

Abstract
A mild, modular, and practical catalytic system for the synthesis of the highly privileged phenethylamine pharmacophore is reported. Using a unique combination of organic catalysts to promote the transfer of electrons and hydrogen atoms, this system performs direct hydroarylation of vinyl amine derivatives with a wide range of aryl halides (including aryl chlorides). This general and highly chemoselective protocol delivers a broad range of arylethylamine products with complete regiocontrol. The utility of this process is highlighted by its scalability and the modular synthesis of an array of bioactive small molecules.
Graphical Abstract

The arylethylamine sca ffold is a key pharmacophore in endogenous neurotransmitters, natural products, and pharmaceuticals that accomplishes a wide range of important functions (a small sampling of which is provided in Figure 1). While this molecular sca ffold can be produced through a range of classical technologies (e.g., amino acid decarboxylation or reductive amination) that utilize functionalized precursors, the ability to access this structural array with modular flexibility remains limited. Specifically, we consider catalytic methods for direct arylethylamine synthesis from readily available synthons where the alteration of the aryl unit, ethyl skeleton, and nitrogen atom would be particularly powerful if they utilized readily available starting materials. Toward this aim, significant progress has been made in intermolecular anti-Markovnikov styrene hydroamination using N,N-dialkylamines,1–4 N-arylamines,5 and sulfonamides.6,7 As a complement to these technologies, we became interested in developing a process for the hydroarylation of vinylamine derivatives, where the flexible substitution of nitrogen would be possible. The utility of this method would be partially driven by its capacity to deliver the desired products from widely available precursors with complete anti-Markovnikov selectivity and good functional group compatibility. We envisioned that a mechanism for the reductive activation of aryl halides to the corresponding radicals and intermolecular addition could be utilized to accomplish these goals.
Figure 1.

Modular catalytic strategies for arylethylamine synthesis.
Aryl radicals are highly reactive intermediates that readily engage a range of unsaturated systems.8,9 As an alternative to arenediazonium salt-based approaches, the single-electron reduction of aryl halides using photoredox catalysts10,11 is a powerful method for aryl radical formation from stable starting materials.12–19 Building on previous results by Stephenson,12 Konig,20,21 and Read de Alaniz and Hawker, we have shown that pyridyl radicals (accessed via pyridyl halide reduction) undergo chemoselective intermolecular coupling with either electron-deficient22,23 or electron-rich olefins,24,25 and Weaver has reported a number of processes involving azolyl or perfluoroaryl radicals.16 However, because aryl halide reduction potentials are very negative and the resulting aryl radicals undergo rapid reduction through hydrogen atom transfer (HAT), a general translation of these findings to aryl systems has yet to materialize.
From the outset, we recognized two elements that would be critical to the success of the proposed transformation: a powerful catalytic reductant (capable of aryl halide activation) and a catalytic hydrogen atom source (such that the rate of aryl radical addition to vinylamines would be competitive with reduction pathways involving HAT). Accordingly, we reasoned that the highly reducing N-phenylphenothiazine (PTH) and cyclohexanethiol (CySH) could operate in concert through transferring electrons and hydrogen atoms, respectively, as illustrated in Figure 2. Specifically, iodobenzene (E1/20 = −1.51 to −2.20 V vs SCE)12 activation via single-electron transfer (SET) from photoexcited PTH (E1/2* = −2.10 V vs SCE)14 would give rise to the corresponding radical anion. Rapid mesolytic fragmentation would expel iodide, thereby delivering the neutral phenyl radical. Regioselective intermolecular addition to the vinylcarbamate substrate would deliver the nucleophilic α-carbamoyl radical, which would undergo polarity-matched HAT from the electrophilic thiol catalyst.26 This event would concurrently furnish desired product 1 and thiyl species CyS•. Finally, the regeneration of both catalysts (via HAT and SET events) would liberate innocuous byproducts CO2 and NaI.
Figure 2.

Proposed dual-catalytic mechanism for intermolecular radical hydroarylation.
In practice, we found that iodobenzene e ffectively reacts with tert-butylvinylcarbamate (2.5 equiv) in the presence of 5 mol % of each catalyst and 3 equiv of sodium formate under irradiation with blue light in 5% H2O/DMSO, a ffording the desired adduct as a single regioisomer (82% isolated yield). Control experiments indicated that all of the reaction components are required for e ffective conversion of the starting materials. This mechanistic proposal is supported by Stern−Volmer experiments and an experiment with alternating light−dark periods. (Optimization and mechanistic experiments are given in the Supporting Information.) To more accurately interrogate the potential contributions of short radical chains in light of the observed results,27 we measured the quantum yield of this process. While Φ = 0.29 is most consistent with a photosensitized mechanism, radical chains may contribute to product formation.
Further evaluation revealed that the aryl iodide scope of this transformation is broad. As shown in Table 1, iodobenzene and derivatives that contained chloride or triflate substituents reacted smoothly with complete retention of the electrophilic cross-coupling handles (1−3, 70−88% yield). Electron-poor arenes were excellent substrates here, and the para-trifluoromethyl and -carboxymethyl groups were not a ffected (4 and 5, 85 and 73% yields, respectively). Electron-releasing substituents (methoxy, carbamate, and hydroxy) were tolerated, although yields of the desired products were slightly lower (6−9, 60−72%). These results are in line with the assertion that the aryl radical SOMO lies within the plane of the arene such that the presence of electron-donating or -withdrawing substituents does not significantly impact the reactivity.28 We also found that substitution is tolerated at various positions around the aryl ring, and as expected, acidic elements did not negatively impact the reaction efficiency.
Table 1.
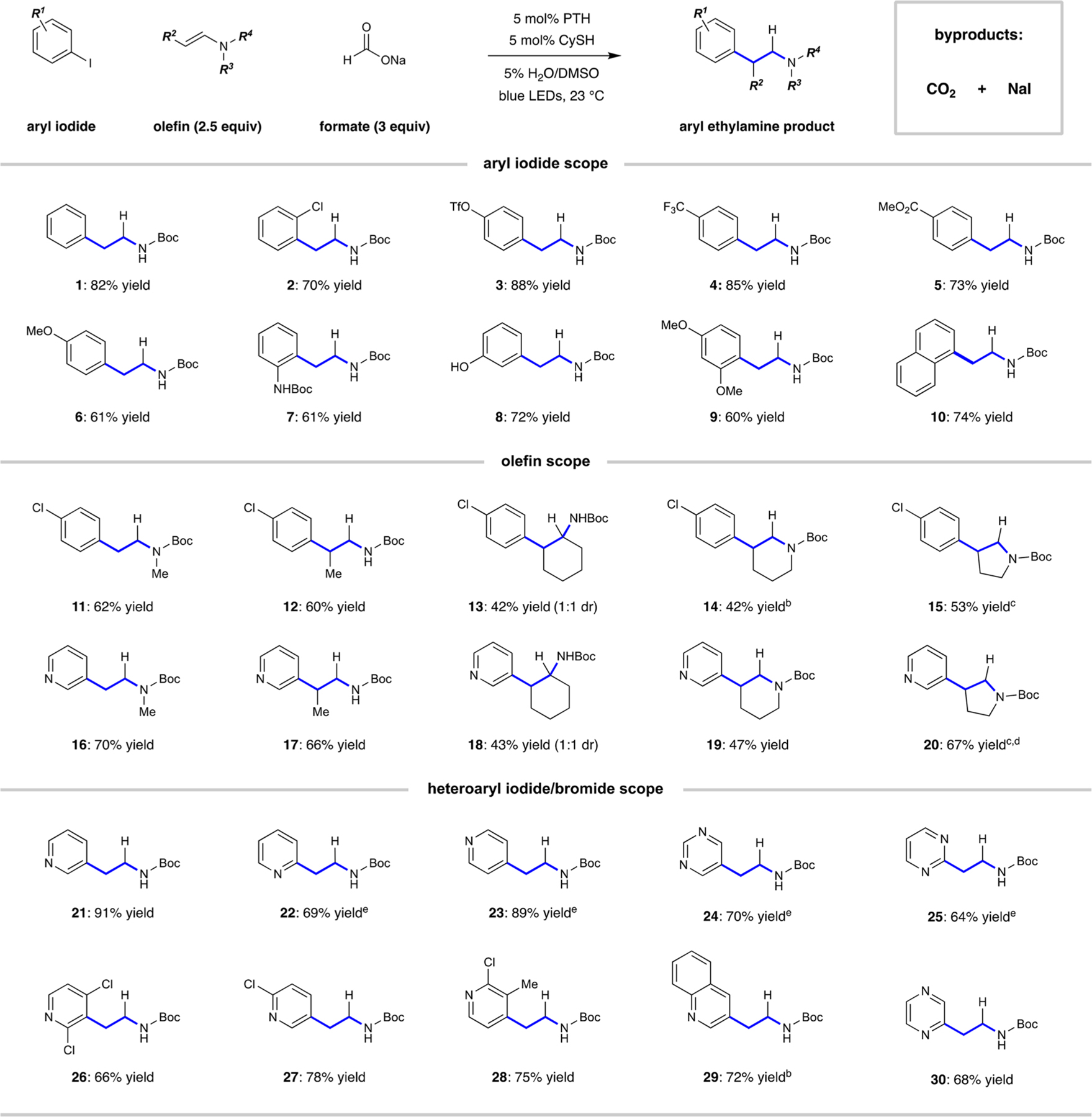 |
Reaction conditions: Iodoarene (1.0 mmol), olefin (2.5 mmol), PTH (5 mol %), CySH (5 mol %), sodium formate (3.0 mmol), 5% (v/v) H2O/ DMSO (10.0 mL), blue light, 16 h; isolated yields given.
The reactions to produce 14 and 29 were conducted on a 0.5 mmol scale (1-chloro-4-iodobenzene).
In the reactions to produce 15 and 20, N-1-naphthylphenothiazine (5 mol %) was used in place of PTH.
In the reaction to produce 20, olefin (1.0 mmol) was used as a limiting reagent with 5.0 mmol of 3-iodopyridine.
In the reactions to produce 22−25, corresponding heteroaryl bromide was used as the aryl substrate.
Also demonstrated in Table 1 is the ability of this mechanistic blueprint to tolerate the substitution of the olefinic partner on nitrogen (11) and at the β-position (12). Hydroarylation of a cyclic encarbamate, derived from cyclohexanone through vinyl triflate formation and subsequent C− N coupling with tert-butylcarbamate, occurred in a moderate yield of 42%, delivering 13 as a mixture of diastereomers (1:1 dr). Furthermore, this strategy is useful for the synthesis of complex saturated nitrogen heterocycles through the transformation of endocyclic enecarbamate substrates. Specifically, the 3-arylpiperidine (14) framework was accessible under this paradigm, albeit in diminished yield (42%). This is noteworthy because this particular motif has proven challenging to access directly, where 3-arylpyridine reduction or a lactam α-arylation and reduction sequence were utilized in the development of Zejula.29,30 Likewise, a dihydropyridine derivative underwent hydroarylation to a fford 3-phenylpyrrolidine (15) in 53% yield.
Given our experience with the complications associated with intermolecular heteroaryl radical coupling, we were pleased to find that halogenated heteroaromatics were good substrates under these conditions. Intermolecular coupling of 3-iodopyridine with the same vinylamine derivatives a fforded pyridylethylamines 16−20 in comparable yields (43−90%). Moreover, these conditions generally activate a range of other halogenated pyridines, where regiospecific radical formation enables alkylation at the 2-, 3-, or 4-position in 66−91% yield. As demonstrated before, iodide cleavage occurs with excellent fidelity, even in the presence of chloride substituents. Pyridylethylamines 26−28 were produced in good yield without overreduction products, thus retaining the ability to perform subsequent cross-coupling or nucleophilic aromatic substitution (SNAr) reactions. In addition, products 26 and 28 further illustrate the ability of this system to tolerate ortho substitution. The incorporation of other nitrogen-containing heterocyclic units was seamless, where the reduction of halogenated pyrimidine (24 and 25), quinoline (29), or pyrazine (30) substrates resulted in acceptable amounts of alkylated products.
The outlined dual catalytic process employs inexpensive organic compounds as catalysts and reagents. Moreover, it is highly tolerant of important functional groups and completely selective for the linear hydroarylation regioisomers. However, we recognize that the requirement for aryl iodide substrates as radical precursors is suboptimal because they are relatively expensive in comparison to the corresponding aryl bromides or chlorides. While these substrate classes are cheaper and more accessible, they are more difficult to reduce (with aryl chloride reduction potentials being the most negative throughout this series). However, as demonstrated earlier by Read de Alaniz and Hawker,14 reductive activation of these classes can be performed using PTH. Indeed, activated chlorobenzene derivatives couple readily with the benchmark vinyl carbamate under standard conditions. As indicated in Scheme 1, 4-chlorobenzonitrile and ethyl-2-chlorobenzoate (E1/20 = −2.00 to −2.10 V vs SCE)31 were e ffectively converted to the corresponding arylethylamines (31 and 32: 86% and 88 yields, respectively), but chlorobenzene (E0 = −2.79 V vs SCE)31 conversion was slower (28% yield after 16 h), where the mass balance was largely composed of unreacted chlorobenzene.
Scheme 1.
Hydroarylation Reactions of Aryl Chlorides and the Basis for Intermolecular Reactivitya,b,c,d a
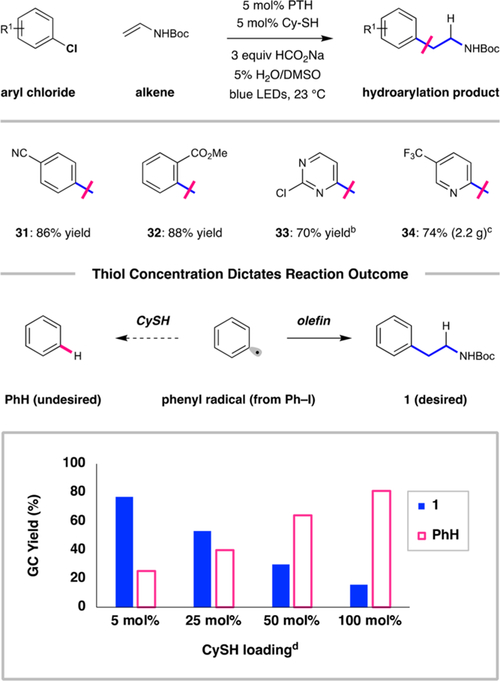
aReactions were conducted as in Table 1 using the indicated aryl chloride. bThe reaction to produce 33 was performed on a 0.5 mmol scale. cThe reaction to produce 34 was performed on a 10 mmol scale. dIn the bar graph, product ratios were determined using GC with an internal standard.
The alkylation of 2,4-dichloropyrimidine occurred exclusively at the 4-position, giving 33 in 70% yield. The selectivity here parallels those which were observed in SNAr or Pd-catalyzed cross-coupling processes. The retention of chloride substituents (in products 16−20, 31−33, and 38) is possible presumably because the installation of the alkyl substituent via hydroarylation would e ffectively increase the reduction potential, thereby protecting the product from subsequent activation. Without an alteration of the standard conditions, this catalytic protocol functioned on a 10 mmol scale from the chloropyridine, a ffording 2.15 g of trifluoromethylpyridine product 34 (74% yield), further illustrating the utility of this protocol (Scheme 1). Importantly, tert-butylvinylcarbamate can be accessed on a large scale (100 mmol) in a single pot from inexpensive reagents.
This design enables the remarkably e ffective intermolecular reactivity of aryl radial species by employing a combination of a thiol HAT catalyst and a stoichiometric formate reductant. As illustrated in Scheme 1, there are two competing pathways for the aryl radical intermediates that are produced here: intermolecular addition to olefins (desired) and reduction by HAT from the electrophilic thiol (undesired). The relative rates of these pathways can be conveniently manipulated by varying the thiol loading, and the yields of the desired product were highest using aliphatic thiol catalysts. (See the SI for details.) Increasing the thiol loading was accompanied by a clear increase in arene reduction through HAT. Optimal conditions (with 5 mol % CySH, Ph−I a as radical precursor) delivered hydroarylation product 1 in 78% yield. In contrast, when a full equivalent of thiol (100 mol %) was utilized, the selectivity was completely overturned, giving benzene (PhH) as the major product (81% yield). While it is understood that radical anion fragmentation rates vary with the halide substituent, we propose that this system operates uniformly via neutral aryl radicals, regardless of the Ar−X substrate class. This hypothesis is supported by thiol loading experiments across a series of halobenzene substrates (using methyl-4-iodo, -bromo, and -chlorobenzoate), where the same thiol-dependent product ratios were observed throughout.
The arylethylamine sca ffold is conserved across a wide range of natural and synthetic neuromodulators. To demonstrate its practical utility, we applied this protocol to the synthesis of endogenous neurotransmitter dopamine as well as to the trace amine-associated receptor (TAAR) agonists32 that are shown in Scheme 2. This system allows for modular substitution of the aryl unit, a ffording phenethylamine (35), all three methylphenethylamine isomers (36−38), and dopamine (39) through a two-step hydroarylation/boc-deprotection sequence (66−75% yield). In addition, this method allows for the flexible substitution of the nitrogen atom, where tyramine (40), N-methyltyramine (41), and hordenine (42) were all accessible in two steps from tert-butyl (4-iodophenyl) carbonate.
Scheme 2.
Flexible Synthesis of Neuromodulatorsa,b
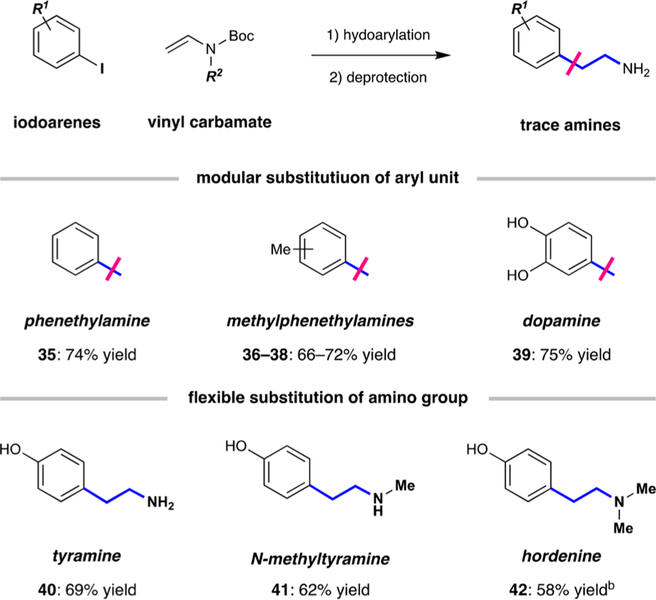
aHydroarylation was accomplished as in Table 1, and Boc deprotection was performed with acid. bHordenine synthesis was accomplished by reduction with LiAlH4.
To illustrate the potential of this process in the early-stage development of medicines and agrochemicals, we reacted 2-bromopyridine (43) with N-vinylbenzamide (44) to directly a fford fungicide Fluopyram (45),33,34 as shown in Scheme 3. Optimal conditions for this transformation involved inverted stoichiometry (using 3 equiv of radical precursor 43). This olefinic partner is available in a single step from commercial materials.
Scheme 3.
Rapid Synthesis of Fluopyram/Analogsa

aReaction conditions: haloarene (3.0 mmol), olefin (1.0 mmol), PTH (5 mol %), CySH (5 mol %), sodium formate (3.0 mmol), 5% (v/v) H2O/ DMSO (10.0 mL), blue light, 16 h; isolated yields are given.
This two-step sequence compares favorably to the patent route of this agrochemical (seven steps). The value of this modular hydroarylation strategy is further highlighted by the expedient preparation of Fluopyram analogs 46−50, where systematic substitution on either side of the sca ffold could be accomplished with excellent fidelity (82−92% yield for the corresponding hydroarylation processes).
In conclusion, we have developed a protocol for the intermolecular addition of aryl and heteroaryl radicals to enecarbamate substrates. This process operates at ambient temperature, mediated by the concerted action of two di fferent catalytic species (PTH and CySH) that accomplish the transfer of electrons and hydrogen atoms, respectively. This system directly a ffords valuable arylethylamine structures with complete regiocontrol with excellent functional group compatibility, and it utilizes stable halogenated arenes as radical precursors. The highly reducing character of the organic photoredox catalyst here allows for e ffective activation of a wide range of aryl halides, including electron-deficient aryl chlorides. We expect that this protocol, founded on the use of a thiol HAT catalyst in combination with a stoichiometric reductant, will enable a range of mild aryl radical-based transformations.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by funds from the National Institutes of Health (GM129495), and NMR data were collected under support of the National Science Foundation (CHE-1531620). Z.X. was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Solar Photochemistry Program under award number DE-FG02–07ER-15906. The authors thank Professors R. Brian Dyer, Jennifer Heemstra, and Tianquan Lian (Emory University) for generous access to instrumentation and Henry Zecca (Emory University) for his assistance in preparing cyclic enecarbamates.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the
ACS Publications website at DOI: 10.1021/jacs.9b01077.
Experimental procedures and spectral data (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Utsunomiya M; Kuwano R; Kawatsura M; Hartwig JF Rhodium-Catalyzed Anti-Markovnikov Hydroamination of Vinylarenes. J. Am. Chem. Soc 2003, 125, 5608–5609. [DOI] [PubMed] [Google Scholar]
- (2).Utsunomiya M; Hartwig JF Ruthenium-Catalyzed AntiMarkovnikov Hydroamination of Vinylarenes. J. Am. Chem. Soc 2004, 126, 2702–2703. [DOI] [PubMed] [Google Scholar]
- (3).Basalov IV; Rosça SC; Lyubov DM; Selikhov AN; Fukin GK; Sarazin Y; Carpentier JF; Trifonov AA. Divalent Heteroleptic Ytterbium Complexes Effective Catalysts for Intermolecular Styrene Hydrophosphination and Hydroamination. Inorg. Chem 2014, 53, 1654–1661. [DOI] [PubMed] [Google Scholar]
- (4).Musacchio AJ; Lainhart BC; Zhang X; Naguib SG; Sherwood TC; Knowles RR Catalytic Intermolecular Hydroaminations of Unactivated Olefins with Secondary Alkyl Amines. Science 2017, 355, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bronner SM; Grubbs RH Formal Anti-Markovnikov Hydroamination of Terminal Olefins. Chem. Sci 2014, 5, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nguyen TM; Manohar N; Nicewicz DA Anti-Markovnikov Hydroamination of Alkenes Catalyzed by a Two-Component Organic Photoredox System: Direct Access to Phenethylamine Derivatives. Angew. Chem., Int. Ed 2014, 53, 6198–6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Monos TM; Mcatee RC; Stephenson CRJ Arylsulfonylacetamides as Bifunctional Reagents for Alkene Aminoarylation. Science 2018, 361, 1369–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Heinrich MR Intermolecular Olefin Functionalisation Involving Aryl Radicals Generated from Arenediazonium Salts. Chem. - Eur. J 2009, 15, 820–833. [DOI] [PubMed] [Google Scholar]
- (9).Kindt S; Heinrich MR Recent Advances in Meerwein Arylation Chemistry. Synthesis 2016, 48, 1597–1606. [Google Scholar]
- (10).Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- (11).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Nguyen JD; D’Amato EM; Narayanam JMR; Stephenson CRJ Engaging Unactivated Alkyl, Alkenyl and Aryl 2012, 4, 854–859. [DOI] [PubMed] [Google Scholar]
- (13).Kim H; Lee C Visible-Light-Induced Photocatalytic Reductive Transformations of Organohalides. Angew. Chem., Int. Ed 2012, 51, 12303–12306. [DOI] [PubMed] [Google Scholar]
- (14).Discekici EH; Treat NJ; Poelma SO; Mattson KM; Hudson ZM; Luo Y; Hawker CJ; de Alaniz JR A Highly Reducing Metal-Free Photoredox Catalyst: Design and Application in Radical Dehalogenations. Chem. Commun 2015, 51, 11705–11708. [DOI] [PubMed] [Google Scholar]
- (15).Poelma SO; Burnett GL; Discekici EH; Mattson KM; Treat NJ; Luo Y; Hudson ZM; Shankel SL; Clark PG; Kramer JW; Hawker CJ; Read de Alaniz J Chemoselective Radical Dehalogenation and C-C Bond Formation on Aryl Halide Substrates Using Organic Photoredox Catalysts. J. Org. Chem 2016, 81, 7155–7160. [DOI] [PubMed] [Google Scholar]
- (16).Arora A; Weaver JD Visible Light Photocatalysis for the Generation and Use of Reactive Azolyl and Polyfluoroaryl Intermediates. Acc. Chem. Res 2016, 49, 2273–2283. [DOI] [PubMed] [Google Scholar]
- (17).Ghosh I; Marzo L; Das A; Shaikh R; Konig B Visible Light Mediated Photoredox Catalytic Arylation Reactions. Acc. Chem. Res 2016, 49, 1566–1577. [DOI] [PubMed] [Google Scholar]
- (18).Naumann R; Kerzig C; Goez M Laboratory-Scale Photoredox Catalysis Using Hydrated Electrons Sustainably Generated with a Single Green Laser. Chem. Sci 2017, 8, 7510–7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Matsubara R; Yabuta T; Idros U; Hayashi M; Ema F; Kobori Y; Sakata K UVA- and Visible-Light-Mediated Generation of Carbon Radicals from Organochlorides Using Nonmetal Photocatalyst. J. Org. Chem 2018, 83, 9381–9390. [DOI] [PubMed] [Google Scholar]
- (20).Ghosh T; Ghosh J; Bardagi I; König B. Reduction of Aryl Halides by Consecutive Visible Light-Induced Electron Transfer Processes. Science 2014, 346, 725–728. [DOI] [PubMed] [Google Scholar]
- (21).Bardagi JI; Ghosh I; Schmalzbauer M; Ghosh T; König B. Anthraquinones as Photoredox Catalysts for the Reductive Activation of Aryl Halides. Eur. J. Org. Chem 2018, 2018, 34–40. [Google Scholar]
- (22).Aycock RA; Wang H; Jui NT A Mild Catalytic System for Radical Conjugate Addition of Nitrogen Heterocycles. Chem. Sci 2017, 8, 3121–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Aycock RA; Vogt D; Jui N A Practical and Scalable System for Heteroaryl Amino Acid Synthesis. Chem. Sci 2017, 8, 7998–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Boyington AJ; Riu MLY; Jui NT Anti-Markovnikov Hydroarylation of Unactivated Olefins via Pyridyl Radical Intermediates. J. Am. Chem. Soc 2017, 139, 6582–6585. [DOI] [PubMed] [Google Scholar]
- (25).Seath CP; Vogt DB; Xu Z; Boyington AJ; Jui NT Radical Hydroarylation of Functionalized Olefins and Mechanistic Investigation of Photocatalytic Pyridyl Radical Reactions. J. Am. Chem. Soc 2018, 140, 15525–15534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Roberts BP Polarity-Reversal Catalysis of Hydrogen-Atom Abstraction Reactions : Concepts and Applications in Organic Chemistry. Chem. Soc. Rev 1999, 28, 25–35. [Google Scholar]
- (27).Cismesia MA; Yoon TP Characterizing Chain Processes in Visible Light Photoredox Catalysis. Chem. Sci 2015, 6, 5426–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Garden SJ; Avila DV; Beckwit ALJ; Bowry VW; Ingold KU; Lusztyk J Absolute Rate Constant for the Reaction of Aryl Radicals with Tri-N-Butyltin Hydride. J. Org. Chem 1996, 61, 805–809. [DOI] [PubMed] [Google Scholar]
- (29).Jones P; Wilcoxen K; Rowley M; Toniatti C Niraparib: A Poly(ADP-Ribose) Polymerase (PARP) Inhibitor for the Treatment of Tumors with Defective Homologous Recombination. J. Med. Chem 2015, 58, 3302–3314. [DOI] [PubMed] [Google Scholar]
- (30).Chung CK; Bulger PG; Kosjek B; Belyk KM; Rivera N; Scott ME; Humphrey GR; Limanto J; Bachert DC; Emerson KM Process Development of C − N Cross-Coupling and Enantioselective Biocatalytic Reactions for the Asymmetric Synthesis of Niraparib. Org. Process Res. Dev 2014, 18, 215–227. [Google Scholar]
- (31).Enemærke RJ; Christensen TB; Jensen H; Daasbjerg K Application of a New Kinetic Method in the Investigation of Cleavage Reactions of Haloaromatic Radical Anions. J. Chem. Soc., Perkin Trans 2001, 2, 1620–1630. [Google Scholar]
- (32).Gainetdinov RR; Hoener MC; Berry MD Trace Amines and Their Receptors. Pharmacol. Rev 2018, 70, 549–620. [DOI] [PubMed] [Google Scholar]
- (33).Lhermitte F; Coqueron P-Y; Desbordes P; Himmlet T WO2006/067103, June 29, 2006.
- (34).Moradi WA; Schnatterer A; Bielefeldt D; Gertzmann R; Havekest D WO2018/114484, June 28, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.