

Abstract
Study 232, an open-label pilot study with an extension phase, evaluated the pharmacokinetics and preliminary safety/tolerability and efficacy of adjunctive perampanel oral suspension (≤0.18 mg/kg/d) in epilepsy patients aged ≥2 to <12 years. Patients were grouped into cohorts 1 (aged ≥7 to <12 years) and 2 (aged ≥2 to <7 years). The Core Study included pretreatment (≤2 weeks) and treatment phases (7-week titration; 4-week maintenance; 4-week follow-up [for those not entering the extension]). The extension phase consisted of 41-week maintenance and 4-week follow-up periods. Pharmacokinetic data were pooled with adolescent pharmacokinetic data from phase II/III studies. Population pharmacokinetic analysis showed that perampanel pharmacokinetics was independent of age, weight, or liver function, suggesting age- or weight-based dosing is not required and that the same dose can be given to adults and children to achieve exposures shown to be efficacious. Perampanel was well tolerated and efficacious for ≤52 weeks.
Keywords: antiepileptic drugs, efficacy, epilepsy, pediatric, seizures
Perampanel, a selective, noncompetitive α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonist, is a once-daily oral antiepileptic drug for focal seizures (previously classified as partial-onset seizures) and primary generalized tonic-clonic seizures.1,2 The safety and efficacy of perampanel in adolescent and adult patients with focal seizures or primary generalized tonic-clonic seizures have been well documented in 4 double-blind, randomized, placebo-controlled phase III studies.3-6 Long-term tolerability and improvements in seizure outcomes have also been demonstrated with perampanel.7 Furthermore, the pharmacokinetic profiles of perampanel (in tablet form) in adolescent (≥12 to <18 years of age) and adult patients are not significantly different.8
Selection of an antiepileptic drug for the treatment of pediatric patients with epilepsy has been a challenge for clinicians, which is largely due to the lack of published clinical studies reflecting efficacy and safety data in these patients.9 Age-related changes in body size and liver function (enzyme activity) in children mean that understanding the pharmacokinetics, identifying appropriate antiepileptic-drug dose levels, and avoiding potential adverse events (AEs) resulting from incorrect dosing constitute significant unmet clinical needs in this patient population,10,11 which suffers from epilepsy to a disproportionate degree.9
The effects of age on pharmacokinetics, pharmacodynamics, and dose requirements are not fully understood.10 However, it is known that, in general, pharmacokinetic processes in children can differ from those in adults including absorption, metabolism, and excretion.11,12 For example, age can influence cytochrome P450 (CYP)-dependent hepatic metabolism, as the activity of these enzymes is different at birth compared with that in a developing child or adult.11,12 It has not been elucidated whether perampanel, a drug with a notably long half-life (approximately 105 hours) that is eliminated primarily via CYP3A4/5 hepatic metabolism,2,8 is influenced by age-related changes in liver function and CYP3A enzyme activity. In turn, this may impact dosing in children relative to achieving plasma concentrations shown to be effective. The present study (Study 232) is an open-label pilot study with an extension phase. Study 232 was designed to evaluate the pharmacokinetics of perampanel following once-daily oral administration of a suspension formulation and to generate preliminary safety, tolerability, and efficacy data when perampanel is administered as an adjunctive therapy in pediatric patients aged ≥2 to <12 years with epilepsy. This is the first study to evaluate perampanel treatment in patients in this age group with heterogeneous diagnoses of epilepsies.
Methods
Standard Protocol Approvals, Registration, and Patient Consent
Study 232 (ClinicalTrials.gov identifier: NCT01527006) was conducted between January 2012 and May 2014 at 15 sites in North America. The extension phase of Study 232 was conducted between January 2012 and February 2015 at 14 sites in North America. Study 232 was performed in accordance with the Declaration of Helsinki, the International Conference on Harmonization (ICH)-E6 Guideline for Good Clinical Practice CPMP/ICH/135/95, and the US Code of Federal Regulations Title 21. The trial protocol, amendments, and informed consent were reviewed by independent ethics committees (IECs) or institutional review boards (IRBs) for each site. A full list of participating IECs and IRBs can be found in Supplementary Table S1. Before trial participation, written informed consent was received from a parent/legal guardian for each young person participating in this study; for patients aged ≥7 years, written or verbal assent, or equivalent, was obtained from the patient.
Patients
Patients included in Study 232 were aged ≥2 to <12 years at the time of study entry, had a diagnosis of epilepsy with any type of seizure at least 6 months prior to visit 1 based on clinical history and electroencephalography, had experienced at least 1 seizure during the 4 weeks prior to visit 1, and were on a stable dose of 1 to 3 antiepileptic drugs for ≥4 weeks before visit 1 and throughout the study. Patients were allowed a maximum of 3 concomitant antiepileptic drugs during the study; use of concomitant enzyme-inducing antiepileptic drugs (EIAEDs; carbamazepine, oxcarbazepine, or phenytoin) was limited to 1 per patient, and the number of patients taking EIAEDs was restricted to ≥1/3 to ≤1/2 of the patients in each study cohort. Changes in baseline antiepileptic drugs (addition, deletion, or dose adjustments) were not allowed during the core study treatment phase.
Study Design
During the core study, patients were enrolled in 1 of 2 cohorts, based on age at study entry: cohort 1 included patients aged ≥7 to <12 years and cohort 2 included patients aged ≥2 to <7 years. The core study comprised 2 phases: a pretreatment phase (≤2 weeks) and a treatment phase consisting of a 7-week titration period and a 4-week maintenance period; a 4-week follow-up period was also included for patients not entering the extension phase (Figure 1). Because of the uncertainty of age-related effects on perampanel metabolism, the daily dose of perampanel oral suspension was determined for this pilot study based on the patient’s weight, using an adult weight of 70 kg as the reference. Furthermore, patients in Study 232 were dosed using a suspension formulation that was bioequivalent to the tablet formulation administered to adolescents and/or adults in Studies 235, 304, 305, and 306.13 At the beginning of the 7-week titration period, patients were initiated on a once-daily dose of 0.015 mg/kg at visit 2 (adult-equivalent dose of 1 mg/70 kg/d). At visit 3 (week 1), patients were up-titrated to 0.03 mg/kg/d (adult-equivalent dose of 2 mg/70 kg/d). Patients were up-titrated by 0.03 mg/kg/wk for a total of 6 titration steps to a maximum dose of 0.18 mg/kg/d (adult-equivalent dose of 12 mg/70 kg/d), or until they reached their maximum tolerated dose (Figure 1). During the 4-week maintenance period, patients continued at the once-daily dose level they had achieved at the end of the titration period.
Figure 1.

Study 232 design. Patients who did not roll over into the extension phase or who discontinued from the study were required to complete the follow-up period.
aFour-week follow-up for patients not enrolling into the extension phase.
Patients participating in the core study who completed all scheduled visits, including the final visit, were eligible to participate in the extension phase. The extension phase consisted of 2 periods: a 41-week maintenance period and a 4-week follow-up period. The final visit of the core study maintenance period was the first visit of the extension phase (Figure 1). During the extension phase, patients continued taking once-daily perampanel at the dose level achieved at the end of the core study treatment phase. Changes in concomitant antiepileptic drugs (addition, deletion, or dose adjustments) were allowed during the extension phase.
Pharmacokinetic Analysis
Pharmacokinetic analysis was the primary objective of Study 232. The Pharmacokinetic Analysis Set included patients who had at least 1 pharmacokinetic assessment and a documented dosing history. The population pharmacokinetic approach, using nonlinear mixed-effect modeling, was used to characterize the pharmacokinetic properties of perampanel. Data from Study 232 were pooled with data from adolescents with inadequately controlled focal seizures from the phase II Study 235 and phase III Studies 304, 305, and 306. For Study 232, 1 blood sample (0.2 mL) was collected during visits 3 (week 1), 5 (week 5), 7 (week 9), and 8 (week 11) for the determination of blood perampanel concentrations. Perampanel blood concentrations were analyzed according to the dried blood spot method. In order to pool data from Study 232 with data from other studies, perampanel blood concentrations and hematocrit values from Study 232 were used to derive plasma concentrations, because, in other studies, blood samples for the determination of plasma perampanel concentrations were collected via venipuncture. A 1-compartment disposition model with first-order elimination parameterized for apparent clearance (CL/F) and volume of distribution adequately described the perampanel pharmacokinetic data.
Safety and Efficacy Endpoints
The safety endpoints for both the core and extension phases of Study 232 included monitoring of treatment-emergent adverse events using Medical Dictionary for Regulatory Activities (MedDRA) search terms, clinical laboratory parameters, vital signs, and 12-lead electrocardiogram (ECG) results. The efficacy endpoints for overall seizures and individual seizure types (focal seizures, generalized seizures, and unclassified seizures) included the percentage change in 28-day seizure frequency compared with baseline, responder rate (defined as the proportion of patients with a ≥50% decrease in 28-day seizure frequency during the maintenance period compared with baseline in the core study), and seizure freedom during the core study maintenance period. The same measurements were conducted at 13-week intervals during the overall treatment duration for the extension phase. For all efficacy endpoints, the baseline 28-day seizure frequency was determined using seizure diary data from the 2-week pretreatment phase of the core study.
Results
During the core study, 50 patients were treated. Of these, 28 were aged ≥7 to <12 years (cohort 1) and 22 were aged ≥2 to <7 years (cohort 2). A total of 42 patients completed the core study and 41 entered the extension phase (Figure 2). There were no remarkable differences in baseline characteristics between the patients in the core study and those who entered the extension phase. The demographic and baseline characteristics of the 41 patients who entered the extension phase are shown in Table 1. Patients were most likely to have experienced focal seizures, followed by generalized seizures. At baseline, 24.4% of patients were taking 1 antiepileptic drug, 58.5% were taking 2 antiepileptic drugs, and 17.1% were taking 3 antiepileptic drugs (Table 1). The proportion of patients taking 2 antiepileptic drugs was similar in cohorts 1 and 2 (59.1% vs 57.9%, respectively), whereas the proportion of patients taking 1 antiepileptic drug was lower in cohort 1 than in cohort 2 (18.2% vs 31.6%, respectively), and a greater proportion of patients in cohort 1 than in cohort 2 were taking 3 antiepileptic drugs (22.7% vs 10.5%, respectively). Nearly one-half (45.5%) of patients in cohort 1 and nearly one-third (31.6%) of patients in cohort 2 were receiving background therapy that included EIAEDs, which are known to induce perampanel metabolism and decrease its exposure.2 The most commonly used concomitant antiepileptic drugs were levetiracetam (39.0%), valproic acid (36.6%), oxcarbazepine (31.7%), lamotrigine (17.1%), topiramate (19.5%), and zonisamide (14.6%).
Figure 2.

Study 232 patient disposition.
Table 1.
Demographic and Baseline Characteristics of Patients Entering the Extension Phase of Study 232.
| Cohort 1: ≥7 to <12 years (n = 22) |
Cohort 2: ≥2 to <7 years (n = 19) |
Total (N = 41) |
|
|---|---|---|---|
| Age, years, mean (SD) | 9.1 (1.36) | 4.5 (1.17) | 7.0 (2.65) |
| Gender, n (%) | |||
| Male | 15 (68.2) | 13 (68.4) | 28 (68.3) |
| Female | 7 (31.8) | 6 (31.6) | 13 (31.7) |
| Weight, kg, mean (SD) | 38.08 (15.43) | 20.07 (6.28) | 29.73 (15.01) |
| Height, cm, mean (SD) | 136.20 (13.03) | 107.49 (16.63) | 123.36 (20.51) |
| BMI, kg/m2, mean(SD) | 19.31 (4.86) | 19.63 (12.02) | 19.45 (8.68) |
| Time since diagnosis, years, mean (SD) | 5.77 (2.89) | 3.80 (1.50) | 4.86 (2.53) |
| Seizure type (past 2 years), n (%) | |||
| Focal seizuresa | 19 (86.4) | 15 (78.9) | 34 (82.9) |
| Generalized seizuresb | 6 (27.3) | 12 (63.2) | 18 (43.9) |
| Unclassified seizures | 1 (4.5) | 1 (5.3) | 2 (4.9) |
| Number of AEDs, n (%) | |||
| 1 AED | 4 (18.2) | 6 (31.6) | 10 (24.4) |
| 2 AEDs | 13 (59.1) | 11 (57.9) | 24 (58.5) |
| 3 AEDs | 5 (22.7) | 2 (10.5) | 7 (17.1) |
Abbreviations: AED, antiepileptic drug; BMI, body mass index; SD, standard deviation.
a Focal seizures include simple focal seizures without motor signs, simple focal seizures with motor signs, complex focal seizures, and focal seizures with secondarily generalized seizures.
b Generalized seizures include absence, myoclonic, clonic, tonic, tonic-clonic, and atonic (astatic).
Pharmacokinetic Outcomes
The pharmacokinetic data set included a total of 845 plasma perampanel observations from 194 patients (maintenance period of Study 232: 83 observations from 42 patients; Studies 235, 304, 305, and 306: 762 observations from 152 adolescent patients). A summary of demographic variables and additional covariates for these patients is presented in Supplementary Table S2. Among these patients, 45.9% were receiving concomitant EIAEDs, all of which exhibited statistically significant and clinically relevant effects on perampanel CL/F by factors of 2.57 for carbamazepine and 1.90 for oxcarbazepine/phenytoin. These findings are consistent with previous population-based analyses of perampanel pharmacokinetic properties in both adolescents and adults. The pharmacokinetic profile of perampanel was independent of both dose and time, and was not significantly affected by age, weight, gender, race (Caucasian vs non-Caucasian), alanine transaminase or aspartate transaminase levels, creatinine clearance, formulation, or coadministration of other non-EIAEDs (ie, valproic acid, lamotrigine, topiramate, or levetiracetam).
In order to facilitate comparison by age group, all data were normalized to an 8-mg dose of perampanel. Patients aged ≥2 to <12 years were already dosed on a milligrams-per-kilogram basis (0.12 mg/kg, corresponding with 8 mg/70 kg in an adult/adolescent), while patients aged ≥12 to <18 years received a dose of 8 mg/d. Dose-normalized, steady-state average concentration during a perampanel dosing interval (Cav,ss) was independent of age for both concomitant EIAEDs and non-EIAEDs, whether considered as a continuous covariate (Figure 3A) or stratified by the following age groups: ≥2 to <7 years, ≥7 to <12 years, and ≥12 to <18 years. A large overlap in dose-normalized exposure to perampanel was also observed across these 3 age groups in the pooled analysis for both EIAEDs and non-EIAEDs. In addition, dose-normalized perampanel Cav,ss was weight-independent across a wide weight range for both EIAEDs and non-EIAEDs (Figure 3B). Table 2 displays a summary of the individual post hoc estimations of CL/F and Cav,ss calculated from the final pharmacokinetic model, and Figure 4 shows the relationship between model-predicted CL/F and age. Although there was a small number of patients in the ≥2 to <7 years and ≥7 to <12 years age groups, there was a relatively high degree of variability in almost all of the age groups, as reflected by the standard deviation. There were no apparent age-dependent metabolism effects on perampanel, based on the observation that the model-predicted CL/F of perampanel was comparable among the 3 age groups for both EIAEDs and non-EIAEDs. Predicted perampanel Cav,ss values based on a 0.12-mg/kg dose (adult-equivalent dose of 8 mg/70 kg) in children in Study 232 were lower than those at the 8-mg dose in adolescents in Studies 235, 304, 305, and 306. However, because the predicted CL/F was comparable among the 3 age groups and was independent of weight, the lower Cav,ss in children is likely due to the lower total dose administered to children compared with adolescents. Notably, the magnitude of the effect of CYP3A4/5 induction by EIAEDs was comparable between children aged ≥2 to <12 years and adolescents aged ≥12 to <18 years.
Figure 3.

Relationship between model-predicted Cav,ss of perampanel dose normalized to 8 mg and age (A) and weight (B) stratified by concomitant use of EIAEDs and non-EIAEDs. The red line is loess smooth. (Cav,ss, steady-state average concentration during a dosing interval; EIAED, enzyme-inducing antiepileptic drug.)
Table 2.
Summary of Individual Predicted Pharmacokinetic Parameters for Perampanel by Age Group.
| Age group | Concomitant EIAEDs | Total daily dose (mg/kg) | Weight (kg) | Total daily dose (mg/body weight) | N | CL/F (L/h) | Dose-normalized Cav,ss a (ng/mL) |
|---|---|---|---|---|---|---|---|
| ≥2 to <7 years | Non-EIAED | 0.12 | 20.7 ± 6.9 | 2.5 ± 0.8 | 14 | 0.7 ± 0.4 | 179 ± 110 |
| EIAEDb | 0.12 | 18.2 ± 5.1 | 2.2 ± 0.6 | 6 | 1.7 ± 1.2 | 97 ± 90 | |
| ≥7 to <12 years | Non-EIAED | 0.12 | 40.4 ± 19.6 | 4.8 ± 2.4 | 12 | 1.0 ± 0.4 | 266 ± 220 |
| EIAEDb | 0.12 | 37.9 ± 12.8 | 4.5 ± 1.5 | 10 | 1.9 ± 0.5 | 105 ± 39 | |
| ≥12 to <18 years | Non-EIAED | – | 55.5 ± 15.0 | 8 | 79 | 0.7 ± 0.4 | 584 ± 367 |
| EIAEDb | – | 56.8 ± 16.5 | 8 | 73 | 1.6 ± 0.8 | 282 ± 184 |
Abbreviations: Cav,ss, steady-state average concentration during a dosing interval; CL/F, apparent clearance; EIAED, enzyme-inducing antiepileptic drug.
a Dose normalized to 0.12 mg/kg in patients ≥2 to <12 years of age and 8 mg in those ≥12 to <18 years of age.
b EIAEDs were defined as carbamazepine, oxcarbazepine, and phenytoin, which showed clinically relevant effects on perampanel CL/F based on population pharmacokinetic analysis using Studies 304, 305, and 306.
Values shown are mean ± standard deviation.
Figure 4.

Relationship between model-predicted CL/F and age. (CL/F, apparent clearance; EIAED, enzyme-inducing antiepileptic drug.)
Safety Assessments
The mean daily dose of perampanel was 0.14 mg/kg during the core study maintenance period and 0.15 mg/kg during the extension phase; the mean maximum dose during the extension phase was 0.17 mg/kg. During the core study, the incidence of treatment-emergent adverse events was similar between the 2 cohorts (cohort 1: n = 27 [96.4%]; cohort 2: n = 22 [100.0%]; total: n=49 [98.0%]). All patients who entered the extension phase experienced at least 1 treatment-emergent adverse event while receiving perampanel treatment (during the core study and/or the extension phase; Table 3). For patients who entered the extension phase, the most common treatment-emergent adverse events reported among patients in cohort 1 were pyrexia, upper respiratory tract infection, vomiting, fatigue, irritability, and upper abdominal pain (Table 3). Similarly, the most common treatment-emergent adverse events reported in cohort 2 were pyrexia, upper respiratory tract infection, aggression, vomiting, lethargy, and cough (Table 3). The treatment-emergent adverse events reported in cohorts 1 and 2 are consistent with illnesses commonly observed in children in these age groups, and also with treatment-emergent adverse events previously reported with perampanel treatment in adolescents and adults.3-7
Table 3.
TEAEsa Occurring in ≥10% of Patients Who Entered the Extension Phase (Safety Analysis Set).
| TEAE category MedDRA preferred term |
Cohort 1 ≥7 to <12 years (n = 22) |
Cohort 2 ≥2 to <7 years (n = 19) |
Total (N = 41) |
|---|---|---|---|
| Any TEAE, n (%) | 22 (100.0) | 19 (100.0) | 41 (100.0) |
| Pyrexia | 7 (31.8) | 8 (42.1) | 15 (36.6) |
| Upper respiratory tract infection | 6 (27.3) | 5 (26.3) | 11 (26.8) |
| Vomiting | 6 (27.3) | 4 (21.1) | 10 (24.4) |
| Irritabilityb | 5 (22.7) | 3 (15.8) | 8 (19.5) |
| Fatigue | 6 (27.3) | 1 (5.3) | 7 (17.1) |
| Ear infection | 4 (18.2) | 3 (15.8) | 7 (17.1) |
| Lethargy | 3 (13.6) | 4 (21.1) | 7 (17.1) |
| Aggressionb | 2 (9.1) | 5 (26.3) | 7 (17.1) |
| Nasopharyngitis | 3 (13.6) | 3 (15.8) | 6 (14.6) |
| Otitis media | 3 (13.6) | 3 (15.8) | 6 (14.6) |
| Somnolence | 3 (13.6) | 3 (15.8) | 6 (14.6) |
| Cough | 2 (9.1) | 4 (21.1) | 6 (14.6) |
| Abdominal pain, upper | 5 (22.7) | 0 (0.0) | 5 (12.2) |
| Increased appetite | 4 (18.2) | 1 (5.3) | 5 (12.2) |
| Weight increased | 4 (18.2) | 1 (5.3) | 5 (12.2) |
| Headache | 3 (13.6) | 2 (10.5) | 5 (12.2) |
| Dizziness | 2 (9.1) | 3 (15.8) | 5 (12.2) |
Abbreviations: MedDRA, Medical Dictionary for Regulatory Activities; TEAE, treatment-emergent adverse event.
a A TEAE is defined as an adverse event with an onset date, or a worsening in severity from baseline (pretreatment), on or after the first dose of study drug up to 30 days following study drug discontinuation. A patient with 2 or more adverse events in the same preferred term is counted only once for that preferred term.
b There was 1 patient who experienced events of both irritability and aggression; the event of irritability occurred on day 17 of the core study and the event of aggression occurred during the extension phase on day 98 of treatment. The event of aggression led to study discontinuation. There were no other patients who experienced both irritability and aggression.
With regard to noteworthy differences in the occurrence of treatment-emergent adverse events between the 2 cohorts, aggression was more common in cohort 2 compared with cohort 1 (26.3% [n = 5] vs 9.1% [n = 2], respectively). Treatment-emergent adverse events that were more common in cohort 1 compared with cohort 2 included upper abdominal pain (22.7% vs 0.0%, respectively), fatigue (27.3% vs 5.3%, respectively), increased appetite (18.2% vs 5.3%, respectively), and increased weight (18.2% vs 5.3%, respectively) (Table 3). The majority of treatment-emergent adverse events in both cohorts were considered as mild or moderate by the investigator; 11 (26.8%) treatment-emergent adverse events were considered severe (cohort 1: n = 5 [22.7%]; cohort 2: n = 6 [31.6%]). Most treatment-emergent adverse events were judged by the investigator to be possibly (63.4%) or probably (26.8%) related to perampanel treatment.
Serious adverse events occurred at a similar rate in the 2 cohorts during both the core study and extension phase. During the entire study, and among patients who entered the extension phase, 23 serious adverse events were reported in 7 (31.8%) patients in cohort 1, and 9 serious adverse events were reported in 6 (31.6%) patients in cohort 2. Those serious adverse events occurring in more than 1 patient included convulsion (cohort 1: n = 1 [4.5%]; cohort 2: n = 2 [10.5%]), status epilepticus (cohort 1: n = 2 [9.1%]), and mental status changes (cohort 2: n = 2 [10.5%]). Three of the serious adverse events reported in 2 patients in cohort 1 and 4 of the serious adverse events reported in 2 patients in cohort 2 were considered by the investigator to be possibly related to the study drug; however, no action was taken with regard to the use of the study drug, and all patients recovered from their serious adverse events with no sequelae. None of the patients who entered the extension phase discontinued perampanel treatment as a result of these serious adverse events.
During the core study, 3 (6.0%) patients discontinued from study drug as a result of treatment-emergent adverse events (cohort 1: n = 2 [7.1%]; cohort 2: n = 1 [4.5%]); none of the treatment-emergent adverse events leading to discontinuation occurred in more than 1 patient (Supplementary Table S3). During the extension phase, 5 (12.2%) patients discontinued from study drug as a result of treatment-emergent adverse events (cohort 1: n = 2 [9.1%]; cohort 2: n = 3 [15.8%]). Aggression was the only treatment-emergent adverse event that resulted in discontinuation during the extension phase in more than 1 patient, occurring in 1 patient from each cohort. No deaths occurred during Study 232.
Treatment-emergent adverse events related to hostility and/or aggression were reported in 9 (32.1%) patients in cohort 1 and 9 (40.9%) patients in cohort 2 during the core study, using both narrow and broad Standardized MedDRA Queries (SMQ) terms (Supplementary Table S4). The most common treatment-emergent adverse events related to hostility and/or aggression were irritability (cohort 1: n = 5 [17.9%]; cohort 2: n = 3 [13.6%]) and aggression (cohort 1: n = 1 [3.6%]; cohort 2: n = 3 [13.6%]). There were 2 patients in cohort 1 who had treatment-emergent adverse events related to hostility and/or aggression that led to discontinuation. One of these patients experienced irritability and psychomotor hyperactivity that were moderate in severity and considered probably related to study drug, and the other patient experienced a severe event of abnormal behavior that was possibly related to study drug; both patients recovered. Two patients (1 from each cohort) had treatment-emergent adverse events of irritability that led to dose reduction, and both were considered possibly related to study drug. In cohort 1, the event of irritability was mild in severity and did not resolve; in cohort 2, the event of irritability was moderate and resolved. There was also 1 patient in cohort 2 who experienced a treatment-emergent adverse event of aggression that led to study drug dose reduction; this event was moderate, probably related to study drug, and resolved. There was 1 patient in cohort 1 who experienced suicidal ideation during the titration phase; this event of suicidal ideation was mild and not related to the study drug according to the investigator, and the patient recovered.
During the extension phase, treatment-emergent adverse events related to hostility and/or aggression were reported in 8 (36.4%) patients in cohort 1 and 9 (47.4%) patients in cohort 2 (Supplementary Table S4). The most common of these treatment-emergent adverse events were irritability (cohort 1: n = 5 [22.7%]; cohort 2: n = 3 [15.8%]) and aggression (cohort 1: n = 2 [9.1%]; cohort 2: n = 5 [26.3%]). There were 2 events of aggression that led to the discontinuation of 2 patients (1 from each cohort) during the extension phase maintenance period. Both events were moderate in severity and considered to be probably related to the study drug; the event of aggression in the patient from cohort 1 did not resolve, while the patient from cohort 2 was reported as recovering/resolving. These events were also identified as treatment-emergent adverse events related to alertness or cognition. In addition to the patient in cohort 1 who experienced suicidal ideation during the core study titration phase, there were 2 further patients (1 from each cohort) who experienced suicidal ideation during the extension phase maintenance period. In cohort 1, the event of suicidal ideation was severe, possibly related to the study drug, and resulted in discontinuation. In cohort 2, the event of suicidal ideation was mild and possibly related to the study drug; the drug was interrupted, but, then, no further action was taken; both patients recovered. There were no events of homicidal ideation reported during either the core or extension phases.
Among patients who entered the extension phase, reports of treatment-emergent adverse events related to laboratory abnormalities with perampanel treatment were infrequent, and none were associated with markedly abnormal liver function tests. No marked changes in systolic or diastolic blood pressure, body temperature, pulse rate, or respiratory rate were noted between baseline and the end of treatment, and no changes of clinical importance in mean ECG parameters over time were observed.
Efficacy Assessments
During the core study treatment phase (titration plus maintenance), the median 28-day seizure frequency decreased relative to baseline for overall seizures in both cohorts, specifically for focal seizures and unclassified seizures (Supplementary Figure S1A). For generalized seizures, there was a decrease in 28-day seizure frequency from baseline in cohort 2 but an increase in cohort 1. However, it should be noted that patient numbers in cohort 1 were relatively low for all generalized seizure subtypes (absence, n = 4; myoclonic, n = 2; clonic, n = 0; tonic, n = 2; tonic-clonic, n = 4; and atonic [astatic], n = 3), and large variations were observed in the data, which may have confounded these results. This subgroup aside, the majority of patients, by age and seizure type, were responders; that is, they experienced a decrease in seizure frequency of at least 50% relative to baseline during the maintenance period (last observation carried forward, with the pretreatment phase plus 4 weeks prior to visit 1 constituting the baseline; Supplementary Figure S1B).
During the extension phase, median seizure frequency decreased relative to baseline during all 13-week intervals for overall seizures (Figure 5A) and focal seizures (Supplementary Figure S2A). For generalized seizures (n = 6), there was an increase in median seizure frequency relative to baseline during weeks 1-13 in cohort 1, which is consistent with results from the core study (Supplementary Figure S2B). There was also an increase in median seizure frequency for unclassified seizures (n = 1) during weeks 27-39 in cohort 1 (data not shown). Median percentage changes in seizure frequency from baseline during each 13-week interval of the extension phase were greater in cohort 2 than in cohort 1 (Figure 5A and Supplementary Figures S2A-B).
Figure 5.
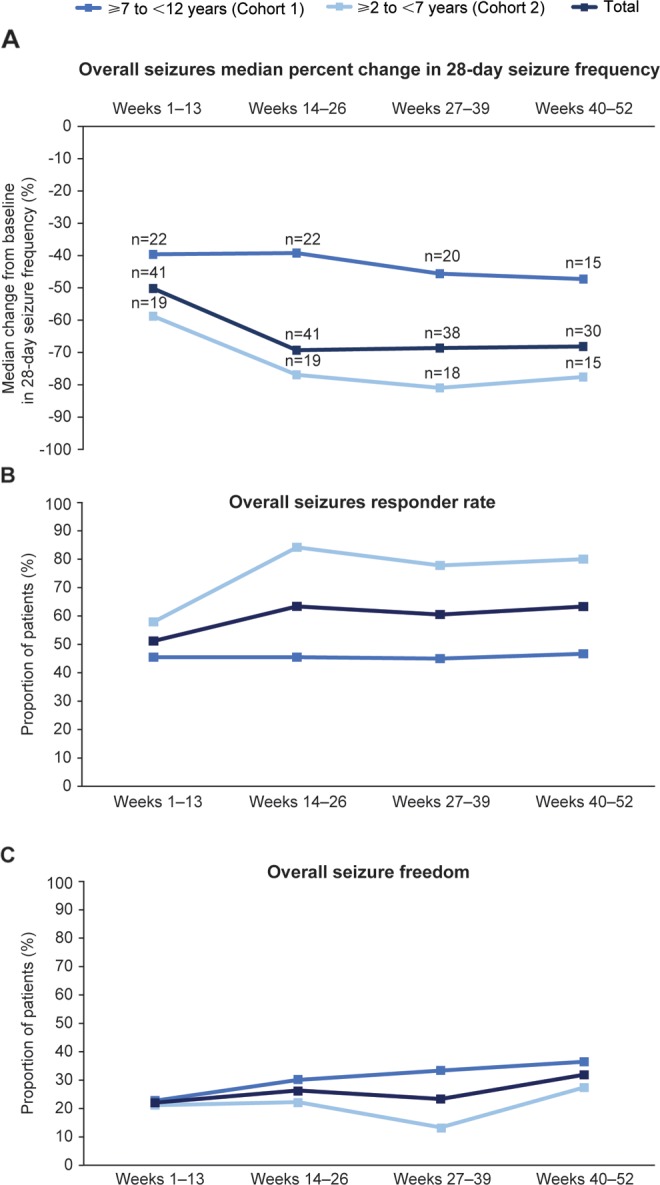
Efficacy outcomes during Study 232 extension: (A) median percentage change from baseline in 28-day overall seizure frequency; (B) overall seizure responder ratea; (C) overall seizure freedom.b
aA responder is a patient who experienced a 50% or greater reduction in seizure frequency per 28 days from baseline (ie, pretreatment phase plus 4 weeks prior to visit 1).
bSeizure freedom is only defined for patients who completed the time interval (eg, week 1-13 seizure freedom only includes patients who completed week 13).
The responder rate from week 1 through each successive 13-week interval was higher in cohort 2 than in cohort 1 for overall seizures (Figure 5B), and for focal and generalized seizures (Supplementary Figure S3A-B). Overall seizure freedom was higher in cohort 1 than in cohort 2 for all 13-week intervals (Figure 5C). By weeks 40-52, seizure freedom for overall seizures was achieved in 36.4% (n = 4) of patients in cohort 1 and 27.3% (n = 3) of patients in cohort 2.
Discussion
The primary objective of Study 232 was to evaluate the pharmacokinetics of perampanel following oral suspension administered as an adjunctive therapy in pediatric patients aged ≥2 to <12 years with epilepsy. Body-weight–corrected dosing was used in order to determine whether there were any potential age-related differences in pharmacokinetics, which could suggest that children would require a different dose regimen compared with adolescents and adults. The results of the present population pharmacokinetic analysis of Study 232, pooled with data from adolescent patients with focal seizures participating in a phase II study (Study 235) and 3 phase III studies (Studies 304, 305, and 306), indicate that the pharmacokinetic properties of perampanel in children aged ≥2 to <12 years are independent of age and weight; that the effects of concomitant use of EIAEDs on perampanel pharmacokinetics are independent of age; and that perampanel exposure is, in general, not influenced by age-related changes in body weight or liver function. Taken together, the pharmacokinetic results suggest that age- or weight-based dosing is not required for adjunctive perampanel therapy, and that the same dose given to adolescents and adults can be given to children to achieve exposures shown to be efficacious.
Making dose adjustments for children based on body weight using an adult weight reference is common in pediatric clinical practice; however, adult dosing regimens cannot always be extrapolated to children.10,14 For example, carbamazepine, which is eliminated by CYP3A4, is metabolized more rapidly in children than in adults, resulting in the need for a higher dose of carbamazepine in younger patients to achieve comparable therapeutic plasma concentrations.14-16 The results of the present study show that unlike carbamazepine, the pharmacokinetics of perampanel does not change with age, despite the fact that perampanel is also eliminated by CYP3A4. This difference may be associated with the very slow rate of metabolism for perampanel, as evidenced by its low clearance and long half-life of approximately 105 hours.2 It is possible, therefore, that age-related differences in hepatic metabolism may impact high-clearance substrates more so than low-clearance substrates. This study demonstrates that a simple extrapolation of adult pharmacokinetic data to children is not sufficient, and that studies in children are essential for characterizing potentially unique pharmacokinetic properties of individual drugs to ensure optimal dosing in children.
It should be noted that although a suspension rather than a tablet formulation of perampanel was given to the children participating in Study 232, the observed pharmacokinetic properties were not notably different from those of the perampanel tablet formulation administered to adolescents and adults participating in Studies 235, 304, 305, and 306. Since the completion of this study, the suspension formulation has been approved for adjunctive therapy for the treatment of focal seizures and primary generalized tonic-clonic seizures in patients with epilepsy aged ≥12 years.13 The oral suspension was found to be bioequivalent and interchangeable with the tablet formulation.17
Safety outcomes from the core and extension phases of Study 232 indicate that once-daily adjunctive therapy with perampanel oral suspension at daily doses up to 0.18 mg/kg was well tolerated over the entire duration of the study (up to 52 weeks) in pediatric patients (aged ≥2 to <12 years) with epilepsy. The majority of serious adverse events and other significant events were transient and manageable, with patients recovering without sequelae. Additionally, the observed safety profile was similar between the 2 age cohorts. The Food and Drug Administration prescribing information for perampanel contains a boxed warning for serious psychiatric and behavioral reactions, including aggression, hostility, irritability, anger, and homicidal ideation, since these treatment-emergent adverse events have been previously reported in adult/adolescent patients treated with perampanel.2 During Study 232, treatment-emergent adverse events relating to hostility and/or aggression were reported at similar rates in cohorts 1 and 2 and across the core and extension phases (32.1%-47.4%); few of these events led to discontinuation (core study: n = 2; extension phase, n = 2). The most common treatment-emergent adverse events relating to hostility and/or aggression were irritability and aggression during both the core and extension phases. In a pooled analysis of safety data from 3 phase III studies in patients aged ≥12 years with focal seizures, 11.8% of perampanel-treated patients had treatment-emergent adverse events related to hostility and/or aggression based on narrow and broad SMQ terms compared with 5.7% of placebo-treated patients; with perampanel, the most common treatment-emergent adverse events were irritability (7.0%) and aggression (1.6%),18 which is consistent with findings in Study 232. Similar rates of treatment-emergent adverse events related to hostility and/or aggression were also observed in a phase III study in patients aged ≥12 years with primary generalized tonic-clonic seizures (perampanel: 18.5%; placebo: 4.9%).6 Overall, there was a higher proportion of pediatric patients in Study 232 who experienced treatment-emergent adverse events relating to hostility and/or aggression following adjunctive perampanel treatment (32.1%-47.4%) compared with the phase III studies in patients aged ≥12 years with focal seizures (11.8%) or primary generalized tonic-clonic seizures (18.5%); however, the small patient numbers in Study 232 may have contributed to the higher rates of these treatment-emergent adverse events (Study 232: n = 50 [core study] and n = 41 [extension phase]; pooled phase III studies in focal seizures: n = 1038; phase III study in primary generalized tonic-clonic seizures: n = 81).6,18 There were no patients who experienced homicidal ideation during Study 232, but there were 3 patients who experienced suicidal ideation across the core and extension phases; none of these events were considered as serious adverse events, and all 3 patients recovered. Taken together, these data provide preliminary evidence to suggest that dosing in pediatric patients aged ≥2 to <12 years should follow the same label recommendations as in adolescent/adult patients in relation to psychiatric and behavioral AEs.
Preliminary efficacy results from the core study showed adjunctive therapy with perampanel oral suspension at daily doses up to 0.18 mg/kg to be efficacious in controlling overall seizures in pediatric patients aged ≥2 to <12 years with epilepsy. This efficacy was maintained during the extension phase, evidenced by the proportion of patients achieving seizure freedom for overall seizures over the 13-week intervals of the extension phase, which ranged from 22.0% to 31.8%. For generalized seizures, there was an increase in 28-day average seizure frequency from baseline in cohort 1 during the core study and weeks 1-13 of the extension phase maintenance period. However, given the low patient numbers for all generalized seizure subtypes and the large variability in these data, firm conclusions cannot be made and additional efficacy analyses may be warranted in patients ≥2 to <12 years of age with generalized seizure types.
Several limitations exist within the present analysis including its open-label design and consequent lack of a control group. Furthermore, the study included a relatively small patient population, a limitation that may have been more significant in the extension phase, in which the total patient population was 41, as well as in the subgroup analyses, where certain subgroups included fewer than 10 patients. The study population consisted of pediatric patients with heterogeneous diagnoses of epilepsies allowing for an overall view of the epilepsy population, but prevented conclusions regarding specific epilepsy types. Finally, the use of concomitant antiepileptic drugs was an inevitable limitation of a study in this patient population, although the study’s analysis of the influence of EIAEDs on perampanel pharmacokinetics was an important outcome and showed the similarity between children (aged ≥2 to <12 years), adolescents (aged ≥12 to <18 years), and adults.
Conclusions
The outcomes of this analysis demonstrate that the pharmacokinetic profile of perampanel is not influenced by age-related changes in body weight or liver function, as the profile was comparable with that observed in adolescents and adults. These data suggest that age- or weight-based dosing for adjunctive perampanel therapy is not required. In addition, the safety and efficacy outcomes from Study 232 are consistent with those observed in previous analyses of perampanel in both adolescent and adult populations.3-6
Supplemental Material
Supplementary_Figure_S1 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplemental Material
Supplementary_Figure_S2 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplemental Material
Supplementary_Figure_S3 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplemental Material
Supplementary_Table_S1 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplemental Material
Supplementary_Table_S2 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplemental Material
Supplementary_Table_S3 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplemental Material
Supplementary_Table_S4 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Acknowledgments
The authors thank Yi Xia, a former employee of Eisai Inc., who provided statistical assistance in the early stages of the study. Editorial support, under the direction of the authors, was provided by Lesley Wassef-Birosik, PhD, of IMPRINT Science, New York, NY, USA, and Adele Blair, PhD, of CMC AFFINITY, a division of McCann Health Medical Communications Ltd., Glasgow, UK, funded by Eisai Inc., in accordance with Good Publication Practice (GPP3) guidelines.
Authors' Note: Part of this work was presented as a poster at the 69th Annual Meeting of the American Epilepsy Society, Philadelphia, PA, USA, December 4-8, 2015. ClinicalTrials.gov identifier: NCT01527006.
Author Contributions: All authors were involved in the study design, interpretation of the results, and the reviewing and approval of the manuscript, and in the decision to submit the article for publication. All authors also confirm accountability for the accuracy and integrity of the work.
Declaration of Conflicting Interests: The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: J.B. Renfroe has received speaker fees for Eisai Inc. and LivaNova, and served as an investigator for Allergan, Eisai Inc., GSK, Sepracor, and UCB Pharma. M. Mintz serves on the editorial boards of the Journal of Child Neurology and Vision Development and Rehabilitation; has conducted research for AlkaliDx Inc, Aquestive Therapeutics, Curemark, Eisai Inc., Impax Laboratories, Neurim Pharmaceuticals, and Nuvelution Pharma Inc. contracted through the Clinical Research Center of New Jersey, LLC (CRCNJ); is a consultant to Philips-Electrical Geodesics contracted through The Center for Neurological and Neurodevelopmental Health, LLC (CNNH); is President, CEO, Founder, and sole proprietor of CNNH and CRCNJ; is President and Founder of NeurAbilities, a 501(c)(3) public charity; and has functioned as an expert witness in various litigation and mitigation cases. R. Davis has received speaker fees from LivaNova, Eisai Inc., and Lundbeck, and has served as an investigator for LivaNova, Eisai Inc., Global Pharmaceuticals, Lundbeck, Pfizer, and UCB Pharma. J. Ferreira has received speaker fees and served as a consultant and investigator for Eisai Inc., S. Dispoto, and J. Ferry are employees of Eisai Inc., USA. Y. Umetsu is an employee of Eisai Co., Ltd., Japan. B. Rege and A. Laurenza are former employees of Eisai Inc., USA. O. Majid and Z. Hussein are employees of Eisai Ltd., UK.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this study was funded by Eisai Inc.
Supplemental Material: Supplemental material for this article is available online.
References
- 1. European Medicines Agency (EMA). Fycompa® Annex I: Summary of Product Characteristics, April 2017. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002434/WC500130815.pdf. Accessed July 05, 2018.
- 2. Food and Drug Administration (FDA). Fycompa® Prescribing Information, September 2018. https://fycompa.com/-/media/Files/Fycompa/Fycompa_Prescribing_Information.pdf. Accessed January 24, 2019.
- 3. French JA, Krauss GL, Biton V, et al. Adjunctive perampanel for refractory partial-onset seizures: randomized phase III study 304. Neurology. 2012;79(6):589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. French JA, Krauss GL, Steinhoff BJ, et al. Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305. Epilepsia. 2013;54(1):117–125. [DOI] [PubMed] [Google Scholar]
- 5. Krauss GL, Serratosa JM, Villanueva V, et al. Randomized phase III study 306: adjunctive perampanel for refractory partial-onset seizures. Neurology. 2012;78(18):1408–1415. [DOI] [PubMed] [Google Scholar]
- 6. French JA, Krauss GL, Wechsler RT, et al. Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy: a randomized trial. Neurology. 2015;85(11):950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krauss GL, Perucca E, Ben-Menachem E, et al. Long-term safety of perampanel and seizure outcomes in refractory partial-onset seizures and secondarily generalized seizures: results from phase III extension study 307. Epilepsia. 2014;55(7):1058–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patsalos PN. The clinical pharmacology profile of the new antiepileptic drug perampanel: a novel noncompetitive AMPA receptor antagonist. Epilepsia. 2015;56(1):12–27. [DOI] [PubMed] [Google Scholar]
- 9. Cormier J, Chu CJ. Safety and efficacy of levetiracetam for the treatment of partial onset seizures in children from one month of age. Neuropsychiatr Dis Treat. 2013;9:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu H, Rosenbaum S. Developmental pharmacokinetics in pediatric populations. J Pediatr Pharmacol Ther. 2014;19(4):262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anderson GD. Children versus adults: pharmacokinetic and adverse-effect differences. Epilepsia. 2002;43(suppl 3):53–59. [DOI] [PubMed] [Google Scholar]
- 12. Fernandez E, Perez R, Hernandez A, et al. Factors and mechanisms for pharmacokinetic differences between pediatric population and adults. Pharmaceutics. 2011;3(1):53–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eisai Launches New Oral Suspension Formulation for Antiepileptic Drug Fycompa® (Perampanel) in the United States. Eisai Inc Press Release 2016. http://www.eisai.com/news/news201642.html. Accessed July 5, 2018.
- 14. Cella M, Knibbe C, Danhof M, Della PO. What is the right dose for children? Br J Clin Pharmacol. 2010;70(4):597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Food and Drug Administration (FDA). Tegretol® Prescribing Information, August 2015. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/016608s097,018281s045,018927s038,020234s026lbl.pdf. Accessed July 5, 2018.
- 16. Riva R, Contin M, Albani F, et al. Free concentration of carbamazepine and carbamazepine-10,11-epoxide in children and adults. Influence of age and phenobarbitone co-medication. Clin Pharmacokinet. 1985;10(6):524–531. [DOI] [PubMed] [Google Scholar]
- 17. Eisai Announces FDA Approval of Fycompa® (Perampanel) Oral Suspension for Adjunctive Therapy in the Treatment of Partial-Onset Seizures and Primary Generalized Tonic-Clonic Seizures. New Formulation Expands Treatment Options for Epilepsy Patients with POS and PGTC Seizures. PR Newswire 2016. http://www.prnewswire.com/news-releases/eisai-announces-fda-approval-of-fycompa-perampanel-oral-suspension-for-adjunctive-therapy-in-the-treatment-of-partial-onset-seizures-and-primary-generalized-tonic-clonic-seizures-300260456.html. Accessed July 5, 2018.
- 18. Ettinger AB, LoPresti A, Yang H, et al. Psychiatric and behavioral adverse events in randomized clinical studies of the noncompetitive AMPA receptor antagonist perampanel. Epilepsia. 2015;56(8):1252–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary_Figure_S1 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplementary_Figure_S2 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplementary_Figure_S3 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplementary_Table_S1 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplementary_Table_S2 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplementary_Table_S3 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology
Supplementary_Table_S4 for Adjunctive Perampanel Oral Suspension in Pediatric Patients From ≥2 to <12 Years of Age With Epilepsy: Pharmacokinetics, Safety, Tolerability, and Efficacy by J. Ben Renfroe, Mark Mintz, Ronald Davis, Jose Ferreira, Sharon Dispoto, Jim Ferry, Yuko Umetsu, Bhaskar Rege, Oneeb Majid, Ziad Hussein, and Antonio Laurenza in Journal of Child Neurology