

Abstract
Background: Adenosine kinase (ADK) is a key regulator of hepatic metabolism. Its deficiency in the liver causes hepatic steatosis and methylation defects. In this study, we investigated whether reduced ADK expression affects the susceptibility of the liver to a carcinogen.
Methods: We investigated ADK expression in samples from 11 liver cancer patients. We used transgenic Adk-tg mice with reduced hepatic ADK to study their susceptibility to a carcinogen. We exposed 45 Adk-tg and 21 wild-type (WT) mice to the carcinogen diethylnitrosamine (DEN) and the tumor promoter phenobarbital (PB) and examined the survival and body weight.
Results: Seven of 11 patients with liver cancer had reduced ADK expression. A Kaplan–Meier survival curve showed a significantly increased mortality rate of DEN/PB-exposed Adk-tg mice compared with WT mice.
Conclusions: Reduced hepatic ADK increases the susceptibility to the acute toxic effects of a carcinogen. Low hepatic ADK might be a risk factor and biomarker for cancer development.
Keywords: adenosine kinase, liver, methylation, metabolism, carcinogen, hepatic toxicity
Introduction
Adenosine kinase (ADK; EC 2.7.1.20) is an evolutionarily conserved ribokinase that has an important role in maintaining cellular metabolism. It regulates intracellular and extracellular adenosine levels through phosphorylation of adenosine into 5′-adenosine monophosphate using 5′-adenosine triphosphate as the phosphate group donor.1,2
Adenosine combines adenosine receptor-dependent and independent activities that include the regulation of energy homeostasis and transmethylation pathways. ADK is a high-affinity, low-capacity enzyme that controls adenosine metabolism under baseline conditions, and therefore plays a critical role in maintaining the homoeostatic and metabolic balance in all living systems.1 ADK has two isoforms derived from alternative splicing and alternative promoter use, a long nuclear isoform (ADK-L) and a short cytoplasmic isoform (ADK-S).3 The nuclear localization of ADK-L suggests a specific function as a global regulator of methylation in the nucleus, which includes DNA methylation, whereas the cytoplasmic form controls metabolic pathways, which includes the methylation of lipids in the liver.4–6
ADK is linked to the transmethylation pathway by removing adenosine, an obligatory product of all S-adenosylmethionine (SAM)-dependent transmethylation reactions. Thereby, high levels of ADK drive the flux of methyl groups through the transmethylation pathway, whereas high levels of adenosine (low levels of ADK expression) provide negative feedback inhibition of the pathway.4,5
Liver is not only the organ with the highest expression levels of ADK in the body, but also 85% of all transmethylation reactions take place in the liver.7–9 Therefore, ADK plays a critical role in the maintenance of transmethylation homeostasis in the liver; consequently homozygous deletion of the ADK gene in mice triggers hepatic steatosis, which is a fatal condition characterized by rapid microvesicular fat infiltration and early postnatal mortality.4 Similarly, ADK-deficient patients show disruption of the transmethylation pathway resulting in major physiological abnormalities of liver metabolism including microvesicular hepatic steatosis and hepatic encephalopathy.10,11 In addition, ADK-deficient patients are characterized by global developmental delay, seizures, and cognitive impairment.10,11
Given the importance of ADK for liver physiology and because disruption of normal liver function has been associated with an increased risk for the development of liver cancers,12–14 we hypothesized that reduced expression of ADK might render the liver more susceptible to the effects of a carcinogen. We studied the expression of ADK in patients with liver cancer, specifically hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC), and showed reduced ADK expression in 7 of 11 patients. In line with those pathological changes, transgenic mice with reduced ADK expression in the liver (Adk-tg)6 were more susceptible to the acute toxic effects of a carcinogen and displayed an increased acute mortality rate. We conclude that reduced ADK expression might render the liver more susceptible to cancer development.
Methodology
Analysis of tumor bank samples
Matched pairs of cancer and adjacent normal control tissues were obtained from the College of American Pathologists-accredited Legacy Tumor Bank after approved protocols and regulatory requirements. We obtained frozen tissues from eight HCC samples and three ICC samples. All samples were subjected to Western blot analysis as described hereunder.
Mouse model of hepatic ADK deficiency
All studies were conducted in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited facility in accordance with protocols approved by the Legacy Institutional Animal Care and Use Committee. All animals were social housed under standardized conditions of light, temperature, humidity, and environmental enrichment and had ad libitum access to food and water. For the Western blot analysis we used 1- to 2-month-old male Adk-tg and C57BL/6 wild-type (WT) mice, which were bred in our vivarium and for carcinogen exposure we used mice at an age of 3 weeks, immediately after weaning. Adk-tg mice were developed by introducing a human ubiquitin promoter-driven Adk-S transgene into an Adk-null background. The function of ADK in the brain of those animals has been studied extensively.6,15,16 Those animals are characterized by complete lack of ADK-L and transgene driven expression of murine ADK-S. All mice were generated and propagated on an identical C57BL/6 background.
Western blot analysis
Tissue was homogenized with radioimmunoprecipitation assay buffer containing a protease inhibitor cocktail. Twenty micrograms of proteins was separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membranes. The blots were probed overnight at 4°C with primary antibody anti-ADK (1:4500; Bethyl Laboratories) and anti-Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:2500; Abcam). The membranes were incubated with secondary antibody anti-rabbit (1:20,000; Life Technologies) conjugated with horseradish peroxidase for 1 hour at room temperature. Chemoluminescence detection was performed with an ECL-PLUS Western blot detection reagent. ImageJ was used to quantify the results, and ADK expression levels were normalized to GAPDH expression that was used as internal standard.
Exposure to a carcinogen
Carcinogen exposure was initiated in juvenile male mice at an age of 3 weeks and an initial body weight between 8 and 13 g for WT and 10 and 14 g for Adk-tg mice. Our carcinogen exposure group consisted of Adk-tg (n = 45) and WT mice (n = 21) injected initially with diethylnitrosamine (DEN; Sigma-Aldrich) in saline (DEN; 50 mg/kg i.p.) twice a week for 4 weeks and subsequently with 25 mg/kg DEN and 100 mg/kg phenobarbital (PB; Sigma-Aldrich) twice a week for an additional 8 weeks; the total observation period was 12 weeks. Our control group consisted of additional Adk-tg (n = 10) and WT (n = 9) mice injected with saline instead of carcinogen during the 12-week period. All mice were checked for survival on a daily basis. Body weight was monitored twice a week.
Histopathological analysis
After conclusion of the experiments at 3 months, surviving animals were killed by 4% paraformaldehyde perfusion fixation. Livers were extracted and subjected to standard histopathology analysis by hematoxylin and eosin staining.
Statistical analysis
All data are presented as mean ± standard deviation. Statistical analysis and graphing of data were performed using GraphPad Prism software version 7 (GraphPad Software, Inc.). For the survival studies (Kaplan–Meier analysis), the log-rank (Mantel–Cox) test was used for evaluating the significance of the difference between groups. Body weights and Western blot data were compared by Student's t-test. Results were considered significant at values of p < 0.05.
Results
ADK expression in liver from patients with HCC and ICC
We investigated the expression of ADK in surgically retrieved liver cancer samples from a total of 11 patients with HCC and ICC. Most of these patients did not receive neoadjuvant therapy before surgery. We obtained samples from eight HCC patients (seven men and one woman) and three ICC patients (two men and one woman). The age at the time of surgery ranged from 53 to 81 years (Table 1). HCC samples were graded from stage I to III, whereas all ICC samples were from stage II cancers. From each surgery we retrieved core cancer tissue (C) and adjacent noncancerous (N) control tissue. Western blot analysis (Fig. 1) showed major changes in ADK expression levels in 7 of 11 patients as assessed by comparing cancer tissue (C) with adjacent noncancerous tissue (N). We observed not only a global overall reduction in ADK but also a shift in the relative expression levels of different isoforms of ADK. We noted that all HCC cases with reduced ADK expression developed recurrence of cancer development, which was not the case in patients with normal ADK levels.
Table 1.
Clinical Characteristics of Patients with Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma
| Cancer type | HCC patients | ICC patients | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| Gender | M | M | M | M | M | F | M | M | F | M | M |
| Tumor grade | I | I | I | I | I | I | I | I | II | II | II |
| Age at death | 65.8 | 83.3 | 81.5 | 51 | 54.2 | 56.9 | 64.4 | 61.6 | |||
| Survival days | 676 | 804 | 32 | 2228 | 2042 | 238 | 1001 | 2858 | 2974 | 498 | 1854 |
| Age at surgery | 64.1 | 81.2 | 81.4 | 67.4 | 45.4 | 53.7 | 54.3 | 56.6 | 56.3 | 60.5 | 70 |
| Age at diagnosis | 64.0 | 81.1 | 81.5 | 67.4 | 45.4 | 53.6 | 54.2 | 56.6 | 56.3 | 60.3 | 70.0 |
| Disease status at last follow-up | Evidence of HCC | No evidence of HCC | No evidence of HCC | No evidence of HCC | No evidence of HCC | Evidence of HCC | Evidence of HCC | Evidence of HCC | Evidence of ICC | No evidence of ICC | No evidence of ICC |
| Vital status at last follow-up | Deceased | Deceased | Deceased | Alive | Deceased | Deceased | Deceased | Deceased | Alive | Deceased | Alive |
| Pretreatment with chemo? | N | N | N | N | N | N | N | N | Y | N | N |
| Pretreatment with radiation? | N | N | N | N | N | N | N | N | N | N | N |
F, female; HCC, hepatocellular carcinoma; ICC, intrahepatic cholangiocarcinoma; M, male; N, no; Y, yes.
FIG. 1.

ADK expression in immunoblots from liver cancer patient samples. (A) Western blot analysis of ADK expression in surgically resected cancer tissue (C) and adjacent noncancerous (N) control tissue from eight HCC and three ICC patients. Western blot shows the bands corresponding to ADK-L, ADK-S, and GAPDH, which was used as loading control. (B) Bar graphs of normalized data showing the ADK-S/GAPDH and the ADK-L/GAPDH ratios. Please note the overall reduction in ADK expression in seven patients (asterisks) in their cancer tissue compared with control tissue. ADK, adenosine kinase; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; HCC, hepatocellular carcinoma; ICC, intrahepatic cholangiocarcinoma.
Mouse model to mimic ADK expression changes in cancer patients
To test the hypothesis that reduced expression of ADK renders the liver more susceptible to hepatotoxic lesions, we mimicked the reduced expression of ADK seen in several of our patients, by using a mouse model characterized by overall reduction of hepatic ADK and lack of the expression of the nuclear isoform ADK-L of the enzyme. ADK-S expression in the liver of Adk-tg mice was significantly lower (sevenfold) than in WT mice (p ≤ 0.01). Adk-tg mice were generated by introduction of a ubiquitously expressed Adk-S transgene (that encodes the cytoplasmic isoform of ADK6) into an Adk-knockout4 background. Western blot analysis of liver samples (Fig. 2) shows a striking similarity of ADK expression in the liver of an Adk-tg mouse with patient number 10.
FIG. 2.

Decreased ADK expression in Adk-tg mice. Western blot analysis of ADK expression in healthy livers from three adult WT and three age-matched Adk-tg mice. Left panel shows Western blot assay, showing the bands corresponding to ADK-L, ADK-S, and GAPDH, which was used as loading control. Right panel shows the histogram analysis of bands. Please note the expression of both isoforms, ADK-L and ADK-S, in the liver from WT mice, but complete lack of ADK-L and reduced ADK-S in liver from Adk-tg mice. Data are represented as mean and SEM; **p ≤ 0.01. SEM, standard error of the mean; WT, wild-type.
ADK deficiency increases the susceptibility to DEN-induced acute mortality
To investigate whether reduced expression of ADK in the liver would render the liver more susceptible to a carcinogen, we analyzed the response of Adk-tg mice to the carcinogen DEN using PB as a tumor promoter. This model is a well-established chemical carcinogenesis model.17–19 A Kaplan–Meier survival curve shows a significant genotype effect (p = 0.0005) between the survival of carcinogen-exposed Adk-tg and WT mice (Fig. 3). Fifty-one percent of all Adk-tg mice died between days 10 and 24 after initiation of the carcinogen exposure, whereas only 10% of WT mice died within the same time span. By day 80, 66% of Adk-tg mice had died compared with 22% in the WT mice. These results show that decreased expression of ADK in the liver renders the mice more susceptible to DEN-induced carcinogenic liver injury.
FIG. 3.
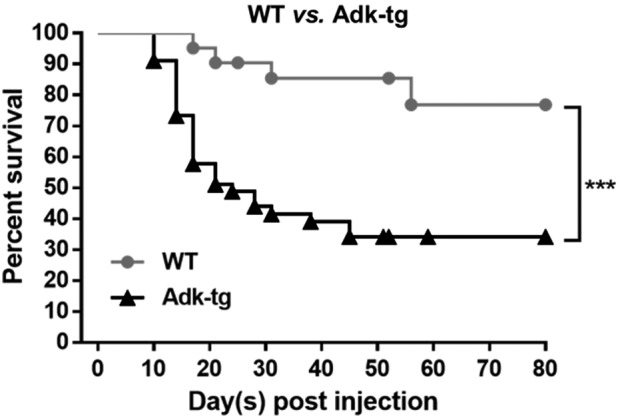
Carcinogen-exposed Adk-tg mice exhibit decreased survival compared with age-matched WT mice. A Kaplan–Meier survival curve for WT mice (DEN and DEN/PB; n = 21) and Adk-tg mice (DEN and DEN/PB; n = 45) shows significant reduction in the survival rate of carcinogen-treated Adk-tg (***p ≤ 0.001) compared with WT mice. Statistical analysis: log–rank (Mantel–Cox) test. DEN, diethylnitrosamine; PB, phenobarbital.
ADK deficiency does not affect the body weight of DEN/PB-exposed animals
Furthermore, we investigated the effect of the DEN/PB treatment on the body weight of WT and Adk-tg mice (Fig. 4). Whereas the DEN/PB administration caused a significant reduction in the body weight of both genotypes during the course of treatment, compared with control mice (p ≤ 0.0001), there was no significant genotype effect (p = 0.07), indicating that reduction in ADK expression does not affect the body weight in DEN/PB-treated animals.
FIG. 4.
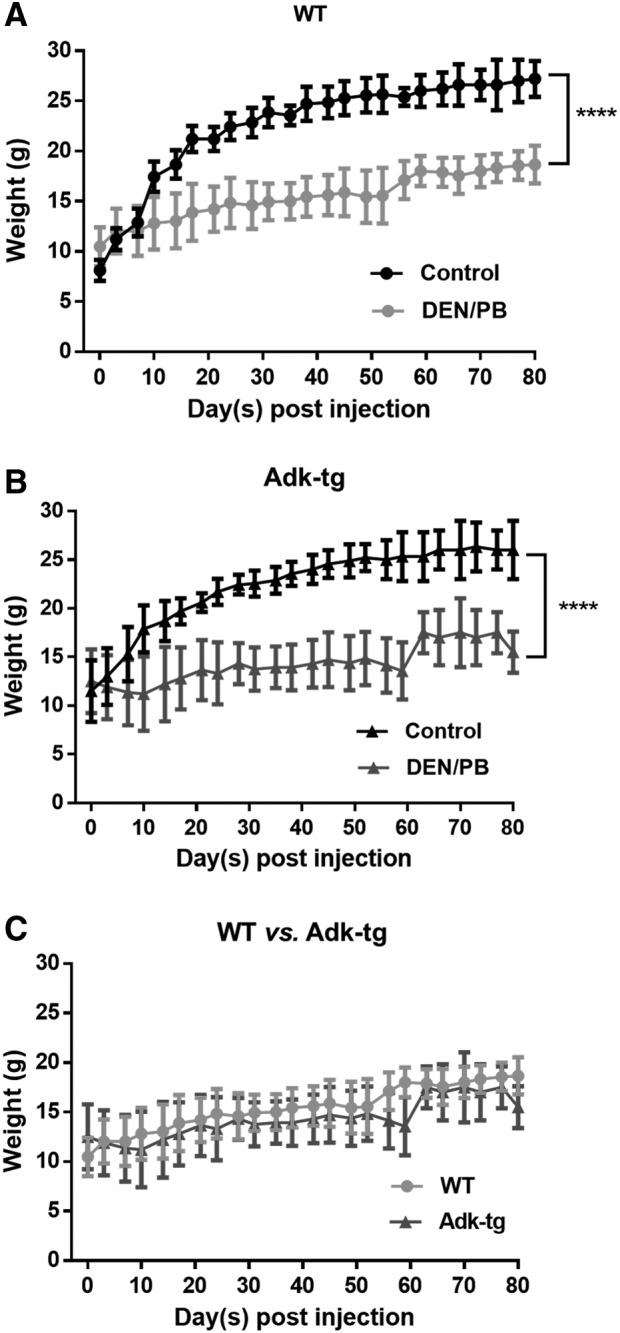
Body weight development of carcinogen-exposed Adk-tg and WT mice. Effect of DEN and DEN/PB treatment on body weight of WT mice (A), Adk-tg mice (B), and body weight comparison of DEN and DEN/PB-treated WT and Adk-tg mice (C). WT mice (control; n = 9, DEN/PB; n = 21) and Adk-tg mice (control; n = 10, DEN/PB; n = 45). Administration of the carcinogen caused a significant reduction in the body weight of both genotypes compared with control mice (****p ≤ 0.0001), but no significant difference between Adk-tg and WT mice. Data are represented as mean and SEM.
ADK deficiency does not affect liver histopathology in DEN/PB-exposed animals
Throughout the course of the experiment we collected livers from moribund animals and at predetermined time points during the course of the experiment ranging from 4 to 12 weeks after the initiation of carcinogen exposure (n = 2 to 4 livers per time point). In both genotypes, we observed gradual changes of liver histopathology comprising large and small cell dysplasia and accumulation of lymphocytes in intrahepatic bile ducts. Other pathological changes included hepatocytes with vacuolated nuclei, nuclear pleomorphism, multinucleation, giant cells, and hepatic cytolysis. Figure 5 gives a representative example of changes seen 12 weeks after the initiation of carcinogen exposure. We did not observe any marked differences between genotypes.
FIG. 5.

Liver histopathology of WT and Adk-tg mice 12 weeks after the initiation of carcinogen exposure. Representative image of liver histopathology at 12 weeks after initiation of carcinogen treatment (bottom) compared with control (top). Liver sections (5 μm) were stained with hematoxylin and eosin. Please note similar histological characteristics including hypertrophic cells and accumulation of lymphocytes in intrahepatic bile ducts in both genotypes (WT vs. Adk-tg). Top panel, scale bar = 200 μm; bottom panel, scale bar = 200 μm.
Discussion
In this study, reduced expression of ADK in the liver was associated with a subset of human liver cancers and with increased susceptibility to the acute toxic effects of a carcinogen in mice. Those findings suggest that ADK expression levels in the liver might play a role in determining the liver's susceptibility to cancer development. Several aspects of our work warrant further discussion.
Role of ADK for hepatic metabolism
The liver has the highest expression levels of ADK in the human body and it is the organ where 85% of all transmethylation reactions take place. This association is in line with a biochemical function of ADK as regulator of the transmethylation pathway.1,4,5,10 Therefore, ADK homeostasis in the liver has broad implications for liver function.
In 2011, a study by Bjursell et al.10 discovered that homozygous missense mutations in the Adk gene in six patients from three unrelated families caused a disruption of the transmethylation cycle leading to hypermethioninemia, abnormalities in hepatic metabolite levels, and hepatic encephalopathy. In 2016, an additional 11 patients with ADK deficiency from 8 families were identified, and associated with microvesicular hepatic steatosis, portal and periportal fibrosis, and chronic cholestasis.11 Microvesicular steatosis may indicate mitochondrial dysfunction and deficiencies in fatty acid oxidation.20,21 Recurrent hypoglycemia, global developmental delay, seizures, and cognitive impairment were other clinical conditions that ADK deficiency caused in those patients.11
Genetically engineered mice with a global deletion of ADK showed a pathological phenotype similar to patients with ADK deficiency. ADK knockout mice developed progressive microvesicular hepatic steatosis within 4 days after birth leading to premature death within 14 days. The animals showed retardation in body weight gain, a delay in the time to eye opening, intermittent periods of apnea, and abnormal temperature regulation. Biochemically, deletion of the Adk gene caused depletion of all three adenine nucleotides, which would affect mitochondrial function and therefore, promote cell degeneration.4 Of importance, and in line with the human cases of ADK deficiency, the lack of ADK in mice induced failure of the transmethylation pathway, an increase in the levels of adenosine, and microvesicular hepatic steatosis. In agreement with defects in the transmethylation pathway, ADK knockout mice were characterized by an increase of S-adenosylhomocysteine (SAH) in the liver. The increase in SAH inhibits SAM-dependent transmethylation reactions.4 Although adenosine levels were not quantified directly in ADK-deficient livers, the depletion of adenine nucleotides and the elevation of SAH are consistent with a rise in hepatic adenosine.
In this study, we have used the chemical carcinogen DEN in combination with PB as tumor promoter in mice, replicating one of the most widely used experimental models to study hepatocarcinogenesis.17–19,22,23 DEN is a potent hepatocarcinogenic nitrosamine present in many products, such as in tobacco smoke, cured and fried meats, cosmetics, and agricultural chemicals. DEN requires metabolic activation by cytochrome P450 (CYP450) in the liver.23–25 CYP450-dependent metabolism of DEN causes the formation of reactive oxygen species (ROS), including hydrogen peroxide,18,26 whereas PB promotes carcinogenesis by increasing the expression of P450.27,28
According to our results, we have seen a higher mortality rate in Adk-tg mice treated with DEN/PB compared with their age-matched WT mice. High concentrations of adenosine as a result of reduced ADK expression in Adk-tg mice would stimulate apoptosis by increasing ROS production and mitochondrial membrane dysfunction.29 Therefore, the high mortality rate in Adk-tg mice could be the result of a combinatorial effect of high levels of adenosine and P450-dependent DEN metabolism both promoting ROS formation and mitochondrial dysfunction.
Taken together, those findings demonstrate an important role of ADK for liver metabolism. Reduced ADK expression as seen in a subset of liver cancer samples would therefore render the liver more susceptible to the adverse effects of potential cancer-promoting triggers.
Consequences of AKD-S reduction
To understand the consequences of the reduced expression of ADK, it is important to consider the specific roles of two isoforms of the enzyme, ADK-S and ADK-L, expressed in the cytoplasm and nucleus of a cell, respectively. Both isoforms are derived from the same gene through alternative splicing and alternative promoter use, with the long-form ADK-L having a specific N-terminal nuclear localization signal.1,3
Adk-tg mice have an 88% reduction of cytoplasmic ADK-S in the liver compared with WT mice (Fig. 2). A robust reduction in ADK-S expression was likewise found in a subset of our cancer samples (Fig. 1). ADK-S regulates the intra- and extracellular levels of adenosine, implying that a reduced expression of ADK-S leads to an increase in adenosine. The levels of adenosine also depend on the expression and activity of the adenosine-producing enzyme CD73,30,31 which is expressed in tumor cells and lymphocytes and on the adenosine-removing enzyme adenosine deaminase (ADA). Therefore, extracellular levels of adenosine and hence adenosine receptor activation depend on the equilibrium of adenosine production (CD39 and CD73)30–32 and metabolism (ADK-S and ADA). A limitation of this study is the unknown state of other members of the purinome. Future work should consider a comprehensive analysis of all adenosine-regulating proteins including ADK, ADA, CD39, CD73, SAH hydrolase, and nucleoside transporters.
There are several studies demonstrating an accumulation of adenosine under hypoxic conditions, as occurs in the tumor microenvironment, which would favor tumor progression by helping cancer cells escape from immune surveillance.32,33–35 Accumulation of adenosine facilitates immunosuppression through activation of A2A receptors by suppressing an effective T cell-mediated response while promoting the activity of suppressive regulatory T cells (Tregs) and T-helper cell 17, and accelerating the conversion of antitumor type 1 macrophages into protumor type 2 macrophages.33
The activation of A2A and A2B receptors by adenosine promotes angiogenesis by enhancing angiogenic factor production by stimulating vascular endothelial growth factor gene expression in microvascular endothelial cells in response to the hypoxia-induced accumulation of adenosine.36 A3 receptor activation stimulates cell proliferation through activation of p44/p42 and p38 mitogen-activated protein kinases.37
In conclusion, increased expression of CD73 and/or reduced expression of ADK-S are expected to impair immune responses and to promote angiogenesis through increased activation of adenosine receptors.
Consequences of ADK-L reduction
Adk-tg mice are also characterized by a complete lack of ADK-L. ADK-L is an epigenetic regulator because it drives the flux of methyl groups through the DNA transmethylation pathway.4,5 A lack of ADK-L blocks methylation reactions through the accumulation of adenosine and resulting formation of SAH, which is a direct metabolic inhibitor of methylation enzymes. The block of methylation leads to global hypomethylation, which is considered to be one of the earliest epigenetic changes involved in the transformation from a normal to a precancerous cell.38,39
Because of the direct role of ADK-L in the regulation of the DNA methylome, and as a result of the contribution of DNA methylation changes for cancer development,5 we expect that ADK-L-deficient mice would have an altered propensity for cancer development, a possibility we could not address further because of the high mortality of DEN/PB-treated Adk-tg mice.
Conclusions and outlook
Our results demonstrated that low hepatic ADK expression in Adk-tg mice increase the susceptibility to the acute toxic effects of a carcinogen. Low hepatic ADK in a subset of human samples was associated with liver cancer and poor prognosis. Therefore, low ADK might be both a risk factor and biomarker for cancer development. Additional studies are needed to unravel the long-term consequences of engineered mutation in hepatic ADK for cancer development.
Acknowledgments
The authors acknowledge the Legacy Tumor Bank for providing frozen tissue samples from liver cancer patients. This work was supported by the Legacy Health Foundation and the Wayne D. and Joan E. Kuni Foundation.
Author Disclosure Statement
Dr. Boison is a co-founder of PrevEp LLC and acts as consultant for F. Hoffmann-La Roche AG.
References
- 1. Boison D. Adenosine kinase: Exploitation for therapeutic gain. Pharmacol Rev. 2013;65:906–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jaoek Park RSG. Adenosine metabolism, adenosine kinase, and evolution. In: Adenosine: A Key Link Between Metabolism and Brain Activity. Susan Masino D.B. (Ed). New York: Springer-Verlag; 2013 [Google Scholar]
- 3. Cui XA, Singh B, Park J, et al. . Subcellular localization of adenosine kinase in mammalian cells: The long isoform of AdK is localized in the nucleus. Biochem Biophys Res Commun. 2009;388:46–50 [DOI] [PubMed] [Google Scholar]
- 4. Boison D, Scheurer L, Zumsteg V, et al. . Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci U S A. 2002;99:6985–6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Williams-Karnesky RL, Sandau US, Lusardi TA, et al. . Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest. 2013;123:3552–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fedele DE, Gouder N, Guttinger M, et al. . Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain. 2005;128:2383–2395 [DOI] [PubMed] [Google Scholar]
- 7. Andres CM, Fox IH. Purification and properties of human placental adenosine kinase. J Biol Chem. 1979;254:11388–11393 [PubMed] [Google Scholar]
- 8. Sahin B, Kansy JW, Nairn AC, et al. . Molecular characterization of recombinant mouse adenosine kinase and evaluation as a target for protein phosphorylation. Eur J Biochem. 2004;271:3547–3555 [DOI] [PubMed] [Google Scholar]
- 9. Sakowicz M, Grden M, Pawelczyk T. Expression level of adenosine kinase in rat tissues. Lack of phosphate effect on the enzyme activity. Acta Biochim Pol. 2001;48:745–754 [PubMed] [Google Scholar]
- 10. Bjursell MK, Blom HJ, Cayuela JA, et al. . Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function. Am J Human Genet. 2011;89:507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Staufner C, Lindner M, Dionisi-Vici C, et al. . Adenosine kinase deficiency: Expanding the clinical spectrum and evaluating therapeutic options. J Inherited Metabol Dis. 2016;39:273–283 [DOI] [PubMed] [Google Scholar]
- 12. Sun X, Han L, Seth P, et al. . Disordered purinergic signaling and abnormal cellular metabolism are associated with development of liver cancer in Cd39/ENTPD1 null mice. Hepatology 2013;57:205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu SC, Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92:1515–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mato JM, Martinez-Chantar ML, Lu SC. S-adenosylmethionine metabolism and liver disease. Ann Hepatol. 2013;12:183–189 [PMC free article] [PubMed] [Google Scholar]
- 15. Li T, Ren G, Lusardi T, et al. . Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest. 2008;118:571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pignataro G, Maysami S, Studer FE, et al. . Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. J Cerebral Blood Flow Metabol. 2008;28:17–23 [DOI] [PubMed] [Google Scholar]
- 17. Aydinlik H, Nguyen TD, Moennikes O, et al. . Selective pressure during tumor promotion by phenobarbital leads to clonal outgrowth of β-catenin-mutated mouse liver tumors. Oncogene. 2001;20:7812. [DOI] [PubMed] [Google Scholar]
- 18. Brown ZJ, Heinrich B, Greten TF. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat Rev Gastroenterol Hepatol. 2018;15:536–554 [DOI] [PubMed] [Google Scholar]
- 19. He G, Karin M. NF-kappaB and STAT3—key players in liver inflammation and cancer. Cell Res. 2011;21:159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tandra S, Yeh MM, Brunt EM, et al. . Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J Hepatol. 2011;55:654–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Urasaki Y, Zhang C, Cheng J-X et al. Quantitative assessment of liver steatosis and affected pathways with molecular imaging and proteomic profiling. Sci Rep. 2018;8:3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aleksic K, Lackner C, Geigl JB, et al. . Evolution of genomic instability in diethylnitrosamine-induced hepatocarcinogenesis in mice. Hepatology 2011;53:895–904 [DOI] [PubMed] [Google Scholar]
- 23. El-Kharrag R, Amin A, Hisaindee S, et al. . Development of a therapeutic model of precancerous liver using crocin-coated magnetite nanoparticles. Int J Oncol. 2017;50:212–222 [DOI] [PubMed] [Google Scholar]
- 24. Shirakami Y, Gottesman ME, Blaner WS. Diethylnitrosamine-induced hepatocarcinogenesis is suppressed in lecithin:retinol acyltransferase-deficient mice primarily through retinoid actions immediately after carcinogen administration. Carcinogenesis. 2012;33:268–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tolba R, Kraus T, Liedtke C, et al. . Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab Anim. 2015;49:59–69 [DOI] [PubMed] [Google Scholar]
- 26. Sakurai T, He G, Matsuzawa A, et al. . Hepatocyte necrosis induced by oxidative stress and IL-1α release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Czekaj P. Phenobarbital-induced expression of cytochrome P450 genes. Acta Biochim Pol. 2000;47:1093–1105 [PubMed] [Google Scholar]
- 28. Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol. 2009;90:367–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ma Y, Zhang J, Zhang Q, et al. . Adenosine induces apoptosis in human liver cancer cells through ROS production and mitochondrial dysfunction. Biochem Biophys Res Commun. 2014;448:8–14 [DOI] [PubMed] [Google Scholar]
- 30. Antonioli L, Pacher P, Vizi ES, et al. . CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunotherapy Cancer. 2018;6:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vijayan D, Young A, Teng MWL, et al. . Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17:709. [DOI] [PubMed] [Google Scholar]
- 33. Jiang T, Xu X, Qiao M, et al. . Comprehensive evaluation of NT5E/CD73 expression and its prognostic significance in distinct types of cancers. BMC Cancer. 2018;18:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Antonioli L, Blandizzi C, Pacher P, et al. . Immunity, inflammation and cancer: A leading role for adenosine. Nat Rev Cancer. 2013;13:842–857 [DOI] [PubMed] [Google Scholar]
- 35. Antonioli L, Novitskiy SV, Sachsenmeier KF, et al. . Switching off CD73: A way to boost the activity of conventional and targeted antineoplastic therapies. Drug Discov Today. 2017;22:1686–1696 [DOI] [PubMed] [Google Scholar]
- 36. Du X, Ou X, Song T, et al. . Adenosine A(2B) receptor stimulates angiogenesis by inducing VEGF and eNOS in human microvascular endothelial cells. Exp Biol Med. 2015;240:1472–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Merighi S, Benini A, Mirandola P, et al. . A(3) adenosine receptors modulate hypoxia-inducible factor-1α expression in human A375 melanoma cells. Neoplasia. 2005;7:894–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guerrero-Preston R, Santella RM, Blanco A, et al. . Global DNA hypomethylation in liver cancer cases and controls: A phase I preclinical biomarker development study. Epigenetics. 2007;2:223–226 [DOI] [PubMed] [Google Scholar]
- 39. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239–259 [DOI] [PMC free article] [PubMed] [Google Scholar]