

Abstract
Introduction:
Although cure rates for acute leukemia have steadily improved over the past decades, leukemia remains a deadly disease. Enhanced risk stratification and new therapies are needed to improve outcome. Extensive genetic analyses have identified many mutations that contribute to the development of leukemia. However, most mutations occur infrequently and most gene alterations have been difficult to target. Most patients have more than one driver mutation in combination with secondary mutations, that result in a leukemic transformation via the alteration of proteins. The proteomics of acute leukemia could more directly identify proteins to facilitate risk stratification, predict chemoresistance and aid selection of therapy.
Areas covered:
This review discusses aberrantly expressed proteins identified by mass spectrometry and reverse phase protein arrays and their relationship to survival. In addition, we will discuss proteins in the context of functionally related protein groups.
Expert opinion/commentary:
Proteomics is a powerful tool to analyze protein abundance and functional alterations simultaneously for large numbers of patients. In the forthcoming years, validation of tools to quickly assess protein levels to enable routine rapid profiling of proteins with differential abundance and functional activation may be used as adjuncts to aid in therapy selection and to provide additional prognostic insights.
Keywords: Leukemia, Reverse Phase Protein Array, RPPA, Mass Spectrometry, MS, risk stratification, Drug target selection
Introduction
1.1. Acute leukemia
Acute leukemias are malignant diseases that involve unchecked clonal proliferation of cells having (or re-acquiring) the unlimited differentiation properties of primitive hematopoietic stem cell and that occur at all ages [1]. Similar to the chronic leukemias such as chronic lymphocytic leukemia (CLL), which is the most common form of leukemia in adults and accounts for 35% of the new diagnoses in adults, acute leukemia predominantly appears later in life, with a median age of 68 in the United States. In children acute leukemia is the most common form of cancer, comprising around 30% of pediatric malignancies, and occurs most commonly in toddlers, with another peak in the teenage years [1]. Although the survival rate for children with acute lymphoblastic leukemia (ALL) is excellent, with a 5-year overall survival (OS) rate over 90% [2], survival for ALL in adults is around 45% [3]. Outcomes for AML are worse with a 25% OS for adult patients below the age of 60 years [4], but only 10% for those above the age of 60 years [5], and survival for children with AML remains at 62% for intermediate-risk patients despite intensive therapy and stem cell transplant (SCT) [6]. Curability after relapse is dismal for most, ranging from <10% in adults and children with AML to 57% in late-relapse ALL in children [7].
Risk stratification has traditionally been based on clinical features (age, performance status, prior myelodysplasia in adults) complemented by cytogenetic abnormalities, and more recently by incorporation of prognostic information related to specific gene mutations according to the World Health Organization (WHO) classification system [8]. The transformation of normal cells to leukemia involves the mutations of many genes, some of which are driver mutations, while others contribute to the leukemia phenotype. Incorporating genetic mutation analysis into risk stratification for both AML and ALL have yielded significant prognostic information and enabled stratification of patients into favorable, intermediate and unfavorable risk groups. However, for the 50% of the adult AML patients with diploid cytogenetics this classification is unspecific and many subgroups based on mutations (NPM1, FLT3, etc.) are now recognized [9]. Moreover, sequencing studies in AML have identified a large number of driver and secondary mutations co-occurring in many possible combinations. Since each patient has the potential to be genetically exclusive it may be difficult to derive accurate prognostic information for the individual patient [10].
The combined consequences of the genetic and epigenetic events, along with influences from the microenvironment, impact the leukemia cell predominantly through proteins by altering protein expression levels and activation states. Therefore, the application of proteomics could potentially measure and assess the summary effects of all mutational events, which could in turn enhance risk stratification and prognostication. In contrast to the very large number of genetic mutation permutations, the far fewer number protein activation state permutations would involve many fewer network nodes, enhancing risk stratification and drug selection. Proteomics may also be predictive of response to targeted drug therapies, aiding in therapy selection by predicting chemoresistance and sensitivity by revealing pathway dependencies. While gene expression profiling (GEP), has the ability to comprehensively define the level of mRNA expression, it cannot serve as a surrogate for protein measurement as transcription has only limited correlation with translation (~45% of mRNA correlate with total protein levels) [11,12]. A given mRNA level can be translated to produce more than one protein due to alternative slicing. Also, since genomic techniques cannot assess post translational modifications (PTM), protein activation states cannot be measured. Proteomic methods can infer protein activation states by assessing proteins using antibodies specific for protein PTMs or in mass spectrometry (MS) through recognition of the mass changes associated with the PTM (e.g. addition of a phosphate group to a tyrosine, serine or threonine, or a sugar molecule etc.). Therefore, the assessment of protein abundance and PTM activation states has the potential to provide information representing the summary of genetic, epigenetic, and environmental actions on the cell. By summarizing the effects of genetic and epigenetic information, proteomics could provide both prognostic information and therapeutic guidance.
2. Proteomic technologies as tool to identify prognostic markers
Over the years many methods have been employed that look at individual proteins (i.e. western blotting, enzyme-linked immunosorbent assays). Proteomics, the study of multiple proteins simultaneously, allows a broader picture of the protein composition. Here, we discuss the two most frequent high-throughput methodologies currently used in leukemia research; MS and the Reverse Phase Protein Array (RPPA). Immunohistochemical analysis of tumor microarrays, commonly used in solid tumor research, are discussed elsewhere [13,14].
2.1. Mass spectrometry
Traditionally, MS-based approaches are used to identify and quantify proteins from highly complex biological samples. MS starts with the ionization of samples which breaks down the sample into charged fragments (ions), that can then be sorted based on their mass-to-charge ratio by accelerating and subjecting them to an electric or magnetic field. It is often used in tandem with liquid chromatography (LC-MS or LC/MS), which combines the physical separation of liquid chromatography together with the mass separation of the mass spectrometer. To enable reproducible, sensitive and selective protein quantitation, the targeted MS approach “selected reaction monitoring” (SRM) was recently implemented in proteomics. This method is able to distinguishing highly similar proteoforms such as isoforms and PTM proteins such as phosphorylation and ubiquination as well as genetic variants that result in amino acid changes such as single nucleotide polymorphism (SNPs) [15]. SRM first filters peptides with a particular specific mass-to-charge ratio (precursor ion) and secondly activates (fragmentates) those ions to generate compound-specific product ions. Those specific products are then filtered and detected. Because this method only analyzes samples with a particular mass-to-charge, it can detect low abundance proteoforms, which are below the limit of detection of standard MS analyses. This decreases the global benefit of MS but increases the ability to detect MS-based phosphorylation and other PTM necessary to study many signal transduction pathways.
Using different MS-based methods, many laboratories have identified proteins that stratify patients based on their prognosis. Kaźmierczak et al. performed proteomic analysis in two subtypes of AML (M1 and M2) from 33 newly diagnosed patients and 17 healthy volunteers and found that, within the AML patients four proteins significantly correlated with treatment results; annexin I, glutathione transferase omega, esterase D and gamma 1 actin [16]. This was irrespective of the traditional clinical characteristics. From those four proteins, esterase D and gamma 1 actin might allow prediction of results of induction therapy: high levels of esterase D were associated with a higher complete remission rate, whereas higher gamma 1 actin levels were related with resistance. Another MS-based proteomic study by Nicolas et al. analyzed 54 newly diagnosed AML patients and identified two protein signatures that had significant variation in OS (p=0.001) and disease-free survival (p=0.0004) [17]. They identified S100A8 as the highest discriminative prognostic marker, associated with poor survival. Bai et al. performed protein profiling in serum samples from different AML disease groups (72 newly diagnosed AML patient samples, 36 samples from AML patients that had attained CR, and 30 serum samples from patients that were refractory and/or had relapsed AML) and from 72 healthy controls [18]. They observed significantly inferior outcome associated with increased ubiquitin-like modifier activating enzyme 1 (UBA1) expression, as well as a superior overall survival (OS) with increased fibrinogen alpha chain precursor isoform 1 (FIBA) and platelet factor 4 (PLF4).
Another study compared protein profiles in adriamycin-resistant (HL-60/ADR) and non-resistant HL-60 cell lines and identified 13 up-regulated and 3 down-regulated proteins in HL-60/ADR [19]. Verification of nucleophosmin/B23 (NPM B23) and nucleolin C23 (C23) (which demonstrated the highest fold change increase of +3.49 fold and +2.42 fold respectively between resistant and non-resistant HL-60 cell line) by western blot in 12 cell lines (3 resistant and 9 non-resistant) showed strong upregulation in the resistant cell lines. Further validation in 80 primary samples of patients with newly diagnosed acute leukemia (58 with AML and 22 with ALL) showed higher expression compared to healthy controls in 43% of the de novo AML patients, versus 69% of the refractory patients and 77% of the relapsed patients. In addition, patients that were both NPM B23 and C23 positive had higher relapse rates, suggesting that NPM B23 and C23 may play a role in drug resistance. Similarly, Jiang et al. studied protein levels in prednisolone sensitive and resistant ALL leukemic cell lines using MS [20]. Between pre and post treatment samples, they identified 17 protein that were differentially expressed in prednisolone resistant ALL cell lines and 77 proteins with a different expression in prednisolone sensitive ALL cell lines. Ten percent of proteins altered in prednisolone sensitive cells were involved in cell cycle regulation and all were down regulated by prednisolone treatment. This suggests that proteins that involved in cell cycle regulation may act as a distinct signature for prednisolone response. Next, they validated proliferating cell nuclear antigen (PCNA), as one of the proteins that down-regulated after treatment in sensitive cell lines and remaining stable in the prednisolone resistant cell lines. Jiang et. Al examined 43 paired pediatric B-ALL bone marrow patient samples taken at day 0 (pretreatment) and at day to determine that PCNA expression was predictive of prednisolone response. Negoro et al. searched for cytarabine resistant factors using 5 cytarabine-resistant leukemic cell lines and their non-resistant parental counterparts and found reduced stathmin 1 protein in the cytarabine resistant cell lines [21].
A study by the group of Serve et.al, performed a global proteome analysis and revealed predictive phosphorylation markers for the treatment of AML with quizartinib [22]. They collected diagnostic bone marrow samples from 21 FLT3-ITD+ patients that participated in a single arm quizartinib phase II clinical trial. Patients were divided into training and test sets and all patients with CR or PR were considered as responders (12/21 = 57%). Super stable isotope labeling with amino acids in cell culture (SILAC) in combination with quantitative MS was used to monitor the phosphor-proteomes. Direct comparison on the proteomes from the responders vs. non-responders identified three significantly different sites; regulation site S160 on endonuclease/exonuclease/phosphatase family domain-containing protein 1 (EEPD1), S630 on B-cell lymphoma/leukemia 11A (BCL11A) and S333 on Ran-binding protein 3 (RANBP3). Secondly, using another supervised learning approach, they were able to identify a phosphor-signature consisting of five phosphorylation sites that strongly separates the responders from the non-responders. Three of the five phosphorylation sites (EEPD1-S160, BCL11A-S630, RANBP3-S333) were already identified with their first approach, the other two sites were S961 on retinitis pigmentosa GTPase regulator (RP3) and S458 on the lamins A/C (LMN1). When they applied their model to the test set (n = 9), they found a predictive accuracy of 78%.
Another study by Alcolea et al. also looked at the phosphorylation profiles in AML. In this study, LC-MS/MS was performed to profile protein phosphorylation in AML cell lines with different sensitivities to kinase inhibitors (MEK, JAK1, PI3K and PKC inhibitors) [23]. They identified several phosphorylation sites that were inhibited by the kinase inhibitors but observed that the intensities of inhibition did not perfectly correlate with sensitivity or resistance. However, many phosphorylation sites that did correlate with sensitivity or resistance to the drugs were not in general inhibited by the drugs. From this, they concluded that protein markers of pathway activity may not always be the best indicators of sensitivity to drugs that target these pathways, because the activity of other parallel kinases can contribute to resistance of the leukemic cell. Brown and colleagues [24] performed functional proteomics with high accuracy MS techniques of patient AML samples collected at time of diagnosis from patients with primary chemotherapy resistance and failure of induction therapy. They identified the Myocyte Enhancer Factor 2C (MEF2C) phosphorylation site S222 as a specific marker of primary chemoresistance in patients with both cytogenetically normal and chromosomally rearranged AML.
2.2. Reverse Phase Protein Array (RPPA)
The RPPA methodology is a contemporary approach that uses validated monoclonal antibodies to measure the relative protein expression in samples. The name “reverse phase” indicates that the antigens (protein lysates) are printed on nitrocellulose slides and subsequently probed with primary and secondary antibodies that target specific proteins in the solid phase lysates. This is the “reverse” of traditional immunoassays in which specific antibodies that are immobilized on the solid phase of the array to capture the antigen of interest. RPPA is a cost-effective technique as it can assess thousands of protein lysate samples on a single slide and requires only a minimal amount of protein samples (approx. 3×105 cells for 400 proteins).
In contrast to MS, RPPA cannot be used as a de novo discovery platform as it is biased to the protein targets of interest. MS can in theory be used to analyze all cell proteins, although in practice methods of protein depletion are often performed to remove highly abundant proteins, such as albumin or immunoglobulins, to facilitate the detection and recognition of low abundance proteins. RPPA antibodies must be validated and shown to have a single, specific high-affinity band by immunoblot.
The advantage of RPPA is the ability to simultaneously measure the expression levels of thousands of samples. Because of its high throughput capacity, RPPA are highly applicable to correlate protein expression in multiple patients with varying clinical characteristics, cytogenetic genotypes, and disease outcome. Accordi et al. used the RPPA technology to profile 92 proteins in a cohort of 118 newly diagnosed pediatric B-cell precursor ALL patients [25]. Their goal was to map pathway activation changes in LCK kinase in patients that responded poorly to prednisolone compared to those with a good response. In addition, they found that Cyclin E1 was an adverse prognostic factor for relapse free survival. Milani et al. [26] performed a RPPA phosphoproteomic screen on 98 pediatric T-cell ALL samples and identified a large variation in protein kinase C alpha (PKCα) phosphorylation at serine 657. Real-time quantitative polymerase chain reaction verified down-regulation of PRKα in a cohort of 173 patients as a prognostic marker for increased cumulative risk of relapse. Another study reported that B-cell lymphoma (BCL)-2-Associated Athanogene 1 (BAG1) was over expressed in 87 of the 99 analyzed adult AML patients compared to healthy controls and that patients with very high BAG1 levels had a decreased 5-year event-free survival [27].
Our group has previously performed RPPA on 511 AML patients and, when compared to normal CD34+ cells, identified Forkhead O Transcription Factor 3A (FOXO3A), Bromodomain Containing 4 (BRD4), Absent, Small, Or Homeotic Discs-like Protein (ASH2L), Tripartite Motif Containing 62 (TRIM62) and Friend Leukemia Virus Integration 1 (FLI1) as prognostic factors that were significantly associated with OS. FOXO3A was found to be expressed within the range of the healthy control samples but was higher at relapse compared to diagnosis. Also, higher FOXO3A phosphorylation, or a higher ratio between phosphorylated and total FOXO3A, were associated with primary chemoresistance and a shorter relapse free survival and OS [28]. Higher BRD4 levels were found to predict shorter OS (ASH 2017 #3794), lower ASH2L was associated with increased OS [29] and loss of TRIM62 related to a shorter CR duration and OS [30]. For FLI1, both increased and decreased expression was associated with a shorter remission duration and OS [31]. Ruvolo et al. found that phosphorylation of Glycogen Synthase Kinase 3 α/β (GSKα/β) correlated with activation of AKT and that GSK activation was prognostic for poor OS [32]. They also found that the protein phosphatase 2A (PP2A) regulatory subunit B55α was lower expressed in AML compared to normal CD34+ cells, and that low levels of B55α were associated with a shorter CR duration [33]. Within adult ALL, cathepsin G protein expression measured by RPPA was also a poor prognostic marker [34].
3. Analyzing proteins using a more network-based approach
Although the majority of the proteomic studies in acute leukemia focus on single proteins as prognostic biomarkers, our group has shifted towards analyzing proteins in the context of functionally related protein groups. This change is based on the assumption that the effects of protein alterations on the cell function are determined by the net consequence of the combined influences of all proteins in a cell, rather than changes in a single protein. As an example, one array of 256 newly diagnosed AML patients assessed expression of 51 total and phospho-proteins. Proteins were clustered based on the absolute value of the Pearson correlation and this suggested the existence of ten protein constellations; proteins that were strongly correlated (negative or positive) with each other and most proteins had a related function supporting the idea that the approach had functional validity. Principal component analysis was then performed on each constellation, retaining the first principal component. This computed a score for each patient and constellation, and from there seven protein signatures were identified based on the extent to which a constellation was present or not in the protein expression pattern of a patient. Protein signatures were correlated with treatment outcome and was irrespective of cytogenetics [35]. Recently, we employed an even newer method, that used a segmented approach, by first analyzing proteins in relation to functionally related proteins, before taking them all together [36]. The rationale for this switch away from the traditional unsupervised hierarchical clustering, which simultaneously analyzes all proteins at once, was that unsupervised clustering ignores known relationships between protein pathways. We hypothesized that if we created subsets of proteins with known functional relationships, we would obtain better, more robust model of protein interactions [31]. In this second AML RPPA, we analyzed 205 adult AML patients for 231 proteins, which were first divided into 31 “Protein Functional Groups” (e.g. cell cycle, apoptosis, differentiation). Within each protein functional group, we utilized mathematical algorithms to divide patients into distinct “Protein Clusters.” A protein cluster was defined as a subset of cases with similar (correlated) expression of core protein functional group components. Correlations between proteins in each cluster could be positively or negatively and some of the individual protein clusters within a functional group were significantly associated with outcome. As an example, we analyzed the “Histone modification” protein functional group that was formed by 20 histone modification proteins in which we observed that five different protein clusters associated with varied expression or activation states relative to normal CD34+ cells existed [37]. Among these clusters, the “on”-state (i.e. expression above that of the normal CD34+ cells) of the histone modifier proteins was associated with a poor outcome compared to AML cells like normal CD34+ and “off” states. Also, we found that FLT3 mutated patients were overrepresented in the “on”-state. Another example showed the nine proteins related to TP53 and identified seven protein clusters which were prognostic for OS and CR duration [38]. Interestingly, we found tight correlation between TP53 mutation status and P53 pathway cluster membership.
Next, when we organized all protein clusters (n=154) from the 31 protein functional groups in a binary matrix, to search for higher order structures, we recognized using the Block Clustering methodology that recurrences between protein clusters did exist [39]. Those correlations were defined as “Protein Constellations”; a group of protein clusters from various protein functional groups that were strongly correlated with each other. Based on constellation membership for each patient, we could identify 13 “Protein Signatures”. A protein signature was defined as a group of patients with similar patterns of protein constellations. The schematic work flow of the method is shown in Figure 1. Protein signatures were significantly correlated with OS and CR duration. Although developed for a large adult AML dataset, this methodology also worked when applied to two pediatric datasets from both AML and acute lymphoblastic leukemia (ALL) patients samples [40,41], as well as to data from patient samples from adult ALL (manuscript in preparation) and the myelodysplastic syndrome (MDS) (manuscript in preparation). In each disease we found similar structures within the datasets with clusters of correlated proteins and the formation of protein constellations and signatures. Similar to the signatures that were found in the adult AML data, some of the pediatric AML protein signatures were associated with relatively favorable and unfavorable outcome. Due to the high overall survival (86%) in the pediatric ALL cohort, no associations between signatures and outcome were found for pediatric ALL. Figures 2 clearly show the patterning found within the separate diseases, including adult AML, pediatric ALL, pediatric AML and combined analysis of pediatric leukemias. Analysis of the combined pediatric AML and ALL patients showed that AML samples (blue, annotation bar on Figure 2D) could be clearly separated from T-ALL (pink, annotation bar on Figure 2D) and B-ALL (yellow, annotation bar on Figure 2D) patient samples based-on proteomics alone, and that only a few patients showed an overlapping protein profile between AML and ALL. Three constellations (C5, C6, and C11) were present in both diseases, suggesting that some shared deregulations exist. Another study from our group, studied proteomics in mesenchymal stromal cells (MSC) from AML patients and healthy control using RPPA [42]. This study found 28 proteins differentially expressed between both cohorts, and clustering based on those proteins revealed three protein constellations dividing samples into four protein signatures. Again, similar to the classification within the AML and ALL patients, signatures were significantly associated with survival. This suggests that there is a two-way crosstalk between the AML blasts and the MSC that affects the protein expression of each and raises the possibility of a new approach of treating AML through manipulation of the MSC. Furthermore, we found variation between MSC protein expression at initial diagnosis, compared to the expression pattern seen at the time of salvage (i.e. relapse/refractory) towards an osteoblastic character, suggesting that this inter-relationship is dynamic over the course of the disease.
Figure 1. Computational workflow.

The foundation for modeling the protein expression was the establishment of 31 “Protein Functional Groups” (PFG); groups of protein pathways known from the existing literature. (A) Samples were collected from bone marrow or peripheral blood and protein lysates prepared from the blast population. (B) Protein clusters were identified within each PFG using the “Progeny clustering” algorithm [55]. Principal component analysis was used to determine if a protein cluster was similar or different from the normal CD34+ samples. Protein clusters were associated with therapy response and patient demographics. (C) Protein clusters from all 31 PFG were then assembled into a large binary dataset; 1 (blue) if a member of a protein cluster, 0 (yellow) if not. Block clustering analysis was used to search for correlation between protein clusters. Protein clusters from various PFG that were strongly correlated with each other were defined as “Protein Constellation” (horizontally). Patients that expressed similar combinations of protein constellations were grouped into patient signatures (vertically). Protein signatures were correlated with outcome and disease and patient characteristics. Within each protein signature and constellation, lists of proteins that were significantly up or down regulated compared to the normal CD34+ samples were identified.
Figure 2.

Functional patterning in (A) 205 adult AML patients, (B) 73 pediatric ALL, (C) 95 pediatric AML patients, and the (D) combination of the 95 AML and 73 ALL patients. Each horizontal line represents a single protein cluster. Each vertical line represents an individual patient. Blue points indicate protein cluster membership of a patient. For each disease group, block clustering identified strong correlation between protein clusters and identified 10–12 “Protein Constellations” (denoted by the colored annotation bar on the left of the figures). Patients that expressed similar patterns of constellation membership were defined as protein signature (denoted by the colored annotation bar on the top of the figures).
4. Using proteomics to guide in selecting the right therapy
Ideally, therapies could directly target only the mutated form of a protein to specifically reverse the biological consequences of a leukemia-specific genetic mutation. However, to date such specificity has only been demonstrated for mutations in IDH1 and IDH2. Other novel targeted therapeutics have been developed that act on both wildtype and mutated forms of a gene such as the FLT3 inhibitors. So far, it has not been possible to directly target many of the genes commonly mutated in AML such as NPM1, DNMT3A, ASXL1, and TET2. Many new targeted therapies are in development that instead target affected proteins downstream of the actions of these so-far undruggable mutations.
Proteomic profiling provides a means to potentially match the targeted therapies to the appropriate patient, via the identification of proteins that could facilitate the rational and effective combination of targeted therapy, perhaps in combination with existing chemotherapy, to improve outcome. For instance, venetoclax, a small molecule inhibitor of the BCL2 protein, approved for use in CLL, has shown promising effects in relapsed and refractory adult AML patients [43]. The BCL2 protein is overexpressed in most hematological diseases and has been implicated in the maintenance and survival of AML cells [44,45]. It is associated with therapeutic resistance and poor overall survival in adult AML BCL2 is an anti-apoptotic protein that acts in the intrinsic pathway, or the mitochondrial pathway. This pathway triggers apoptosis via the mitochondria outer membrane permeabilization (MOMP), which allows proteins located in the intermembrane space of the mitochondria to be released into the cytosol. As BCL2 has an anti-apoptotic function, it prevents MOMP. Venetoclax binds and neutralizes BCL2 with a sub nanomolar affinity to induce cell death. Therefore, if we can identify patients with high BCL2 expression at time of diagnosis, or with high levels of other anti-apoptotic proteins BCL-xL and MCL1, which suggest resistance to venetoclax, we can potentially rationally select patients that might benefit from combinational therapy including venetoclax. Also, bromodomain (BRD) and bromodomain and ExtraTerminal (BET) inhibitors that target the BET protein family are being tested in clinical trials. The BET protein family is defined by the presence of two acetyl-histone reading bromodomains and an ET domain and includes BRD2, BRD3, BRD4, and BRDT. BET inhibitors bind to the bromodomain of the BET proteins, preventing the protein-protein interactions between the BET proteins and acetylated histones and transcription factors. If they appear to be safe and effective, a potential application of proteomics could be to identify patients with high levels of BRD4 that might benefit from combinational therapy with BET inhibitors (ASH 2017 #3794). Mahmud et al. suggested the potential use of the inhibitor erlotinib in a subset of adult AML patients that expressed high epidermal growth factor receptor (EGFR) and EGFR phosphorylation levels relative to normal CD34+ cells measured by RPPA [47], and the study by Quintas-Cardama et al. showed increased sensitivity to nutlin-3 (MDM2 inhibitor) in patients that had MDM2 overexpression and a reduction of the wildtype protein TP53 [38]. Although MDM2 inhibitors are currently only been tested in phase I clinical trials as combinational therapy [48], this study shows the potential clinical application of proteomics in selecting patients most likely to respond to the addition of MDM2 inhibition [36].
Besides risk stratification, this approach could potentially aid in selecting rational combinatorial therapy. Within each constellation and signature, we identified lists of proteins that were aberrantly expressed compared to the normal CD34+ cells (i.e. statistically significantly higher or lower expressed) (Figure 3). While most protein expressions are in agreement with previous literature, some proteins, such as BCL2, are increased in many patients with poor prognosis, but are down regulated in patients with a good prognosis, such as those in Signature #2 (Figure 3). Moreover, we could identify proteins that were universally changed in the same direction in every signature, as well as proteins that were universally changed in both AML and ALL, or in both pediatric and adult AML, including GATA1, NR4A1, TCF4 and STAT1.pY701 (all universally low). Selecting proteins that were universally changed would therefore, if validated, be superior therapeutic leads, as they would be applicable to both AML and ALL and could be used across age groups and acute leukemia subgroups. Proteins significantly decreased in AML could function as targets for restoration, whereas upregulated proteins could be inhibited. These targeted agents could be combined with standard therapy. Formal validation of this hypothesis requires future experiments.
Figure 3.
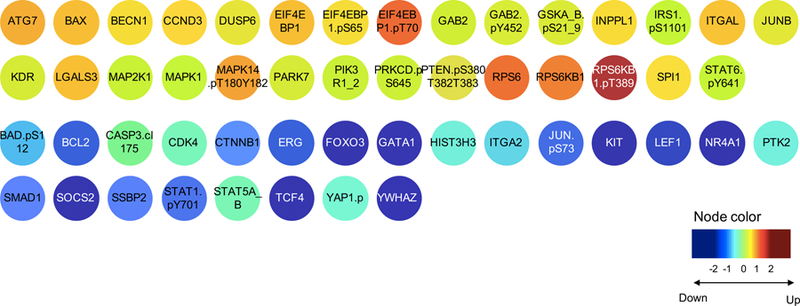
Example of the protein target identification in adult AML patients. Proteins that were statistically differentially expressed compared to the healthy CD34+ samples were identified for signature 2. Colors represent the median expression within that particular signature compared to the normal CD34+ samples. Yellow-orange-red proteins were significantly overexpressed versus those in cyan-blue-navy that were significantly under expressed.
Conclusion
Proteomics in acute leukemia have traditionally been studied using either MS or RPPA methodology. Both methods have proven to be very useful tools to analyze proteins on a large scale in acute leukemia and both have identified several proteins that were associated with outcome, chemoresistance, and disease state (e.g. diagnosis vs. relapse/refractory). Since the function of the majority of proteins occurs in the context of other proteins, in networks and pathways, rather than individually determining a functional state of the cell, we recently switched to analyzing individual proteins in the context of protein networks. Current leukemia classification system cannot fully classify all patients, we feel that protein network analysis can improve risk stratification by identifying prognostic features unique to proteins. We have also learned that the most (cyto)genetic events are difficult to drug, and proteomics could identify pathways that may be more easily impacted using our current arsenal of developmental therapeutics agents. Proof of principle has been demonstrated for a few agents that target aberrantly expressed proteins in acute leukemia clinical trials, making rapid assessment of protein levels in leukemia cells crucial in the process of therapy selection. Finally, direct comparison of pediatric AML and ALL has shown that despite large uniqueness of both diseases, overlap exists in the proteomic landscape, suggesting some shared deregulations in disease pathophysiology that might be similarly targetable.
Expert commentary
Analysis of the proteome could enhance our knowledge of leukemia and its treatment in several ways. There is strong evidence that analysis of proteins and alterations in proteins pathways could be identified as prognostic markers. Examples include high BCL2 expression in those with more resistant disease and high BRD4 associated with a shorter OS. It is also possible that protein expression could act as a predictive marker by identifying pathways targeted by current drugs and predicting chemoresistance to these agents. Finally, it is also possible that a protein detection kit could be developed in the future to facilitate the recognition of protein expression patterns and that this could be used as an aid in the selection of combination therapies for subsets of patients.
Although having great promise, certain studies could help optimize the use of proteomics in the future. Proteomics would benefit from a fuller characterization of PTM in addition to total protein abundance. There is also much to be gained from studying the proteome in different cell types. It has been postulated that hematopoietic stem cells (HSCs) or leukemic stem-like cells (LSCs) will have different patterns of gene/ protein expression and activation when compared to more differentiated progeny. As an illustration, Noverhstern et al. profiled gene expression of 211 human samples from 38 hematopoietic compartments and identified specific profiles of which some were cell-type specific while others showed overlap between multiple hematopoietic populations [50]. A study by Majeti et al. performed a genome-wide expression analysis that compared the expression profiles of highly enriched HSCs (n=7) and LSCs (n=16) from AML patients and identified 3005 differentially expressed genes, including aberrantly expressed genes that are involved in Wnt-signaling and MAP kinase signaling [51]. Gal et al. isolated CD34+CD38- and CD34+CD38+ cell fractions from AML patients and identified 409 genes using microarrays that were differentially expressed between the two cell populations [52]. Proteomic analysis of different cell types can help identify the best proteins to target or the best proteins to predict outcome. Our work has previously shown that protein expression patterns in leukemic-stem cell population (CD34+/CD38-) significantly differ from bulk (CD3-/CD19-) leukemic cells and other CD34+ populations [53], and another study suggested that the identification of specific populations of mesenchymal stem cells (MSC) in AML patients may be an important determinant of therapeutic response [42]. MSCs are essential for the normal hematopoiesis and several studies have shown that MSCs could protect leukemia cells from chemotherapy; since LSCs reside in the lymph nodes and the bone marrow, they could take refuge with the leukemia MSC niche during chemotherapy, which eventually contributes to relapse of the disease.
Despite the ability of chemotherapy to kill the vast majority of leukemic cells and put most patients into remission, the substantial number that relapse confirms the need to identify chemoresistant cells. Evidence suggest that these are the rare leukemic stem cell that survives. Proteomic analysis of these rare, slowly-dividing chemoresistant stem cells might be more informative than the analysis of the bulk leukemia population [54]. In addition, as individual leukemias contain very heterogeneous cell populations and studying single cells proteomics, rather than average leukemia cell expression, might provide useful information about cell to cell heterogeneity and changes to therapy over time. This information is likely to raise new biological questions requiring further investigations. Despite those challenging considerations, the use of proteomics has definite clinical potential, as measuring protein levels has a far shorter turnaround time compared to the traditional cytogenetic and mutation sequencing analysis and could be made available shortly after diagnosis. This could provide additional information for initial treatment decisions and could aid in risk stratification following induction chemotherapy.
Five-year view
Several prognostic factors are used in risk stratification for leukemia, including white cell count, age, cytogenetics and minimal residual disease. However, these markers are imperfect, with many patient relapses coming from those in low and standard risk groups. Proteomics holds the promise of being able to improve risk stratification by identifying risk groups that are not identified using traditional cytogenetic information. Proteomic studies, combined with genetic and clinical analyses could increase the accuracy of current risk stratification regimens, allowing patients with low-risk disease to be less intensively treated, whereas high-risk patients could be treated with more intensive treatment protocols. Development of protein assessment kits that quickly evaluate protein levels of specific prognostic proteins, could also help guide personalized therapy by rational selecting the combinational agents, something that is already emerging with treatment protocols.
Key issues.
Acute leukemia is a deadly disease for both children and adults, and tools to enhance risk stratification of patients into low, intermediate and high-risk patients are needed.
Proteomics are a promising tool to identify prognostic proteins.
Identification of proteins could potentially guide rational selection of the right combinational treatment for subgroups of patients.
Analyzing individual proteins in a more network-based fashion provides more insights about what is going on in a cell.
Post-translational modifications (PTM) are important to study as PTM are responsible for the activity of the protein
Studying the right cell fractions might provide superior information about prediction of therapy response.
Acknowledgments
Funding
This article was funded by the R01 CA164024 to TMH.
Footnotes
Declaration of interest
The authors declare no conflict of interest.
References
- [1].Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA: A Cancer Journal for Clinicians 64(2), 83–103 (2014). [DOI] [PubMed] [Google Scholar]
- [2].Hunger SP, Loh ML, Whitlock JA et al. Children’s Oncology Group’s 2013 blueprint for research: acute lymphoblastic leukemia. Pediatric Blood & Cancer 60(6), 957–963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Thomas D, O’Brien S, Faderl S et al. Anthracycline dose intensification in adult acute lymphoblastic leukemia: Lack of benefit in the context of the fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone regimen. Cancer 116(19), 4580–4589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fernandez HF, Sun Z, Yao X et al. Anthracycline dose intensification in acute myeloid leukemia. N. Engl. J. Med 361(13), 1249–59 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lwenberg B, Ossenkoppele GJ, van Putten W et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N. Engl. J. Med 361(13), 1235–48 (2009). [DOI] [PubMed] [Google Scholar]
- [6].Gamis AS, Alonzo TA, Perentesis JP, Meshinchi S. Children’s Oncology Group’s 2013 blueprint for research: Acute myeloid leukemia. Pediatric Blood & Cancer 60(6), 964–971 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Oskarsson T, Sderhll S, Arvidson J et al. Relapsed childhood acute lymphoblastic leukemia in the Nordic countries: prognostic factors, treatment and outcome. Haematologica 101(1), 68–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Arber DA, Orazi A, Hasserjian R et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20), 2391–405 (2016). [DOI] [PubMed] [Google Scholar]
- [9].Grimwade D, Hills RK, Moorman AV et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 116(3), 354–65 (2010). [DOI] [PubMed] [Google Scholar]
- [10].Papaemmanuil E, Gerstung M, Bullinger L et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med 374(23), 2209–2221 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol 19(3), 1720–1730 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Maier T, Gell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett 583(24), 3966–73 (2009). [DOI] [PubMed] [Google Scholar]
- [13].Mueller C, Liotta LA, Espina V. Reverse phase protein microarrays advance to use in clinical trials. Molecular Oncology 4(6), 461–481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Akbani R, Becker KF, Carragher N et al. Realizing the promise of reverse phase protein arrays for clinical, translational, and basic research: a workshop report: the RPPA (Reverse Phase Protein Array) society. Mol Cell Proteomics 13(7), 1625–1643 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vidova V, Spacil Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal. Chim. Acta 964, 7–23 (2017). [DOI] [PubMed] [Google Scholar]
- [16].Kaźmierczak M, Luczak M, Lewandowski K et al. Esterase D and gamma 1 actin level might predict results of induction therapy in patients with acute myeloid leukemia without and with maturation. Med. Oncol 30(4), 725 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nicolas E, Ramus C, Berthier S et al. Expression of S100A8 in leukemic cells predicts poor survival in de novo AML patients. Leukemia 25(1), 57–65 (2011). [DOI] [PubMed] [Google Scholar]
- [18].Bai J, He A, Zhang W et al. Potential biomarkers for adult acute myeloid leukemia minimal residual disease assessment searched by serum peptidome profiling. Proteome Science 11(1), 1–17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hu J, Lin M, Liu T, Li J, Chen B, Chen Y. DIGE-based proteomic analysis identifies nucleophosmin/B23 and nucleolin C23 as over-expressed proteins in relapsed/refractory acute leukemia. Leuk. Res 35(8), 1087–1092 (2011). [DOI] [PubMed] [Google Scholar]
- [20].Jiang N, Kham SKY, Koh GS et al. Identification of prognostic protein biomarkers in childhood acute lymphoblastic leukemia (ALL). Journal of Proteomics 74(6), 843–857 (2011). [DOI] [PubMed] [Google Scholar]
- [21].Negoro E, Yamauchi T, Urasaki Y, Nishi R, Hori H, Ueda T. Characterization of cytarabine-resistant leukemic cell lines established from five different blood cell lineages using gene expression and proteomic analyses. International journal of oncology 38(4), 911 (2011). [DOI] [PubMed] [Google Scholar]
- [22].Schaab C, Oppermann FS, Klammer M et al. Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia 28(3), 716–719 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Alcolea MP, Casado P, Rodrguez-Prados JC, Vanhaesebroeck B, Cutillas PR. Phosphoproteomic analysis of leukemia cells under basal and drug-treated conditions identifies markers of kinase pathway activation and mechanisms of resistance. Molecular & cellular proteomics : MCP 11(8), 453–66 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Brown FC, Still E, Koche RP et al. MEF2C Phosphorylation Is Required for Chemotherapy Resistance in Acute Myeloid Leukemia. Cancer discovery 8(4), 478–497 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Accordi B, Espina V, Giordan M et al. Functional protein network activation mapping reveals new potential molecular drug targets for poor prognosis pediatric BCP-ALL. PloS One 5(10), e13552 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Milani G, Rebora P, Accordi B et al. Low PKC? expression within the MRD-HR stratum defines a new subgroup of childhood T-ALL with very poor outcome. Oncotarget 5(14), 5234–45 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Aveic S, Viola G, Accordi B et al. Targeting BAG-1: a novel strategy to increase drug efficacy in acute myeloid leukemia. Exp. Hematol 43(3), 190.e6 (2015). [DOI] [PubMed] [Google Scholar]
- [28].Kornblau SM, Singh N, Qiu Y, Chen W, Zhang N, Coombes KR. Highly phosphorylated FOXO3A is an adverse prognostic factor in acute myeloid leukemia. Clin. Cancer Res 16(6), 1865–1874 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Butler JS, Qiu YH, Zhang N et al. Low expression of ASH2L protein correlates with a favorable outcome in acute myeloid leukemia. Leuk. Lymphoma 58(5), 1207–1218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Quintas-Cardama A, Zhang N, Qiu YH et al. Loss of TRIM62 expression is an independent adverse prognostic factor in acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk 15(2), 127.e15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kornblau SM, Qiu YH, Zhang N et al. Abnormal expression of FLI1 protein is an adverse prognostic factor in acute myeloid leukemia. Blood 118(20), 5604–5612 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ruvolo PP, Qiu Y, Coombes KR et al. Phosphorylation of GSK3?/? correlates with activation of AKT and is prognostic for poor overall survival in acute myeloid leukemia patients. BBA clinical 4, 59–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ruvolo PP, Qui YH, Coombes KR et al. Low expression of PP2A regulatory subunit B55? is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia 25(11), 1711–1717 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Khan M, Carmona S, Sukhumalchandra P et al. Cathepsin G Is Expressed by Acute Lymphoblastic Leukemia and Is a Potential Immunotherapeutic Target. Frontiers in immunology 8, 1975 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kornblau SM, Tibes R, Qiu YH et al. Functional proteomic profiling of AML predicts response and survival. Blood 113(1), 154–164 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hu CW, Qiu Y, Yoo SY et al. Quantifying Proteomic Heterogeneities and Hallmarks in Acute Myelogenous Leukemia (AML). . Manuscript under review
- [37].van Dijk AD, Hu CW, de Bont E et al. Histone Modification Patterns using RPPA-based Profiling Predict Outcome in Acute Myeloid Leukemia Patients. Proteomics (2018). [DOI] [PubMed]
- [38].Quintas-Cardama A, Hu C, Qutub A et al. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia (2016). [DOI] [PubMed]
- [39].Govaert G, Nadif M. Clustering with block mixture models. Pattern Recognit 36(2), 463–473 (2003). [Google Scholar]
- [40].Hoff FW, Hu CW, Qiu Y et al. Recognition of Recurrent Protein Expression Patterns in Pediatric Acute Myeloid Leukemia Identified New Therapeutic Targets. Molecular Cancer Research (2018). [DOI] [PMC free article] [PubMed]
- [41].Hoff FW, Hu CW, Qiu Y et al. Recurrent Patterns of Protein Expression Signatures in Pediatric Acute Lymphoblastic Leukemia: Recognition and Therapeutic Guidance. Molecular Cancer Research (2018). [DOI] [PMC free article] [PubMed]
- [42].Kornblau SM, Ruvolo PP, Wang RY et al. Distinct protein signatures of acute myeloid leukemia bone marrow-derived stromal cells are prognostic for patient survival. Haematologica (2018). [DOI] [PMC free article] [PubMed]
- [43].DiNardo CD, Rausch CR, Benton C et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am. J. Hematol 93(3), 401–407 (2018). [DOI] [PubMed] [Google Scholar]
- [44].Kornblau SM, Thall PF, Estrov Z et al. The prognostic impact of BCL2 protein expression in acute myelogenous leukemia varies with cytogenetics. Clinical Cancer Research 5(7), 1758–66 (1999). [PubMed] [Google Scholar]
- [45].Kornblau SM, Vu HT, Ruvolo P et al. BAX and PKCalpha modulate the prognostic impact of BCL2 expression in acute myelogenous leukemia. Clinical Cancer Research 6(4), 1401–9 (2000). [PubMed] [Google Scholar]
- [46].Delmore JE, Issa GC, Lemieux ME et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146(6), 904–17 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mahmud H, Kornblau SM, ter Elst A et al. Epidermal growth factor receptor is expressed and active in a subset of acute myeloid leukemia. Journal of Hematology & Oncology 9, 64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Andrew eBurgess, Kee Ming Chia, Sue eHaupt, David eThomas, Ygal eHaupt, Elgene eLim. Clinical overview of MDM2/X targeted therapies. frontiers in oncology 6 (2016). [Google Scholar]
- [49].Doron B, Abdelhamed S, Butler JT, Hashmi SK, Horton TM, Kurre P. Transmissible ER Stress Reconfigures the AML Bone Marrow Stroma. Leukemia, In Press. [DOI] [PMC free article] [PubMed]
- [50].Novershtern N, Subramanian A, Lawton LN et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144(2), 296–309 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Majeti R, Becker MW, Tian Q et al. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc. Natl. Acad. Sci. U. S. A 106(9), 3396–401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gal H, Amariglio N, Trakhtenbrot L et al. Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia 20(12), 2147–54 (2006). [DOI] [PubMed] [Google Scholar]
- [53].Kornblau SM, Qutub A, Yao H et al. Proteomic profiling identifies distinct protein patterns in acute myelogenous leukemia CD34+CD38- stem-like cells. PLoS One 8(10), e78453 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kornblau SM, Qiu YH, Bekele BN et al. Studying the right cell in acute myelogenous leukemia: dynamic changes of apoptosis and signal transduction pathway protein expression in chemotherapy resistant ex-vivo selected “survivor cells”. Cell cycle 5(23), 2769–77 (2006). [DOI] [PubMed] [Google Scholar]
- [55].Hu CW, Kornblau SM, Slater JH, Qutub AA. Progeny Clustering: A Method to Identify Biological Phenotypes. Sci. Rep 5, 12894 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]