genes (ZmJMJ2, ZmJMJ4, ZmJMJ7, ZmJMJ11, ZmJMJ12 and ZmJMJ16) were greatly down-regulated (< 0.5-fold) during the heat stress treatment. These results demonstrated that these predicted genes of the ZmJMJ gene family were responsive to heat stress treatment, suggesting their potential roles in heat stress response. Additionally, to further demonstrate that these predicted genes of the ZmJMJ gene family were responsive to heat stress treatment, we also constructed the expression profile of 22 newly-identified heat shock protein genes (ZmHSP70–1~ZmHSP70–22) (unpublished), which may play a central role in the heat stress response (Additional file 9: Figure S7). The results revealed that most of the identified heat shock protein genes in maize exhibited significantly up-regulated expression levels.
Fig. 7.

Relative gene expression of ZmJMJs analyzed by qRT-PCR response to heat stress treatment. QRT-PCR data was normalized using maize Actin gene and are shown relative to CK (normal condition). X-axes show different treatments (CK, normal condition and HTP, heat stress condition) and y-axes are scales of relative expression level. Bars represent standard deviations (SD) of three technical replicates. A significant difference is indicated by asterisks according to the t test (*P < 0.05, **P < 0.01)
Discussion
Histone methylation plays an important role in epigenetic regulation of gene expression, which is determined by adjusting the homeostasis of histone methylation through histone methyltransferase and demethylase. JmjC domain-containing proteins represent a large family of histone demethylases in both animals and plants and play essential roles in histone modification, which is a significant part of epigenetics [7, 30]. In the present study, a comprehensive set of 19 no redundant ZmJMJ genes were identified and characterized from the latest version of the maize (B73) genome database, including their phylogenetic relationships, gene structure, domain architecture, chromosome location, duplication patterns, and expression profiles.
Initially, phylogenetic analysis provided insights into the evolution of gene family members and gene multiplicity in maize. In our investigation of JHDM proteins, we found that maize JHDM genes were mainly categorized into three distinct subclasses, which is also present in Arabidopsis and rice. However, in the JHDM6 or JmjC-only subfamily, no any JHDM gene was found in maize, indicating that the evolution of the JHDM genes in these two groups may occur after divergence of monocots and dicots. Moreover, although the maize genome size (2300Mbp) is larger than that of Arabidopsis (125Mbp) and rice (389Mbp), only 19 JHDM genes were identified and characterized in maize. The number of ZmJMJ genes is similar to previous studies in Arabidopsis (21) and rice (20). This phenomenon may result from less gene duplications in maize genome or experiencing a large gene loss during maize genome duplication. This further demonstrates that the JHDM proteins are highly conserved in evolution. Moreover, the structure features of ZmJMJ proteins were revealed preliminarily by analyzing the structures of ZmJMJ proteins. All of the ZmJMJ proteins are mainly sorted into three different subclasses and each contains the conserved JmjC domain of ZmJMJ protein. Remarkably, the majority of members of the JARID1 subfamily share FYRN and FYRC domains, which may possess chromatin-binding activity [11] or contribute to JmjC domain function by collaborating with other proteins. For instance, the functional specificity of AtJMJ14 in flowering time control has been demonstrated to be on account of the specificity of its interaction with transcription factors through the FYRC domain [31]. Moreover, the ARID domain (AT-rich interaction domain) contained in ZmJMJ10 has been revealed to be implicated in sequence-specific DNA binding in Drosophila melanogaster [32].
Furthermore, to further investigate the possible functions of maize JHDM genes in growth and development and stress-responsiveness of these JHDM genes to environmental stress, several distinct kinds of digital expression pattern analyses were conducted by applying different databases. Firstly, a comprehensive expression analysis was accomplished based on whole-genome coverage according to the microarray data released by Sekhon et al. (2011) [29]. As shown from the heat map in Fig. 6, the maize JHDM genes showed apparent differential expression patterns. Some genes such as ZmJMJ1, ZmJMJ11 and ZmJMJ12 appear to be invariable or lowly expressed among all tissues. By contrast, ZmJMJ2, ZmJMJ16 and ZmJMJ17 exhibited remarkably different expression levels in different tissues. For instance, the expression of ZmJMJ2 in silk and ZmJMJ17 in shoot tips was remarkably more than any other tissues. Furthermore, the expression level of ZmJMJs was similar between seedling and stems, leaves and tassels or husks and endosperms. However, in the same tissues of maize, different ZmJMJs exhibited huge diverse expression profiles. For example, in silks and shoot tips, some genes such as ZmJMJ2 and ZmJMJ17 showed a high expression level, whereas other genes like ZmJMJ7 and ZmJMJ18 exhibited a lower expression level. Taken together, most of ZmJMJs exhibited apparent differential expression patterns. Interestingly, some genes with close relationship, such as ZmJMJ8/ ZmJMJ10 and ZmJMJ11/ZmJMJ12, have similar expression patterns in different tissues, suggesting functional redundancy among these genes. Secondly, a comprehensive expression profile analysis of the maize JHDM gene family by using GENEVESTIGATOR database (Additional file 6: Figure S4) was further performed to demonstrate that the maize JHDM gene family displayed different expression patterns at diverse developmental stages of maize. Additionally, to further confirm stress-responsiveness of JHDM genes to environmental stress, we carried out a comprehensive expression profile analysis of the maize JHDM gene family using GENEVESTIGATOR database (Additional file 7: Figure S5), indicating that the maize JHDM gene family displayed different expression patterns under diverse environmental stress conditions, suggesting that these genes were responsive to stress treatments.
Eventually, to confirm these gene models and their stress-responsiveness to abiotic stress, the transcripts of the 19 genes were firstly detected in the leaves by heat treatment using RT-PCR (Additional file 8: Figure S6). Moreover, the expression analysis of quantitative RT-PCR (Fig. 7) also showed that maize JHDM genes exhibit different expression levels by heat stress. Thus, the results of semi-quantitative PCR and quantitative RT-PCR revealed the authenticity of these genes and their potential roles in heat stress response. In this study, we detected the majority of members from the JARID1 subfamily and the JHDM3 subfamily with significantly higher expression, when subjected to heat stress, indicating that these two subfamilies was closely correlated with maize heat stress response. Moreover, seven genes (ZmJMJ3, ZmJMJ5, ZmJMJ6, ZmJMJ8, ZmJMJ9, ZmJMJ10 and ZmJMJ19) of the ZmJMJ gene family were remarkably up-regulated under heat stress condition, which suggested specific roles for these genes in maize during heat stress. It is note-worthy that two duplicated pairs (ZmJMJ5/ZmJMJ6, ZmJMJ3/ZmjMJ17) in this study exhibited similar expression patterns under heat stress condition. It implied that the expression patterns among the surviving duplicated pairs are relatively conserved, suggesting little functional differentiation has occurred following the WGD/segmental events. Furthermore, heat shock proteins usually play a central role in the heat stress response and the accumulation of heat shock proteins might be a useful indicator for the heat stress treatment. Therefore, to further demonstrate that these predicted genes of the ZmJMJ gene family were responsive to heat stress treatment, a comprehensive expression profile analysis of 22 newly-identified heat shock protein genes (ZmHSP70–1~ZmHSP70–22) was detected in the leaves by heat treatment using RT-PCR (Additional file 9: Figure S7), indicating that these predicated genes may share potential roles in heat stress response. Above all, the analysis of these differentially expressed genes provides some novel insights into plants response to heat stress. However, the functions of targets genes still require further investigation.
Conclusion
In our study, a total of 19 JHDM genes were identified in maize, distributing on nine chromosomes at different densities. These JHDM proteins were mainly categorized into three subfamilies based on the similarities of the amino acid sequences. The exon/intron composition, conserved domain, and amino acid residues were considerably conserved among the members of the same group or subgroup. The close phylogenetic relationship among ZmJMJs, OsJMJs and AtJMJs in the same subgroup provided insights into their putative functions. A comprehensive expression profile of all members of JHDM gene family in maize suggest that many maize JHDM genes play functional developmental roles in multiple tissues. Furthermore, the expression profiles by RT-PCR and quantitative real-time PCR revealed that the majority of identified ZmJMJs most likely are expressed in maize and these genes are induced by heat stress with differential induction levels in leaves. Taken together, all of these results provided valuable clues in future efforts to identify specific gene functions of this gene family and gene diversity among different genotype of maize and other plants in monocots.
Methods
Identification of the members of JHDM gene family in maize
To identify JmjC domain-containing proteins in maize, the maize genomic database (http://www.maizesequence.org/index.html) was searched using Basic Local Alignment Search Tool algorithms (BLASTP) (P value = 0.001), with the published 21 Arabidopsis and 20 rice JmjC domain-containing protein sequences and their JmjC domain used as initial query sequences, respectively. Redundant sequences were then removed manually, and the Hidden Markov Model (HMM) profile of the JmjC domain (PF02373, SM00558) was obtained from Pfam database [33] (http://pfam.xfam.org/) and SMART database [34] (http://smart.embl-heidelberg.de/) to determine each candidate protein as a member of the ZmJMJ gene family [35]. The newly identified genes were designated on the basis of their phylogenetic relatedness to other members of the same gene family in Arabidopsis and rice. Maize JHDM gene basic information including the number of amino acids, ORF lengths and chromosome locations was obtained from the B73 maize sequence database. Physicochemical parameters including the molecular weight (kDa) and isoelectric point (pI) of each gene product were calculated using compute pI/Mw tool from ExPASy (http://www.expasy.org/tools/) and parameter was set to average [36]. The exon and intron structures of individual maize JHDM genes were determined using GSDS (Gene structure display server; http://gsds.cbi.pku.edu.cn/) via alignment of the cDNAs with their corresponding genomic DNA sequences. The conserved domains in the JmjC domain-containing proteins encoded by ZmJMJ genes were investigated by using Pfam (http://pfam.xfam.org/) and SMART (http://smart.embl-heidelberg.de/) with default parameters. Moreover, the conserved amino acid residues for interaction with co-factors were also deduced by using Clustal-W software using default parameters [37].
Phylogenetic analysis of maize JHDM proteins
Multiple sequence alignment of all predicted maize JmjC domain-containing protein sequences and the orthologs from Arabidopsis and rice was performed with Clustal-W software using default parameters. All protein sequences were downloaded from National Center for Biotechnology Information (NCBI). Then, based on this alignment, the analysis of phylogenetic relationship from three different species was conducted using the MEGA 6.0 program [38] with the neighbor-joining method and 1000 bootstrap replicates were performed.
Chromosomal localization, synteny analysis and gene duplication of maize JHDM genes
The chromosomal location image of ZmJMJ genes was generated by MapInspect (https://mapinspect.software.informer.com/) according to their starting positions in maize chromosomes. To compare the genomic context of maize JHDM genes with that in other grass species, information on their patterns of microsynteny was retrieved from the database Phytozome v12.1 (www.phytozome.net). Orthologs of maize JHDM genes in other grasses were investigated in the CoGe database [29]. Duplication analysis of maize JHDM genes was carried out in the Synteny Mapping and Analysis Program (SyMAP) v4.2 [27] and WGMapping program in the PLAZA 4.0 platform [28]. To identify tandem and segmental duplications, two genes in the same species located in the same clade of the phylogenetic tree were defined as paralogs according to described previously [39, 40]. Two JHDMs placed on the synteny blocks in maize genome were designated as segmental duplicated paralogs, and two JHDMs separated by five or fewer gene loci were regarded as tandem duplicated paralogs as described [41, 42].
Prediction of cis-acting elements in maize JHDM genes
Stress response and hormone-related cis-acting elements in the promoter sequences of the JHDM genes were surveyed using the PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html) [43] by means of subjected the 1.5 kb sequences upstream of the initiation codon (ATG) of each JHDM genes.
Plant materials and stress treatment
Maize (Zea mays L. inbred line B73) plants were grown in a growth chamber at 28 °C with a photoperiod of 15-h light and 9-h dark and a relative humidity of 60%. Three-week-old seedlings (five-leaf stage) of maize were used to examine the expression patterns of ZmJMJ genes under heat stress. For heat treatment, seedling were incubated in an incubator at 42 °C in the dark for 8 h. Seedlings maintained with enough water at 28 °C in the dark were used as controls, and seedling leaves were sampled after heat treatment, frozen in liquid nitrogen, and stored at − 80 °C.
Expression profile analysis of maize JHDM genes
To determine the expression patterns of the JHDM genes in maize tissues, a comprehensive expression analysis was initially performed based on whole-genome coverage according to the microarray data released by Sekhon et al. (2011) [29], which was publically available at MaizeGDB (http://www.maizegdb.org). Microarray analysis was accomplished to identify expression patterns in representative tissues, including seed, root, seedling, stem, shoot tip, silks, leaf, tassel, husk and endosperm. By using Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm) and Treeview software (http://jtreeview.sourceforge.net/), we carried out the hierarchical clustering analysis based on the Pearson coefficients with average linkage and analyzed the corresponding gene expression patterns in multiple different tissues or organs, respectively. Moreover, a comprehensive expression profile analysis of the maize JHDM gene family using GENEVESTIGATOR database (https://genevestigator.com/gv/index.jsp) was also conducted to further confirm the expression patterns of 19 JHDM genes in maize. Furthermore, a semi-quantitative expression profile was firstly analyzed in the leaves by heat stress treatment and a quantitative real-time PCR analysis of expression profile was further performed in the leaves by heat stress treatment to reveal the impact of external environmental factor on expression of ZmJMJ genes in maize. Total RNA was isolated from the collected samples using Trizol reagent (Invitrogen, USA), followed by DNase I treatment to remove any genomic DNA contamination. The first-strand cDNA was synthesized from 1 μg of total RNA using QuantiTect Rev.Transcription Kit (Qiagen, Germany). Gene-specific RT-PCR and qRT-PCR primers were designed according to their CDSs (Additional file 10: Table S3 and Additional file 11: Table S4) and then synthesized commercially (Generay), respectively. Quantitative RT-PCR was carried out using an ABI PRISM 7300 real-time PCR system (Applied Biosystems, USA). The qRT-PCR machine was set with 40 cycles and an annealing temperature of 60 °C. At least three biological replicates were performed per cDNA sample. The maize Actin gene was used as internal control for normalization. The relative mRNA level for each gene was calculated as 2-ΔΔCT method to calculate the fold change in the expression level of the relevant genes. Statistical analyses were performed using SDS software version 1.3.1 (Applied Biosystems, USA). Subsequently, significant difference analysis was performed by Student’s t test (http://www.physics.csbsju.edu/stats/t-test_bulk_form.html), at significance level of P < 0.05.
Additional files
Table S1. Basic information of JHDM genes identified in maize. (DOC 48 kb)
{kind=link}
Figure S1. KDM5/JARID1 group proteins contain potential H3K4 demethylases in Arabidopsis, rice and maize. (JPG 375 kb)
{kind=link}
Figure S2. KDM4/JHDM3 group proteins are potential active histone demethylases in Arabidopsis, rice and maize. (JPG 396 kb)
{kind=link}
Figure S3. KDM3/JHDM2 group proteins contain potential H3K9 demethylases in Arabidopsis, rice and maize. (JPG 668 kb)
Table S2. KDM3/JHDM2 group proteins contain potential H3K9 demethylases in Arabidopsis, rice and maize. The original data of expression profiles of ZmJMJ1-19 genes (except ZmJMJ15) in 10 tissues. (DOC 76 kb)
{kind=link}
Figure S4. Hierarchical clustering of developmental expression patterns of maize ZmJMJ genes by Genevestigator database. (JPG 89 kb)
{kind=link}
Figure S5. Hierarchical clustering of expression patterns of maize ZmJMJ genes under diverse environmental stress conditions by Genevestigator database. (JPG 1.79 kb)
{kind=link}
Figure S6. Semi-quantitative RT-PCR analysis of ZmJMJ genes under heat stress treatment. (JPG 57.9 kb)
{kind=link}
Figure S7. Semi-quantitative RT-PCR analysis of ZmHsp70 genes under heat stress treatment. (JPG 71 kb)
Table S3. RT-PCR primers used in this study. (DOC 43 kb)
Table S4. QRT-PCR primers used in this study. (DOC 42 kb)
Acknowledgements
We thank the editors and reviewers for their work on promoting the manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (NO.31571673) and the Anhui Provincial University Excellent Youth Talent Support Program (No. 06146007).
Availability of data and materials
All data used and/or analyzed during the present study are available from the corresponding author if requested.
Abbreviations
- ARID
AT-rich interaction domain
- BR
Brassinosteroid
- ELF6
Early flowering 6
- FAD
Flavin adenine dinucleotide
- FLC
Flowering locus C
- FYRC
FY-rich domain C-terminal
- FYRN
FY-rich domain N-terminal
- IBM1
Increase in bonsai methylation 1
- JARID
Jumonji, AT-rich interactive domain
- JHDM
JmjC domain-containing histone demethylase
- JMJ
JmjC domain protein
- JmjC
Jumonji C
- KDM
lysine demethylase
- LSD
lysine-specific demethylase
- LSDs
lysine-specific demethylases
- PHD
plant home box domain
- PRMT
Protein arginine methyltransferase
- qRT-PCR
quantitative real-time PCR
- REF6
Relative of early flowering 6
- SET
Su (var) 3–9, Enhancer-of-zeste and Trithorax
- а-KG
а-ketoglutarate
Authors’ contributions
These studies were designed by YQ. LJ, CC, QR, and JZ carried out all the experimental analyses and prepared all figures and tables. The manuscript was drafted by YQ. QR and JZ assisted in explaining the results and revised the final manuscript. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yexiong Qian, Phone: +86-553-3869571, Email: qyx2011@ahnu.edu.cn.
Changle Chen, Email: ccl198528@163.com.
Lingyu Jiang, Email: jianglingyu10@163.com.
Jing Zhang, Email: zhangjing1837533@163.com.
Qiaoyu Ren, Email: 18196543798@163.com.
References
- 1.Egger GLG, Aparicio A, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 2.Holliday R. DNA methylation and epigenetic defects in carcinogenesis. Mutat Res. 1987;181(2):215–217. doi: 10.1016/0027-5107(87)90098-4. [DOI] [PubMed] [Google Scholar]
- 3.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 4.Han Y, Li X, Cheng L, Liu Y, Wang H, Ke D, Yuan H, Zhang L, Wang L. Genome-wide analysis of soybean JmjC domain-containing proteins suggests evolutionary conservation following whole-genome duplication. Front Plant Sci. 2016;7:1800. doi: 10.3389/fpls.2016.01800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu C, Lu F, Cui X, Cao X. Histone methylation in higher plants. Annu Rev Plant Biol. 2010;61:395–420. doi: 10.1146/annurev.arplant.043008.091939. [DOI] [PubMed] [Google Scholar]
- 6.Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8(4):307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 7.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7(9):715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 8.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131(4):633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 9.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 11.Lu F, Li G, Cui X, Liu C, Wang XJ, Cao X. Comparative analysis of JmjC domain-containing proteins reveals the potential histone demethylases in Arabidopsis and rice. J Integr Plant Biol. 2008;50(7):886–896. doi: 10.1111/j.1744-7909.2008.00692.x. [DOI] [PubMed] [Google Scholar]
- 12.Trewick SC, McLaughlin PJ, Allshire RC. Methylation: lost in hydroxylation? EMBO Rep. 2005;6(4):315–320. doi: 10.1038/sj.embor.7400379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Lu F, Cui X, Zhang S, Liu C, Cao X. JMJ14 is an H3K4 demethylase regulating flowering time in Arabidopsis. Cell Res. 2010;20(3):387–390. doi: 10.1038/cr.2010.27. [DOI] [PubMed] [Google Scholar]
- 15.Lu F, Cui X, Zhang S, Jenuwein T, Cao X. Arabidopsis REF6 is a histone H3 lysine 27 demethylase. Nat Genet. 2011;43(7):715–719. doi: 10.1038/ng.854. [DOI] [PubMed] [Google Scholar]
- 16.Yu X, Li L, Li L, Guo M, Chory J, Yin Y. Modulation of brassinosteroid-regulated gene expression by Jumonji domain-containing proteins ELF6 and REF6 in Arabidopsis. Proc Natl Acad Sci U S A. 2008;105(21):7618–7623. doi: 10.1073/pnas.0802254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen Y, Conde ESN, Audonnet L, Servet C, Wei W, Zhou DX. Over-expression of histone H3K4 demethylase gene JMJ15 enhances salt tolerance in Arabidopsis. Front Plant Sci. 2014;5:290. doi: 10.3389/fpls.2014.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Duan CG, Tang K, Wang B, Zhang H, Lei M, Lu K, Mangrauthia SK, Wang P, Zhu G, et al. RNA-binding protein regulates plant DNA methylation by controlling mRNA processing at the intronic heterochromatin-containing gene IBM1. Proc Natl Acad Sci U S A. 2013;110(38):15467–15472. doi: 10.1073/pnas.1315399110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inagaki S, Miura-Kamio A, Nakamura Y, Lu F, Cui X, Cao X, Kimura H, Saze H, Kakutani T. Autocatalytic differentiation of epigenetic modifications within the Arabidopsis genome. EMBO J. 2010;29(20):3496–3506. doi: 10.1038/emboj.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saze HSA, Miura A, Kakutani T. Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana. Science. 2008;319:462–465. doi: 10.1126/science.1150987. [DOI] [PubMed] [Google Scholar]
- 21.Lu SX, Knowles SM, Webb CJ, Celaya RB, Cha C, Siu JP, Tobin EM. The Jumonji C domain-containing protein JMJ30 regulates period length in the Arabidopsis circadian clock. Plant Physiol. 2011;155(2):906–915. doi: 10.1104/pp.110.167015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones MA, Covington MF, DiTacchio L, Vollmers C, Panda S, Harmer SL. Jumonji domain protein JMJD5 functions in both the plant and human circadian systems. Proc Natl Acad Sci U S A. 2011;107(50):21623–21628. doi: 10.1073/pnas.1014204108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michael TP, Mockler TC, Breton G, McEntee C, Byer A, Trout JD, Hazen SP, Shen R, Priest HD, Sullivan CM, et al. Network discovery pipeline elucidates conserved time-of-day-specific cis-regulatory modules. PLoS Genet. 2008;4(2):e14. doi: 10.1371/journal.pgen.0040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mockler TCMT, Priest HD, Shen R, Sullivan CM, Givan SA, et al. The DIURNAL project:DIURNAL and circadian expression profiling, model-based pattern matching, and promoter analysis. Cold Spring Harb Symp Quant Biol. 2007;72:353–363. doi: 10.1101/sqb.2007.72.006. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, et al. Structural insights into histone demethylation by JMJD2 family members. Cell. 2006;125(4):691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, Chen D, Liu C, Shen W, Ruan Y. Evolution and conservation of JmjC domain proteins in the green lineage. Mol Gen Genomics. 2016;291(1):33–49. doi: 10.1007/s00438-015-1089-4. [DOI] [PubMed] [Google Scholar]
- 27.Soderlund C, Bomhoff M, Nelson WM. SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 2011;39(10):e68. doi: 10.1093/nar/gkr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Bel M, Proost S, Wischnitzki E, Movahedi S, Scheerlinck C, Van de Peer Y, Vandepoele K. Dissecting plant genomes with the PLAZA comparative genomics platform. Plant Physiol. 2012;158(2):590–600. doi: 10.1104/pp.111.189514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sekhon RS, Lin H, Childs KL, Hansey CN, Buell CR, de Leon N, Kaeppler SM. Genome-wide atlas of transcription during maize development. Plant J. 2011;66(4):553–563. doi: 10.1111/j.1365-313X.2011.04527.x. [DOI] [PubMed] [Google Scholar]
- 30.Chen X, Hu Y, Zhou DX. Epigenetic gene regulation by plant Jumonji group of histone demethylase. Biochim Biophys Acta. 2011;1809(8):421–426. doi: 10.1016/j.bbagrm.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Ning YQ, Ma ZY, Huang HW, Mo H, Zhao TT, Li L, Cai T, Chen S, Ma L, He XJ. Two novel NAC transcription factors regulate gene expression and flowering time by associating with the histone demethylase JMJ14. Nucleic Acids Res. 2015;43(3):1469–1484. doi: 10.1093/nar/gku1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gregory SLKR, Kalionis B, Saint R. Characterization of the dead ringer gene identifies a novel, highly conserved family of sequence-specific DNA-binding proteins. Mol Cell Biol. 1996;16:792–799. doi: 10.1128/MCB.16.3.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34(Database issue):D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Letunic ICR, Schmidt S, Ciccarelli FD, Doerks T, et al. SMART 4.0_ towards genomic data integration. Nucleic Acids Res. 2004;32:D142–D144. doi: 10.1093/nar/gkh088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bateman ACL, Durbin R, Finn RD, Hollich V, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gasteiger E. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31(13):3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson JHD, Gibson T. CLUSTAL W_ improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang SZX, Yue J, et al. Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol Gen Genomics. 2008;280:187–198. doi: 10.1007/s00438-008-0355-0. [DOI] [PubMed] [Google Scholar]
- 40.Gu ZCA, Chen FC, Bouman P, Li WH. Extent of gene duplication in the genomes of drosophila, nematode, and yeast. Mol Biol Evol. 2002;19:256–262. doi: 10.1093/oxfordjournals.molbev.a004079. [DOI] [PubMed] [Google Scholar]
- 41.Wei F, Coe E, Nelson W, Bharti AK, Engler F, Butler E, Kim H, Goicoechea JL, Chen M, Lee S, et al. Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet. 2007;3(7):e123. doi: 10.1371/journal.pgen.0030123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Guo K, Li Y, Tu Y, Hu H, Wang B, Cui X, Peng L. Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol. 2010;10:282. doi: 10.1186/1471-2229-10-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lescot MDP, Thijs G, Marchal K, Moreau Y, Van de Peer Y, Rouzé P, Rombauts S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30:325–327. doi: 10.1093/nar/30.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Basic information of JHDM genes identified in maize. (DOC 48 kb)
Figure S1. KDM5/JARID1 group proteins contain potential H3K4 demethylases in Arabidopsis, rice and maize. (JPG 375 kb)
Figure S2. KDM4/JHDM3 group proteins are potential active histone demethylases in Arabidopsis, rice and maize. (JPG 396 kb)
Figure S3. KDM3/JHDM2 group proteins contain potential H3K9 demethylases in Arabidopsis, rice and maize. (JPG 668 kb)
Table S2. KDM3/JHDM2 group proteins contain potential H3K9 demethylases in Arabidopsis, rice and maize. The original data of expression profiles of ZmJMJ1-19 genes (except ZmJMJ15) in 10 tissues. (DOC 76 kb)
Figure S4. Hierarchical clustering of developmental expression patterns of maize ZmJMJ genes by Genevestigator database. (JPG 89 kb)
Figure S5. Hierarchical clustering of expression patterns of maize ZmJMJ genes under diverse environmental stress conditions by Genevestigator database. (JPG 1.79 kb)
Figure S6. Semi-quantitative RT-PCR analysis of ZmJMJ genes under heat stress treatment. (JPG 57.9 kb)
Figure S7. Semi-quantitative RT-PCR analysis of ZmHsp70 genes under heat stress treatment. (JPG 71 kb)
Table S3. RT-PCR primers used in this study. (DOC 43 kb)
Table S4. QRT-PCR primers used in this study. (DOC 42 kb)
Data Availability Statement
All data used and/or analyzed during the present study are available from the corresponding author if requested.