

Abstract
In recent decades, the majority of ligands developed for the vitamin D receptor (VDR) bind at its deeply buried genomic ligand binding pocket. Theses ligands can be categorized into agonists and partial agonists/antagonists. A limited number of ligands, most of them peptides, bind the VDR–coactivator binding site that is formed in the presence of an agonist and inhibit coactivator recruitment, and therefore transcription. Another solvent exposed VDR–ligand binding pocket was identified for lithocholic acid, improving the overall stability of the VDR complex. Additional proposed interactions with VDR are discussed herein that include the alternative VDR–ligand binding pocket that may mediate both non-genomic cellular responses and binding function 3 that was identified for the androgen receptor. Many VDR ligands increase blood calcium levels at therapeutic concentrations in vivo, thus the identification of alternative VDR–ligand binding pockets might be crucial to develop non-calcemic and potent ligands for VDR to treat cancer and inflammatory disease.
Keywords: vitamin D receptor; ligand; alternative VDR–ligand binding pocket; binding function 3; VDR–coactivator inhibitor; 1,25(OH)2D3
1. Introduction
The vitamin D receptor (VDR) is a ligand-activated transcription factor and belongs to the superfamily of nuclear receptors, as recently reviewed (Pike et al., 2018). The receptor is expressed primarily in the epithelia of endocrine organs (e.g. parathyroid gland, mammary gland), digestive system, bronchi, kidneys, and thymus (Wang et al., 2012). In addition, VDR can be found in leukocytes and bone cells. Many natural occurring VDR ligands have been identified including vitamin D metabolites with a secosteroid structure and bile acids that partially control VDR function in the intestine. For a recent review see (Makishima and Yamada, 2018). In the absence of a genomic agonist, VDR can be found in the cytosol or attached to the cell membrane (Huhtakangas et al., 2004). Ligand binding will induce nuclear localization of VDR and promote VDR–DNA complexation in conjunction with the retinoid X receptor (RXR) (Figure 1) (Orlov et al., 2012). Specific VDR response elements have been identified in the promoter sequences of genes that are induced or repressed by VDR (Haussler et al., 2011). VDR-mediated gene regulation is dependent on interactions with coregulators (coactivators and corepressors). These proteins are part of the transcriptional complex that interacts with RNA polymerase II and other binding proteins in addition to DNA, as reviewed by (White et al., 2018). Chromatin remodeling and specific gene transcription are nuclear functions of these transcriptional VDR complexes, which have been summarized in (Nurminen et al., 2018).
Figure 1.
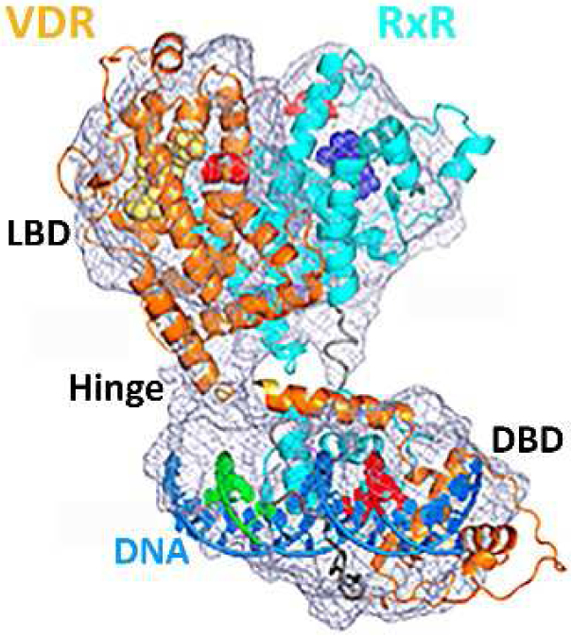
Cryo-electron microscopy structure of liganded human VDR and RXR, bound to DNA and coactivator peptides. (Redrawn after Orlov et al., 2012)
2. VDR and its genomic ligand binding pocket
VDR can be divided into the following domains: N-terminus, DNA binding domain (DBD), a hinge region that enables flexibility for dimerization, DNA binding domain, and a ligand binding domain (LBD) (Figure 2a). The LBD consists of 12 helices and forms a compact three dimensional structure, especially when bound to a ligand (Figure 2b). The ligand binding pocket is buried deeply in the receptor, which enables very specific interactions with natural ligands, most prominently 1,25-dihydroxy vitamin D3 (1,25(OH)2D3) (Figure 2c). Thousands of VDR ligands have been synthesized in recent decades that bind at this particular ligand binding pocket (reviewed by (Verlinden et al., 2018). It has been shown that these ligands influence the shape of the binding pocket, which was elegantly shown with the Gemini ligand bearing two alkyl side chain substituents (Norman et al., 2000). The occupation of an additional 25% volume in comparison to 1,25(OH)2D3 resulted in higher transcriptional activity and pronounced anticancer activity of Gemini. Other recently reviewed VDR ligands include (Belorusova and Rochel, 2018) and antagonists (Saitoh, 2018). Furthermore, VDR ligands with multiple aromatic rings have been developed that, in contrast to 1,25(OH)2D3, did not elevate blood calcium levels (Stites et al., 2018). Commercialized VDR ligands include tacalcitol and calcipotriol for psoriasis, doxercalciferol, falecalcitriol, and maxacacitol for secondary hyperparathyroidism, and eldecalcitol for osteoporosis (Leyssens et al., 2014). During the last three decades, anti-cancer properties of vitamin D analogs have been demonstrated in vivo, however, one of the most promising vitamin D analogs, seocalcitol, showed insufficient efficacy in phase II clinical trials (Dalhoff et al., 2003). The biochemical changes induced by VDR ligands in cancer cells include gene regulation, activation and inhibition of enzymatic pathways, and overall changes in cell differentiation and proliferation (Van Driel et al., 2018). The specific changes for different cancers were recently reviewed for leukemia (Studzinski et al., 2018), breast cancer (Beaudin and Welsh, 2018), prostate, renal, and bladder cancer (Trump, 2018), colon cancer (Barbchano et al., 2018), skin cancer (Ransohoff et al., 2018), and lung cancer (Shaurova et al., 2018). In addition, some vitamin D analogs have anti-inflammatory properties that have prompted detailed investigations on the role VDR and its ligands play in inflammation and immunity summarized by (Mann et al., 2018).
Figure 2.
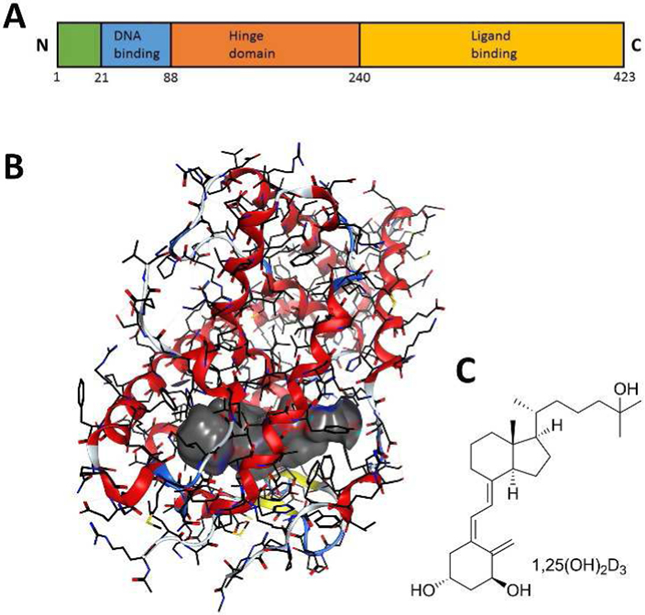
A) VDR domains; B) X-ray crystal structure of VDR-LBD bound to 1,25(OH)2D3 (gray surface) [1DB1]; C) Structure of 1,25(OH)2D3.
3. VDR and its predicted alternative (non-genomic) binding site
In addition to genomic actions mediated by VDR, many non-genomic fast cellular processes are triggered by vitamin D analogs and have been reviewed by (Hii and Ferrante, 2016). This includes activation of phospholipases (Bourdeau et al., 1990), phosphatases (Bettoun et al., 2004), kinases (de Boland and Norman, 1998), and voltage-gated ion channels (Zanello and Norman, 2004). VDR ligands such as 1,25(OH)2 lumisterol D3 (Figure 3B) weakly inhibit the VDR–1,25(OH)2D3 interaction, but potentiate chlorine ion channels in Sertoli cells at 1 nM, more effectively than 1,25(OH)2D3 (Menegaz et al., 2010). This prompted the hypothesis of an alternative VDR ligand binding pocket (A pocket) mediating non-genomic processes in cells (Mizwicki et al., 2004). Molecular modeling identified additional hydrophobic space in VDR-LBD by reversing the donor-acceptor interaction of H229 and Y295 (Figure 3A). Subsequent docking revealed that 1,25(OH)2 lumisterol D3 (Figure 3B) has a high affinity for the VDR “A pocket”, possibly supporting non-genomic pathways at very low concentrations. Molecular modeling also confirmed binding of a high energy β-chair conformation of 1,25(OH)2D3 to the “A pocket”, providing a possible explanation for non-genomic effects of 1,25(OH)2D3 in addition to its genomic effects (Mizwicki et al., 2004). The ability of VDR to bind different biomolecules, such as lipids (Jurutka et al., 2007), supported the hypothesis that the VDR “A pocket” might accommodate phosphatidyl-inositol (3,4,5)-trisphosphate and mediate the attachment of VDR to the cell membrane. Structural evidence for nuclear receptor–lipid binding was observed for nuclear receptor SF-1 (steroidogenic factor 1) and LRH-1 (liver receptor homolog 1) (Krylova et al., 2005). High affinity genomic agonists are expected to compete for VDR binding and enable nuclear localization. To support this hypothesis, specific binding assays are needed to identify and confirm molecular binding to the “A pocket” of VDR.
Figure 3.
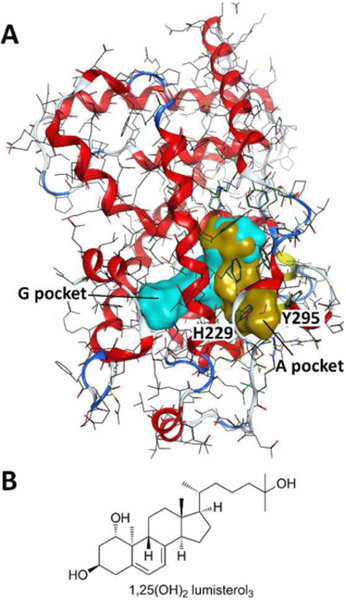
A) VDR-LBD with genomic pocket (cyan) and alternative pocket (gold) recreated using crystal structure [1DB1] and instruction from (Mizwicki et al. 2004); B) Structure of 1,25(OH)2 lumisterol D3
4. VDR and its coactivator binding site
The VDR coactivator binding site is formed when a VDR ligand is bound to the ligand binding pocket (genomic pocket or G pocket) of VDR. The agonistic ligand forms hydrogen bond interactions with VDR’s H305 located on the loop between helix 6 and helix 7 and H397 located on helix 11 (Figure 4A). This triggers a conformational change whereby helix 12 moves towards VDR-LBD and forms hydrogen bond interactions with I414, T415 and E420 (Figure 4A). F422, located on helix 12, interacts with H305 via a π-H interaction. The movement of helix 12 provides a hydrogen bond acceptor (E420) in the relative vicinity of hydrogen bond donor K246 (Figure 4B). These “charge clamp” residues form hydrogen bond interactions with coactivators and enable activation of VDR-mediated transcription by recruitment of RNA polymerase II to the transcription start site. Details of this process have been summarized by (Pike et al., 2018).
Figure 4.
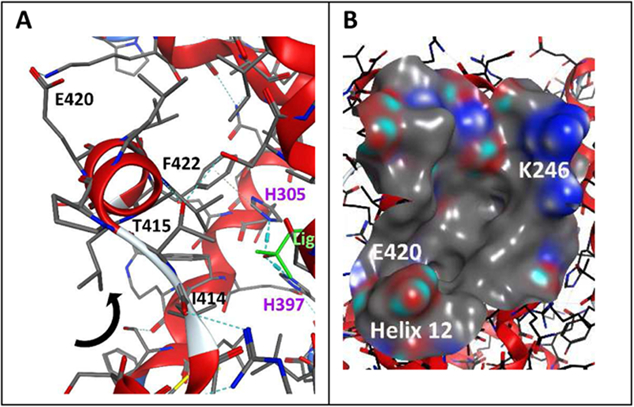
A) Interaction between ligand and VDR-LBD with helix 12 that form the coactivator binding pocket; B) Surface of the coactivator binding site with clamp charged residues K246 and E420 [1DB1, crystal structure].
5. Peptide-based ligands that bind the VDR coactivator pocket
Crystal structures of VDR-LBD in the presence of agonists and short coactivator peptides reveal the possibility of developing peptide-based ligands to inhibit the interaction between coactivators and VDR (Figure 5) (Vanhooke et al., 2004). To bind the predominately hydrophobic VDR coactivator binding site, successful peptide ligands have hydrophobic residues such as leucine. Proteomic mutation identified the most effective spacing of leucine residues to be LxxLL (L = leucine and X = any amino acid) with flanking residues to stabilize its helical structure (McInerney et al., 1998). Binding between synthetic peptides derived from coactivator amino acid sequences and VDR was determined by fluorescence polarization and alpha screen (Teichert et al., 2009). Peptides with strong VDR affinity have a general sequence of A/QLLRYLLDK/R. Furthermore, interactions between VDR and coactivator peptides were influenced by the structure of the VDR agonist (Zhang et al., 2010). To further improve VDR binding, especially with the support of flanking residues, a phage library (1.5 × 108 members) was created in E. coli and the corresponding peptides were pulled-down with 1,25(OH)2D3 liganded VDR (Chang et al., 1999). Peptides C33 and D47 interacted tightly with VDR as determined by a mammalian two-hybrid assay (Table 1, entries 2 and 3).
Figure 5.
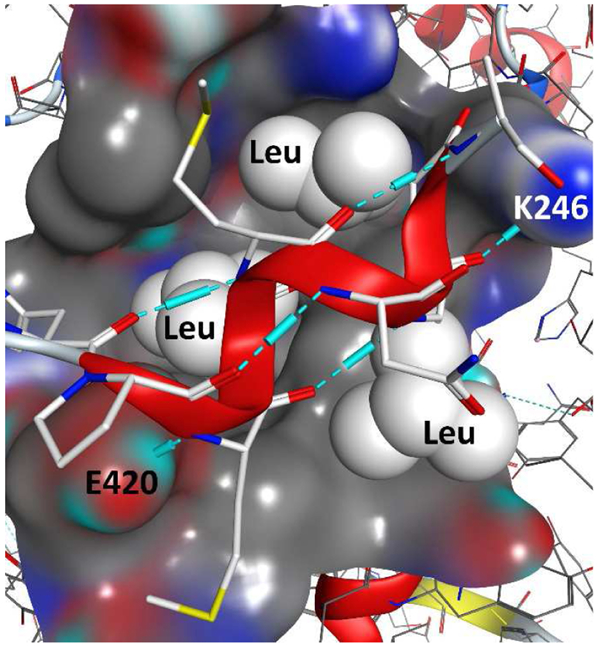
Interaction between VDR-LBD and coactivator peptide DRIP2 (LxxLL) in the presence of 1,25(OH)2D3.[1RKH, crystal structure]
Table 1.
Amino acid sequences of peptides that inhibit the interaction between VDR and coactivators.
| Entry | name | Amino acid sequence | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | DRIP2 | N | T | K | N | H | P | M | L | M | N | L | L | K | D | N | P | A | Q | D |
| 2 | C33 | H | V | E | M | H | P | L | L | M | G | L | L | M | E | S | Q | W | G | A |
| 3 | D47 | H | V | Y | Q | H | P | L | L | L | S | L | L | S | S | E | H | E | S | G |
| 4 | EBIP-37 | T | G | G | G | V | S | L | L | L | H | L | L | N | T | E | Q | G | E | S |
| 5 | EBIP-51 | F | P | A | E | F | P | L | L | T | Y | L | L | E | R | Q | G | M | D | E |
| 6 | EBIP-70 | V | M | G | N | N | P | I | L | V | S | L | L | E | E | P | S | E | E | P |
| 7 | EBIP-96 | V | E | S | E | F | P | Y | L | L | S | L | L | G | E | V | S | P | Q | P |
| 8 | 3 | L | S | E | T | H | P | L | L | W | T | L | L | S | S | E | G | D | S | M |
| 9 | 4 | M | Q | E | R | F | P | M | L | W | D | L | L | D | L | P | S | P | T | S |
| 10 | 5 | L | G | E | S | H | P | L | L | M | Q | L | L | T | E | N | V | G | T | H |
Investigations with other nuclear receptors indicated that the binding between C33 and D47 with VDR is not selective. Cell-based assays showed a reduction of 1,25(OH)2D3-induced transcription for a luciferase gene under control of a bone gamma carboxyglutamate protein (BGLAP) promoter in the presence of transcriptionally expressed C33 and D47 (Pathrose et al., 2002). In addition to VDR, further research identified peptides EBIP-37, EBIP-51, EBIP-70, and EBIP-96 interacted with estrogen receptor β (ERβ) in the presence of estradiol (Table 1, entries 4-7) (Hall et al., 2000). Identified by a similar method, peptides 3, 4, and 5, introduced by the Pike group, exhibited stong VDR binding in the presence of 1,25(OH)2D3 (Table 1, entries 8-10) (Zella et al., 2007). All three peptides were able to inhibit transcription induced by 1,25(OH)2D3 of a luciferase gene under control of an BGLAP promoter when transcriptionally expressed in cells. Based on these results, the initial LxxLL motif can be better described as a PL/MLxxLL motif for peptides with strong VDR binding. The investigation of focused phage peptide libraries has confirmed that specific flanking residues adjacent to the LxxLL motif promote VDR binding. Interestingly, the random approach identified peptides with sequences very similar to natural occurring coactivators such as DRIP205 (Table 1, entry 1). When expressed in cells, these peptides can inhibit 1,25(OH)2D3-induced transcription, demonstrating that coactivator binding is essential for VDR-meditated transcription. Although these peptides found applications as fluorescent probes to identify new ligands for VDR (Nandhikonda et al., 2013), they lack the ability to cross cell membranes.
Recently, the Kurihara group introduced stapled, helical peptides that bind liganded VDR (Demizu et al., 2013). The peptides have a central LLxxLL motif and a covalent crosslinker in the i and i+3 position (Figure 6). Furthermore, 2-aminoiso-butyric acid was introduced to promote peptide helicity (Demizu et al., 2016). The introduction of hydrophilic hydroxyl groups attached to the linker significantly increased the affinity to VDR, as shown for DPI-06 and DPI-07, with IC50 values for inhibiting the VDR–coactivator interaction at 220 μM and 3.2 μM, respectively. The introduction of α-hydroxymethylserine, rather than 2-aminoiso-butyric acid, also increased the affinity to VDR, which lead to DPI-10 with an IC50 of 20 μM (Misawa et al., 2015).
Figure 6:
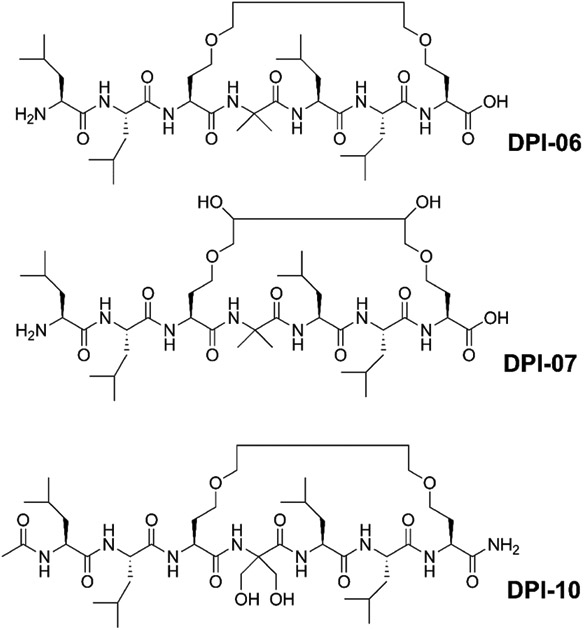
Structures of cyclic, peptide-based VDR–coactivator inhibitors.
6. Small molecules that bind the VDR coactivator pocket
Rational design led to the development of a series of benzodiazepines in 2010 (Mita et al., 2010). Compound 1 is a bicyclic compound bearing three hydrophobic groups that mimics the i, i+3, and i+4 leucine position of coactivators (Figure 7A). Structure-activity relationship studies revealed that the electron donating amine function in position 8 is preferable to diamines or a guanine structure. In the absence of a crystal structure, molecular modeling confirms the possibility that compound 1 binds both charge clamp residues E420 and K246 (Figure 7B). A later study exploring substituents in the 7 position identified compound 2, which inhibited the interaction between VDR and coactivator peptides at 14 μM (Figure 7B) (Mita et al., 2013). Compound 1 showed activity in cells and inhibited 1,25(OH)2D3-induced transcription with an IC50 of 17 μM. Cellular activity for compound 2 was not reported.
Figure 7.
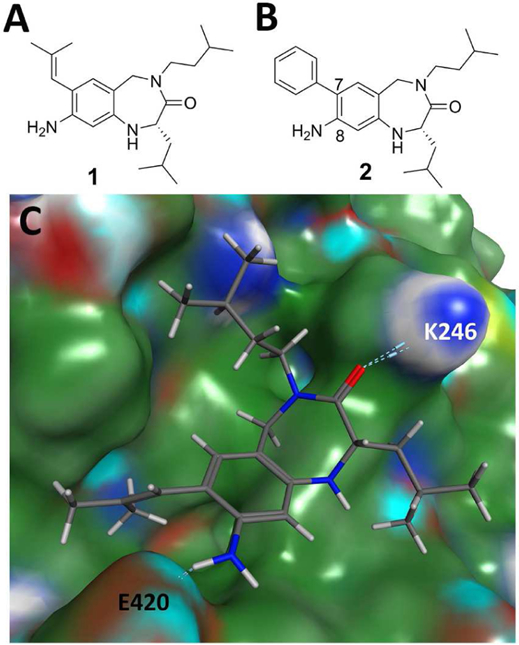
A&B) Small molecule inhibitors of the VDR-coactivator interaction; C) docking structure of 1 bond to the VDR coactivator site using 1RKH crystal structure.
High-throughput screening of 275,000 molecules identified a new class of VDR–coactivator inhibitors, 3-indolylmethanamines (Nandhikonda et al., 2012). These compounds have cellular activity and irreversibly inhibited the interaction between VDR and coactivator SRC2, with an IC50 of 4.2 μM (Figure 8). A free energy relationship study confirmed the slow formation of an electrophilic species in aqueous buffer (pH = 7.2). Binding studies in the presence of 2-mercaptoethanol confirmed the ability of 3-indolylmethanamines to react with nucleophiles in aqueous media. Because of the unique position of VDR cysteine residues, compounds such as 31B selectively inhibited VDR–coactivator interactions among other nuclear receptor–coactivator interactions. In respect to different coactivators, 31B preferably inhibited the interaction between VDR and SRC2. Interestingly, 31B induced apoptosis in cisplatin-resistant ovarian cancer cells SKOV3 (Guthrie et al., 2015). Further experiments in vivo confirmed the ability of 31B to reduce the growth of SKOV3-derived tumors. Mechanistic studies showed a reduction of glycose and lipid metabolism, which was mediated partially by VDR. Additional work resulted in the identification of PS121912 with sub-micromolar affinity for VDR (Figure 8) (Sidhu et al., 2014). PS121912 exhibited high selectivity toward VDR among other nuclear receptors. Chromatin immunoprecipitation studies showed the recruitment of NCoR to DNA bound VDR when treated with PS121912 (Sidhu et al., 2014). PS121912 induced apoptosis in leukemic HL-60 cell at 4.7 μM (EC50) and regulated genes that are involved in the cell cycle and apoptosis, such as CASP3 and CASP7. Like 31B, PS121912 was active in vivo and reduced rapid growth of HL-60 derived tumors in mice (Guthrie et al., 2015).
Figure 8:
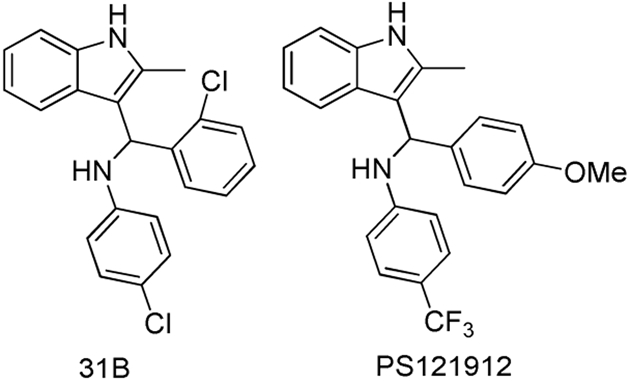
Structure of VDR–coactivator inhibitors 31B and PS121912.
7. The VDR corepressor binding site
The VDR corepressor binding site is assumed to be formed either in the absence of VDR ligand or when bound to a VDR antagonist, which suppresses the relocation of helix 12 (Anami et al., 2016) (Figure 9). In contrast to other nuclear receptors, X-ray crystal structures of VDR bound to corepressor peptides have not been reported. However, in vitro studies have confirmed interactions between VDR and corepressors (Meyer and Pike, 2013). When bound to VDR, antagonist ML 3-452 decreased SRC3 binding to CYP24A1 promoter-bound VDR and increased the recruitment of corepressor NCoR (Lamblin et al., 2010). The conformational change of VDR helix 12 was confirmed by hydrogen/deuterium exchange experiments when bound to antagonists/partial agonists (Zhang et al., 2010). The absence of helix 12 creates a large interaction surface between VDR and corepressors (Figure 9). Therefore, truncated corepressor peptides have very weak affinities for VDR-LBD (Teichert et al., 2009). Phage peptide libraries were used to identify novel peptide ligands for the VDR–antagonist complex (Zella et al., 2007). Although several peptides were isolated by interacting with VDR bound to antagonist/partial agonist ZK159222, none of them were able to bind VDR using a two-hybrid assay. Currently, no peptides or small molecules are known to bind the corepressor pocket of VDR.
Figure 9.
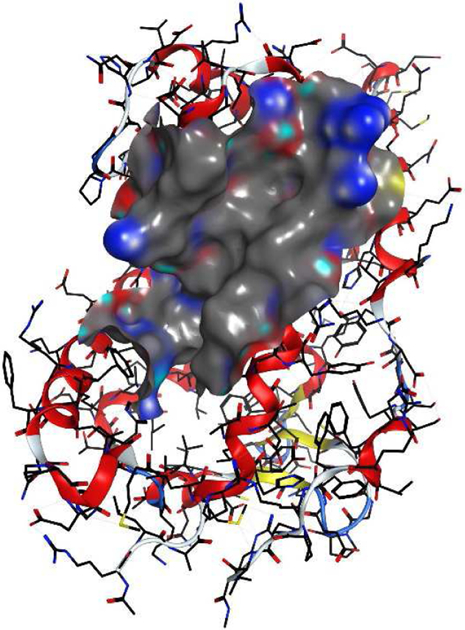
VDR corepressor binding site is visible when H12 was deleted from the crystal structure 1RKH.
8. A second binding site for lithocholic acid
Recent crystallization of zVDR and lithocholic acid (LCA) in the presence of coactivator peptide SRC2 resulted in two LCA molecules bound to zVDR (Figure 10) (Belorusova et al., 2014). Although this phenomenon is new for VDR, other nuclear receptors have been crystallized with two ligands bound, e.g. ERβ (Wang et al., 2006). The location, however, differs from ERβ in the sense that coactivator peptide SRC2 can bind VDR*2xLCA, whereas helix 12 blocks the coactivator binding site for the ERβ*2xhydroxytamoxifen complex. Thus, the chemical structure of the ligand influences the nuclear receptor conformation, which in turn influences the location and shape of the second binding site. For zVDR and LCA, the second LCA binding pocket is partially solvent exposed, has a relative large hydrophobic area, and offers interactions with S263 and K268 (Figure 10).
Figure 10.
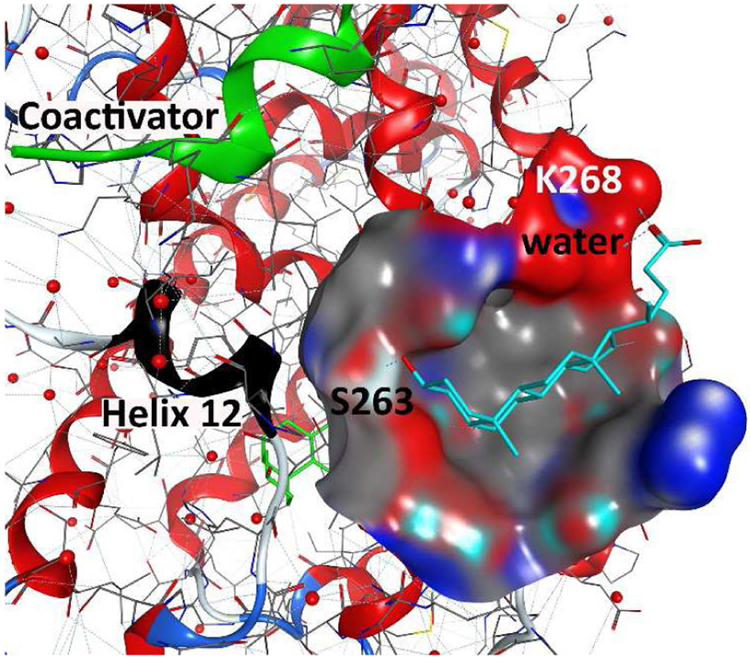
Novel binding site for lithocholic acid in addition to the genomic VDR–ligand binding pocket based on crystal structure [4Q0A].
9. VDR and binding function 3
An alternative binding site for the androgen receptor (AR) called binding function 3 (BF3) was introduced in 2007 (Estebanez-Perpina et al., 2007). The analogous binding function 3 for VDR would be formed by helix 4 and helix 9 (Figure 11A). The first molecules that bound to AR-BF3 were identified by high throughput screening (Estebanez-Perpina et al., 2007). A later study applied virtual screening to identify additional molecules with high docking scores that inhibited AR-mediated transcription in cells (Lack et al., 2011). Interestingly, these molecules might also exhibit a good affinity for VDR-BF3, which is supported by the fact that molecular modeling give good docking scores for select molecules (Figure 11B).
Figure 11.
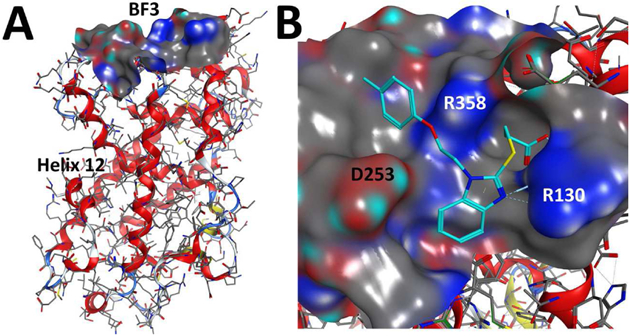
A) Proposed binding function 3 (BF3) of VDR [1DB1]. B) Molecular docking of an AR-BF3 binder to VDR-BF3 using 1DB1 structure.
10. Possible medical application of alternative VDR ligands
The clinical development of alternative VDR ligands is still underexplored, in contrast to ligands that bind the genomic VDR pocket (Figure 12). Ligands, including 1,25(OH)2D3, bind cytosolic VDR and mediate non-genomic effects such as regulating ion-channel, phospholipase, and kinase activities. Anti-inflammatory effects have been shown to be mediated by direct interaction between liganded VDR and IKkβ (Chen et al., 2013). Thus, further ligand design might lead to the development of highly anti-inflammatory ligands. Inhibition of VDR–RXR dimerization and inhibition of VDR–coactivator interactions have antagonizing effects in transcription. Ligands that inhibit this interaction have been shown to exhibit anti-tumor activities (Guthrie et al., 2015). These ligand were non-calcemic, thus inhibiting VDR–coactivator interactions that mediate transcription of genes involved in calcium homeostasis (Sidhu et al., 2014). The activation of VDR by minimizing interactions with corepressors has great potential because this mode would enhance vitamin D action, which in turn might be applicable to hyperparathyroidism. Unfortunately, ligands inhibiting this interaction have not yet been developed.
Figure 12.
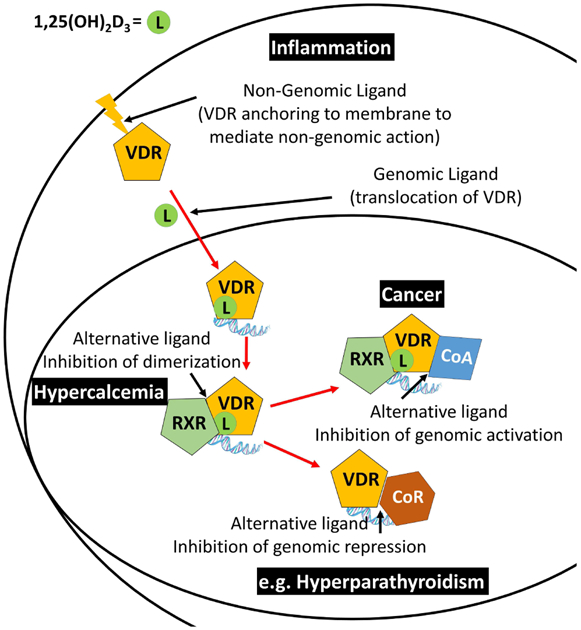
Cellular interaction of VDR with endogenous ligand 1,25(OH)2D3 and proteins that enable interception by alternative VDR ligands and their possible therapeutic applications.
11. Conclusion and future directions
Protein crystal structures and molecular modeling have enabled the identification of new binding sites for small molecules (Figure 12). The majority of ligands bind the genomic VDR ligand binding pocket and induce small changes in the overall VDR structure. The alternative ligand binding pocket and BF3 pocket have been proposed for VDR, but have yet to be confirmed by X-ray crystallography. In contrast, peptides and a small molecule have been shown to bind the coactivator and the LCA second binding site, respectively. The VDR corepressor site is still elusive because of the absence of an apoVDR structure and the large number of naturally occurring VDR ligands that promote interactions with coactivators rather than corepressors. To accelerate the identification of alternative VDR binders, new assays have to be developed because current, commercially available binding assays only report the detection of small molecule binding to the genomic ligand and coactivator binding site. Many VDR ligands have been investigated for VDR binding and modulation of transcription. Rapid cellular effects were only investigated with a limited number of VDR ligands. A more concise battery of assays to characterize new VDR ligands would be helpful to identify classes of ligands with similar biological activity. This in turn will help to identify unique VDR ligands that interact with VDR at novel binding pockets.
Figure 13.
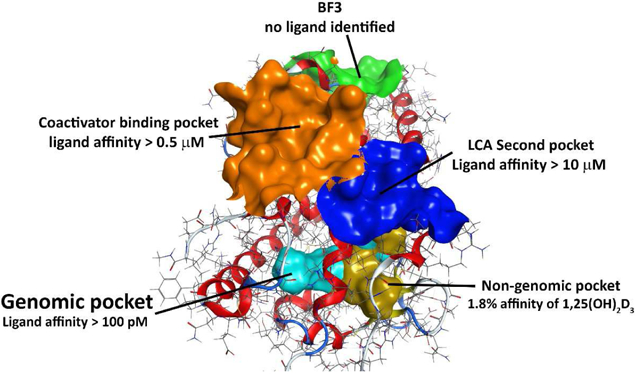
Currently identified and proposed binding sites for VDR ligands.
Highlights:
Ligands with strong VDR interactions bind the genomic binding pocket
The majority of biological effects mediated by VDR are based on gene transcription
Ligands that bind alternative VDR pockets have a low affinity for VDR
12.
Funding
This work was supported by the University of Wisconsin–Milwaukee, the Milwaukee Institute for Drug Discovery, the UWM Research Growth Initiative, NIH R03DA031090, the UWM Research Foundation, the Lynde and Harry Bradley Foundation, and the Richard and Ethel Herzfeld Foundation.
Abbreviations:
- VDR
vitamin D receptor
- RXR
retinoid X receptor
- DBD
DNA binding domain
- LBD
ligand binding domain
- 1,25(OH)2D3
1,25-dihydroxy vitamin D3
- SF-1
steroidogenic factor 1
- LRH-1
liver receptor homolog 1
- OC
osteocalcin
- ERβ
estrogen receptor
- DRIP205
vitamin D-interacting protein 205
- SRC2
steroid receptor coactivator 2
- NCoR
Nuclear receptor co-repressor 1
- zVDR
zebrafish vitamin D receptor
- LCA
lithocholic acid
- AR
androgen receptor
- BF3
binding function 3
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
13. References
- Anami Y, Shimizu N, Ekimoto T, Egawa D, Itoh T, Ikeguchi M and Yamamoto K, 2016. Apo- and Antagonist-Binding Structures of Vitamin D Receptor Ligand-Binding Domain Revealed by Hybrid Approach Combining Small-Angle X-ray Scattering and Molecular Dynamics. J Med Chem. 59, 7888–900. 10.1021/acs.jmedchem.6b00682. [DOI] [PubMed] [Google Scholar]
- Barbchano A, Larriba MJ, Ferrer-Mayorga G, Gonzalez-Sancho JM and Munoz A, 2018. Vitamin D and Colon Cancer, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 838–855. [Google Scholar]
- Beaudin S and Welsh J, 2018. Vitamin D action in Mammary Gland and Breast Cancer: Genomics, Metabolism, and Stem Cells, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 802–814. [Google Scholar]
- Belorusova AY, Eberhardt J, Potier N, Stote RH, Dejaegere A and Rochel N, 2014. Structural Insights into the Molecular Mechanism of Vitamin D Receptor Activation by Lithocholic Acid Involving a New Mode of Ligand Recognition. Journal of Medicinal Chemistry. 57, 4710–4719. 10.1021/jm5002524. [DOI] [PubMed] [Google Scholar]
- Belorusova AY and Rochel N, 2018. Strucutral Basis for Ligand Activity in Vitamin D Receptor, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 189–206. [Google Scholar]
- Bettoun DJ, Lu J, Khalifa B, Yee Y, Chin WW and Nagpal S, 2004. Ligand modulates VDR-Ser/Thr protein phosphatase interaction and p70S6 kinase phosphorylation in a cell-context-dependent manner. J Steroid Biochem Mol Biol. 89–90, 195-8. 10.1016/j.jsbmb.2004.03.087. [DOI] [PubMed] [Google Scholar]
- Bourdeau A, Atmani F, Grosse B and Lieberherr M, 1990. Rapid effects of 1,25-dihydroxyvitamin D3 and extracellular Ca2+ on phospholipid metabolism in dispersed porcine parathyroid cells. Endocrinology. 127, 2738–43. 10.1210/endo-127-6-2738. [DOI] [PubMed] [Google Scholar]
- Chang C, Norris JD, Gron H, Paige LA, Hamilton PT, Kenan DJ, Fowlkes D and McDonnell DP, 1999. Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors alpha and beta. Mol Cell Biol. 19, 8226–39. https://doi.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang J, Ge X, Du J, Deb DK and Li YC, 2013. Vitamin D Receptor Inhibits Nuclear Factor kappa B Activation by Interacting with I kappa B Kinase beta Protein. Journal of Biological Chemistry. 288, 19450–19458. 10.1074/jbc.M113.467670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalhoff K, Dancey J, Astrup L, Skovsgaard T, Hamberg KJ, Lofts FJ, Rosmorduc O, Erlinger S, Bach Hansen J, Steward WP, Skov T, Burcharth F and Evans TR, 2003. A phase II study of the vitamin D analogue Seocalcitol in patients with inoperable hepatocellular carcinoma. Br J Cancer. 89, 252–7. 10.1038/sj.bjc.6601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boland AR and Norman AW, 1998. 1alpha,25(OH)2-vitamin D3 signaling in chick enterocytes: enhancement of tyrosine phosphorylation and rapid stimulation of mitogen- activated protein (MAP) kinase. J Cell Biochem. 69, 470–82. https://doi.org/ [DOI] [PubMed] [Google Scholar]
- Demizu Y, Nagoya S, Shirakawa M, Kawamura M, Yamagata N, Sato Y, Doi M and Kurihara M, 2013. Development of stapled short helical peptides capable of inhibiting vitamin D receptor (VDR)-coactivator interactions. Bioorg Med Chem Lett. 23, 4292–6. 10.1016/j.bmcl.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Demizu Y, Okitsu K, Yamashita H, Doi M, Misawa T, Oba M, Tanaka M and Kurihara M, 2016. alpha-Helical Structures of Oligopeptides with an Alternating L-Leu-Aib Segment. European Journal of Organic Chemistry. 2815–2820. 10.1002/ejoc.201600327. [DOI] [Google Scholar]
- Estebanez-Perpina E, Arnold AA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, Webb P and Fletterick RJ, 2007. A surface on the androgen receptor that allosterically regulates coactivator binding. Proceedings of the National Academy of Sciences of the United States of America. 104, 16074–16079. 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie ML, Sidhu PS, Hill EK, Horan TC, Nandhikonda P, Teske KA, Yuan NY, Sidorko M, Rodali R, Cook JM, Han L, Silvaggi NR, Bikle DD, Moore RG, Singh RK and Arnold LA, 2015. Antitumor Activity of 3-Indolylmethanamines 31B and PS121912. Anticancer Res. 35, 6001–7. https://doi.org/ [PMC free article] [PubMed] [Google Scholar]
- Hall JM, Chang CY and McDonnell DP, 2000. Development of peptide antagonists that target estrogen receptor beta-coactivator interactions. Molecular Endocrinology. 14, 2010–2023. [DOI] [PubMed] [Google Scholar]
- Haussler MR, Jurutka PW, Mizwicki M and Norman AW, 2011. Vitamin D receptor (VDR)-mediated actions of 1 alpha,25(OH)(2)vitarnin D-3: Genomic and non-genomic mechanisms. Best Practice & Research Clinical Endocrinology & Metabolism. 25, 543–559. 10.1016/j.beem.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Hii CS and Ferrante A, 2016. The Non-Genomic Actions of Vitamin D. Nutrients. 8, 135 10.3390/nu8030135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhtakangas JA, Olivera CJ, Bishop JE, Zanello LP and Norman AW, 2004. The vitamin D receptor is present in caveolae-enriched plasma membranes and binds 1 alpha,25(OH)(2)-vitamin D-3 in vivo and in vitro. Molecular Endocrinology. 18, 2660–2671. 10.1210/me.2004-0116. [DOI] [PubMed] [Google Scholar]
- Jurutka PW, Bartik L, Whitfield GK, Mathern DR, Barthel TK, Gurevich M, Hsieh JC, Kaczmarska M, Haussler CA and Haussler MR, 2007. Vitamin D receptor: key roles in bone mineral pathophysiology, molecular mechanism of action, and novel nutritional ligands. J Bone Miner Res. 22 Suppl 2, V2–10. 10.1359/jbmr.07s216. [DOI] [PubMed] [Google Scholar]
- Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, Lebedeva L, Suzawa M, Williams JD, Williams SP, Guy RK, Thornton JW, Fletterick RJ, Willson TM and Ingraham HA, 2005. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 120, 343–55. 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Lack NA, Axerio-Cilies P, Tavassoli P, Han FQ, Chan KH, Feau C, LeBlanc E, Guns ET, Guy RK, Rennie PS and Cherkasov A, 2011. Targeting the Binding Function 3 (BF3) Site of the Human Androgen Receptor through Virtual Screening. Journal of Medicinal Chemistry. 54, 8563–8573. 10.1021/jm201098n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamblin M, Spingarn R, Wang TT, Burger MC, Dabbas B, Moitessier N, White JH and Gleason JL, 2010. An o-Aminoanilide Analogue of 1 alpha,25-Dihydroxyvitamin D-3 Functions as a Strong Vitamin D Receptor Antagonist. Journal of Medicinal Chemistry. 53, 7461–7465. 10.1021/jm1007159. [DOI] [PubMed] [Google Scholar]
- Leyssens C, Verlinden L and Verstuyf A, 2014. The future of vitamin D analogs. Front Physiol. 5, 122 10.3389/fphys.2014.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M and Yamada S, 2018. Bile Acid-Derived Vitamin D Receptor Ligands, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 629–641. [Google Scholar]
- Mann EH, Pfeffer PE and Hawrylowicz CM, 2018. Vitamin D and Adaptive Immunology in Health and Disease, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 938–945. [Google Scholar]
- McInerney EM, Rose DW, Flynn SE, Westin S, Mullen TM, Krones A, Inostroza J, Torchia J, Nolte RT, Assa-Munt N, Milburn MV, Glass CK and Rosenfeld MG, 1998. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 12, 3357–68. https://doi.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegaz D, Barrientos-Duran A, Kline A, Silva FR, Norman AW, Mizwicki MT and Zanello LP, 2010. 1alpha,25(OH)2-Vitamin D3 stimulation of secretion via chloride channel activation in Sertoli cells. J Steroid Biochem Mol Biol. 119, 127–34. 10.1016/j.jsbmb.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Meyer MB and Pike JW, 2013. Corepressors (NCoR and SMRT) as well as coactivators are recruited to positively regulated 1 alpha,25-dihydroxyvitamin D-3-responsive genes. Journal of Steroid Biochemistry and Molecular Biology. 136, 120–124. 10.1016/j.jsbmb.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa T, Demizu Y, Kawamura M, Yamagata N and Kurihara M, 2015. Structural development of stapled short helical peptides as vitamin D receptor (VDR)-coactivator interaction inhibitors. Bioorg Med Chem. 23, 1055–61. 10.1016/j.bmc.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Mita Y, Dodo K, Noguchi-Yachide T, Hashimoto Y and Ishikawa M, 2013. Structure-activity relationship of benzodiazepine derivatives as LXXLL peptide mimetics that inhibit the interaction of vitamin D receptor with coactivators. Bioorg Med Chem. 21, 993–1005. https://doi.org/S0968-0896(12)00935-2 [pii] 10.1016/j.bmc.2012.11.042. [DOI] [PubMed] [Google Scholar]
- Mita Y, Dodo K, Noguchi-Yachide T, Miyachi H, Makishima M, Hashimoto Y and Ishikawa M, 2010. LXXLL peptide mimetics as inhibitors of the interaction of vitamin D receptor with coactivators. Bioorg Med Chem Lett. 20, 1712–7. https://doi.org/S0960-894X(10)00091-0 [pii] 10.1016/j.bmcl.2010.01.079. [DOI] [PubMed] [Google Scholar]
- Mizwicki MT, Keidel D, Bula CM, Bishop JE, Zanello LP, Wurtz JM, Moras D and Norman AW, 2004. Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1alpha,25(OH)2-vitamin D3 signaling. Proc Natl Acad Sci U S A. 101, 12876–81. 10.1073/pnas.0403606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandhikonda P, Lynt WZ, McCallum MM, Ara T, Baranowski AM, Yuan NY, Pearson D, Bikle DD, Guy RK and Arnold LA, 2012. Discovery of the first irreversible small molecule inhibitors of the interaction between the vitamin D receptor and coactivators. J Med Chem. 55, 4640–51. 10.1021/jm300460c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandhikonda P, Yasgar A, Baranowski AM, Sidhu PS, McCallum MM, Pawlak AJ, Teske K, Feleke B, Yuan NY, Kevin C, Bikle DD, Ayers SD, Webb P, Rai G, Simeonov A, Jadhav A, Maloney D and Arnold LA, 2013. Peroxisome proliferation-activated receptor delta agonist GW0742 interacts weakly with multiple nuclear receptors, including the vitamin D receptor. Biochemistry. 52, 4193–203. 10.1021/bi400321p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman AW, Manchand PS, Uskokovic MR, Okamura WH, Takeuchi JA, Bishop JE, Hisatake JI, Koeffler HP and Peleg S, 2000. Characterization of a novel analogue of 1 alpha,25(OH)(2)-vitamin D-3 with two side chains: Interaction with its nuclear receptor and cellular actions. Journal of Medicinal Chemistry. 43, 2719–2730. 10.1021/jm0000160. [DOI] [PubMed] [Google Scholar]
- Nurminen V, Neme A, Seuter S and Carlberg C, 2018. The impact of the vitamin D-modulated epigenome on VDR target gene regulation. Biochim Biophys Acta. 1861, 697–705. 10.1016/j.bbagrm.2018.05.006. [DOI] [PubMed] [Google Scholar]
- Orlov I, Rochel N, Moras D and Klaholz BP, 2012. Structure of the full human RXR/VDR nuclear receptor heterodimer complex with its DR3 target DNA. Embo Journal. 31, 291–300. 10.1038/emboj.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathrose P, Barmina O, Chang CY, McDonnell DP, Shevde NK and Pike JW, 2002. Inhibition of 1,25-dihydroxyvitamin D3-dependent transcription by synthetic LXXLL peptide antagonists that target the activation domains of the vitamin D and retinoid X receptors. J Bone Miner Res. 17, 2196–205. 10.1359/jbmr.2002.17.12.2196. [DOI] [PubMed] [Google Scholar]
- Pike JW, Meyer MB, Lee SM, Onal M and Benkusky NA, 2018. Genone-Wide Perspectives on Vitamin D Receptor-Mediated Control of Gene Expression in Target Cells, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 142–167. [Google Scholar]
- Ransohoff KJ, Epstein EH and Tang JY, 2018. Vitamin D and Skin Cancer, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 863–870. [Google Scholar]
- Saitoh H, 2018. Vitamin D Receptor Antagonists, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 679–691. [Google Scholar]
- Shaurova T, Seshadri M and Hershberger PA, 2018. Vitamin D and Lung Cancer, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 876–885. [Google Scholar]
- Sidhu PS, Nassif N, McCallum MM, Teske K, Feleke B, Yuan NY, Nandhikonda P, Cook JM, Singh RK, Bikle DD and Arnold LA, 2014. Development of novel Vitamin D Receptor-Coactivator Inhibitors ACS Med Chem Lett. 5, 199–204. 10.1021/ml400462j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu PS, Teske K, Feleke B, Yuan NY, Guthrie ML, Fernstrum GB, Vyas ND, Han L, Preston J, Bogart JW, Silvaggi NR, Cook JM, Singh RK, Bikle DD and Arnold LA, 2014. Anticancer activity of VDR-coregulator inhibitor PS121912. Cancer Chemother Pharmacol. 74, 787–98. 10.1007/s00280-014-2549-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stites RE, MacKrell JG and Stayrook KR, 2018. Nonsecosteroial LIgands and Modulators of Vitamin D Receptor, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 615–625. [Google Scholar]
- Studzinski GP, Gocek E, Coffman F and M. D, 2018. Effects of Vitamin D Derivaties on Differentiation, Cell Cycle, and Apoptosis in Hematological Malignances, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 762–787. [Google Scholar]
- Teichert A, Arnold LA, Otieno S, Oda Y, Augustinaite I, Geistlinger TR, Kriwacki RW, Guy RK and Bikle DD, 2009. Quantification of the vitamin D receptor-coregulator interaction. Biochemistry. 48, 1454–61. 10.1021/bi801874n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trump DL, 2018. The Antitumor Effect of Vitamin D in Genitourinary Cancer, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 821–836. [Google Scholar]
- Van Driel M, Van Leeuwen JPTM, Munoz A and Feldman D, 2018. Overview of Vitamin D Actions in Cancer, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 711–728. [Google Scholar]
- Vanhooke JL, Benning MM, Bauer CB, Pike JW and DeLuca HF, 2004. Molecular structure of the rat vitamin D receptor ligand binding domain complexed with 2-carbon-substituted vitamin D3 hormone analogues and a LXXLL-containing coactivator peptide. Biochemistry. 43, 4101–10. 10.1021/bi036056y. [DOI] [PubMed] [Google Scholar]
- Verlinden L, Bouillon R, De Clercq P and Verstuyf A, 2018. Analogs of Calcitriol, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 583–611. [Google Scholar]
- Wang Y, Chirgadze NY, Briggs SL, Khan S, Jensen EV and Burris TP, 2006. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor beta. Proceedings of the National Academy of Sciences of the United States of America. 103, 9908–9911. 10.1073/pnas.0510596103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Zhu JG and DeLuca HF, 2012. Where is the vitamin D receptor? Archives of Biochemistry and Biophysics. 523, 123–133. 10.1016/j.abb.2012.04.001. [DOI] [PubMed] [Google Scholar]
- White JH, Salehi-Tabar R, Dimitrow V and Bouttier M, 2018. Diverse mechanism of transcriptional regulation by the vitamin D receptor, in: Feldman D (Ed.), Vitamin D. Elsevier, London, pp. 175–187. [Google Scholar]
- Zanello LP and Norman AW, 2004. Rapid modulation of osteoblast ion channel responses by 1alpha,25(OH)2-vitamin D3 requires the presence of a functional vitamin D nuclear receptor. Proc Natl Acad Sci U S A. 101, 1589–94. 10.1073/pnas.0305802101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zella LA, Chang CY, McDonnell DP and Pike JW, 2007. The vitamin D receptor interacts preferentially with DRIP205-like LxxLL motifs. Arch Biochem Biophys. 460, 206–12. 10.1016/j.abb.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chalmers MJ, Stayrook KR, Burris LL, Garcia-Ordonez RD, Pascal BD, Burris TP, Dodge JA and Griffin PR, 2010. Hydrogen/deuterium exchange reveals distinct agonist/partial agonist receptor dynamics within vitamin D receptor/retinoid X receptor heterodimer. Structure. 18, 1332–41. 10.1016/j.str.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]