

THE HYPOTHALAMIC–NEUROHYPOPHYSEAL–RENAL AXIS NORMALLY MAIN-tains water balance during variations in water intake and nonrenal losses of water. Failure of this mechanism is common in hospitalized patients, and it results in a variety of water-balance disorders. In this article, we begin by reviewingthe classic, integrative principles of water balance in mammals and then usethis classic model as a framework to discuss the genes and gene products (proteins)involved in water balance. In so doing, our goal is to provide clinicians witha mechanistic basis for decisions regarding the diagnosis and treatment of waterbalance disorders.
The regulation of water balance is governed by a high-gain feedback mechanism involving the hypothalamus, the neurohypophysis, and the kidneys (Fig. 1).Osmoreceptors in the hypothalamus, which originally were described by Verney,1 sense plasma osmolality. The molecular mechanism of “osmosensing” has recently been described by Danziger and Zeidel.2 It is, in part, dependent on activationof nonselective calcium-permeable cation channels in osmosensing neuronsthat can serve as stretch receptors.
Figure 1. Feedback Loop Governing Regulation of PlasmaOsmolality through Control of Arginine VasopressinSecretion and Thirst.
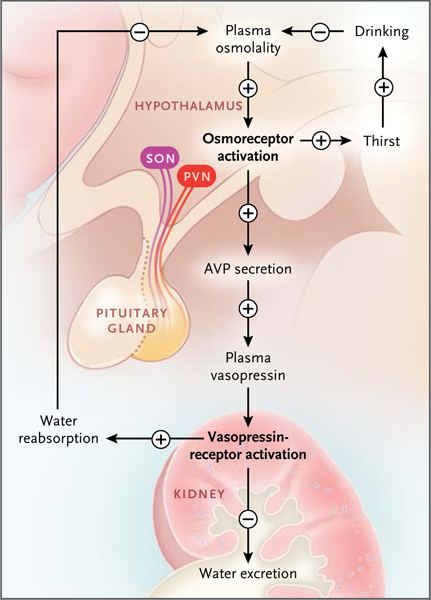
An increase in plasma osmolality activates hypothalamicosmoreceptors to stimulate vasopressin secretionby the posterior pituitary gland. The resulting increasein the level of plasma vasopressin leads to anincrease in renal water reabsorption and a decrease inwater excretion. Increased water reabsorption reducesplasma osmolality. Osmosensing in the hypothalamusalso stimulates thirst and drinking to help restore plasmaosmolality. AVP denotes arginine vasopressin, PVNparaventricular nucleus, and SON supraoptic nucleus.
When plasma osmolality increases to levels above a physiologic threshold (290 to 295 mOsm per kilogram of water in most persons), there is increased secretion of the peptide hormone vasopressin from vasopressinergic nerve endings in the neurohypophysis. High osmolality also triggers thirst. Vasopressin binds to receptors in the kidney that decrease excretion of water (Fig. 2), and a greater fraction of filtered water is returned to the blood. The rate of water excretion can vary over a broad range in response to changes in plasma vasopressin levels without substantial changes in net solute excretion (osmolar clearance). This independent control of water and solute excretion is the result of specialized urinary concentrating and diluting mechanisms; these mechanisms are reviewed elsewhere.3
Figure 2. Relationships among Plasma Vasopressin Concentration, Rate of Water Excretion, and Solute Excretion (Osmolar Clearance).
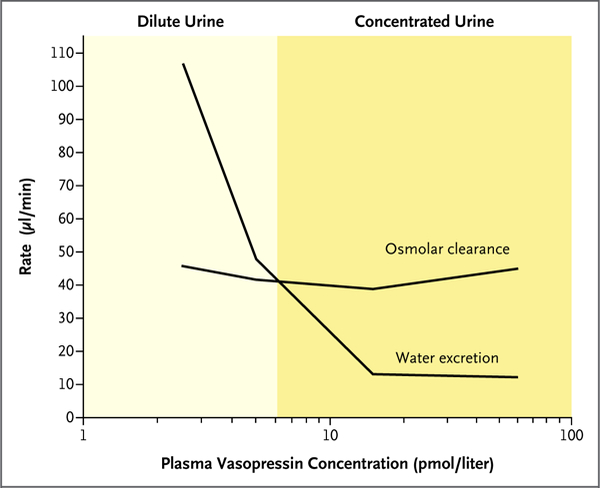
Water excretion decreases with increased levels of plasma vasopressin, whereas solute excretion remains relatively constant. This results in concentrated urine at a high vasopressin concentration and dilute urine at a low vasopressin concentration.
Increased renal reabsorption of water in response to vasopressin lowers plasma osmolality, thereby reducing the stimulus for vasopressin secretion and thirst and completing the feedback loop (Fig. 1). Table 1 provides a list of the major proteins that are responsible for components of the integrative model shown in Figure 1. These proteins are the focus of this review.
Table 1.
Key Proteins Involved in Regulation of Water Balance.
| Protein | Gene | Structure or Cell Type Relevant to Water Balance |
Manifestation of Loss of Function* |
Drugs That Target Protein |
|---|---|---|---|---|
| Arginine vasopressin | AVP | NeuNeurons of supraoptic nucleus and paraventricular nucleus | Central diabetes insipidus | None |
| Vasopressin receptor | ||||
| V2 | AVPR2 | RenRenal thick ascending limb of the loop of Henle, distal convoluted tubule, connecting tubule, collecting duct | X-linked nephrogenic diabetes insipidus | DesmDesmopressin acetate (agonist), tolvap- tan (antagonist) |
| V1a | AVPRlA | RenRenal medullary vasculature (vasa recta) | None | Conivaptan (nonselec- tive V1a and V2 antagonist) |
| BumeBumetanide-sensitive sodi- um-potassium–chlo–ride cotransporter | SLCl2Al | RenRenal thick ascending limb of the loop of Henle | Type I Bartter’s syndrome | Loop Loopdiuretics |
| ThiaziThiazide-sensitive sodium- chloride cotransporter | SLCl2A3 | RenRenal distal convoluted tubule | Gitelman’s syndrome | Thiazide diuretics |
| Aquaporin | ||||
| Aquaporin-1 | AQPl | Renal proximal tubule, thin descending limb of the loop of Henle, erythrocyte | Colton blood group-null | None |
| Aquaporin-2 | AQP2 | RenRenal connecting tubule, collecting duct | Autosomal nephrogenic diabetes insipidus | None |
| Aquaporin-3 | AQP3 | RenRenal connecting tubule, collecting duct, erythrocyte | GIL blood group-null | None |
| Aquaporin-4 | AQP4 | RenRenal connecting tubule, collecting duct | None | None |
| Vasopressin-regulated urea channel | SLCl4A2 | RenRenal inner medullary collecting duct, thin descending limb of the loop of Henle | None | None |
| Epithelial sodium channel | ||||
| Beta subunit | SCNNlB | RenRenal connecting tubule, collecting duct | Type I pseudohypoaldoster- onism | Amiloride |
| Gamma subunit | SCNNlG | RenRenal connecting tubule, collecting duct | Type I pseudohypoaldoster- onism | Amiloride |
Data are from the Online Mendelian Inheritance in Man database.
ARGININE VASOPRESSIN
The gene coding for arginine vasopressin (AVP) is expressed in neurons of the supraoptic and paraventricular nuclei of the hypothalamus. Arginine vasopressin is a typical neuropeptide, since its gene codes for a prohormone that must undergo specific proteolytic processing to produce the active hormone. Thus, AVP codes for three peptides — the 9–amino acid peptide arginine vasopressin, a car-rier protein called neurophysin-2, and a small glycoprotein called copeptin. Because vasopressin itself is difficult to measure in plasma samples, some investigators are using measurements of copeptin in plasma as a surrogate for arginine vasopressin.4 Mutations in the arginine vasopressin gene that interfere with the processing and release of arginine vasopressin are associated with central diabetes insipidus.
The oxytocin gene has a structure that is very similar to that of the arginine vasopressin gene. It is expressed in distinct oxytocinergic cells in the supraoptic and paraventricular nuclei of the hypothalamus and, like vasopressin, its secretion is increased by osmotic stimuli.5 It binds to vasopressin receptors in the kidney and produces similar, although weaker, responses than arginine vasopressin.6 Consequently, oxytocin is sometimes considered to be a second “antidiuretic hormone.” Rarely, in the third trimester of pregnancy, a syndrome called transient vasopressin-resistant diabetes insipidus of pregnancy occurs as a result of placental secretion of vaso-pressinase (also called oxytocinase), which hydrolyzes circulating vasopressin and oxytocin.7 Affected patients have a response to desmopressin acetate, which is resistant to this enzyme.
VASOPRESSIN RECEPTORS
After secretion into the general circulation from the posterior pituitary gland (neurohypophysis) (Fig. 1), arginine vasopressin is delivered to the kidney, where it exerts regulatory actions through the V2 receptor (gene symbol, AVPR2). The V2 vasopressin receptor is a G protein–coupled receptor with physiologic functions that are mediated largely by the heterotrimeric G-protein Gs, resulting in activation of adenylyl cyclases to increase the intracellular level of cyclic AMP (cAMP).3 Mutations in AVPR2 are responsible for X-linked nephrogenic diabetes insipidus.8
The kidney also expresses the V1a vasopressin receptor, largely in the vasculature of the renal medulla9; this receptor mediates the effects of vasopressin on renal blood flow.10 The V1a vasopressin receptor signals chiefly through the hetero-trimeric G-protein Gq/11; this G protein activates phospholipase C and stimulates calcium mobilization. The V1a receptor is widely expressed throughout the body, whereas the V2 receptor is located chiefly in renal epithelia. A variety of localization studies and corresponding functional studies have shown that the V2 receptor acts chiefly in the principal cells of the renal collecting duct, the connecting tubule cells, the distal convoluted tubule cells, and the cells of the thick ascending limb of Henle (Fig. 3).
Figure 3. Renal Tubule.
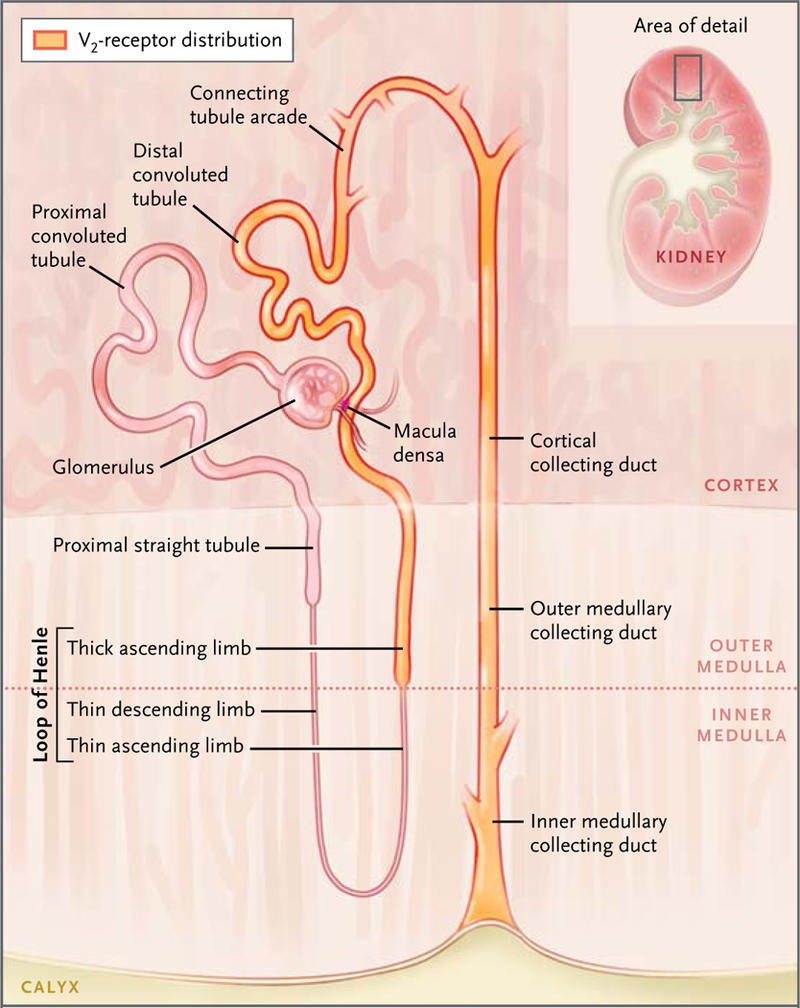
The segments shown in orange are targets for vasopressin regulation through the V2 receptor. Loop of Henle segments generate a corticomedullary osmolality gradient through the process of countercurrent multiplication. Connecting-tubule and collecting-duct segments are those that manifest regulated osmotic water transport through the action of vasopressin to regulate the water channel aquaporin-2. The macula densa is the point along the nephron where contact is made with the glomerulus of the same nephron. It provides a feedback signal (luminal sodium chloride concentration) that regulates the glomerular filtration rate to stabilize the sodium chloride concentration in the luminal fluid delivered to the distal convoluted tubule.
BUMETANIDE-SENSITIVE SODIUM–POTASSIUM–CHLORIDE COTRANSPORTER
Vasopressin increases the rate of active absorption of sodium chloride in the medullary thick ascending limbs of Henle,11,12 enhancing counter-current multiplication, which is the process responsible for the medullary accumulation of solutes.3 The high concentration of solutes in the medullary interstitium furnishes the osmotic gradient needed to drive reabsorption of water from the renal collecting ducts. Consequently, the up-regulation of medullary interstitial accumulation of solutes by vasopressin contributes to the regulation of water excretion.
In the thick ascending limbs of Henle, the transport of sodium and chloride from the lumen is mediated by the bumetanide-sensitive sodium–potassium–chloride cotransporter.13 Vasopressin up-regulates this cotransporter in at least two ways: short-term regulation that is a consequence of vesicular trafficking14 and long-term regulation that is a consequence of an increase in the expression of SLC12A1, which codes for the cotransporter protein.15 Up-regulation of the sodium–potassium–chloride cotransporter generally does not affect salt excretion, primarily because the thick ascending limb is upstream from the macula densa (Fig. 3), which compensates for changes in salt delivery by adjusting the glomerular filtration rate. This compensatory process is called glomerulotubular feedback.
The diuretic bumetanide and other loop diuretics increase salt excretion because they inhibit the sodium–potassium–chloride cotransporter in the macula densa, thereby blocking the feedback to the glomerulus.16 Similarly, type I Bartter’s syndrome (loss-of-function mutations in SLC12A1) is manifested by a salt-losing syndrome rather than by a simple defect in water balance.
THIAZIDE-SENSITIVE SODIUM–CHLORIDE COTRANSPORTER
Vasopressin also regulates salt transport in the distal convoluted tubule, which is present in each nephron a short distance downstream from the macula densa. It transports salt at a high rate but is impermeable to water and thereby contributes to dilution of the tubular fluid. The classic studies of Gottschalk and Mylle showed that the luminal fluid is dilute relative to blood plasma in this segment (independently of whether vasopressin levels are high or low),17 thereby establishing that vasopressin increases water reabsorption downstream from this site in the collecting ducts. Vasopressin-induced increases in salt transport out of the distal convoluted tubule can, in principle, increase the extent of luminal dilution, thereby increasing the driving force for reabsorption of water downstream.
Vasopressin exerts its effects on salt transport in the distal convoluted tubule by up-regulating the apical thiazide-sensitive sodium–chloride cotransporter, in part through effects on protein phosphorylation.18,19 This cotransporter is also regulated by aldosterone, which increases the abundance of the cotransporter protein in the distal convoluted tubule cells.20 As a result, the thiazide-sensitive sodium–chloride cotransporter also plays a critical role in the regulation of sodium and chloride balance.
Inactivating mutations in the thiazide-sensitive sodium–chloride cotransporter cause Gitelman’s syndrome, which is manifested by hypotension, hypokalemia, hypomagnesemia, hypocalciuria, and metabolic alkalosis. Polyuria, which also occurs in patients with this syndrome, is primarily the result of hypokalemia21 rather than a direct effect of the loss of active sodium-chloride cotransport in the distal convoluted tubule.
AQUAPORINS
AQUAPORIN-1
In the early 1990s, Agre and colleagues identified the first molecular water channel, aquaporin-1, which was found to be ubiquitously expressed.22 In the kidney, this water channel is expressed in the proximal tubule and thin descending limb of the loop of Henle.23 In the thin descending limb, it plays an important role in the countercurrent multiplier mechanism, allowing a rapid osmoti- cally driven exit of water from the lumen, thereby concentrating the luminal fluid.
In mice24 and humans,25 a lack of aquaporin-1 results in a near inability to concentrate urine.Aquaporin-1 appears to be constitutively expressed in the kidney and is not regulated by vasopressin.
AQUAPORIN-2
Another aquaporin, aquaporin-2, is expressed throughout the collecting-duct system26 (i.e., in the region of the renal tubule where vasopressin regulates osmotic transport of water). Aquapo-rin-2 mediates the apical component of transepithelial water transport, and its regulation by vasopressin controls the overall rate of water permeation across the collecting-duct epithelium. Most patients with non–X-linked nephrogenic diabetes insipidus have mutations in AQP2.8
Extensive physiological studies involving ratsand mice have revealed two basic forms of vasopressin-mediated regulation of the aquaporin-2 water channel. Short-term regulation occurs over a period of a few minutes, and long-term regulation occurs over a period of hours to days.27
The short-term regulation of aquaporin-2 occurs as a result of membrane trafficking.28 Immunoelectron microscopy showed that in the absence of vasopressin, aquaporin-2 water channels were located predominantly in intracellular vesicles (endosomes), but when vasopressin was added to isolated collecting ducts, water channels were seen predominantly in the luminal plasma membrane. Other studies showed that the translocation of aquaporin-2 occurs as a result of both stimulated exocytosis and inhibited endocytosis29,30 (Fig. 4).
Figure 4. Collecting-Duct Principal Cell.
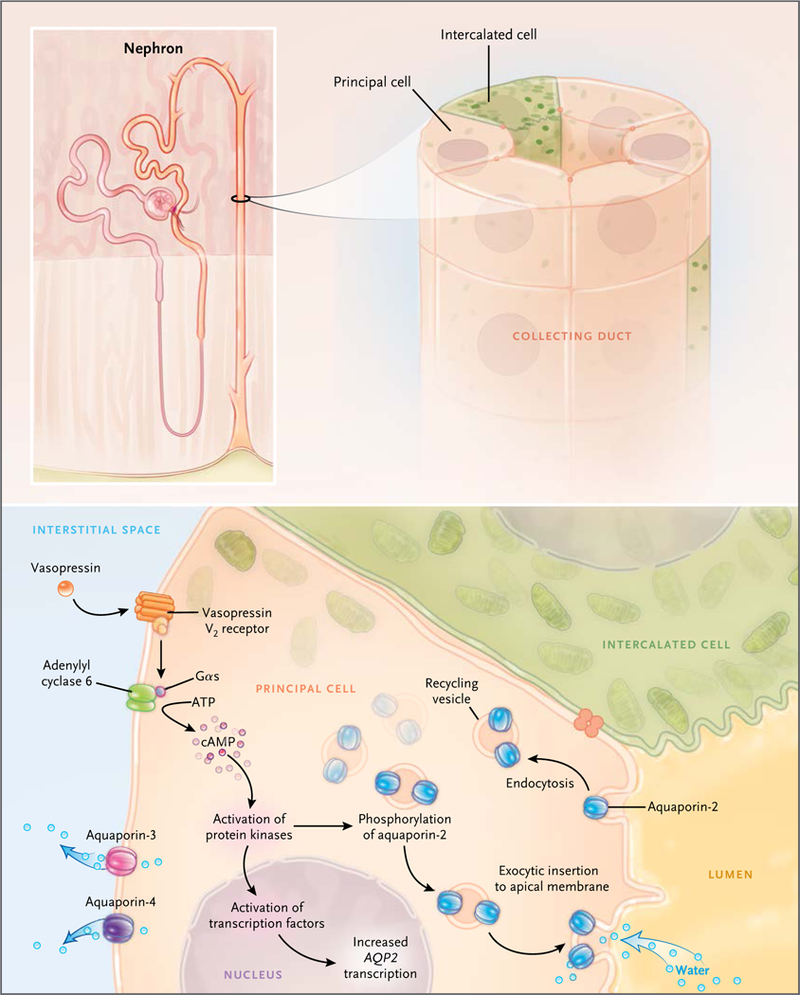
In the presence of vasopressin, water enters the principal cell from the lumen through aquaporin-2 (right) and exitsto the interstitium through aquaporin-3 and aquaporin-4. Regulation of aquaporin-2 by vasopressin is a result ofcyclic AMP (cAMP)–dependent activation of a protein kinase network that causes increased transcription of AQP2and redistribution of aquaporin-2 to the luminal membrane. The redistribution results from inhibition of aquaporin-2 endocytosis and stimulation of aquaporin-2 exocytosis. Intercalated cells mediate acid–base transport andare thought to be water-impermeable. Gαs denotes heterotrimeric G-protein alpha subunit.
These actions of vasopressin are associated with changes in phosphorylation of the aquaporin-2 protein at four sites near the carboxyl terminus.31–33 Exocytosis of aquaporin-2 appears to require phosphorylation at serine 256.34–36 Vasopressin also markedly increases aquaporin-2 phosphorylation at serine 269.33 This phosphorylation event inhibits aquaporin-2 endocytosis.33,37,38 Vasopressin decreases phosphorylation of serine 261 by reducing the activity of one or more MAP kinases.39,40 Phosphorylation at this site appears to decrease the stability of the aquaporin-2 protein,40 but it was not found to affect aquaporin-2 trafficking.41 In addition to its requirement for phosphorylation, vasopressin-induced redistribution to the apical plasma membrane has been shown to be dependent on actin depolymerization in the apical region of collecting-duct cells, secondary to inhibition of the small guanosine triphosphate–binding protein RhoA42 and binding of aquaporin-2 to tropomyosin.43
The long-term regulation of aquaporin-2 occurs as a result of a vasopressin-induced increase in the total abundance of the aquaporin-2 protein in collecting-duct cells.44 At least two independent processes are involved. First, the half-life of the aquaporin-2 protein is increased by vasopressin.40,45 One study showed that in cultured mpkCCD cells, the half-life increased from 9 to 14 hours.45 The process of endocytosis and degradation of aquaporin-2 is regulated in part through ubiquitylation of the C-terminal tail of the aquaporin-2 protein.46 Second, transcription of the aquaporin-2 gene is markedly increased by vasopressin,47 resulting in increased aquaporin-2 translation rates.46
The identification of the transcription factors involved in this long-term regulation of aquaporin-2 transcription is under investigation.48 Typically, transcriptional regulation is combinatorial and involves two or more transcription factors acting simultaneously.49 In the 5-flanking region of AQP2, there are two conserved clusters of putative transcriptional-regulator binding elements centered at –513 bp from the transcription start site (corresponding to the SF1, NFAT, and FKHD transcription factor families) and at –224 bp (corresponding to the AP2, SRF, CREB, GATA, and HOX transcription factor families).50
Studies of animal models of disease have shown that dysregulation of aquaporin-2 plays a central role in both polyuric disorders and disorders associated with dilutional hyponatremia.27 Polyuric disorders due to abnormalities in the regulation of water transport that are intrinsic to the kidney are referred to as nephrogenic diabetes insipidus syndromes. Heritable nephrogenic diabetes insipidus syndromes have been reviewed by Fujiwara and Bichet.8 Acquired nephrogenic diabetes insipidus syndromes are much more common in clinical practice and can occur in patients who have hypokalemia, hypercalcemia, or partial urinary tract obstruction, as well as in patients who receive certain drugs such as lithium carbonate27 (see the case reports in the Supplementary Appendix, available with the full text of this article at NEJM.org). Animal models of each of these syndromes have shown a marked reduction in the abundance of the aquaporin-2 protein, presumably because of abnormalities in the normal long-term regulatory mechanisms described above.
Aquaporin-2 dysregulation also occurs in a number of syndromes associated with renal water retention and dilutional hyponatremia, chiefly severe congestive heart failure, hepatic cirrhosis, and the syndrome of inappropriate antidiuretic hormone secretion (SIADH)27 (see the case reports in the Supplementary Appendix). In these states, increased levels of circulating vasopressin occur “inappropriately” (i.e., independently of regulation through the hypothalamic osmoreceptors) (Fig. 1). The pathophysiological mechanisms that are involved have been reviewed by Schrier.51 SIADH is the most common cause of hyponatremia in hospitalized patients.52
Animal models of SIADH have shown marked increases in aquaporin-2 protein abundance.53,54 However, these increases are attenuated by a counterregulatory process called “vasopressin escape.”54 This escape phenomenon is associated with resistance to vasopressin in the collecting duct owing to decoupling of the liganded V2 receptor from cAMP production.55 Thus, despite high levels of circulating vasopressin, the renal collecting ducts become relatively impermeable to water,55 thereby limiting the decrease in the serum sodium to a concentration typically in the range of 120 to 129 mmol per liter. Although data from studies are lacking, it seems likely that similar mechanisms limit the hyponatremia seen in severe congestive heart failure.
The development of a class of orally available drugs that block the V2 vasopressin receptor — the vaptans — offers a new type of therapy for the treatment of chronic, symptomatic dilutional hyponatremia. However, the use of these drugs is limited by high cost.56
AQUAPORIN-3
A third aquaporin, aquaporin-3, which is constitutively localized to the basolateral plasma membrane (Fig. 4) of collecting-duct principal cells, connecting-tubule cells, and inner medullary collecting-duct cells,57 provides an exit pathway for the water that enters across the apical plasma membrane through aquaporin-2. Unlike aquaporin-1, aquaporin-2, and aquaporin-4, aquaporin-3 conducts glycerol in addition to water and may have a role in the regulation of metabolism. Like aquaporin-2, its abundance is regulated over a period of hours to days by vasopressin57 through changes in its messenger RNA (mRNA) levels.54
Aquaporin-3 is widely expressed throughout the body. In erythrocytes, it is responsible for the GIL blood-group antigen. AQP3-null persons and GIL-negative persons have no obvious clinical manifestations.58 In contrast, AQP3-null mice have severe polyuria.59
AQUAPORIN-4
The water channel aquaporin-4 is localized to the basolateral plasma membrane in the collecting-duct system (i.e., the same cells that express aquaporin-2 and aquaporin-3).27 In contrast to aquaporin-3, its abundance is not regulated by vasopressin.27
The genetic deletion of aquaporin-4 in mice results in a modest concentrating defect.60 This result is in contrast to the much more severe phenotype seen with the genetic deletion of aquaporin-3.
VASOPRESSIN-REGULATED UREA CHANNEL
In the inner medullary collecting duct, vasopressin rapidly and reversibly increases transepithelialurea permeability, allowing urea to exit and become trapped within the countercurrent exchange system.3 Accumulation of urea in the medullary interstitium by this mechanism contributes to the high osmolality in the inner medulla. The high urea permeability of the inner medullary collecting duct is attributable to two urea-channel proteins (UT-A1 and UT-A3) that are produced from the same gene, SLC14A2.3
As seen with aquaporin-2, the regulation of SLC14A2 proteins by vasopressin is dependent on phosphorylation at multiple sites.61,62 These phosphorylation events are associated with increases in the number of the urea channels that are present in the apical plasma membrane.61
Genetic deletion of the UT-A1 and UT-A3 urea channels in mice markedly reduces the urea permeability of the inner medullary collecting duct63 and eliminates the corticomedullary urea gradient.63,64 Despite elimination of urea accumulation in the inner medulla, accumulation of sodium and chloride is unaffected. This finding seemingly ruled out concentrating models that predict that the accumulation of sodium chloride in the inner medulla is dependent on urea efflux from the inner medullary collecting duct.65
A third isoform of SLC14A2, called UT-A2, is produced by transcription from a different promoter and is expressed in the descending limbs of the loop of Henle. Urea transport in this segment is thought to be important for the recycling of the urea that is reabsorbed from the collecting duct back into the renal tubule.3
EPITHELIAL SODIUM CHANNEL
Transport of sodium ions out of the lumen of the cortical collecting duct is strongly and rapidly up-regulated by vasopressin.66,67 Transepithelial sodium transport in this segment is mediated both by an electroneutral mechanism68,69 and by an electrogenic mechanism that is regulated by vasopressin. The electrogenic component depends on apical entry of sodium ions through the epithelial sodium channel. The epithelial sodium channel is a heterotrimeric complex consisting of alpha, beta, and gamma subunits.
Studies by Snyder70 suggest that the rapid regulation by vasopressin is due to membrane trafficking of the epithelial sodium channel, which results from regulation of the ubiquitin ligase Nedd4–2. In addition, there appears to be long-term regulation of the epithelial sodium channel in the collecting duct in response to vasopressin. Specifically, the abundances of the beta and gamma subunits of the epithelial sodium channel are increased by vasopressin in a period of hours to days.71 The increases in protein abundance are associated with increases in beta and gamma subunit mRNA levels; this association points to pre-translational mechanisms of regulation.72
In contrast to vasopressin, aldosterone selectively increases the abundance of the alpha subunit protein without affecting levels of the beta and gamma subunits.73 Thus, the overall regulation of electrogenic transport in the cortical collecting duct appears to be synergistically dependent on both vasopressin and aldosterone. As shown in Figure 2, however, increases in vasopressin concentrations do not generally result in reduced excretion of total solute because of compensatory effects of the renin–angiotensin–aldosterone system.
In mice, selective deletion of the epithelial sodium channel in the connecting tubule and collecting duct is associated predominantly with abnormalities in the regulation of extracellular fluid volume (i.e., salt balance), rather than with abnormalities of water balance.74 Likewise, in humans, type I pseudohypoaldosteronism is due to loss-of-function mutations in the subunit genes of the epithelial sodium channel.75
CONCLUSIONS
In this brief review, we have described recent progress in understanding the roles of selected gene products in the regulation of water balance, with an emphasis on aspects relevant to the water-balance disorders that are most common in clinical practice. In addition, we have described a compendium of protein targets for pharmacologic agents that are useful in the treatment of disorders of salt and water balance (Table 1).
Agents that block water channels (aquaporins) or urea channels are not currently available. However, the important roles of these channels in normal water balance suggest that such agents (which are currently under development) may be useful in the treatment of water-balance disorders.
Supplementary Material
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
REFERENCES
- 1.Verney EB. The antidiuretic hormone and the factors which determine its release. Proc R Soc Lond B Biol Sci 1947; 135:25–106. [PubMed] [Google Scholar]
- 2.Danziger J, Zeidel ML. Osmotic homeostasis. Clin J Am Soc Nephrol 2014. July 30 (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fenton RA, Knepper MA. Mouse models and the urinary concentrating mechanism in the new millennium. Physiol Rev 2007;87:1083–112. [DOI] [PubMed] [Google Scholar]
- 4.Fenske WK, Christ-Crain M, Horning A, et al. A copeptin-based classification of the osmoregulatory defects in the syndrome of inappropriate antidiuresis. J Am Soc Nephrol 2014;25:2376–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Windle RJ, Forsling ML, Smith CP, Balment RJ. Patterns of neurohypophysial hormone release during dehydration in the rat. J Endocrinol 1993;137:311–9. [DOI] [PubMed] [Google Scholar]
- 6.Chou CL, DiGiovanni SR, Mejia R, Nielsen S, Knepper MA. Oxytocin as an antidiuretic hormone. I. Concentration dependence of action. Am J Physiol 1995; 269:F70–77. [DOI] [PubMed] [Google Scholar]
- 7.Brewster UC, Hayslett JP. Diabetes insipidus in the third trimester of pregnancy. Obstet Gynecol 2005;105:1173–6. [DOI] [PubMed] [Google Scholar]
- 8.Fujiwara TM, Bichet DG. Molecular biology of hereditary diabetes insipidus. J Am Soc Nephrol 2005;16:2836–46. [DOI] [PubMed] [Google Scholar]
- 9.Ostrowski NL, Young WS III, Knepper MA, Lolait SJ. Expression of vasopressin V1a and V2 receptor messenger ribonucleic acid in the liver and kidney of embryonic, developing, and adult rats. Endocrinology 1993;133:1849–59. [DOI] [PubMed] [Google Scholar]
- 10.Nakanishi K, Mattson DL, Gross V, Roman RJ, Cowley AWJ Jr. Control of renal medullary blood flow by vasopressin V1 and V2 receptors. Am J Physiol 1995; 269:R193–R200. [DOI] [PubMed] [Google Scholar]
- 11.Hall DA, Varney DM. Effect of vasopressin on electrical potential difference and chloride transport in mouse medullary thick ascending limb of Henle’s loop. J Clin Invest 1980;66:792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sasaki S, Imai M. Effects of vasopressin on water and NaCl transport across the in vitro perfused medullary thick ascending limb of Henle’s loop of mouse, rat, and rabbit kidneys. Pflugers Arch 1980;383:215–21. [DOI] [PubMed] [Google Scholar]
- 13.Greger R, Schlatter E, Lang F. Evidence for electroneutral sodium chloride cotransport in the cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflugers Arch 1983;396:308–14. [DOI] [PubMed] [Google Scholar]
- 14.Ortiz PA. cAMP increases surface expression of NKCC2 in rat thick ascending limbs: role of VAMP. Am J Physiol Renal Physiol 2006;290:F608–F616. [DOI] [PubMed] [Google Scholar]
- 15.Kim GH, Ecelbarger CA, Mitchell C, Packer RK, Wade JB, Knepper MA. Vasopressin increases Na-K-2Cl cotransporter expression in thick ascending limb of Henle’s loop. Am J Physiol 1999;276:F96–F103. [DOI] [PubMed] [Google Scholar]
- 16.Schnermann J, Briggs JP. Tubuloglo-merular feedback: mechanistic insights from gene-manipulated mice. Kidney Int 2008;74:418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gottschalk CW, Mylle M. Micropuncture study of the mammalian urinary concentrating mechanism: evidence for the countercurrent hypothesis. Am J Physiol 1959;196:927–36. [DOI] [PubMed] [Google Scholar]
- 18.Mutig K, Saritas T, Uchida S, et al. Short-term stimulation of the thiazide-sensitive Na+-Cl-cotransporter by vasopressin involves phosphorylation and membrane translocation. Am J Physiol Renal Physiol 2010;298:F502–F509. [DOI] [PubMed] [Google Scholar]
- 19.Pedersen NB, Hofmeister MV, Rosen-baek LL, Nielsen J, Fenton RA. Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int 2010;78:160–9. [DOI] [PubMed] [Google Scholar]
- 20.Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, Knepper MA. The thiazide-sensitive Na–Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci U S A 1998;95:14552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris RG, Hoorn EJ, Knepper MA. Hypokalemia in a mouse model of Gitel-man’s syndrome. Am J Physiol Renal Physiol 2006;290:F1416–F1420. [DOI] [PubMed] [Google Scholar]
- 22.Agre P, Preston GM, Smith BL, et al. Aquaporin CHIP: the archetypal molecular water channel. Am J Physiol 1993;265: F463–F76. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen S, Smith BL, Christensen EI, Knepper MA, Agre P. CHIP28 water channels are localized in constitutively water-permeable segments of the nephron. J Cell Biol 1993;120:371–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Severely impaired urinary concentrating ability in transgenic mice lacking aquaporin-1 water channels. J Biol Chem 1998;273:4296–9. [DOI] [PubMed] [Google Scholar]
- 25.King LS, Choi M, Fernandez PC, Car-tron JP, Agre P. Defective urinary-concentrating ability due to a complete deficiency of aquaporin-1. N Engl J Med 2001;345: 175–9. [DOI] [PubMed] [Google Scholar]
- 26.Fushimi K, Uchida S, Hara Y, Hirata Y, Marumo F, Sasaki S. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature 1993;361:549–52. [DOI] [PubMed] [Google Scholar]
- 27.Nielsen S, Fr0kiaer J, Marples D, Kwon TH, Agre P, Kneppe MA. Aquapo-rins in the kidney: from molecules to medicine. Physiol Rev 2002;82:205–44. [DOI] [PubMed] [Google Scholar]
- 28.Nielsen S, Chou CL, Marples D, Christensen EI, Kishore BK, Knepper MA. Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-CD water channels to plasma membrane. Proc Natl Acad Sci U S A 1995;92:1013–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nielsen S, Knepper MA. Vasopressin activates collecting duct urea transporters and water channels by distinct physical processes. Am J Physiol 1993;265:F204–F213. [DOI] [PubMed] [Google Scholar]
- 30.Brown D The ins and outs of aquaporin-2 trafficking. Am J Physiol Renal Physiol 2003;284:F893–F901. [DOI] [PubMed] [Google Scholar]
- 31.Nishimoto G, Zelenina M, Li D, et al. Arginine vasopressin stimulates phosphorylation of aquaporin-2 in rat renal tissue. Am J Physiol 1999;276:F254–F259. [DOI] [PubMed] [Google Scholar]
- 32.Hoffert JD, Pisitkun T, Wang G, Shen RF, Knepper MA. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: regulation of aquaporin-2 phosphorylation at two sites. Proc Natl Acad Sci U S A 2006;103:7159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffert JD, Fenton RA, Moeller HB, et al. Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J Biol Chem 2008;283:24617–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katsura T, Gustafson CE, Ausiello DA, Brown D. Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. Am J Physiol 1997;272:F817–F822. [PubMed] [Google Scholar]
- 35.Fushimi K, Sasaki S, Marumo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem 1997;272:14800–4. [DOI] [PubMed] [Google Scholar]
- 36.van Balkom BW, Savelkoul PJ, Markovich D, et al. The role of putative phosphorylation sites in the targeting and shuttling of the aquaporin-2 water channel. J Biol Chem 2002;277:41473–9. [DOI] [PubMed] [Google Scholar]
- 37.Moeller HB, Knepper MA, Fenton RA. Serine 269 phosphorylated aquaporin-2 is targeted to the apical membrane of collecting duct principal cells. Kidney Int 2009;75:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moeller HB, Praetorius J, Rützler MR, Fenton RA. Phosphorylation of aquaporin-2 regulates its endocytosis and protein-protein interactions. Proc Natl Acad Sci U S A 2010;107:424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rinschen MM, Yu MJ, Wang G, et al. Quantitative phosphoproteomic analysis reveals vasopressin V2-receptor-depen- dent signaling pathways in renal collecting duct cells. Proc Natl Acad Sci U S A 2010;107:3882–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nedvetsky PI, Tabor V, Tamma G, et al. Reciprocal regulation of aquaporin-2 abundance and degradation by protein kinase A and p38-MAP kinase. J Am Soc Nephrol 2010;21:1645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu HJ, Matsuzaki T, Bouley R, Hasler U, Qin QH, Brown D. The phosphorylation state of serine 256 is dominant over that of serine 261 in the regulation of AQP2 trafficking in renal epithelial cells. Am J Physiol Renal Physiol 2008;295: F290–F294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valenti G, Procino G, Tamma G, Carmosino M, Svelto M. Minireview: aquaporin 2 trafficking. Endocrinology 2005; 146:5063–70. [DOI] [PubMed] [Google Scholar]
- 43.Noda Y, Horikawa S, Kanda E, et al. Reciprocal interaction with G-actin and tropomyosin is essential for aquaporin-2 trafficking. J Cell Biol 2008;182:587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DiGiovanni SR, Nielsen S, Christensen EI, Knepper MA. Regulation of collecting duct water channel expression by vasopressin in Brattleboro rat. Proc Natl Acad Sci U S A 1994;91:8984–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandoval PC, Slentz DH, Pisitkun T, et al. Proteome-wide measurement of protein half-lives and translation rates in vasopressin-sensitive collecting duct cells. J Am Soc Nephrol 2013;24:1793–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kamsteeg EJ, Hendriks G, Boone M, et al. Short-chain ubiquitination mediates the regulated endocytosis of the aquapo- rin-2 water channel. Proc Natl Acad Sci U S A 2006;103:18344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsumura Y, Uchida S, Rai T, Sasaki S, Marumo F. Transcriptional regulation of aquaporin-2 water channel gene by cAMP. J Am Soc Nephrol 1997;8:861–7. [DOI] [PubMed] [Google Scholar]
- 48.Wilson JL, Miranda CA, Knepper MA. Vasopressin and the regulation of aquaporin-2. Clin Exp Nephrol 2013;17:751–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weingarten-Gabbay S, Segal E. The grammar of transcriptional regulation. Hum Genet 2014;133:701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu MJ, Miller RL, Uawithya P, et al. Systems-level analysis of cell-specific AQP2 gene expression in renal collecting duct. Proc Natl Acad Sci U S A 2009;106: 2441–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schrier RW. Vasopressin and aquaporin 2 in clinical disorders of water homeostasis. Semin Nephrol 2008;28:289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson C, Hoorn EJ. Hyponatraemia: an overview of frequency, clinical presentation and complications. Best Pract Res Clin Endocrinol Metab 2012;26:Suppl 1:S1–S6. [DOI] [PubMed] [Google Scholar]
- 53.Fujita N, Ishikawa SE, Sasaki S, et al. Role of water channel AQP-CD in water retention in SIADH and cirrhotic rats. Am J Physiol 1995;269:F926–F931. [DOI] [PubMed] [Google Scholar]
- 54.Ecelbarger CA, Nielsen S, Olson BR, et al. Role of renal aquaporins in escape from vasopressin-induced antidiuresis in rat. J Clin Invest 1997;99:1852–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ecelbarger CA, Chou CL, Lee AJ, Di-Giovanni SR, Verbalis JG, Knepper MA. Escape from vasopressin-induced antidiuresis: role of vasopressin resistance of the collecting duct. Am J Physiol 1998; 274:F1161–F1166. [DOI] [PubMed] [Google Scholar]
- 56.Jovanovich AJ, Berl T. Where vaptans do and do not fit in the treatment of hyponatremia. Kidney Int 2013;83:563–7. [DOI] [PubMed] [Google Scholar]
- 57.Ecelbarger CA, Terris J, Frindt G, et al. Aquaporin-3 water channel localization and regulation in rat kidney. Am J Physiol 1995;269:F663–F672. [DOI] [PubMed] [Google Scholar]
- 58.Roudier N, Ripoche P, Gane P, et al. AQP3 deficiency in humans and the molecular basis of a novel blood group system, GIL. J Biol Chem 2002;277:45854–9. [DOI] [PubMed] [Google Scholar]
- 59.Ma T, Song Y, Yang B, et al. Nephrogenic diabetes insipidus in mice lacking aquaporin-3 water channels. Proc Natl Acad Sci U S A 2000;97:4386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chou CL, Ma T, Yang B, Knepper MA, Verkman AS. Fourfold reduction of water permeability in inner medullary collecting duct of aquaporin-4 knockout mice. Am J Physiol 1998;274:C549–C554. [DOI] [PubMed] [Google Scholar]
- 61.Blount MA, Mistry AC, Fröhlich O, et al. Phosphorylation of UT-A1 urea transporter at serines 486 and 499 is important for vasopressin-regulated activity and membrane accumulation. Am J Physiol Renal Physiol 2008;295:F295–F299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bansal AD, Hoffert JD, Pisitkun T, et al. Phosphoproteomic profiling reveals vasopressin-regulated phosphorylation sites in collecting duct. J Am Soc Nephrol 2010; 21:303–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fenton RA, Chou CL, Stewart GS, Smith CP, Knepper MA. Urinary concentrating defect in mice with selective deletion of phloretin-sensitive urea transporters in the renal collecting duct. Proc Natl Acad Sci U S A 2004;101:7469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fenton RA, Flynn A, Shodeinde A, Smith CP, Schnermann J, Knepper MA. Renal phenotype of UT-A urea transporter knockout mice. J Am Soc Nephrol 2005; 16:1583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kokko JP, Rector FC Jr. Countercurrent multiplication system without active transport in inner medulla. Kidney Int 1972;2:214–23. [DOI] [PubMed] [Google Scholar]
- 66.Tomita K, Pisano JJ, Knepper MA. Control of sodium and potassium transport in the cortical collecting duct of the rat: effects of bradykinin, vasopressin, and deoxycorticosterone. J Clin Invest 1985;76:132–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reif MC, Troutman SL, Schafer JA. Sodium transport by rat cortical collecting tubule: effects of vasopressin and des-oxycorticosterone. J Clin Invest 1986;77: 1291–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tomita K, Pisano JJ, Burg MB, Knep-per MA. Effects of vasopressin and bradykinin on anion transport by the rat cortical collecting duct: evidence for an electroneutral sodium chloride transport pathway. J Clin Invest 1986;77:136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leviel F, Hübner CA, Houillier P, et al. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 2010;120:1627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Snyder PM. Minireview: regulation of epithelial Na+ channel trafficking. Endocrinology 2005;146:5079–85. [DOI] [PubMed] [Google Scholar]
- 71.Ecelbarger CA, Kim GH, Terris J, et al. Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. Am J Physiol Renal Physiol 2000; 279:F46–F53. [DOI] [PubMed] [Google Scholar]
- 72.Nicco C, Wittner M, DiStefano A, Jounier S, Bankir L, Bouby N. Chronic exposure to vasopressin upregulates ENaC and sodium transport in the rat renal collecting duct and lung. Hypertension 2001; 38:1143–9. [DOI] [PubMed] [Google Scholar]
- 73.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 1999;104:R19–R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Christensen BM, Perrier R, Wang Q, et al. Sodium and potassium balance depends on aENaC expression in connecting tubule. J Am Soc Nephrol 2010;21: 1942–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Furgeson SB, Linas S. Mechanisms of type I and type II pseudohypoaldosteron-ism. J Am Soc Nephrol 2010;21:1842–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.