

Abstract
The highly enantioselective addition of terminal ynamides to a variety of isatins, catalyzed by a bisoxazolidine copper complex under mild, base-free reaction conditions, is described. The reaction is broad in scope, scalable, applicable to unprotected isatins and provides efficient access to 3-hydroxyoxindoles carrying a tetrasubstituted chirality center with excellent yields and enantioselectivities. Synthetically versatile, multifunctional 3-hydroxyindolinones are obtained via hydration, partial hydrogenation or hydroxyacyloxylation of the ynamide moiety at room temperature and exhaustive hydrogenation followed by reductive detosylation and spontaneous cyclization affords cinchonamidine alkaloids.
Keywords: Asymmetric catalysis, ynamides, chimonamidine, bisoxazolidines, isatins
Graphical Abstract
Terminal ynamide additions to isatins are accomplished with high yields and enantioselectivities via base-free Cu(I)-bisoxazolidine catalysis. This approach provides efficient access to chimonamidines and 3-hydroxyindolinones carrying a tetrasubstituted chirality center with a synthetically versatile ynamide moiety.

The natural occurrence and pharmaceutical relevance of chiral oxindoles displaying a tetrasubstituted stereogenic carbon center at the 3-position has received considerable attention from medicinal and synthetic chemists in recent years.1 This is particularly the case for the family of 3-substituted 3-hydroxyindolin-2-ones, which share an important structural motif commonly found in alkaloids such as chimonamidine and various emerging drug candidates.2 To this end, we noticed that few reports on the asymmetric synthesis of chimonamidine I, all suffering from low overall yields, have appeared in the literature.2cgi Retrosynthetic analysis pointed toward an unprecedented 3-(aminoethynyl)-3-hydroxyindolin-2-one scaffold II that could be generated via catalytic enantioselective addition of terminal ynamides to isatins III of which a wide variety is commercially available (Figure 1). In comparison to alkyne additions,3 incorporation of an ynamide moiety into readily available isatins could generate unique synthetic opportunities via late stage functionalization of the highly polarized N-substituted triple bond, for example to 3-substituted 3-hydroxyindolin-2-ones carrying 1,3-amino alcohol, β-hydroxy amide, β-hydroxy enamine or α-acyloxyenamide functionalities. We envisioned a catalytic asymmetric method producing the key scaffold II which would ultimately streamline access to multifunctional 3-hydroxyoxindoles IV-VII and chimonamidine or other important alkaloids.
Figure 1.

Synthetic utility of ynamide catalysis with isatins.
Internal ynamides have been used in a wide range of cycloadditions, cycloisomerizations, cross-couplings, ring-closing metathesis and other valuable transformations.4 Although terminal ynamides are readily available building blocks,5 they remain underutilized compared to both internal ynamides and regular alkynes which have been exploited in countless asymmetric 1,2-addition reactions with carbonyl electrophiles.6 This might be attributed to the difficulty with the enamide like reactivity of ynamides and the formation of keteniminium intermediates which is conceptually different from the generally accepted C-C bond formation pathways of alkynes.7 Catalytic enantioselective additions with terminal ynamides which are poor nucleophiles under neutral conditions have neither been accomplished with isatins nor applied in natural product synthesis to date.
We began our search for a catalytic asymmetric method that affords the key 3-(aminoethynyl)-3-hydroxyindolin-2-one scaffold II using N-ethynyl-N-butylbenzenesulfonamide, 1, and the N-protected isatins 2 and 3 as test compounds. At the onset of our reaction screening efforts, we investigated the suitability of previously reported catalytic enantioselective alkynylation protocols.3,6 The zinc triflate catalyzed ynamide addition using amino alcohols or bisoxazolines L1-L4 as ligands and stoichiometric amounts of diisopropylethylamine as base gave the desired alkynylated oxindole products 4 and 5 in high yields but with negligible asymmetric induction (Table 1, entries 1-4). Results did not improve with copper iodide or under organocatalytic conditions with thiourea L5 (entries 5-7). We then turned our attention to bisoxazolidines, a broadly useful class of chiral ligands developed in our laboratory.8 The fully saturated oxazolidine moieties in this ligand type combines a unique ring topology with distinct steric and electronic properties that are remarkably different from bisoxazoline analogues and very useful for asymmetric catalysis.9 We were very pleased to find that copper(I) catalysis with bisoxazolidine L6 gave 4 in quantitative yields and 79% ee under base free conditions (entry 8). Additional screening of various copper and zinc catalysts did not further improve results (entries 9-12 and SI). After optimization of the reaction temperature, however, we isolated 4 in 93% yield and 90% ee (entry 16). Solvent screening using N-phenyl isatin, 3, as substrate showed that chloroform gives superior results and we obtained 5 in 99% yield and with 98% ee (entries 17-21). Interestingly, excellent results were also obtained when bisoxazolidine ligands L7-L9 were employed in the optimized protocol (entries 22-24).
Table 1.
Optimization of the asymmetric addition of ynamides to isatins.a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | Catalyst | Ligand | Conditions | Yield (%)b | Ee (%)d |
| 1 | Me | Zn(OTf)2 | L1 | C6H6, DIPEA, 25 °C | 99 | <1 |
| 2 | Me | Zn(OTf)2 | L2 | DCE, DIPEA, 25 °C | 86 | <1 |
| 3 | Me | Zn(OTf)2 | L3 | DCE, DIPEA, 25 °C | 94 | 2 |
| 4 | Me | Zn(OTf)2 | L4 | DCE, DIPEA, 25 °C | 89 | 1 |
| 5 | Me | CuI | L3 | DCM, DIPEA, 25 °C | 64 | 16 |
| 6 | Me | CuI | L4 | DCM, DIPEA, 25 °C | 51 | 17 |
| 7 | Me | / | L5 | DCE, DIPEA, 25 °C | 54 | 4 |
| 8 | Me | (CuOTf)2·Tol | L6 | CHCl3, 25 °C | 99 c | 79 |
| 9 | Me | (CuOTf)2·Tol | L4 | CHCl3, 25 °C | <5 c | n.d. |
| 10 | Me | CuI | L6 | CHCl3, 25 °C | <50 c | 80 |
| 11 | Me | CuI | L3 | CHCl3, 25 °C | <50 c | 9 |
| 12 | Me | Zn(OTf)2 | L6 | CHCl3, 25 °C | <5 c | n.d. |
| 13 | Me | / | / | CHCl3, DIPEA, 25 °C | n.r. | n.d. |
| 14 | Me | (CuOTf)2·Tol | / | CHCl3, DIPEA, 25 °C | n.r. | n.d. |
| 15 | Ph | (CuOTf)2·Tol | L6 | CHCl3, Et3N, 50 °C | 27 | 2 |
| 16 | Me | (CuOTf)2·Tol | L6 | CHCl3, 0 °C | 93 | 90 |
| 17 | Ph | (CuOTf)2·Tol | L6 | CH2Cl2, 0 °C | 97 | 90 |
| 18 | Ph | (CuOTf)2·Tol | L6 | THF, 0 °C | 36 | 96 |
| 19 | Ph | (CuOTf)2·Tol | L6 | Et2O, 0 °C | 42 | 68 |
| 20 | Ph | (CuOTf)2·Tol | L6 | MTBE, 0 °C | 61 | 66 |
| 21 | Ph | (CuOTf)2·Tol | L6 | CHCl3, 0 °C | 99 | 98 |
| 22 | Ph | (CuOTf)2·Tol | L7 | CHCl3, 0 °C | 98 | 85 |
| 23 | Ph | (CuOTf)2·Tol | L8 | CHCl3, 0 °C | 99 | 91 |
| 24 | Ph | (CuOTf)2·Tol | L9 | CHCl3, 0 °C | 93 | 90 |
Reaction Conditions: Ynamide (0.3 mmol, 1.5 equiv.), isatin (0.2 mmol, 1 equiv.), catalyst (5 mol%), ligand (15 mol%), base (1.1 equiv.) in 0.1 mL of solvent.
Isolated yield.
Conversion determined by HPLC.
Ee determined by chiral HPLC on a Phenomenex Lux 5μ Cellulose-3 column (mobile phase = 85:15 hexanes-EtOH, flow rate = 1.0 mL/min, λ = 254 nm), n.r. = no reaction, n.d. = not determined.
The catalysis is ligand-accelerated and no reaction was observed after 48 hours in the absence of L6 (entries 13 and 14). Further analysis revealed that the ynamide addition does not occur in the presence of Hünig’s base unless a catalyst is added and the replacement of the ynamide with phenylacetylene in our base-free protocol did not give the corresponding alkynylation product. The ynamide addition proceeded sluggishly when we added two equivalents of Et3N, producing almost racemic 5 in 27% yield at 50 °C (entry 15).10 These observations are in agreement with the enamide-like nucleophilicity of ynamides mentioned above, i.e. a base-free C-C bond formation course via a keteniminium intermediate followed by tautomerization toward the 3-(aminoethynyl)-3-hydroxyindolin-2-one scaffold.11 Several examples of enamide-like additions of ynamides to Lewis acid activated carbonyl electrophiles under base-free conditions have been described.12 A plausible reaction mechanism that is in agreement with our observations and these reports is shown in Scheme 1.13 This reaction course is fundamentally different from the generally agreed alkynylation pathway which is initiated by base-promoted deprotonation and generation of an end-on metal-acetylide complex prior to the attack at a carbonyl electrophile.
Scheme 1.

Plausible base-free reaction mechanism.
The optimized protocol was then applied to a range of isatins (Scheme 2). The presence of halogens in various positions does not interfere with the reaction and the 3-(aminoethynyl)-3-hydroxyindolin-2-ones 6-9 were obtained in 87-97% yield and 95-96% ee. Similar results were obtained with isatins carrying a methyl, methoxy or trifluoromethoxy group. The corresponding products 10-12 were produced in 93-97% yield and with high enantioselectivity. The reaction also proceeds smoothly with N-aryl ynamides. When N-ethynyl-N-phenyltolylsulfonamide was applied in the same protocol we obtained 13 in quantitative amounts and in 99% ee. Importantly, the reaction tolerates the easily removable PMP group at the isatin nitrogen which further widens the general usefulness. The catalytic ynamide addition to N-PMP isatin gave 14 in high yield and with 97% ee. Retrosynthetic analysis of chimonamidine revealed that the ynamide must bear a methyl group rather than the n-butyl substituent used in the preceding examples. The protocol was therefore modified using N-ethynyl-N-methyltolylsulfonamide as ynamide to afford 15 and 16. Importantly, 15 can be produced with our Cu(I)-bisoxazolidine catalyzed method in 99% yield and 96% ee on the gram scale. Finally, we found that the nitrogen protection is not a requirement for this reaction and free isatin was converted toward 17 with 75% yield and 91% ee.
Scheme 2.
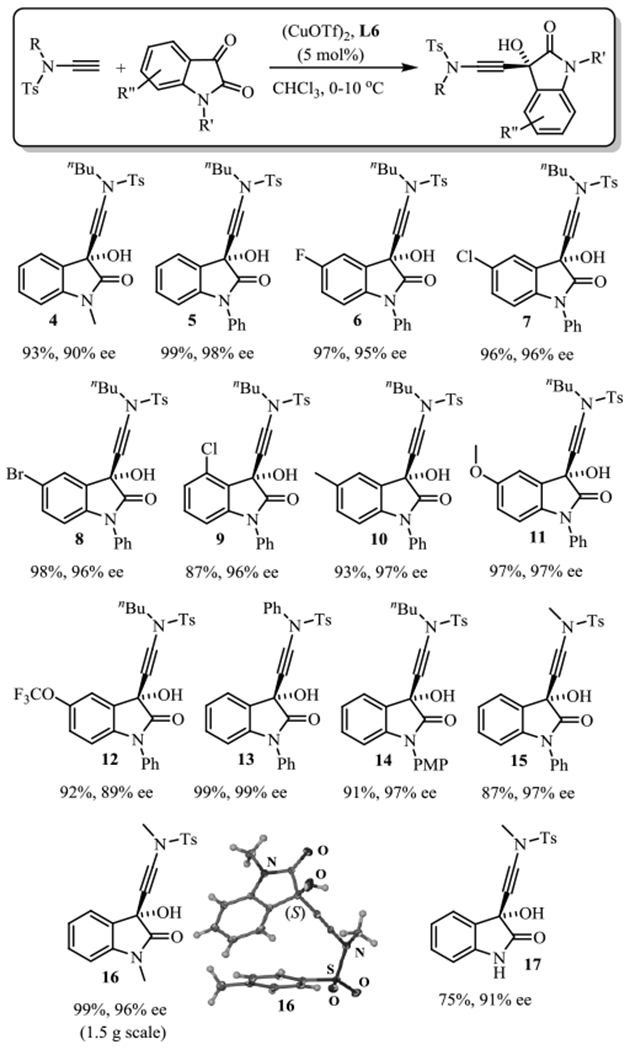
Catalytic enantioselective synthesis of 3-aminoalkynyl-3-hydroxyoxindoles. The absolute configuration of 16 was determined by crystallographic analysis of a single crystal obtained by slow evaporation of a hexanes/ethyl acetate solution (SI). The other configurations were assigned by analogy.
Having established the wide substrate scope of the catalytic enantioselective addition of terminal sulfonynamides to isatins we continued with the investigation of the synthetic versatility of the highly functionalized 3-(aminoethynyl)-3-hydroxyindolin-2-one motif. The incorporation of the chiral propargylic alcohol unit exhibiting a polarized triple bond into the biologically important 3-hydroxyoxindole scaffold enables unique modifications that are distinct from traditional alkyne chemistry, and this affords several new synthetic opportunities (Scheme 3). Complete Pd catalyzed hydrogenation of 5 gave the saturated sulfonamide 18 in 81% yield which was encouraging for our efforts to develop a streamlined asymmetric route toward chimonamidines (vide infra). During optimization of this procedure, we found that careful control of reaction time and catalyst loading allows highly diastereoselective formation of (Z)-β-hydroxy enamines which are important precursors for the synthesis of aminocyclopropyl carbinols, 1,3-amino alcohols and other multifunctional compounds.14 The partial hydrogenation of 5 using 2 mol% of Pd/C generated (Z)-19 in almost quantitative yield.15 Alternatively, the N-tosyl ynamine moiety can be exploited to install a terminal amide bond via hydration under acidic conditions or be converted to α-acyloxyenamides, which are sought-after N,O-ketene acetals with important applications in rearrangement reactions,16 via hydroxyacyloxylation with a carboxylic acid. The amide 20 and the α-acyloxyenamide 21 were obtained in excellent yields from 16 with either aqueous HCl or benzoic acid at room temperature. It is noteworthy that the practical 2-step synthesis of 18-21 from isatins would not be possible via traditional nucleophilic additions with alkynes which underscores the significance of our copper(I)-bisoxazolidine catalyzed ynamide addition method.
Scheme 3.

Selective modifications of the 3-(aminoethynyl)-3-hydroxyindolin-2-one motif.
The synthesis of chimonamidines required deprotection of the sulfonamide group after triple bond saturation and subsequent transamidation with the nascent secondary amino function. First, we prepared 22 by hydrogenation of 16 in 92% yield following the procedure described above (Scheme 4). The deprotection step was very challenging because sulfonamides are notoriously difficult to cleave and this was further complicated by the presence of the acid-sensitive chiral alcohol and the isatin lactam ring. Several literature methods proved unsuccessful or produced low yields and considerable amounts of by-products. After extensive screening and reaction optimization we found that reductive removal of the sulfonamide group in 22 with magnesium turnings in methanol occurs smoothly and with concomitant transamidation in a single step yielding 85% of (S)-chimonamidine, 23, with 96% ee. Altogether, this furnishes 23 in high enantiopurity and in 77% yield over three reactions starting from N-methyl isatin, 2. We were successful in growing a single crystal of chimonamidine 23 by slow evaporation of a hexane/ethyl acetate solution (SI).17 To the best of our knowledge, this is the first example of a natural product synthesis utilizing a terminal ynamide in a catalytic enantioselective nucleophilic addition reaction. This approach to oxindole alkaloids is very efficient and it compares favorably with previous reports on asymmetric chimonamidine synthesis.2c,g,i The reductive tosyl deprotection was also successfully applied in the synthesis of the chimonamidine derivative 24, which demonstrates that the ynamide chemistry is well suited for the production of a variety of medicinally interesting alkaloids.
Scheme 4.

Total synthesis of chimonamidines and X-ray structure of 23. The absolute configuration of 16 was determined by crystallographic analysis of a single crystal obtained by slow evaporation of a hexane/ethyl acetate solution (SI). The other configurations were assigned by analogy.
In conclusion, the first catalytic enantioselective addition of terminal ynamides to isatins has been developed and successfully applied in the total synthesis of chimonamidine. This method uses (CuOTf)2·Tol and a commercially available bisoxazolidine ligand under base-free conditions, and it affords a series of synthetically versatile 3-(aminoethynyl)-3-hydroxyindolin-2-ones in high yields and enantiomeric excess from readily available starting materials. The introduction of the ynamide moiety provides a unique entry to a variety of multifunctional chiral oxindole building blocks including 1,3-amino alcohols, β-hydroxy amides, β-hydroxy enamines and α-acyloxyenamides which were produced in high yields and without erosion of enantiopurity. As a result, the synthesis of chimonamidines can now be achieved by highly enantioselective, upscalable ynamide additions to isatins followed by hydrogenation and tosyl deprotection under mild conditions. This 3-step approach compares favorably with literature protocols and establishes efficient access to important oxindole alkaloids.
Supplementary Material
Acknowledgements
We are grateful for financial support from the NIH (GM106260).
Footnotes
Conflict of interests
The Wolf bisoxazolidine ligand L6 is commercially available at Strem Chemicals (#07-0488) under license from Georgetown University. C. Wolf is one of the listed inventors on US patent 7,763,734.
References
- [1].a) Ishimaru T, Shibata N, Horikawa T, Yasuda N, Nakamura S, Toru T, Shiro M, Angew. Chem. Int. Ed 2008, 47, 4157; [DOI] [PubMed] [Google Scholar]; b) Ma S, Han X, Krishnan S, Virgil SC, Stoltz BM, Angew. Chem. Int. Ed 2009, 48, 8037; [DOI] [PubMed] [Google Scholar]; c) Bui T, Syed S, Barbas CF III, J. Am. Chem. Soc 2009, 131, 8758; [DOI] [PubMed] [Google Scholar]; d) Antonchick AP, Gerding-Reimers C, Catarinella M, Schürmann M, Preut H, Ziegler S, Rauh D, Waldmann H, Nat. Chem 2010, 2, 735; [DOI] [PubMed] [Google Scholar]; e) Tan B, Candeias NR, Barbas CF, Nat. Chem 2011, 3, 473; [DOI] [PubMed] [Google Scholar]; f) Guo C, Song J, Huang J-Z, Chen P-H, Luo S-W, Gong L-Z, Angew. Chem. Int. Ed 2012, 51, 1046; [DOI] [PubMed] [Google Scholar]; g) Wu L, Falivene L, Drinkel E, Grant S, Linden A, Cavallo L, Dorta R, Angew. Chem. Int. Ed 2012, 51, 2870; [DOI] [PubMed] [Google Scholar]; h) Xie W, Jiang G, Liu H, Hu J, Pan X, Zhang H, Wan X, Lai Y, Mae D, Angew. Chem. Int. Ed 2013, 52, 12924; [DOI] [PubMed] [Google Scholar]; i) Mitsunuma H, Shibasaki M, Kanai M, Matsunaga S, Angew. Chem. Int. Ed 2012, 51, 5217; [DOI] [PubMed] [Google Scholar]; j) Zong L, Du S, Chin KF, Wang C, Tan C-H, Angew. Chem. Int. Ed 2015, 54, 9390; [DOI] [PubMed] [Google Scholar]; k) Zhao J, Fang B, Luo W, Hao X, Liu X, Lin L, Feng X, Angew. Chem. Int. Ed 2015, 54, 241; [DOI] [PubMed] [Google Scholar]; l) Yu J-S, Liao F-M, Gao W-M, Liao K, Zuo R-L, Zhou J, Angew. Chem. Int. Ed 2015, 54, 7381; [DOI] [PubMed] [Google Scholar]; m) Engl OD, Fritz SP, Wennemers H, Angew. Chem. Int. Ed 2015, 54, 8193; [DOI] [PubMed] [Google Scholar]; n) Wu M-Y, He W-W, Liu X-Y, Tan B, Angew. Chem. Int. Ed 2015, 54, 9409; [DOI] [PubMed] [Google Scholar]; o) Biswas P, Paul S, Guin J, Angew. Chem. Int. Ed 2016, 55, 7756; [DOI] [PubMed] [Google Scholar]; p) Sankar MG, Garcia-Castro M, Golz C, Strohmann C, Kumar K, Angew. Chem. Int. Ed 2016, 55, 9709; [DOI] [PubMed] [Google Scholar]; q) Kong W, Wang Q, Zhu J, Angew. Chem. Int. Ed 2016, 55, 9714; [DOI] [PubMed] [Google Scholar]; r) Balaraman K, Wolf C, Angew. Chem. Int. Ed 2017, 56, 1390; [DOI] [PMC free article] [PubMed] [Google Scholar]; s) Balaraman K, Ding R, Wolf C, Adv. Synth. Catal 2017, 359, 4165; [DOI] [PMC free article] [PubMed] [Google Scholar]; t) Ding R, Wolf C, Org. Lett 2018, 20, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Selected examples:; a) Sano D, Nagata K, Itoh T, Org. Lett 2008, 10, 1593; [DOI] [PubMed] [Google Scholar]; b) Itoh T, Ishikawa H, Hayashi Y, Org. Lett 2009, 11, 3854; [DOI] [PubMed] [Google Scholar]; c) Chen W-B, Du X-L, Cun L-F, Zhang X-M, Yuan W-C, Tetrahedron 2010, 66, 1441; [Google Scholar]; d) Guo Q, Bhanushali M, Zhao C-G, Angew. Chem. Int. Ed 2010, 49, 9460; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hara N, Nakamura S, Funahashi Y, Shibata N, Adv. Synth. Catal 2011, 353, 2976; [Google Scholar]; f) Yang Y, Moinodeen F, Chin W, Ma T, Jiang Z, Tan C-H, Org. Lett 2012, 14, 4762; [DOI] [PubMed] [Google Scholar]; g) Zhu B, Zhang W, Lee R, Han Z, Yang W, Tan D, Huang K-W, Jiang Z, Angew. Chem. Int. Ed 2013, 52, 6666; [DOI] [PubMed] [Google Scholar]; h) Ding R, Wolf C, J. Org. Chem 2017, 82, 1273; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Reddy UVS, Chennapuram M, Seki K, Ski C, Anusha B, Kwon E, Okuyama Y, Uwai K, Tokiwa M, Takeshita M, Nakano H, Eur. J. Org. Chem 2017, 3874; [Google Scholar]; j) Moskowitz M, Balaraman K, Wolf C, J. Org. Chem 2018, 83, 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]; For asymmetric synthesis of isoindolinones, see; k) Li T, Zhou C, Yan X, Wang J, Angew. Chem. Int. Ed 2018, 57, 4048. [DOI] [PubMed] [Google Scholar]
- [3].a) Xu N, Gu D-W, Zi J, Wu X-Y, Guo X-X, Org. Lett 2016, 18, 2439; [DOI] [PubMed] [Google Scholar]; b) Chen Q, Tang Y, Huang T, Liu X, Lin L, Feng X, Angew. Chem. Int. Ed 2016, 55, 5286; [DOI] [PubMed] [Google Scholar]; c) Chen L, Huang G, Liu M, Huang Z, Chen F-R, Adv. Synth. Catal 2018, 360, 3497. [Google Scholar]
- [4].a) Zificsak CA, Mulder JA, Hsung RP, Rameshkumar C, Wei L-L, Tetrahedron 2001, 57, 7575; [Google Scholar]; b) Evano G, Coste A, Jouvin K, Angew. Chem. Int. Ed 2010, 49, 2840; [DOI] [PubMed] [Google Scholar]; c) DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP, Chem. Rev 2010, 110, 5064; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wang X-N, Yeom H-S, Fang L-C, He S, Ma Z-X, Kedrowski BL, Hsung RP, Acc. Chem. Res 2014, 47, 560; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Adcock HV, Chatzopoulou E, Davies PW, Angew. Chem. Int. Ed 2015, 54, 15525; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Straker RN, Majhail MK, Willis MC, Chem. Sci 2017, 8, 7963; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Zeng X, Li J, Ng CK, Hammond GB, Xu B, Angew. Chem. Int. Ed 2018, 57, 2924; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Gao Y, Wu G, Zhou Q, Wang J, Angew. Chem. Int. Ed 2018, 57, 2716; [DOI] [PubMed] [Google Scholar]; i) Mansfield SJ, Christensen KE, Thompson AL, Ma K, Jones MW, Mekareeya A, Anderson EA, Angew. Chem. Int. Ed 2017, 56, 14428; [DOI] [PubMed] [Google Scholar]; k) Marien N, Reddy BN, De Vleeschouwer F, Goderis S, Van Hecke K, Verniest G, Angew. Chem. Int. Ed 2018, 57, 5660. [DOI] [PubMed] [Google Scholar]
- [5].a) Hamada T, Ye X, Stahl SS, J. Am. Chem. Soc 2008, 130, 833; [DOI] [PubMed] [Google Scholar]; b) Mansfield SJ, Campbell CD, Jones MW, Anderson EA, Chem. Commun 2015, 51, 3316; [DOI] [PubMed] [Google Scholar]; c) Tu Y, Zeng X, Wang H, Zhao J, Org. Lett 2018, 20, 280. [DOI] [PubMed] [Google Scholar]
- [6].a) Wang X-N, Hsung RP, Qi R, Fox SK, Lv M-C, Org. Lett 2013, 15, 2514; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang X-N, Hsung RP, Fox SK, Lv M-C, Qi R, Heterocycles 2014, 88, 1233; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cook AM, Wolf C, Chem. Commun 2014, 50, 3151; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cook AM, Wolf C, Tetrahedron Lett 2015, 56, 2377; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Cook AM, Wolf C, Angew. Chem. Int. Ed 2016, 55, 2929; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zhao J, Yang J, Wang C, Xu S, Angew. Chem. Int. Ed 2018, 57, 10.1002/anie.201811586. [DOI] [Google Scholar]
- [7].Laub HA, Evano G, Mayr H, Angew. Chem. Int. Ed 2014, 53, 4968. [DOI] [PubMed] [Google Scholar]
- [8].a) Wolf C, Liu S, J. Am. Chem. Soc 2006, 128, 10996; [DOI] [PubMed] [Google Scholar]; b) Liu S, Wolf C, Org. Lett 2007, 9, 2965; [DOI] [PubMed] [Google Scholar]; c) Liu S, Wolf C, Org. Lett 2008, 10, 1831; [DOI] [PubMed] [Google Scholar]; d) Yearick Spangler K, Wolf C, Org. Lett 2009, 11, 4724; [DOI] [PubMed] [Google Scholar]; e) Xu H, Wolf C, Synlett 2010, 2765; [Google Scholar]; f) Wolf C, Moskowitz M, J. Org. Chem 2011, 76, 6372; [DOI] [PubMed] [Google Scholar]; g) Xu H, Wolf C, Angew. Chem. Int. Ed 2011, 50, 12249; [DOI] [PubMed] [Google Scholar]; h) Xu H, Wolf C, Chem. Commun 2010, 46, 8026; [DOI] [PubMed] [Google Scholar]; i) Wolf C, Zhang P, Adv. Synth. Catal 2011, 353, 760. [Google Scholar]
- [9].Wolf C, Xu H, Chem. Commun 2011, 47, 3339. [DOI] [PubMed] [Google Scholar]
- [10].Under these conditions, phenylacetylene addition to isatin 3 occurs smoothly to give 99% yield of racemic product.
- [11].For examples of ynamide transformations via keteniminium ion intermediates, see; a) Lecomte M, Evano G, Angew. Chem. Int. Ed 2016, 55, 4547; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Baldassari LL, de la Torre A, Li J, Lüdtke DS, Maulide N, Angew. Chem. Int. Ed 2017, 56, 15723; [DOI] [PubMed] [Google Scholar]; c) Patil DV, Kim SW, Nguyen QH, Kim H, Wang S, Hoang T, Shin S, Angew. Chem. Int. Ed 2017, 56, 3670; [DOI] [PubMed] [Google Scholar]; d) Pinto A, Kaiser D, Maryasin B, Di Mauro G, Gonzalez L, Maulide N, Chem. Eur. J 2017, 24, 2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Kurtz KCM, Hsung RP, Zhang Y, Org. Lett 2006, 8, 231. [DOI] [PubMed] [Google Scholar]; b) You L, Al-Rashid ZF, Ghosh SK, Li G, Hsung RP Synlett 2007, 1656. [Google Scholar]; c) Wang X-N, Ma Z-X, Deng J, Hsung RP, Tetrahedron Lett 2015, 56, 3463. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yabuuchi Y, Kuzuguchi T, Yoshimura T, Matsuo J.-i., Org. Lett 2016, 18, 4951. [DOI] [PubMed] [Google Scholar]; For asymmetric catalysis with enamides:; e) Matsubara R, Kobayashi S, Acc. Chem. Res 2008, 41, 292. [DOI] [PubMed] [Google Scholar]
- [13].The bisoxazolidine ligand undergoes N,N’-coordination to Cu(I).
- [14].a) Valenta P, Carroll PJ, Walsh PJ, J. Am. Chem. Soc 2010, 132, 14179; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gopalaiah K, Kagan HB, Chem. Rev 2011, 111, 4599. [DOI] [PubMed] [Google Scholar]
- [15].Reddy AS, Swamy KCK, Angew. Chem. Int. Ed 2017, 56, 6984. [Google Scholar]
- [16].a) Smith DL, Goundry WRF, Lam HW, Chem. Commun 2012, 48, 1505; [DOI] [PubMed] [Google Scholar]; b) Xu S, Liu J, Hu D, Bi X, Green Chem 2015, 17, 184. [Google Scholar]
- [17].The CCDC numbers for the compounds 16 and 23 are 1882888 and 1882889, respectively. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.