

SUMMARY
Body and organ size regulation in mammals involves multiple signaling pathways and remains largely enigmatic. Here, we report that Pum1 and Pum2, which encode highly conserved PUF RNA-binding proteins, regulate mouse body and organ size by post-transcriptional repression of the cell cycle inhibitor Cdkn1b. Binding of PUM1 or PUM2 to Pumilio binding elements (PBEs) in the 3’ UTR of Cdkn1b inhibits translation, promoting G1-S transition and cell proliferation. Mice with null mutations in Pum1 and Pum2 exhibit gene dosage-dependent reductions in body and organ size, and deficiency for Cdkn1b partially rescues postnatal growth defects in Pum1−/− mice. We propose that coordinated tissue-specific expression of Pum1 and Pum2, which involves auto-regulatory and reciprocal post-transcriptional repression, contributes to the precise regulation of body and organ size. Hence PUM-mediated post-transcriptional control of cell cycle regulators represents an additional layer of control in the genetic regulation of organ and body size.
Graphical Abstract
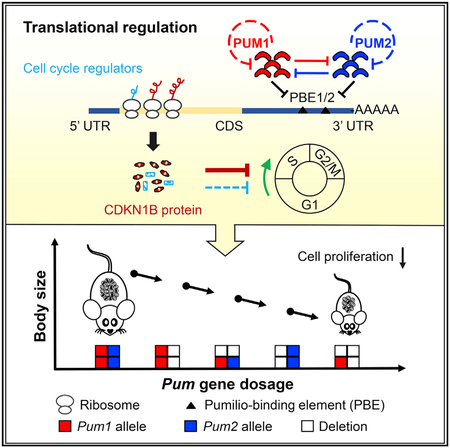
In Brief
Lin et al. show that the RNA-binding proteins PUM1 and PUM2 regulate translation of cell cycle proteins such as CDKN1B by binding to their 3’ UTR and achieve precise control of organ and body size in a gene dosage-sensitive manner via auto and reciprocal gene expression regulation.
INTRODUCTION
Body size is one of the most prominent forms of diversity in nature. Yet many key mechanisms of body size control remain insufficiently understood, in particular how coordinated and proportional regulation of organ size is achieved. Body and organ size control is highly complex and involves the interplay of systemic factors such as growth hormones and local signaling molecules such as insulin-like growth factors (IGFs) (Efstratiadis, 1998; Horikoshi et al., 2016; Pan, 2010; Stanger, 2008; Yu et al., 2015). These signaling pathways then activate the expression of transcription factors and cofactors (effectors) at the cellular level to control cell proliferation and apoptosis and, ultimately, body size (Stanger, 2008; Trumpp et al., 2001; Yu et al., 2015). Disruption in any of the known pathways involved in growth regulation results in severe growth defects or lethality (Efstratiadis, 1998). However, variations in body size are more extensive and continuous than changes associated with known mutations in hormonal and other signaling pathways. The mechanisms for such highly tuned and discrete control of body size remain largely unknown. Increasing evidence indicates that RNA-binding proteins control fundamental cellular functions such as stem cell maintenance, cell differentiation, and cell proliferation at the post-transcriptional level (Blackinton and Keene, 2014; Cannell et al., 2015; Kronja and Orr-Weaver, 2011; Ye and Blelloch, 2014). Nevertheless, the role of post-transcriptional regulation in body size control remains unexplored.
RNA-binding proteins of the PUF family (Pumilio and FBF) play key roles in development and disease (Chen et al., 2012; Crittenden et al., 2002; Gennarino et al., 2015; Lehmann and Nusslein-Volhard, 1987; Lin and Spradling, 1997; Xu et al., 2007; Zamore et al., 1997). PUF-family proteins contain evolutionarily conserved RNA-binding domains and function by binding to Pumilio binding elements (PBEs) on the 3’ UTR of target mRNAs (Chen et al., 2012; Edwards et al., 2001; Lee et al., 2007; Wickens et al., 2002). In multiple species, homologs of Pumilio are required for germline development and differentiation, in particular germline stem cell maintenance (Cao et al., 2010; Crittenden et al., 2002; Forbes and Lehmann, 1998; Lin and Spradling, 1997; Parisi and Lin, 2000; Wickens et al., 2002). The function of the two mammalian members of the PUF family, Pumilio RNA-binding family members 1 and 2 (PUM1 and PUM2), has been investigated in genetic studies in the mouse (Chen et al., 2012; Gennarino et al., 2015; Lin et al., 2018b; Mak et al., 2016; Xu et al., 2007; Zhang et al., 2017). Pum-mutant mice have reduced body weight, suggesting that PUF proteins may function outside the gonads and may be required for growth in mammals (Chen et al., 2012; Siemen et al., 2011). Here, we report that Pum1 and Pum2 control mouse body size by post-transcriptional repression of Cdkn1b expression, uncovering a mechanism allowing precise regulation and incremental control of body and organ size in mammalian growth.
RESULTS
Pum1-Null Mice Exhibit Prenatal and Postnatal Growth Reduction
To investigate the function of Pumilio 1 (Pum1) gene in mammalian growth, we generated a conditional null allele by deleting exons 9 and 10 (Figure S1A). Similar to previously reported PUF-family mouse mutants (Chen et al., 2012; Xu et al., 2007), Pum1−/− mutant mice were viable and fertile and did not exhibit any gross abnormalities, except smaller body weight, compared with wild-type littermates (Figures 1A and 1C). Postnatal body weight curves of female and male Pum1−/− mice largely followed the pattern of those of wild-type littermates but with a significantly lower average weight at all time points assessed (Figures 1B and 1D). On average, Pum1−/− female and male mice weighed 20% and 28% less than their littermates at the age of 2 weeks and 40% and 41% less at the age of 60 weeks. Heterozygous Pum1+/− mice were slightly smaller than wild-type mice, and male Pum1+/− mice weighed significantly less from post-natal week 10 onward, suggesting a potential dosage effect of the Pum1 mutation (Figure 1D). Measuring body length as another relevant parameter of body growth, we found that both male and female Pum1−/− mice were significantly shorter than wild-type and Pum1+/− littermates (Figures 1E and 1F), suggesting an overall reduction in body size.
Figure 1. Pum1−/− Mice Exhibit Prenatal and Postnatal Growth Reduction.

(A and C) Representative images of (A) female and (C) male Pum1−/−, Pum1+/−, and wild-type mice.
(B and D) Postnatal body weight curves (weeks 2–96) of (B) female (17 Pum1−/−, 27 Pum1+/−, and 20 Pum1+/+) and (D) male mice (11 Pum1−/−, 23 Pum1+/−, and 11 Pum1+/+).
(E and F) Body length of 2-week-old (E) females (9 Pum1−/−, 24 Pum1+/−, and 13 Pum1+/+) and (F) males (16 Pum1−/−, 24 Pum1+/−, and 9 Pum1+/+).
(G and H) Representative images of Pum1+/+, Pum1+/−, and Pum1−/− mice on postnatal day 1 (G) and embryonic day 14.5 (H).
(I and J) Growth curve of neonatal (I) female (6 Pum1−/−, 13 Pum1+/−, and 8 Pum1+/+) and (J) male mice (5 Pum1−/−, 12 Pum1+/−, and 11 Pum1+/+).
(K) Body weight of fetuses at E13.5, E14.5, and E16.5 for Pum1−/− (7), Pum1+/− (18), and Pum1+/+ (10) mice.
Data are presented as mean ± SD. Significant p values are indicated by asterisks and pound signs. Significant differences between Pum1+/− or Pum1−/− and wild-type (Pum1+/+) are marked by asterisks, and significant differences between Pum1−/− and Pum1+/− by pound signs (***p < 0.001 and ###p < 0.001, **p < 0.01 and ##p < 0.01, and *p < 0.05).
We next investigated the developmental course of body weight reduction in Pum1 mutant mice. Pum1 homozygotes had significantly lower birth weights than wild-type littermates and remained significantly smaller throughout the first postnatal week (Figures 1G, 1I, and 1J). These findings indicated that the observed reduced postnatal growth reflected a developmental defect rather than growth retardation resulting from poor feeding or growth hormone deficiency. Furthermore, we found that inter-crosses of Pum1+/− mice produced significantly fewer Pum1−/− mice than predicted by the normal Mendelian ratio, suggesting that a proportion of Pum1−/− animals died during embryonic development (Figure S1F). Evaluation of fetuses from heterozygous intercrosses revealed that embryonic day (E) 13.5 and E16.5 Pum1−/− fetuses were significantly smaller than wild-type and heterozygous littermates, and the weight difference increased between E13.5 and E16.5 (Figures 1H and 1K). These findings confirm that Pum1-null mutant embryos exhibit reduced body weight at or before E13.5. We conclude that knockout of Pum1 results in defects in body size control during embryonic and postnatal growth.
Global Reduction of Organ Weight, Tissue Weight, and Fluid in Pum1 Mutant Mice
We next measured organ weight in postnatal Pum1−/− mice, assessing two different age groups: 3-week-old mice, which are undergoing a phase of rapid growth and in which the body weight difference between mutants and wild-type littermates was largest, and adult mice (3 months or older). Major organs from 3-week-old Pum1-mutant mice weighed significantly less than those from wild-type littermates (Figure 2A) but did not exhibit any visible abnormalities other than smaller size (Figure 2B). The organ weight appeared to be reduced proportionately relative to the total body weight within a small range, suggesting a uniform body weight reduction in absence of Pum1 (Figure S2A). With the exception of spleen and liver, organs from adult Pum1−/− mice were significantly smaller than those from controls (Figure 2C) but of normal gross morphology (Figure 2D). Histological examination of organs from Pum1−/− mice confirmed normal tissue architecture and histology (Figure S2C), suggesting that organ size reduction was the primary defect caused by Pum1 deficiency, implying Pum1 in global control of body weight at the organ level.
Figure 2. Global Organ Size and Cell Number Reduction in Pum1−/−-Mutant Mice.

(A) Organ weights were measured in Pum1+/+ (n = 7) and Pum1−/− (n = 12) males at 3 weeks of age.
(B) Representative images of organs from 3-week-old mice.
(C) Organ weights of adult Pum1+/+ (n = 5) and Pum1−/− (n = 5) males.
(D) Representative images of adult organs.
(E–H) Analysis of cell size distribution in testis (E) and bone marrow (G) by flow cytometry. Three distinct cell populations were gated in a FSC-A by FL2-A dot plot and sorted in a FSC-A histogram. FSC, forward scatter; G1, gate 1; G2, gate 2; G3, gate 3. Cell size distribution analysis using median fluorescence values (FSC-A) of testicular (F) and bone marrow (H) cells for adult Pum1+/+ (n = 3) and Pum1−/− (n = 3) males.
(I) Total cell count of testis and thymus in Pum1+/+ (n = 5) and Pum1−/− (n = 6) males at 3 weeks of age.
(J) Total cell count of testis and bone marrow in adult Pum1+/+ (n = 5) and Pum1−/− (n = 5) males.
Br, brain; He, heart; Ki, kidney; Li, liver; Lu, lung; Sp, spleen; Te, testis; Th, thymus; Ti, thigh. Data are presented as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001.
To further evaluate the impact of loss of Pum1 on body size, we determined the body composition of live wild-type, heterozygote, and homozygote mice at 9 weeks of age and in adults by measuring their lean mass, fat, and fluid with nuclear magnetic resonance (NMR). We found that at 9 weeks of age, the significantly reduced body weight in the homozygotes resulted from reduction in lean mass and fluid but not in fat weight (Figures S2D and S2F). The adult body weight reduction was significant for both homozygotes and heterozygotes and resulted from reduction in fat, lean mass, and fluids (Figures S2E and S2G). The impact of body weight reduction on body composition appeared proportional, supporting a role of PUM1 in the regulation of not just organ size but also overall body size. The only disproportional reduction was adult fat weight, and it could be attributed to significant accumulation of fat in older wild-type female mice but not in the Pum1-mutant mice.
Given the overall body size reduction, we then measured the serum IGF-1 level and found a lower level of serum IGF-1 in Pum1 homozygote mice than in wild-type mice and heterozygote mice (Figure S2B), suggesting a potential systemic growth effect on adult mice from loss of Pum1. However, given the embryonic onset of the body size reduction and reduced weight but not IGF-1 in heterozygotes, such systemic growth effect from growth hormone is likely to be secondary and limited to the post-natal stage of growth. Indeed, 3-week-old heterozygotes showed significant weight reduction compared with wild-type but more than homozygotes for all organs examined (Figures S3A and S3B), indicating a clear gene dosage effect at the organ weight level.
Reduction in Cell Number, but Not Cell Size, of Pum1−/−-Mutant Organs
We next assessed if organ size reduction in Pum1 mutants resulted from reduced cell size and/or number. Flow cytometric analyses of bone marrow and testicular cells found a similar distribution of cells with respect to size and relative proportion of cells in mutant and wild-type organs (Figures 2E–2H). However, comparison of the total cell number revealed that mutant organs contained significantly fewer cells: consistent with a reduction in weight (57% for testis and 54% for thymus), testes and thymi from 3-week-old Pum1−/− mice contained 44% and 64% fewer cells than organs from wild-type littermates (Figure 2I). Similarly, the cell counts of adult Pum1−/− testis (39% reduction in organ weight) and bone marrow were 50% and 31% lower than wild-type, respectively (Figure 2J). Interestingly, heterozygotes also showed intermediate cell number between wild-type and homo-zygotes, consistent with organ weight reduction (Figure S3C).
We next expanded our analysis to two other organs, brain and spleen, with the goal of determining if organ weight reduction in Pum1-mutant tissues resulted from reduced cell number and cell proliferation. Pum1 homozygotes consistently have smaller brains, with all parts of the brain proportionally reduced, including the forebrain (Figure S3D). We then compared the weight, cell number, and cell proliferation of the forebrain from neonatal mutant, heterozygotes, and wild-type at postnatal day 7 when the forebrain weight reduction in the homozygotes became significant. The body weight and forebrain weight showed a similar trend of reduction from wild-type, heterozygotes to homozygotes (Figures S3E and S3F). Cellularity of the forebrain exhibited a similar trend of reduction in the total cell number, suggesting that cell number reduction may account for the forebrain weight reduction (Figure S3G). To determine if the mutant neuronal stem cell might exhibit reduced cell proliferation, we cultured neuronal stem cells from neonatal forebrain and found that homozygote mutants produced significantly fewer and smaller neuro-spheres (Figures S3G and S3H), supporting reduced cell proliferation in the absence of PUM1.
We next examined the different cell types from homozygote spleen reduced in weight and found that the ratios of the four cell types were similar to those in the wild-type spleen, but the absolute numbers of B cells, CD8+ T cells, and CD4+ T cells were reduced, with the former two cell types being significant, suggesting that cell number reduction contributed to weight reduction (Figures S3J and S3K). Thus, reduction in cell number but not cell size causes organ and body weight reductions in Pum1-null homozygotes.
Depletion of PUM1 Reduces Cell Proliferation In Vitro and In Vivo
To investigate potential causes of the reduction in cell number in Pum1-mutant animals, we first evaluated the effects of Pum1 depletion on cell proliferation in vitro. RNAi-mediated knockdown of Pum1 expression in NIH 3T3 cells significantly reduced growth rate and DNA synthesis (measured by incorporation of EdU [5-ethynyl-2’-deoxyuridine]) (Figures 3A and 3B). Mouse embryonic fibroblasts (MEFs) from Pum1−/−-mutant mice exhibited a slower growth rate than wild-type MEFs after the sec ond passage (Figure 3C), resulting in a significantly longer doubling time (Figure S4A). To validate the effects of Pum1 depletion on cell proliferation in vivo, we evaluated cell proliferation in spermatogonial cells in the testes of 3-week-old mice and found that the proportion of BrdU-positive cells was reduced by 43% in Pum1−/− testes compared with wild-type (Figures 3D and 3E). The proportion of cells positive for the mitotic cell marker phospho-H3, which represent dividing cells, was similarly reduced in the mutant (Figures 3F and 3G).
Figure 3. Decreased Cell Proliferation in Pum1-Depleted Cells and Mutant Animals.

(A) Proliferation of NIH 3T3 cells following lentiviral transduction with a small hairpin RNA against Pum1 (sh-Pum1KD) or control (sh-Con). Cell proliferation rates were determined by CCK8 assay.
(B) Pum1 knockdown (sh-Pum1KD) and control (sh-Con) NIH 3T3 cells were incubated with EdU to quantify DNA synthesis.
(C) Growth curves of Pum1+/+ (n = 3) and Pum1−/− (n = 3) mouse embryonic fibroblasts (MEFs) from passage 2 to 6 (P2–P6).
(D and E) Immunostaining for BrdU (D) and statistical analysis (E) of BrdU-positive (BrdU+) cells in the testis of 3-week-old Pum1+/+ (n = 5) and Pum1−/− (n = 5) mice.
(F and G) Immunostaining for phospho-Histone 3 (P-H3) (F) and statistical analysis of P-H3-positive (P-H3+) cells (G) in Pum1+/+ (n = 4) and Pum1−/− (n = 5) testes at 3 weeks of age. P-H3+ cells are presented as number per ten tubules. Scale bar, 50 μm.
(H) Annexin V-FITC/propidium iodide (PI) co-staining for apoptotic cells in Pum1 knockdown (sh-Pum1KD) and control (sh-Con) NIH 3T3 cells.
(I and J) Apoptotic cells in passage 3 Pum1+/+ (n = 3) and Pum1−/− (n = 3) MEF by annexin V-FITC/PI co-staining in bar graph (I) and two-dimensional dot blot (J) from flow cytometry analysis.
(K and L) Typical images of TUNEL staining (K) and quantification of apoptotic cells (L) in testes from 3-week-old Pum1+/+ (n = 5) and Pum1−/− (n = 5) mice. TUNEL-positive cells are presented as number per ten tubules. Scale bar, 50 μm.
Data are presented as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001 (t test).
PUM1 depletion has been previously associated with increased apoptosis in the testis (Chen et al., 2012). Although NIH 3T3 cells with Pum1 knockdown and Pum1−/− MEFs contained an increased proportion of apoptotic cells versus controls (Figures 3H–3J), we did not detect a significant difference in the number of apoptotic cells in testicular tubules from the testes of 3-week-old Pum1−/− and wild-type mice (Figures 3K and 3L), suggesting that apoptosis may contribute but does not represent the main cause of smaller cell numbers in Pum1−/− mice. Because PUM1 represses upstream activators of the p53 pathway (Chen et al., 2012), which may affect body weight, we generated Pum1 and p53 (Jacks et al., 1994) double-null mutant mice. Like Pum1−/− single-knockout mice, Pum1−/−;p53−/− mice were smaller in size than wild-type mice, whereas p53−/− mice were not (Figure S4B), suggesting that the smaller body weight of Pum1-mutant mice did not result solely from an unregulated activation of p53. In summary, in vitro and in vivo findings indicate that loss of Pum1 is associated with a defect in cell proliferation, causing decreased organ and body weight in Pum1−/− mice.
Cells Deficient for PUM1 Exhibit a Delay in G1-S Transition and Cell Cycle
Fluorescence-activated cell sorting (FACS)-based cell cycle analysis of PUM1-depleted cells revealed that RNAi-mediated Pum1 knockdown in NIH 3T3 cells significantly increased G0/G1 phase fraction by 22% while reducing S phase fraction by 30% compared with scrambled RNAi control (Figure 4A). Similarly, Pum1−/− MEFs contained 13% more cells in G0/G1 and 34% fewer cells in S phase than wild-type MEFs (Figure 4B), suggesting that depletion of PUM1 caused a delay in the G1-S transition.
Figure 4. Pum1-Depleted Cells and Mutant Tissues Exhibited Cell Cycle Defects and Increased Expression of Cdkn1b.

(A) Cell cycle analysis of control (sh-Con) and Pum1-knockdown (sh-Pum1KD) NIH 3T3 cells by FACS.
(B) Cell cycle analysis of MEF cells from E13.5 Pum1+/+ (n = 3) and Pum1−/− (n = 3) fetuses.
(C) Cell cycle progression analysis of three pairs of wild-type and mutant MEFs cells with EdU pulse labeling at different time points after resumption of cell cycle from G0 phase.
(D) Western blot analysis of cell cycle and apoptosis regulators in Pum1+/+ and Pum1−/− MEFs.
(E) Western blot analysis of G1-S transition regulators (CDK1, CDK2, Cyc E2, and CDKN1B) in the adult testis of Pum1+/+ and Pum1−/− mice.
(F) Immunostaining for PUM1 and CDKN1B in tissue sections from the testis of 3-week-old Pum1+/+ and Pum1−/− mice. Scale bar, 50 μm.
(G) Western blot analysis of PUM1 and CDKN1B in Pum1 knockdown (sh-Pum1KD) and control (sh-Con) NIH 3T3 cells. Extracts from Pum1−/− and Pum1+/+ were loaded for comparison.
(H) Western blot analysis of PUM1 and CDKN1B in NIH 3T3 cells overexpressing wild-type mouse Pum1 and mutant Pum1.
(I) Cdkn1b expression in Pum1−/− relative to Pum1+/+ tissues by densitometric analysis of western signal using ImageJ software (NIH). All tissues were performed from at least two individual samples and are reported as mean ± SD. The mean intensity value of Pum1+/+ mice was set at 100%.
(J) Western blot analysis of PUM1 and CDKN1B protein levels of different tissues from Pum1+/+ and Pum1−/− mice at 3 weeks of age. For each tissue, left lane is from wild-type and right lane is from knockout tissue.
Data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
In order to directly determine if cell cycle progression, specifically G1-S transition, is affected by loss of Pum1, we synchronized wild-type and Pum1-knockout MEF cells by serum starvation and compared the progression of cell cycle by harvesting cells at different time points after replenishment of serum. By labeling MEF cells at 16, 18, 20, 24, and 28 h after serum replenishment with EdU, we observed a delay of Pum1-knockout MEF cells in entry into S phase and progression from G1-S phase. At 16 h, both wild-type and Pum1-mutant MEFs were similar in proportion of EdU+ cells. However, in the next 2 h the proportion of EdU+ cells remained the same in Pum1-mutant MEFs, whereas wild-type MEF increased EdU+ cells to an average of 6%. By 20 h, wild-type MEFs contained twice as many EdU+ cells as mutant MEFs, reaching a significant difference (Figure 4C). Hence a delay in G1-S transition contributes to increased cell doubling time and reduced cell proliferation in Pum1-null animals.
G1-S Cell Cycle Regulators Are Enriched Targets of PUM Proteins
Given the observed G1-S transition delay in the cells of Pum1 knockout or knockdown, we investigated how PUM1 protein might regulate G1-S transition.
We first asked if PUM1 protein expression change during the cell cycle using synchronized MEF cells. We found that PUM1 protein gradually increased after the resumption of cell cycle with serum addition, suggesting that PUM1 might directly regulate cell cycle regulators (Figure S4). Indeed, the 8 nt consensus motif (UGUAHAUA) of PUM binding elements (PBE) is present in the 3’ UTR of many cell cycle regulators, with a significant enrichment among G1-S transition regulators (Figure S4C). Furthermore, testis RNA immunoprecipitation (RIP) experiment showed that mouse PUM1 binding to cell cycle regulators of G1-S transition was significantly enriched (Chen et al., 2012). Examination of eCLIP database of PUM2 binding in human cell culture further established that human PUM2 proteins bind directly to the 3’ UTR of many G1-S transition regulators, such as CDK1, CDK2, CDKN1A(p21), CDKN1B(p27), E2F3, and others (Figure S4D) (Van Nostrand et al., 2016; Zhang et al., 2017). Consistent with alterations in the cell cycle, we detected increased levels of CDK1, CDK2, and CDKN1B among a number of the cell cycle regulatory proteins screened in Pum1−/− MEFs (Figures 4D and S4E). Such an increase in protein expression of cell cycle regulators became more apparent in synchronized Pum1−/− MEFs. In synchronized mutant MEF cell culture, we observed increases in protein expression of both cell cycle activators (CDK1, CDK2, CCNE2, and E2F3) and cell cycle inhibitors (CDKN1b/p27 and CDKN1a/p21) (Figure S4F). Hence PUM1 appeared to be an important post-transcriptional regulator controlling the expression of a number of cell cycle regulators and regulating cell cycle progression on a global level.
Pum1 Deficiency Led to Increased Levels of the Cell Cycle Inhibitor CDKN1B
If PUM1 is a general cell cycle regulator at the post-transcriptional level, how does PUM1 deficiency lead to a delay in G1-S transition, reduced cell proliferation, and ultimately smaller mice? Unlike the general increase of protein expression of multiple cell cycle regulators (PUM1 targets) in the synchronized cell culture, only CDKN1B was consistently upregulated in the Pum1-knockout mouse tissues among the cell cycle regulators examined (Figures 4E and S4F). We hence hypothesized that Cdkn1b may be one of the major cell cycle targets of PUM1 in vivo and that increased level of CDKN1B may contribute to reduced cell proliferation in mutant organs and ultimately in mouse body size.
Immunostaining for PUM1 and CDKN1B in mouse testis sections revealed that cells with high PUM1 expression were largely devoid of CDKN1B, suggesting that CDKN1B may be one of the major downstream targets of PUM1 in the tissues. In normal testis, high levels of PUM1 were found in germ cells, which are highly proliferative, whereas CDKN1B was largely restricted to Sertoli cells and not detectable in germ cells (Figure 4F). In contrast, Pum1−/− testis tissue exhibited a marked increase in CDKN1B in germ cells. An inverse correlation of PUM1 and CDKN1B was observed in other tissues at various time points in development (Figure S4G and data not shown). The inverse and sometimes exclusive expression pattern of these two proteins in the same tissues implied cell-autonomous repression of CDKN1B expression by PUM1. Consistent with this, Pum1 knockdown in NIH 3T3 cells was associated with upregulation of CDKN1B, which was also higher in Pum1−/− than in wild-type MEFs (Figure 4G). Given the inhibitory role of CDKN1B in the cell cycle, we hypothesized that PUM1 might affect cell proliferation through translational repression of CDKN1B expression. To validate this, we transfected NIH 3T3 cells with expression vectors for wild-type PUM1 or a PUF repeat 7 mutant PUM1 (Pum1-mutR7) that was incapable of RNA binding (Weidmann and Goldstrohm, 2012). Overexpression of wild-type PUM1 resulted in downregulation of CDKN1B, whereas similar levels of mutant PUM1 attenuating RNA binding capacity did not affect CDKN1B levels (Figure 4H). Compared with controls, NIH 3T3 cells overexpressing wild-type PUM1 exhibited increased cell proliferation and upregulation of the G1-S transition (Figures S5A and S5B). The small organ and body weight in Pum1−/− mice may therefore be caused by global loss of PUM1-mediated CDKN1B repression, which would delay the progression of cell cycle, resulting in cell number reduction. Consistent with this, we detected elevated levels of CDKN1B in total organ extracts from Pum1−/− mice compared with wild-type, in particular, in extracts from testis, ovary, brain, liver, kidney, spleen, and thymus (Figures 4I and 4J).
Pum1 Represses Cdkn1b Translation by Binding to the 3’ UTR of Cdkn1b
To determine if PUM1 regulates CDKN1B expression by binding to Cdkn1b mRNA, we performed PUM1 RIP. Cdkn1b transcripts were significantly enriched in PUM1 RIP from NIH 3T3 cells (Figure 5A) and mouse testes (Figure 5B), confirming a physical association of PUM1 with Cdkn1b mRNAs. Indeed, there are two PBEs in the 3’ UTR of mouse Cdkn1b (Figure 5C). We next performed EMSA (electrophoretic mobility shift assay) with either the wild-type Cdkn1b 3’ UTR or a mutant 3’ UTR in which thePUM1 binding motif was disrupted (Figure 5C). Purified PUM1 HD (homology domain, conserved RNA binding region) proteins bound to both PBE1 and PBE2 at as low as 50 nM but not to mutant PBEs, supporting direct binding by PUM1 to Cdkn1b. We further evaluated the contribution of PBE1 and PBE2 in PUM1-mediated Cdkn1b regulation via dual luciferase reporter assays. 3’ UTR with two mutated PBEs or only one mutated PBE all resulted in a significant increase of luciferase signal over wild-type 3’ UTR, in both the presence and absence of Pum1 expression construct (Figure 5D). These findings indicate that PUM1 interacts with both PBE motifs in the Cdkn1b 3’ UTR (Kedde et al., 2010).
Figure 5. Pum1 Binds the 3′ UTR of Cdkn1b mRNA to Repress Its Translation.

(A) PUM1 RIP (RNA immunoprecipitation) from NIH 3T3 cell lysate (left). qRT-PCR demonstrates significantly increased levels of Cdkn1b mRNA in Pum1 IP of NIH 3T3 cells in comparison with IgG precipitates.
(B) Enrichment of Cdkn1b mRNAs in PUM1 RIP of testis lysates from 3-week-old mice. Input refers to total protein or total tissue RNA, and Pum1 and IgG refer to protein extract or RNA present in the anti-Pum1 and IgG immune complex.
(C) Diagram of mouse Cdkn1b-3′ UTR constructs (shown as boxes) containing two PBEs (filled boxes). Numbers correspond to positions of PBEs in the mouse Cdkn1b 3′ UTR. The mutated nucleotides are highlighted in lowercase in red. EMSA using different concentration of PUM1 HD domain with wild-type or mutant PBE1 (171 bp) and PBE2 (156 bp), respectively, showed direct binding of PUM1 to wild-type PBE.
(D) Bar graph results from dual-luciferase assay on NIH 3T3 cells expressing the reporter constructs containing either wild-type or mutated Cdkn1b 3′ UTR, PBE1, or PBE2. The cells were also co-transfected with mouse Pum1 vector (Pum1) or the same vector without Pum1 (empty). Cdkn1b 3′ UTR used for each assay contained both wild-type PBE1 and PBE2 or mutated PBE1 and PBE2 or one mutated and one wild-type PBE, as indicated under the graph.
(E and F) Cdkn1b translation was reduced in absence of PUM1. (E) Polysome profiles from fractionation experiments of NIH 3T3 cells lysate of Pum1 knockdown (sh-PUM1KD) and control (sh-Con). Peak position of free RNP (1), 40S (2), 60S (3), and 80S ribosome (4) and polysomes (5–9) are indicated. Wild-type (black line) and Pum1−/−-mutant (red line) profiles are overlaid. The values were normalized against β-actin. The experiments were performed in triplicate. (F) Cdkn1b mRNA distribution among fractions were determined by qRT-PCR using beta-actin as internal control.
Data are presented as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001 (t test).
To test if PUM1 represses the translation of endogenous CDKN1B, we examined translation efficiency of Cdkn1b mRNA in Pum1-depleted cells and tissues by polysome fractionation assay. Following RNA-mediated knockdown of Pum1 in NIH 3T3 cells, Cdkn1b mRNA was increased in all polysome fractions but not in the free RNP fraction (Figures 5E and 5F). We further confirmed that Cdkn1b mRNA levels were significantly increased in one or multiple polysome fractions in Pum1-mutant tissues (Figures S5C–S5F). These results indicate that depletion of Pum1 results in de-repression of translation of the cell cycle inhibitor CDKN1B, resulting in a delay in G1-S transition, reduced cell proliferation, and ultimately reduced overall size of the animal.
Disruption of Cdkn1b Partially Rescues the Smaller Body Weight Phenotype of Pum1-Mutant Mice
If CDKN1B were one of primary effectors of PUM1 in body weight control, its absence would likely affect the phenotype of Pum1 mutants. We therefore intercrossed Pum1−/− and Cdkn1b−/− mice to obtain double-null mutant mice and confirmed the loss of the respective proteins in brain tissues from Cdkn1b−/− (Fero et al., 1998), Pum1−/−, and Pum1−/−;Cdkn1b−/− mice (Figure 6A). As previously reported, Cdkn1b−/− mice were heavier than wild-type littermates after postnatal week 3 (Kiyokawa et al., 1996; Nakayama et al., 1996). The postnatal body weight of Pum1−/−;Cdkn1b−/− mice was consistently higher than that of Pum1−/− mice (Figures 6B–6E), with significant weight differences in females after post-natal week 4. Organs from Pum1−/−;Cdkn1b−/− mice were significantly larger than those from Pum1−/− mice but smaller than those from wild-type or Cdkn1b mutant mice (Figure 6F and data not shown). The partial restoration of body and organ size in Pum1−/−-mutant mice lacking Cdkn1b validates Cdkn1b as a major target of Pum1 in postnatal development and supports our hypothesis that Pum1-mediated post-transcriptional regulation of Cdkn1b contributes substantially to body weight control. However, similar birth weight and partial rescue of body weight defects in double mutants indicates that PUM1 controls other factors contributing to body weight, including other cell cycle regulators during embryonic development (Figures S4C and S4F). Nonetheless our data clearly showed that mammalian PUM1 is a growth regulator.
Figure 6. Cdkn1b Mutation Partially Rescued the Smaller Body Weight Phenotype of Pum1−/−-Mutant Mice.

(A) Western blot analysis of PUM1 and CDKN1B protein expression of brain tissues from adult single- or double-mutant mice validated the loss of function nature of all three mutants.
(B and C) Body weight of female (B) and male (C) mice was measured from postnatal week 3 to week 12. The body weights of double-knockout females were significantly higher than those of Pum1 single-knockout mice but smaller than wild-type. Data are presented as mean ± SD (N = 5–19 mice/genotype).
(D and E) Representative images of age-matched wild-type, Pum1−/−, Cdkn1b−/−, and Pum1−/−;Cdkn1b−/− female (D) and male (E) mice at the age of 2–3 months.
(F) Testis weight (total weight of both testes) was compared among adult male wild-type, Pum1−/−, Cdkn1b−/−, and Pum1−/−;Cdkn1b−/− mice. Double-mutant testes were again significantly bigger than those of Pum1 mutants but smaller than those of wild-type, indicating a partial rescue in organ weight. N = 7–11 mice/genotype. *p < 0.05, **p < 0.01, and ***p < 0.001 (t test).
Similar Tissue Expression Profile of Pum1 and Pum2 in Mice
Because PUM1 and PUM2 share highly conserved RNA binding domains (PUF repeat) and may have redundant function (Spassov and Jurecic, 2003; White et al., 2001), we evaluated Pum1 and Pum2 expression patterns at the mRNA and protein level. Analysis of Pum1 and Pum2 transcript levels in various tissues and embryonic stem cells (ESCs) using qRT-PCR analysis uncovered a remarkably similar expression profile (Figures S6A and S6B): Pum1 and Pum2 transcripts were highly expressed in the testis and brain, and the highest expression of both transcripts was found in 3-week-old testis. Tissue expression profiles did not differ between rapidly growing 3-week-old mice and adults, suggesting that Pum gene expression is organ specific and not globally correlated with growth rate. Western blot analysis confirmed protein expression patterns that were similar to mRNA distribution, with highest expression of PUM1 and PUM2 in the testis and whole brain in 3-week-old and adult mice. In adult mice, both PUM1 and PUM2 were detected at moderate levels in ovary, lung, thymus, cerebellum, and cerebrum, were detected at lower levels in colon, skin, epididymis, and intestine, and were barely detectable in liver, spleen, and kidney (Figure S6C). On the basis of their similar mRNA and protein distribution patterns, we hypothesize that mouse Pum1 and Pum2 may have coordinated and overlapping function and physiological roles.
We tested the hypothesis by inducing double-knockout MEF cells from Pum1F/F;Pum2−/−;RosaERT2Cre. PUM1 proteins in the Pum1F/F;Pum2−/−;RosaERT2Cre were drastically reduced after 4OHT (4-hydroxy tamoxifen) treatment (Figure S6D). The double-knockout MEF cells exhibited significantly reduced growth compared with Pum1−/− or Pum2−/− alone and a similar G1-S transition delay to that of Pum1-knockout cells (Figures S6E and S6F). To determine if the cell proliferation and cell cycle defect were mediated through Cdkn1b, we first compared the BrdU-positive cells in Pum1 and Cdkn1b double-knockout testes between wild-type and Pum1-knockout testes. Indeed, knockout of Cdkn1b not only partially rescued the testis size and cell number but also resulted in an increase in BrdU+ cells relative to Pum1-knockout mice, arguing for a direct involvement of Cdkn1b in PUM1-mediated cell proliferation and cell cycle regulation (Figure S6G). Furthermore, we knocked down Cdkn1b in Pum1−/− or Pum2−/− MEF cells and found that knockdown Cdkn1b increased cell proliferation and mitigated cell cycle defects in the cells depleted for PUM1 or PUM2 (Figures S6H and S6I), supporting coordinated PUM1- and PUM2-mediated regulation of cell proliferation and cell cycle via Cdkn1b.
Depletion of Pum2 Led to Upregulated CDKN1B, G1-S Transition Delay, and Reductions in Cell Proliferation and Body Weight
Knockout of Pum2 in mice resulted in reduced body weight (Figures 7A and 7B), consistent with previous studies (Lin et al., 2018a; Siemen et al., 2011). Knockdown Pum2 in NIH 3T3 cells also reduced cell proliferation, with a significant delay in G1-S transition (Figures S5G–S5I) and little effect on apoptosis rate (data not shown), supporting a role of PUM2-mediated Cdkn1B repression in the cell proliferation and body size. Pum2-depleted NIH 3T3 cells (Figure S5G) contained higher levels of CDKN1B than controls, suggesting that Cdkn1b mRNA is a downstream target of both PUM1 and PUM2. However, Cdkn1b mRNA and pre-mRNA levels did not increase in either Pum2- or Pum1-depleted NIH 3T3 cells or MEF cells (Figures S5J and S5K), suggesting that, like PUM1, PUM2 also regulates cell proliferation and body size via translational regulation of Cdkn1b.
Figure 7. Pum1 and Pum2 Double Mutants Exhibited Discrete Body Weight Reduction, and Reciprocal and Auto-Regulation of PUM Gene Expression May Be Responsible for Such a Precise Control of Body Weight.

(A–C) Body weight was measured at the age of 3 weeks for female (A) and male (B) mice of various combinations of Pum1 and Pum2 single or double mutations. A picture of one representative litter showing a remarkable discrete effect of loss of Pum genes at one gene copy interval is shown (C).
(D) Western blot analyses of key cell cycle regulators in 3-week-old testis of Pum1−/− and Pum2−/−. Left-pointing arrow represents chimeric PUM2-βgeo fusion protein in Pum2−/− mice.
(E and F) Both Pum2 (E) and Pum1 (F) mRNAs were enriched in PUM1 immunoprecipitates. Total cellular RNA (input) and RNA present in the immune complex (anti-Pum1 and anti-IgG, respectively) were used.
(G) Western blot of testis PUM2 RNA immunoprecipitation indicated that PUM2 proteins could be pulled down at high efficiency.
(H) Pum1, Pum2, and Cdkn1b mRNAs were significantly enriched in the PUM2 immunoprecipitates.
(I and J) The relative Pum2 mRNA levels in total RNA (T) and polysome fractions (number) were measured using qRT-PCR on fractionation extracts from adult testis (I) and NIH 3T3 (J). The values are normalized to β-actin. The experiments were performed in triplicate; 1 represents free RNP, and 5–9 represent polysome fractions.
(K) Proposed model of PUM expression feedback loop. Expression of Pum1 and Pum2 is co-expressed in most tissues and precisely regulated by auto and reciprocal translation repression via binding of PBEs on their 3′ UTR.
Dosage-Sensitive and Coordinated Control of Body Size by Pum1 and Pum2
Given similar roles of Pum1 and Pum2 in cell proliferation, we therefore propose that Pum1 and Pum2 play a coordinated role in the control of body size. To test this hypothesis, we generated mouse mutants with different combinations of Pum1- and Pum2-null mutant alleles, with the exception of Pum1−/− and Pum2−/− double homozygotes, which failed during early embryonic development (Lin et al., 2018b). We noted a remarkable dosage effect of Pum genes on body weight (Figures 7A and 7B). Loss of one copy of Pum2 (Pum1+/−+;Pum2+/−) did not affect adult body weight significantly. Pum2-knockout mice (Pum1+/−+;Pum2−/−) exhibited a significant reduction in body weight (the average weights of 3-week-old males and females were 77% and 80% those of wild-type littermates, respectively), and mice lacking one copy of each gene (Pum+/−;Pum2+/−) were smaller than Pum2−/− homozygotes. Homozygous loss of Pum1 (Pum1−/−;Pum2+/−+) was associated with a larger reduction in weight than heterozygotes for both Pum1 and Pum2-null alleles, and mice with only a single Pum1 but no Pum2 allele (Pum1+/−;Pum2−/−) exhibited a further reduction in weight (Figure 7C). The smallest Pum mutants recovered were those lacking Pum1 and heterozygous for Pum2 (Pum1−/−;Pum2+/−) (Figure 7B). Therefore, the effects of single and double compound Pum mutants on body weight reduction follow a discrete decreasing order in Pum wild-type alleles (wild-type R Pum1+/−+;Pum2+/− > Pum1+/−+;Pum2−/− > Pum1+/−; Pum2+/− > Pum1−/−;Pum2+/−+ > Pum1+/−; Pum2−/− > Pum1−/−;Pum2+/−). Furthermore, absence of Pum1 alleles had a stronger effect on the body weight than that of Pum2 alleles. The reduction in body weight correlated with body size in age-matched mice with different combinations of Pum1- and/or Pum2-null alleles (Figure 7C). The difference in body size between Pum2−/− and Pum1 and Pum2 double heterozygotes was subtle but in alignment with the average body weight. The two smallest mice from this litter had the same genotype (Pum1+/−; Pum2−/−), together providing an example of incremental reduction of body weight and body size depending on gene dosage. We also observed a dosage-dependent effect of Pum1 alleles on organ weight and cell number. With the exception of heart, all organs from 3-week-old Pum1+/− mice differed significantly in weight from both wild-type and Pum1−/− organs (Figures S3A and S3B). Testis and thymus also exhibited a dosage-dependent correlation of Pum1 gene dosage and total organ cell number (Figure S3C). Similar effects of Pum1 dosage on mouse organ weight were observed at other postnatal time points (data not shown). Thus we propose that mammalian PUF genes are post-transcriptional growth regulators that govern animal body size through dosage-sensitive post-transcriptional control of cell proliferation.
Reciprocal and Auto-Regulatory Regulation of Pum1 and Pum2 Expression
We detected upregulation of Pum2 expression in the testes from 3-week-old and adult Pum1−/− mice (Figures 7D and S7A) and in Pum1-knockdown cells (Figure S5G). Similar to Cdkn1b mRNA, both Pum1 and Pum2 3′ UTR contain highly conserved PBE motifs (Figures S7B and S7C), suggesting that both PUM1 and PUM2 may repress Pum1 and Pum2 expression post-transcriptionally. PUM1 RIP from cell culture and mouse testis confirmed a physical association of PUM1 with Pum2 mRNA (Figure 7E). Pum1 mRNA is also enriched in PUM1 immune complex (Figure 7F). Similarly, both Pum1 and Pum2 mRNA were highly enriched in PUM2 immune complex (Figures 7G and 7H). Consistent with upregulated expression of CDKN1B in Pum2-knockout tissues (Figure 7D), Cdkn1b mRNAs were also enriched significantly in PUM2 immune complex (Figure 7H). Polysome fractions from Pum1-depleted NIH 3T3 cells or tissues from Pum1−/− mice contained significantly increased Pum2 transcript levels compared with controls (Figures 7I and 7J), revealing increased translation of Pum2 mRNA in the absence of PUM1. To further determine if such auto and reciprocal regulation is also conserved in human cells, we searched the database on whole-genome target identification of PUM2 targets by eCLIP (enhanced UV crosslinking immunoprecipitation) in K562 cells and in neonatal mouse brain (Van Nostrand et al., 2016; Zhang et al., 2017). Significantly enriched peaks were found on the 3’ UTR of both PUM1 and PUM2 genes, confirming direct binding of human and mouse PUM proteins to 3’ UTR of both PUM1 and PUM2 transcripts (Figure S7D) (Van Nostrand et al., 2016; Zhang et al., 2017). Among the known PUM targets involved in cell cycle regulation, CDKN1B and CDK1 showed increased protein expression in the absence of Pum1 or Pum2 and specific enrichment of their 3’ UTR reads in PUM eCLIP-seq (Figures 7D and S4D), suggesting a conserved role of PUM-mediated CDKN1B translation regulation in human cell cycle and growth. These findings led us to propose that precise expression levels and ratio of PUM1 and PUM2 are determined by auto-regulatory and reciprocal post-transcriptional repression (Figure 7K). Such an auto and reciprocal feedback loop mechanism likely established a balanced and fine-tuned PUM protein level in each tissue to ensure precise control of cell proliferation, organ size, and body size.
DISCUSSION
How body size is controlled is a fundamental biological question. Here we demonstrate that mouse Pum1 and Pum2, which encode evolutionarily conserved RNA-binding proteins, regulate body size in a dosage-dependent manner via post-transcriptional repression of the cell cycle regulator CDKN1B. These data unveil a new layer of control in the regulatory hierarchy governing body size. Translational regulation of cell cycle regulators such as CDKN1B by PUM proteins may represent a mechanism for precise control of body size and growth in mammals including human.
PUMILIO Proteins Are Post-transcriptional Regulators of Mammalian Growth
PUM1 has been previously identified to safeguard spermatogenesis by repressing upstream activators of the p53 pathway (Chen et al., 2012). Loss of Pum1 also causes progressive motor neuron degeneration (Gennarino et al., 2015, 2018). Although these studies reported a body weight reduction in Pum1-mutant mice, it was not clear if this phenotype was secondary to the pathological defects, such as over-activation of p53 or neuronal degeneration. We generated a Pum1-null allele and found significant decreases in overall organ weight and tissue cell proliferation in our Pum1-null mice. Although we could not exclude the possibility that p53-mediated apoptosis may contribute to body size reduction during embryonic and neonatal development, reduced cell proliferation appeared to be the primary factor contributing to smaller body size of Pum1-null mice.
Knockdown or knockout of Pum1 reduced cell proliferation in culture and in animals, while overexpression of Pum1 led to increased growth in cell culture, establishing Pum1 as a growth regulator. PUM1 protein is highly conserved, and we observed that PUM1 bound to the 3′ UTR of Cdkn1b and that knockdown of human PUM1 also reduced cell proliferation (unpublished data). Human PUM2 plays a role in adipose stem cell proliferation (Shigunov et al., 2012). We proposed that human PUM-family genes are also growth regulators and may contribute to human height and body weight regulation. Given that four alleles of PUMs govern the body size in a dosage-dependent level, the association between single PUM allele and human height may be difficult to detect. Indeed, examination of current genome-wide association study (GWAS) data on human height and SNPs from human PUM1 or PUM2 locus failed to identify any signifi-cant association (unpublished data). However, in a recent study, two patients carrying de novo human PUM1 missense pathogenic mutations were developmentally delayed, with one being only at 5% growth of her age group, supporting PUM1’s role in human growth (Gennarino et al., 2018).
Pum Genes Regulate Body Size in a Gene Dosage-Dependent Manner
We found that Pum1−/−-mutant mice were not only of lower body weight but also of reduced body length, indicating an overall body size reduction. Pum1−/− and Pum2−/− mice were smaller throughout postnatal development, with a largely proportional size reduction among organs. A gene dosage effect of Pum alleles on body weight has been previously suggested (Gennarino et al., 2015; Siemen et al., 2011), consistent with the overall dosage-sensitive behavior of PUM proteins (Gennarino et al., 2018; Lee et al., 2016; Lin et al., 2018b). In this study, we demonstrated a clear effect of gene dosage for Pum1 and, more interestingly, a synergistic effect of Pum1 and Pum2 on body and organ size. The gene dosage-dependent body size reduction in Pum single and double mutants could be correlated with differences in PUM1 and/or PUM2 expression. Indeed, heterozygote Pum1 mutants exhibited intermediate levels of Pum1 expression level, suggesting that protein expression levels of PUM1 and PUM2 regulate organ size and body size. Interestingly, PUM1-depleted cells exhibited a higher increase in CDKN1B levels than PUM2-depleted cells, suggesting a stronger effect of PUM1 on CDKN1B repression in the NIH 3T3 cell line. This finding is consistent with the observation that the Pum1-null mutation has a more prominent effect on mouse body size than Pum2 knockout, but because relative expression levels of these proteins vary between cell types, the effect of Pum1 and Pum2 on cellular proliferation may differ between cell types.
Both Pum1 and Pum2 are coexpressed in diverse organs at remarkably similar mRNA and protein levels, suggesting their expression may be strictly regulated and coordinated. In addition, upregulation of protein expression in the absence of the other family member, association of Pum transcripts with PUM immunoprecipitate, and the presence of conserved PBEs in the 3’ UTR of Pum1 and Pum2 suggest that expression of PUM proteins is tightly controlled by auto-regulatory and reciprocal translational repression by PUM1 and PUM2. Such a layered regulatory mechanism of PUM protein expression level could constitute a mechanism to ensure the precise control of cell proliferation, body size, and growth. Similar mechanism has been observed for C. elegans PUF members in patterning (Lamont et al., 2004), leading us to propose a conserved fine-tuning mechanism during development and growth via auto and reciprocal regulation of PUF-family proteins in mammals.
PUM-Mediated Post-transcriptional Regulation of Cdkn1b Is Required for Mouse Body Size Control
Our results show that the smaller size of Pum1−/− mice results from a decrease in cell number but not cell size. Previously, Kedde et al. (2010) reported that binding of PUM1 to PBE in CDKN1B 3′ UTR enhances the micro-RNA (miRNA)-mediated inhibition of CDKN1B translation in HeLa cells. The physiological significance of this inhibition was unknown. mRNA targets of PUM proteins have been identified by RIP or eCLIP, and a number of cell cycle regulators, such as CDK1, CDK2, CCNE2, and CDKN1B, were significantly enriched in PUM 1 RIP or CLIP (Chen et al., 2012; Hafner et al., 2010; Morris et al., 2008; Van Nostrand et al., 2016; Vourekas et al., 2015; Zhang et al., 2017). However, it is not known which targets are physiologically relevant to cell proliferation and how such binding of cell cycle regulators affected cell growth and animals. Our results show that loss of PUM1 leads to the upregulation of CDKN1B, which normally inhibits the G1-S transition, through de-repression of Cdkn1b translation. As genetic removal of Cdkn1b can partially rescue the Pum1 knockout phenotype, PUM-mediated post-transcriptional control of Cdkn1b is essential for achieving discrete control of body size. Our study has therefore identified post-transcriptional regulation of cell cycle regulators as an additional layer of regulation of mammalian organ and body size control. At the same time, null mutation of Cdkn1b did not restore the body size of Pum1−/− mice, indicating that there are other targets of PUM proteins in animal body size control.
Although we provide compelling evidence for a cell-autonomous role of PUM1 and PUM2 in cell proliferation, organ size, and body size, a systemic or metabolic effect due to loss of Pum1 may also contribute to body size reduction. Indeed, serum IGF levels in mutant mice are reduced (Figure S2B), but this is unlikely to be the major cause of smaller body size, as Pum1 mutants are smaller prior to the action time of the growth hormone/IGF-1 effect, and heterozygotes with normal amounts of serum IGF-1 also exhibited body weight reduction to some extent. It remains to be determined if the reduced serum IGF-1 level resulted from reduced size of hormone-secreting gland.
Pum genes are involved in the regulation of pluripotency genes in mouse ESCs and in cell proliferation and genomic stability in cancer cell lines (Lee et al., 2016; Leeb et al., 2014; Miles et al., 2012, 2016; Tichon et al., 2016). Accordingly, deregulation of PUMs may result in proliferative and degenerative diseases such as cancers or neuromuscular disorders. In the present study, we did not address how Pum1 and Pum2 are regulated at the transcriptional level and if they regulate organ size through stem cells or all proliferative cells. Further study of other targets of PUM proteins and transcriptional regulation of Pums in different cell types is required to fully explore the contribution of Pum to body size control. Such knowledge may also help us understand the roles of PUM-family proteins in human physiology and disease.
STAR⋆METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eugene Yujun Xu (xuyujun@njmu.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Pum1 null mice were produced by Northwestern University Transgenic and Targeted Mutagenesis Laboratory. Cdkn1b deficient mice (B6; Cdkn1btm1Mlf) were obtained from Model Animal Research Center of Nanjing University and crossed with Pum1 mice. Mutant mice used were on a mixed genetic background of C57BL/6J and 129SvJ. Animals were maintained under standard conditions in the animal facilities of the Nanjing Medical University, Nanjing, China and/or of Northwestern University. All mouse procedures and protocols were approved by the Institutional Animal Care and Use Committees (IACUC) of Nanjing Medical University and Northwestern University and conducted in accordance to institution guidelines for the care and use of animals.
Generation of Pum mutant mice and genotyping
The Pum1 sequence obtained from the NCBI mouse genomic database was used to generate a Pum1-targeting construct in which exons 9 and 10 of the Pum1 gene were flanked by loxP elements. CRE recombinase mediated excision of exons 9 and 10 by CRE recombinase produces a nonsense mutation resulting in premature termination, and truncated transcripts present at very low level. The detailed strategy and confirmation of homologous recombination of each targeting arm is presented in Figure S1. The targeting vector, which includes exons 9 and 10 flanked by loxP elements and a Neo cassette flanked with FRT sequences, was linearized and electroporated into 129 Sv/J mouse ES cells. After selection with G418, ES cells containing the targeted Pum1 allele were identified by Southern blot using BamHI and PstI digestion of ES cell genomic DNA. The positive ES cells heterozygous for the allele containing the integrated homologous recombinant were injected into wild-type mouse blastocysts. Resulting chimeric males were mated with C57 BL/6 females producing targeted (Pum1ta/wt) mice. Pum1ta/wt mice were crossed with a germline-specific Flp transgenic mouse to remove the Neo cassette and generate mice heterozygous for a floxed Pum1 allele (Pum1flox/wt). These mice were bred to homozygosity to generate homozygous Pum1flox/flox mice on a hybrid (C57BL/6J and 129Sv/J) background. Pum1flox/flox male mice were crossbred with Vasa-Cre mice in which CRE expression is restricted to germ cells (Gallardo et al., 2007). Mice with ubiquitous null mutation of Pum1 (Pum1−/−) were obtained from intercrosses of Pum1+/− mice. This new Pum1 mutant allele is a complete loss-of-function allele based on three criteria: Wild-type intact Pum1 transcripts are completely absent in Pum1 homozygote mutants by Northern blot hybridization and RT-PCR, residual transcripts lacking exons 9 and 10 were barely detectable by RT-PCR, and western blot analysis using antibodies against N-terminal of PUM1 failed to detect any truncated peptides from homozygote tissues. Cdkn1b deficient mice (B6; Cdkn1btm1Mlf) and p53 knockout mice (B6; Trp53tm1Tyj) were obtained from Model Animal Research Center of Nanjing University. Pum1+/− male mice were crossbred with Cdkn1b+/− or p53+/− female mice, respectively, to generate double heterozygotes, which were then interbred to produce Pum1−/−;Cdkn1b−/− or Pum1−/−;p53−/− mice for analysis. Primers used for Pum1 genotyping were listed in Table S1, producing a 361 bp fragment for the Pum1-WT allele, a 453 bp fragment for the Pum1-floxed allele and a 557 bp for the Pum1 mutant allele. Genotyping for Cdkn1b and p53 transgenic mice was performed as described previously (Fero et al., 1996; Jacks et al., 1994). A minimum of six pairs of primers spanning the RRM region and other regions were used to confirm the expression of the Pum1 gene.
RosaErt2-Cre mice was crossed into Pum1f/f;Pum2−/− to generate RosaErt2-cre; Pum1f/f;Pum2−/− stock. RosaErt2-cre;Pum1f/f; Pum2−/− MEF cells were generated from E13.5 embryos of the stock.
METHOD DETAILS
Size phenotyping
Mice were weighed daily or weekly and prior to sacrifice for tissue collection. Animal length was defined as the distance from the tip of the nose to the base of the tail. Embryonic day (E) 13.5, E14.5 and E16.5 embryos were dissected from timed mattings of Pum1+/− female and male mice.
Single cell preparation for organ cell count and cell size assay
Testicular cells were prepared by sequential enzymatic digestion. Decapsulated testes from 3-week-old and adult males were first digested in 1 mg/mL collagenase Type IA (Sigma) in DMEM (GIBCO) at 37°C for 20 minutes. Single tubules were allowed to settle by gravity and enzyme-containing medium was removed. Cord fragments were then digested for 15 min at 37° C in 0.25% trypsin (Invitrogen) and 7mg/mL DNase (Sigma), followed by addition of 10% FBS to inhibit enzymatic digestion. Cells were pelleted by centrifugation at 500 g for 5 minutes at 4°C and resuspended in 1 mL DMEM with 0.5% FBS, followed by cell count using a hemocytometer. Bone marrow was flushed from the femora and tibia using 5 mL DMEM (containing 0.5% FBS) and filtered through a 40 μm mesh screen to obtain a single cell suspension. The cell number was determined using a hemocytometer. Thymus cells were collected and counted as previously described (Hu et al., 2012). Cell size was determined by flow cytometry (BD FACSVerse). Per sample, 50,000 events were collected and three distinct cell populations gated in FAC-A by FL2-A dot plots followed by evaluating in FSC-A histograms. Median mean fluorescence intensity values were collected using Flowjo and analyzed by Graphpad Prism (Graphpad, La Jolla, CA).
Lentiviral packaging
The shRNA constructs targeting mouse Pum1 (TRCN0000294814) were designed by the RNAi Consortium (Broad Institute, Cambridge, MA). Lentiviral shRNA vectors were constructed by cloning specific targeting sequences into the pLKO.1 backbone (Addgene 10878). Mouse Pum1 (NM_030722) was cloned from CMV-Pum1 vector and inserted into a lentivirus vector backbone (Addgene 16579). The coding sequence of RNA binding defective mouse Pum1 contained S1082A, N1083A and E1086A mutations in the 7th PUF repeat (Weidmann and Goldstrohm, 2012). 293T cells were cultured in DMEM supplemented with 10%FBS. Lentiviral particles were produced by transfecting 293T cells with the appropriate expression and packaging plasmids using Lentiviral Packaging Systems (Genecopeia), followed by filtering cultured supernatants through a 0.45 mM filter. The shRNA constructs targeting mouse Pum2 (TRCN0000233378, TRCN0000233378, TRCN0000233378) were obtained from the RNAi Consortium (Broad Institute, Cambridge, MA).
Cell culture and EdU labeling
MEFs were isolated from E13.5 embryos. MEFs and NIH 3T3 cells were cultured in DMEM supplemented with 10% FBS, penicillin/streptomycin, and 2 mM L-glutamine following standard methods. MEF cells were synchronized by deprivation of serum over 72 hr, and then refeed with DMEM containing 10%FBS for EdU assay. shRNA-mediated knock-down of Pum1 was carried out by infecting NIH 3T3 cells with lentivirus and transduced cells were cultured under puromycin (2ug/ml) selection for three days. Cell counting or cell cycle analyses was performed on cells harvested during the exponential growth phase. For EdU incorporation experiments, NIH 3T3 cells were treated with 10 μM EdU for 2hr. EdU incorporation was determined using Click-iT EdU Flow Cytometry Assay Kits (Invitrogen) following the manufacturer’s instructions.
Cell proliferation analysis
Cells were plated in 96-well plates at 2,000 cells per well after transfection with lentivirus. Cells were cultured for 24, 48, 72 and 96 hr. The absorbance at 450 nm was measured after incubation with 10 μl of Cell Counting Kit 8 (CCK8, Yeasen) for 2 hr. Results were combined from three independent experiments.
Cell cycle and apoptosis analysis
Cells were seeded in 6-well plates at a density of 3×105 cells per well. For cell cycle analysis, cells were harvested 20 hr after seeding and fixed in 70% ethanol overnight at 4°C. Fixed cells were washed twice with PBS and stained in PI/RNase Staining Buffer (BD Biosciences) at 0.5 to 2.5 × 106 cells per mL for 30 min at room temperature. At least 10,000 cells were counted for each sample, and data were analyzed with MODfit LT 4.0 software (Verity Software House). For cell apoptosis analysis, 1 × 105 cells were collected and washed twice with ice-cold PBS, resuspended in binding buffer (100 ml), treated with Annexin V-FITC and PI (BD Biosciences) and incubated in the dark for 15 min. Another 100 μL of binding buffer was then added, and flow cytometry analysis was performed within 1 h to measure Annexin V-FITC positive cells for apoptosis (BD FACVerse).
Histology, Immunohistochemistry and TUNEL assay
Tissues were fixed overnight in Hartman’s fixative (Sigma) and processed for H&E staining and immunohistochemistry according to standard protocols (VanGompel and Xu, 2010). Immunostaining for PUM1, CDKN1B, phospho-Histone H3 were performed, following citrate buffer antigen retrieval, by incubation with anti-PUM1(1/50, Abcam), anti-CDKN1B (1/50, Abcam), and anti-phospho-H3 (1/50, CST) primary antibodies and detected using Biotin-Streptavidin HRP Detection Systems (ZSGB-BIO). For BrdU incorporation experiments, mice were injected intraperitoneally with 50 mg/kg BrdU in PBS and sacrificed 2 hr later, and tissue sections were analyzed by immunohistochemistry with anti-BrdU antibody (Invitrogen). TUNEL analysis was performed using the In Situ Cell Death Detection Kit from Roche according to the manufacturer’s instructions. A minimum of three randomly chosen discontinuous sections were used to determine positive cells in tubules.
Western blotting
Protein extracts from tissues were prepared using RIPA lysis buffer (Beyotime). Tissues or cells were harvested and lysed in RIPA Buffer (50mM Tris (pH 7.4), 150mM NaCl, 1% Triton X-100, 1% sodium deoxycholate) containing proteinase inhibitors cocktail (Roche) on ice for 40 minutes. Lysates were centrifuged 40 minutes at 4°C at maximum speed (20000 rcf.) and resulting protein extracts separated by SDS–PAGE (12%). After transfer of proteins to PVDF membranes (Bio-rad), membranes were blocked with 5% skim milk in TBS containing 0.1% Tween 20 (TBS-T) for 1 hr at room temperature, followed by two washes with TBS-T. Membranes were incubated overnight at 4°C with primary antibodies followed by three 10-min washes in TBS-T, exposure to HRP-conjugated secondary antibody for 1 hr at room temperature, and 3 washes with TBS-T. Detection of HRP conjugated secondary antibody was performed with ECL (PerkinElmer). Antibodies used in supplemental experiment were anti-E2F3 (ABclonal Technology, A8811), anti-GSK3B (ABclonal Technology, A0479), Anti-P107 (Proteintech, 13354–1-AP).
Northern blot analysis
Total RNA was prepared using TRIZOL reagent (Invitrogen Life Technologies, USA). Per sample, 30 μg RNA was separated by denaturing 1% agarose formaldehyde electrophoresis and transferred to a nylon membrane. Hybridization with 32P–labeled probes was performed at 42°C overnight in hybridization solution containing formamide. The membranes were washed twice with 0.2 × SSC, 0.1% SDS at room temperature for 30 min, twice with 0.2 × SSC, 0.1% SDS at 42°C for 30 min, and then exposed to X-ray film at −80°C with an intensifying screen for 72 h.
Pre-mRNA detection
Total RNA was extracted from NIH 3T3 or MEF cells. After reverse transcribed, qRT-PCR was conducted to detect the transcription of Cdkn1b. PCR primers were listed in Table S1 (5’ to 3’).
Serum IGF assay
Serum was prepared from 2-month-old Pum1+/+, Pum1+/− and Pum1−/− mice. Total IGF-I concentration in serum was measured by Mouse/Rat ELISA kit (DSL-10–29200, Diagnostic Systems Laboratories, Webster, TX).
Body composition analysis
Analysis of body fat and lean body mass (LBM) in Pum1−/− and wild-type controls was performed by nuclear magnetic resonance (NMR) to measure the volume of lean mass, fat and fluid.
Neurosphere assay
For primary culture of neural stem cells (NSCs), the forebrain was microdissected from wild-type and Pum1−/− brain of P7(post-natal day) mice. single cell suspensions were prepared and the dissociated neuronal cells were seeded on glass-based 96-well plate coated with poly-D-lysine(Sigma) in Neurobasal medium (Invitrogen) in the presence of B27 (Invitrogen) and 100 mM L-glutamine, and kept at 37°C under 5% CO2.
Dual-luciferase report system assay
A segment covering bp 123 through 1280 of the mouse Cdkn1b 3’ UTR (NM_009875.4) containing Pumilio-binding elements was subcloned into psiCHECK-2 vector (Promega) by using XhoI and PmeI restriction enzymes (New England Biolabs). Wild-type and mutant PBE sequences were as follows: wt PBE, 5’-TGTATATA-3’; PBE mutant, 5’-acaATATA-3’ (Weidmann and Goldstrohm, 2012). Mutations were introduced using by ClonExpress MultiS One Step Cloning Kit (Vazyme Biotech). NIH 3T3 cells in 12-well plates were transfected with 100 ng of psi-CHECK-2 construct carrying either wild-type or mutant Cdkn1b 3’ UTR plus 400 ng of CMV-Pum1 or control pCMV using FUGENE HD Transfection Reagent (Promega). After 24 hr, Firefly expression and Renilla lucif-erase expression were measured using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions.
EMSA
PUM1 homology domain (PUM1HD, aa 1091 to 1428) of mouse PUM1 protein fused to 6 × His-SUMO was subcloned into V14 vector for protein expression in bacteria. Plasmids were transformed into BL21(DE3) competent cells (Vazyme Biotech) and recombinant proteins were induced with 0.2 mM IPTG at 18°C. Bacteria were lysed in lysis buffer (400 mM NaCl, 50 mM Tris-Cl, 10% glycerol, pH 7.0) and bound proteins were recovered on Ni-NTA agarose resin, washed with lysis buffer at pH 7.0, and eluted with 100–250 mM imidazole at pH 7.0. The concentration of purified proteins was determined by electrophoresis alongside a serial dilution of BSA standards (Pierce) with Coomassie staining. PBE1 or PBE2-contained fragments was PCR-amplified from psiCHECK-2 vector (described above) with each upstream primer containing the T7 promoter sequence and subcloned into pUC57 vector. Linearized PBE(wt) or PBE(mut) DNA templates were obtained by NdeI (NBE) digestion. RNA was generated from plasmids pUC57(described above) using the mMESSAGE mMACHINE T7 Ultra kit (Invitrogen) followed by purification using MEGAclear Transcription Cleanup kit (Invitrogen). And RNA Biotinylation was conducted using Pierce RNA 3’ End Biotinylation Kit (Invitrogen) according to the manufacturer’s instructions. Cdkn1b 3′ UTR PBE1 or PBE2 RNA and Pum1HD protein interactions were detected using LightShift Chemiluminescent RNA EMSA Kit (Invitrogen) according to the manufacturer’s instructions. The RNA and protein binding reactions were added to a native 6% polyacrylamide gel, running in 0.5X TBE on ice for 1 hr, and then transferred to a Nylon Membrane. The membrane was UV cross-linked to fixate transferred RNA to the membrane. After incubation with Stabilizedation Streptavidin-Horseradish Peroxidase Conjugate buffer, the membrane will be exposed under CCD camera to detect the signal.
RNA immunoprecipitation (RIP)
RNA immunoprecipitation was performed as described previously (Keene et al., 2006). NIH 3T3 cells were grown to 90% confluence for collection. 100 mg testis or 1.0 × 107 NIH 3T3 cells were lysed in polysome lysis buffer. Goat anti-Pum1 antibody (Bethyl Lab) and Goat IgG (Beyotime) were coupled to Protein A Agarose (Invitrogen), respectively. NIH 3T3 cell lysate or total testicular lysate were added to antibody-coupled beads to immunoprecipitate PUM1 proteins. RNA associated PUM1 proteins were extracted with Trizol Reagent (Invitrogen) for qPCR analysis of candidate target RNAs.
Polysome profile analysis
Polysome profile analysis was carried out as previously described (del Prete et al., 2007). 50–200 mg tissue or 0.5–1.0 3 107 cells were treated with ice-cold PBS or culture medium containing 100 mg/mL cycloheximide for 10 min followed by lysis in polysome lysis buffer (100 mM KCl, 0.1%Triton X-100, 50mM HEPES, 2mM MgCl2, 10% glycerol, 100 μg/mL cycloheximide, 1mM DTT, 20unit/mL RNase Inhibitor (EDTA free) and 1 3 proteinase inhibitor cocktail (EDTA free). Lysates were loaded onto 20%–50% (w/v) sucrose density gradients (10 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 100 mM NaCl and 1mM DTT) and centrifuged at 38,000 rpm for 2.5 hr at 4°C in a Beckman SW41 Ti rotor. Gradients were fractionated and the absorbance at 254 nm was continuously recorded using Gradient Fractionator (BioComp, Canada).
RNA extraction and quantitative RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. RNAs from polysome fractions were isolated by extraction with 3-fold volume of Trizol. Total RNA (1 μg) was reverse transcribed (RT) with PrimeScript RT Master Mix (Takara). Per PCR reaction, 100 ng of RT template were amplified using primers listed below (Table S1) and Taq Polymerase (Takara) according to the product manual. The number of PCR cycles ranged from 22 to 35 depending on the linearity of the reaction.
Real-time PCR results were analyzed using the comparative Ct method normalized against the housekeeping genes β-Actin or Gapdh. PCR reactions were optimized to measure the exponential phase on the amplification curve.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data in bar and line graphs are expressed as means ± SD (Standard deviation of the mean). All experiments were repeated at least three times unless noted. Statistical significance between two groups of data was evaluated by Student’s t test (two-tailed) comparison using GraphPad Prism software 5 or Excel t test function. Statistical significance is indicated by * for p < 0.05, ** for p < 0.01, *** for p < 0.001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat Polyclonal anti-Pum1 | Bethyl Laboratories | Cat#A300–201A; RRID: AB_2253218 |
| Rabbit Polyclonal anti-Pum2 | Bethyl Laboratories | Cat#A300–202A; RRID: AB_2173752 |
| Rabbit monoclonal anti-Pum1 | Abcam | Cat#ab92545; RRID: AB_10563695 |
| Rabbit monoclonal anti-p27 KIP 1 | Abcam | Cat#92741; RRID: AB_10561762 |
| Mouse monoclonal anti-Actin | Sigma-Aldrich | Cat#A1978; RRID: AB_476692 |
| Mouse monoclonal anti-α Tubulin | Santa Cruz Biotechnology | Cat#sc-8035; RRID: AB_628408 |
| Rabbit monoclonal anti-GAPDH (D16H11) | Cell Signaling Technology | Cat#5174; RRID: AB_10622025 |
| Rabbit Polyclonal anti-Cleaved Caspase-9 | Cell Signaling Technology | Cat#9505; RRID: AB_2290727 |
| Rabbit monoclonal anti-Cleaved Caspase-3 | Cell Signaling Technology | Cat#9664; RRID: AB_2070042 |
| Rabbit monoclonal anti-Phospho-Histone H3 (Ser10) (D2C8) | Cell Signaling Technology | Cat#3377; RRID: AB_1549592 |
| Mouse monoclonal anti-cdc2 (POH1) | Cell Signaling Technology | Cat#9116; RRID: AB_2074795 |
| Rabbit monoclonal anti-CDK2 (78B2) | Cell Signaling Technology | Cat#2546; RRID: AB_2276129 |
| Mouse monoclonal anti-CDK4 (DCS156) | Cell Signaling Technology | Cat#2906; RRID: AB_2078399 |
| Mouse monoclonal anti-CDK6 (DCS83) | Cell Signaling Technology | Cat#3136; RRID: AB_2229289 |
| Mouse monoclonal anti-Cyclin D1 (DCS6) | Cell Signaling Technology | Cat#2926; RRID: AB_2070400 |
| Mouse monoclonal anti-Cyclin D3 (DCS22) | Cell Signaling Technology | Cat#2936; RRID: AB_2070801 |
| Rabbit Polyclonal anti-Cyclin E2 | Cell Signaling Technology | Cat#4132; RRID: AB_2071197 |
| Mouse monoclonal anti-BrdU (ZBU30) | Life Technologies | Cat#03–3940; RRID: AB_2532919 |
| Goat IgG | Beyotime Biotechnology | Cat#A7007; |
| Rabbit IgG | Beyotime Biotechnology | Cat#A7016; |
| Rabbit monoclonal anti-CDKN1A | Abclonal | Cat#A1483; RRID: AB_2761709 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat#7074; RRID: AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat#7076; RRID: AB_330924 |
| Bacterial and Virus Strains | ||
| pLKO.1 - TRC cloning vector | (Sarbassov et al., 2005) | Addgene #10878 |
| Scramble control shRNA | (Moffat et al., 2006) | Addgene #1864 |
| Pumi shRNA | This paper | N/A |
| pSin-EF2-Pum1-Pur | This paper | N/A |
| pSin-EF2-Pum1(mutR7)-Pur | This paper | N/A |
| CMV-Pumi | This paper | N/A |
| V14–6 × His-SUMO | Laboratory of Ming Lei | N/A |
| pUC57 | Genewiz | N/A |
| psiCHECK-2 Vector | Promega | Cat#C8021 |
| Chemicals, Reptides, and Recombinant Proteins | ||
| Hartman’s Fixative | Sigma-Aldrich | Cat#H0290 |
| DMEM | GIBCO | Cat#11995073 |
| Fetal Bovine Serum | GIBCO | Cat#10099141 |
| 0.25%Trypsin | Invitrogen | Cat# 25300062 |
| Collagenase | Sigma-Aldrich | Cat# C9891 |
| Deoxyribonuclease I | Sigma-Aldrich | Cat# D4527 |
| BrdU | Roche | Cat# 10280879001 |
| FuGENE HD Transfection Reagent | Rromega | Cat# E2311 |
| PI/RNase Staining Buffer | BD Biosciences | Cat# 550825 |
| Protein A Agarose | Invitrogen | Cat# 15918014 |
| TRIzol Reagent | Invitrogen | Cat# 15596018 |
| RNasin Ribonuclease Inhibitors | Rromega | Cat#N2511 |
| EDTA-free Protease Inhibitor Cocktail | Roche | Cat#1187358001 |
| Cycloheximide | Sigma-Aldrich | Cat# C7698 |
| Criticai Commerciai Assays | ||
| Lenti-Pac HIV Expression Packaging Kit | GeneCopoeia | Cat# HRK-LvTR-20 |
| Click-iT EdU Alexa Fluor® 647 Flow Cytometry Assay Kit | Invitrogen | Cat# C10419 |
| Dual-Luciferase Reporter Assay System | Rromega | Cat# E1910 |
| ClonExpress II One Step Cloning Kit | Vazyme Biotech | Cat# C112–01 |
| In Situ Cell Death Detection Kit | Roche | Cat# 11684817910 |
| FITC Annexin V Apoptosis Detection Kit | BD Biosciences | Cat# 556547 |
| Biotin-Streptavidin HRP Detection Systems | ZSGB-BIO | Cat# SR-9001 |
| LightShift Chemiluminescent RNA EMSA Kit | Thermo scientific | Cat# 20158 |
| Pierce RNA 3′ End Biotinylation Kit | Thermo scientific | Cat# 20160 |
| mMESSAGE mMACHINE® T7 Ultra Kit | Ambion | Cat# AM1345 |
| MEGAclear Transcription Clean-Up Kit | Ambion | Cat#AM1908 |
| PrimeScript RT Reagent Kit | Takara | Cat#RR037A |
| TB Green Premix Ex Taq II | Takara | Cat#RR820A |
| Experimental Models: Cell Lines | ||
| Mouse: MEF | Established by our lab | N/A |
| Mouse: NIH 3T3 | Laboratory of Yan Cheng | N/A |
| Human: 293T | Laboratory of Xin Wu | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Pum1−/−: B6.129 | Estabiished by our lab | N/A |
| Mouse: 129-Cdkn1btm1Mlf/J | The Jackson Laboratory | JAX: 003122 |
| Oligonucleotides | ||
| shRNA targeting sequence: TRCN0000294814: TGA TCTCAAACTGGCATTTAA | This paper | N/A |
| Primers for PCR and RT-qPCR, see EXPERIMENTAL MODEL AND SUBJECT DETAILS | This paper | N/A |
| Software and Algorithms | ||
| FiowJo 7.6.1 | Ashland | https://www.flowjo.com/solutions/flowjo |
| Illustrator CC | Adobe | http://www.adobe.com/products/illustrator.html |
| Photoshop CC | Adobe | http://www.adobe.com/products/photoshop.html |
| MODfit LT 4.0 | Verity Software House | https://www.vsh.com/products/mflt/index.asp |
| GraphPad Prism software 5 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
Mouse mutations Pum1/Pum2 cause a gene dosage-dependent global reduction in body size
PUM promotes G1-S transition by repressing translation of Cdkn1b via binding its 3’ UTR
Cdkn1b mutation partially rescues reduced cell proliferation and body size in Pum1 mice
Auto- and reciprocally regulated PUM expression contributes to precise control of body size
ACKNOWLEDGMENTS
We would like to thank Dr. Jian Yang from the University of Queensland for examining the association of PUM locus with human height from the GWAS database; Dr. Xiaoming Wang for flow cytometry analysis of spleen cells; Dr. Ming Lei from the Precision Medicine Research Institute at Shanghai 9th People’s Hospital for facilitating protein purification; Dr. Caiyin Guo and Dr. Lynn Doglio at the Northwestern Transgenic and Targeted Mutagenesis Laboratory (TTML) for their assistance in the generation of mutant mice; Drs. Alec Xiaozhong Wang, Kehkooi Kee, Hiroaki Kiyokawa, Jun Yan, Shiyuan Cheng, Liuze Gao, and Haixin Li for discussion and/or comments on our manuscript; and Dr. Gyorgy Paragh, Wenjuan Xia, Mingyue Jiang, Chao Chen, and Baobao Geng for technical assistance. This work was supported by the National Basic Research Program of China (973 program, 2015CB943002 and 2013CB945201), the National Science Foundation of China (31771652 and 81270737), the Natural Science Foundation of Jiangsu Province (BK2012838), a Provincial Innovation and Entrepreneurship grant, and the NIH (U01 HD045871). Funding for the open access charge was provided by the Provincial Shuangchuang Program.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.01.111.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Blackinton JG, and Keene JD (2014). Post-transcriptional RNA regulons affecting cell cycle and proliferation. Semin. Cell Dev. Biol 34, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell IG, Merrick KA, Morandell S, Zhu CQ, Braun CJ, Grant RA, Cameron ER, Tsao MS, Hemann MT, and Yaffe MB (2015). A pleio-tropic RNA-binding protein controls distinct cell cycle checkpoints to drive resistance of p53-defective tumors to chemotherapy. Cancer Cell 28, 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Padmanabhan K, and Richter JD (2010). Pumilio 2 controls translation by competing with eIF4E for 7-methyl guanosine cap recognition. RNA 16, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Zheng W, Lin A, Uyhazi K, Zhao H, and Lin H (2012). Pumilio 1 suppresses multiple activators of p53 to safeguard spermatogenesis. Curr. Biol 22, 420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden SL, Bernstein DS, Bachorik JL, Thompson BE, Gallegos M, Petcherski AG, Moulder G, Barstead R, Wickens M, and Kimble J (2002). A conserved RNA-binding protein controls germline stem cells in Caenorhabditis elegans. Nature 417, 660–663. [DOI] [PubMed] [Google Scholar]
- del Prete MJ, Vernal R, Dolznig H, Müllner EW, and Garcia-Sanz JA (2007). Isolation of polysome-bound mRNA from solid tissues amenable for RT-PCR and profiling experiments. RNA 13, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TA, Pyle SE, Wharton RP, and Aggarwal AK (2001). Structure of Pumilio reveals similarity between RNA and peptide binding motifs. Cell 105, 281–289. [DOI] [PubMed] [Google Scholar]
- Efstratiadis A (1998). Genetics of mouse growth. Int. J. Dev. Biol 42, 955–976. [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, et al. (1996). A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 85, 733–744. [DOI] [PubMed] [Google Scholar]
- Fero ML, Randel E, Gurley KE, Roberts JM, and Kemp CJ (1998). The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 396, 177–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes A, and Lehmann R (1998). Nanos and Pumilio have critical roles in the development and function of Drosophila germline stem cells. Development 125, 679–690. [DOI] [PubMed] [Google Scholar]
- Gallardo T, Shirley L, John GB, and Castrillon DH (2007). Generation of a germ cell-specific mouse transgenic Cre line, Vasa-Cre. Genesis 45, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarino VA, Singh RK, White JJ, De Maio A, Han K, Kim JY, Jafar-Nejad P, di Ronza A, Kang H, Sayegh LS, et al. (2015). Pumilio1 haploinsufficiency leads to SCA1-like neurodegeneration by increasing wild-type Ataxin1 levels. Cell 160, 1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarino VA, Palmer EE, McDonell LM, Wang L, Adamski CJ, Koire A, See L, Chen CA, Schaaf CP, Rosenfeld JA, et al. (2018). A mild PUM1 mutation is associated with adult-onset ataxia, whereas haploinsufficiency causes developmental delay and seizures. Cell 172, 924–936.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr., Jungkamp AC, Munschauer M, et al. (2010). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi M, Beaumont RN, Day FR, Warrington NM, Kooijman MN, Fernandez-Tajes J, Feenstra B, van Zuydam NR, Gaulton KJ, Grarup N, et al. ; CHARGE Consortium Hematology Working Group; Early Growth Genetics (EGG) Consortium (2016). Genome-wide associations for birth weight and correlations with adult disease. Nature 538, 248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Nicol SA, Suen AY, and Baldwin TA (2012). Examination of thymic positive and negative selection by flow cytometry. J. Vis. Exp (68), 4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, and Weinberg RA (1994). Tumor spectrum analysis in p53-mutant mice. Curr. Biol 4, 1–7. [DOI] [PubMed] [Google Scholar]
- Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JA, Elkon R, and Agami R (2010). A Pumilio-induced RNA structure switch in p27–3′ UTR controls miR-221 and miR-222 accessibility. Nat. Cell Biol 12, 1014–1020. [DOI] [PubMed] [Google Scholar]
- Keene JD, Komisarow JM, and Friedersdorf MB (2006). RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc 1, 302–307. [DOI] [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, and Koff A (1996). Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 85, 721–732. [DOI] [PubMed] [Google Scholar]
- Kronja I, and Orr-Weaver TL (2011). Translational regulation of the cell cycle: when, where, how and why? Philos. Trans. R. Soc. Lond. B Biol. Sci 366, 3638–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont LB, Crittenden SL, Bernstein D, Wickens M, and Kimble J (2004). FBF-1 and FBF-2 regulate the size of the mitotic region in the C. elegans germline. Dev. Cell 7, 697–707. [DOI] [PubMed] [Google Scholar]
- Lee MH, Hook B, Pan G, Kershner AM, Merritt C, Seydoux G, Thomson JA, Wickens M, and Kimble J (2007). Conserved regulation of MAP kinase expression by PUF RNA-binding proteins. PLoS Genet. 3, e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kopp F, Chang TC, Sataluri A, Chen B, Sivakumar S, Yu H, Xie Y, and Mendell JT (2016). Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell 164, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M, Dietmann S, Paramor M, Niwa H, and Smith A (2014). Genetic exploration of the exit from self-renewal using haploid embryonic stem cells. Cell Stem Cell 14, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann R, and Nusslein-Volhard C (1987). Involvement of the pumilio gene in the transport of an abdominal signal in the Drosophila embryo. Nature 329, 167. [Google Scholar]
- Lin H, and Spradling AC (1997). A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development 124, 2463–2476. [DOI] [PubMed] [Google Scholar]
- Lin KB, Zhang SK, Chen JL, Yang D, Zhu MY, and Yujun Xu E (2018a). Generation and functional characterization of a conditional Pumilio2 null allele. J. Biomed. Res 32, 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Zhang S, Shi Q, Zhu M, Gao L, Xia W, Geng B, Zheng Z, and Xu EY (2018b). Essential requirement of mammalian Pumilio family in embryonic development. Mol. Biol. Cell 29, 2922–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak W, Fang C, Holden T, Dratver MB, and Lin H (2016). An important role of Pumilio 1 in regulating the development of the mammalian female germ-line. Biol. Reprod 94, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles WO, Tschöp K, Herr A, Ji JY, and Dyson NJ (2012). Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev. 26, 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles WO, Lembo A, Volorio A, Brachtel E, Tian B, Sgroi D, Provero P, and Dyson N (2016). Alternative polyadenylation in triple-negative breast tumors allows NRAS and c-JUN to bypass PUMILIO post-transcriptional regulation. Cancer Res. 76, 7231–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Morris AR, Mukherjee N, and Keene JD (2008). Ribonomic analysis of human Pum1 reveals cis-trans conservation across species despite evolution of diverse mRNA target sets. Mol. Cell. Biol 28, 4093–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, and Nakayama K (1996). Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85, 707–720. [DOI] [PubMed] [Google Scholar]
- Pan D (2010). The hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, and Lin H (2000). Translational repression: a duet of Nanos and Pumilio. Curr. Biol 10, R81–R83. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphor-ylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Shigunov P, Sotelo-Silveira J, Kuligovski C, de Aguiar AM, Rebelatto CK, Moutinho JA, Brofman PS, Krieger MA, Goldenberg S, Munroe D, et al. (2012). PUMILIO-2 is involved in the positive regulation of cellular proliferation in human adipose-derived stem cells. Stem Cells Dev. 21, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemen H, Colas D, Heller HC, Brüstle O, and Pera RA (2011). Pumilio-2 function in the mouse nervous system. PLoS ONE 6, e25932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassov DS, and Jurecic R (2003). Mouse Pum1 and Pum2 genes, members of the Pumilio family of RNA-binding proteins, show differential expression in fetal and adult hematopoietic stem cells and progenitors. Blood Cells Mol. Dis 30, 55–69. [DOI] [PubMed] [Google Scholar]
- Stanger BZ (2008). Organ size determination and the limits of regulation. Cell Cycle 7, 318–324. [DOI] [PubMed] [Google Scholar]
- Tichon A, Gil N, Lubelsky Y, Havkin Solomon T, Lemze D, Itzkovitz S, Stern-Ginossar N, and Ulitsky I (2016). A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat. Commun 7, 12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, and Bishop JM (2001). c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414, 768–773. [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGompel MJ, and Xu EY (2010). A novel requirement in mammalian spermatid differentiation for the DAZ-family protein Boule. Hum. Mol. Genet 19, 2360–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vourekas A, Zheng K, Fu Q, Maragkakis M, Alexiou P, Ma J, Pillai RS, Mourelatos Z, and Wang PJ (2015). The RNA helicase MOV10L1 binds piRNA precursors to initiate piRNA processing. Genes Dev. 29, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidmann CA, and Goldstrohm AC (2012). Drosophila Pumilio protein contains multiple autonomous repression domains that regulate mRNAs independently of Nanos and brain tumor. Mol. Cell. Biol 32, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EK, Moore-Jarrett T, and Ruley HE (2001). PUM2, a novel murine puf protein, and its consensus RNA-binding site. RNA 7, 1855–1866. [PMC free article] [PubMed] [Google Scholar]
- Wickens M, Bernstein DS, Kimble J, and Parker R (2002). A PUF family portrait: 3’UTR regulation as a way of life. Trends Genet. 18, 150–157. [DOI] [PubMed] [Google Scholar]
- Xu EY, Chang R, Salmon NA, and Reijo Pera RA (2007). A gene trap mutation of a murine homolog of the Drosophila stem cell factor Pumilio results in smaller testes but does not affect litter size or fertility. Mol. Reprod. Dev 74, 912–921. [DOI] [PubMed] [Google Scholar]
- Ye J, and Blelloch R (2014). Regulation of pluripotency by RNA binding proteins. Cell Stem Cell 15, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, and Guan KL (2015). Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Williamson JR, and Lehmann R (1997). The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA 3, 1421–1433. [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chen D, Xia J, Han W, Cui X, Neuenkirchen N, Hermes G, Sestan N, and Lin H (2017). Post-transcriptional regulation of mouse neuro-genesis by Pumilio proteins. Genes Dev. 31, 1354–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.