

Abstract
Caspase-6 is a cysteine protease that plays essential roles in programmed cell death, axonal degeneration, and development. The excess neuronal activity of Caspase-6 is associated with Alzheimer disease neuropathology and age-dependent cognitive impairment. Caspase-6 inhibition is a promising strategy to stop early stage neurodegenerative events, yet finding potent and selective Caspase-6 inhibitors has been a challenging task due to the overlapping structural and functional similarities between caspase family members. Here, we investigated how four rare non-synonymous missense single-nucleotide polymorphisms (SNPs), resulting in amino acid substitutions outside human Caspase-6 active site, affect enzyme structure and catalytic efficiency. Three investigated SNPs were found to align with a putative allosteric pocket with low sequence conservation among human caspases. Virtual screening of 57,700 compounds against the putative Caspase-6 allosteric pocket, followed by in vitro testing of the best virtual hits in recombinant human Caspase-6 activity assays identified novel allosteric Caspase-6 inhibitors with IC50 and Ki values ranging from ~2 to 13 µM. This report may pave the way towards the development and optimisation of novel small molecule allosteric Caspase-6 inhibitors and illustrates that functional characterisation of rare natural variants holds promise for the identification of allosteric sites on other therapeutic targets in drug discovery.
Introduction
Caspases belong to a highly conserved family of cysteine proteases that have essential roles in programmed cell death, inflammation, differentiation, neuronal development and axon pruning1. Although apoptotic caspases are traditionally classified as initiators (Caspase-2, -8, -9 and -10) and effectors (Caspase-3, -6, and -7), data from Caspase-6 (Casp6) has challenged this classification. In addition to being activated by initiator and effector caspases, Casp6 can be activated by a unique intramolecular self-cleavage mechanism due to its longer intersubunit linker2. Moreover, the peptide substrate specificity of Casp6 is distinct from Caspase-3 (Casp3) and Caspase-7 (Casp7)3–5. Furthermore, inhibitors of apoptosis proteins that suppress Casp3 and Casp7 activity do not suppress Casp6 activity6,7. Finally, Casp3 or Casp7 activation in human embryonic kidney cells induces apoptosis whereas Casp6 activation does not8,9. These findings indicate that Casp6 displays unique structural features, substrate preference, and function compared to the other effector caspases.
Excess neuronal active Casp6 is associated with Alzheimer disease (AD) neuropathology and age-dependent cognitive impairment. Whereas active Casp6 is undetected in normal brains, it is highly abundant in post-mortem AD brains. Active Casp6 and Tau cleaved by Casp6 are present in neurofibrillary tangles (NFT), neuropil threads and neuritic plaques in the brain of mild, moderate, severe and very severe sporadic AD and in familial AD individuals10–12. Neuronal activation of Casp6 is associated with an increased production of amyloid beta peptide and caspase cleavage of Golgi-localised γ-ear-containing ARF binding protein 3 (GGA3), an inhibitor of the beta secretase13–15. In addition, Casp6 cleaves numerous substrates such as amyloid precursor protein14,16–18, tau10, α-tubulin19, valosin-containing protein20; a facilitator of misfolded ubiquitinated protein degradation, and actin-regulating postsynaptic density proteins spinophilin, α-actinin-1 and -4, and drebrin19, that may lead to neuronal dysfunction. Furthermore, Casp6 has been implicated in age-dependent cognitive impairment since levels of active Casp6 in the entorhinal cortex, the first region to be affected by AD21, inversely correlate with global cognitive, episodic, and semantic memory scores in individuals within normal cognitive range22. Knock-in mice that express a self-activated form of human Casp6 in the CA1 region of the hippocampus display age-dependent spatial and episodic memory impairment, in the absence of NFTs and amyloid plaques23. Furthermore, in nerve growth factor deprived mouse primary neuron cultures24–26, activation of Casp6 is associated with axonal degeneration, while Casp3 activation induces cell death. In wild type or mutant amyloid precursor protein-transfected human primary CNS neurons, Casp6 is activated in absence of Casp3 activation and leads to neuritic degeneration but not cell death27, consistent with observations in AD brains that Casp6 activity is present in neurons that do not have an apoptotic morphology10. Therefore, Casp6 represents a novel early therapeutic target in age-dependent cognitive impairment and AD.
Casp6 is a viable drug target. Casp6 null mice develop normally without any noticeable apoptotic defects28. Due to the role of Casp6 in B cell activation and plasma cell differentiation, these mice display increased antigen-stimulated production of antibodies; thus, Casp6 inhibition in AD patients may increase their immunity29. In humans, pro-Casp6 is not significantly expressed in foetal or adult brains30. Casp6 expression in peripheral tissues declines with age, but Casp6 expression remains relatively high in the gastrointestinal system30 raising concern that Casp6 inhibition may promote cancer. However, Casp6 involvement in tumour progression has been excluded in an inflammation-associated model of colon carcinogenesis31. Hence, Casp6 inhibition looks promising as a potential treatment for age-dependent cognitive impairment and AD.
Many competitive Casp6 inhibitors have been reported. Substrate mimetic peptide inhibitors were derived from the Casp6 cleavage site in transcription factor activator protein 2 isoform alpha32, lamin A33, Huntingtin (Htt)34, and synthetic caspase peptide substrates35. Most of these inhibitors contain an electrophilic warhead, aldehyde, fluoromethylketone or methyl vinyl sulfone36, which reacts with a catalytic cysteine. In addition, aza peptide epoxides37, Michael acceptors38, non-peptide inhibitors, sulfon amide isatin Michael acceptors39, 1,2,3-triazoles coupled to 2,3,5,6-tetrafluorophenoxy methyl ketone40, methylene blue and its derivatives41, have been shown to inhibit Casp6. Although most of these inhibitors display low nanomolar to micromolar potency against Casp6, some are toxic in mammals, have low blood brain barrier permeability, or lack selectivity. The catalytic site is highly conserved among the caspases, which challenges the development of highly selective Casp6 active site inhibitors.
A more promising approach is to target non-conserved allosteric sites42. Casp6 inhibition via zinc binding at exosite E (K36, E244, and H287)43, and phosphorylation of S257 at exosite D by ARK5 kinase44–46 represent two examples of natural Casp6 allosteric regulation. However, only exosite E has low conservation across the caspase family constituting an attractive allosteric site for Casp6 specific drug design. Despite the growing knowledge in the field of caspase allosteric regulation42,47, very few synthetic allosteric Casp6 inhibitors have been developed to date. A noncompetitive small peptide inhibitor identified through phage display screen against pro-Casp6 selectively inhibits Casp6 activity by trapping Casp6 in a tetrameric zymogen-like form48 but has poor cell penetration properties. Small molecule Casp6 inhibitors that bind at the dimer interface and stabilise the zymogen conformation49 do not bind to active Casp6. In addition, selective uncompetitive Casp6 inhibitors have been reported, but their action depends upon binding of an artificial peptide substrate limiting their therapeutic potential50. Active Casp6 inhibition is desirable because it accumulates before the onset of AD symptoms11. To our knowledge, no synthetic small molecule noncompetitive allosteric inhibitors of active Casp6 have been reported.
By investigating the activity of four rare natural Casp6 variants (Casp6-A34E, Casp6-E35K, Casp6-A109T, and Casp6-T182S) and employing X-ray crystallography along with molecular modelling, we have identified a putative allosteric pocket on mature active Casp6. An in silico screen of a diverse small molecules collection against this allosteric site identified two hits, S10G (IC50 = 4.2 µM) and C13 (IC50 = 13.2 µM) that inhibited Casp6 activity in vitro through a noncompetitive mechanism. Our data serves as a starting point for the development of a small molecule allosteric Casp6 inhibitors and also illustrates that screening of rare natural variants holds promise for the identification of allosteric sites on other therapeutic targets in drug discovery.
Results
Rare Casp6 variants display lower activity
In this study, we investigated how naturally occurring rare missense variants of human CASP6 affect Casp6 activity and structure. Following our previous work, which described rare natural Casp6-R65W and Casp6-G66R variants with significantly impaired activity51, we examined four additional missense rare variants with single amino acid substitutions located remotely from Casp6 active site: Casp6-A34E, Casp6-E35K, Casp6-A109T, and Casp6-T182S (Table S1, Fig. S1). A34, E35, A109, and T182 are not conserved in 12 human caspases and in Casp6 from different species (Fig. 1a,b). Recombinant Casp6-WT and the four variants were expressed in E. coli, purified, and active site titrated (Fig. S2) for functional analyses. Like Casp6-WT, Casp6 variants, underwent complete self-processing at TETD23, and TEVD193 sites, and slight self-cleavage at the DVVD179 site, generating mainly large subunit with a linker (LS-L), small subunit (SS), and a minor amount of large subunit (LS) (Fig. 1c,d). Casp6 variants were catalytically active against Ac-VEID-AFC but Casp6-A34E, Casp6-E35K, and Casp6-T182S, displayed lower activity than Casp6-WT in enzyme concentration-dependent manner (Fig. 1e) whereas the activity of Casp6-A109T was comparable to Casp6-WT. Casp6-A34E, Casp6-A109T, and Casp6-T182S did not show significant changes in kinetic parameters, KM, and kcat, for Ac-VEID-AFC hydrolysis compared with Casp6-WT, whereas Casp6-E35K displayed slightly reduced kcat and consequently showed ~2-fold reduced catalytic efficiency kcat/KM (Fig. 1f and Table 1). These results suggest that E35K rare natural Casp6 substitution negatively affects Casp6 activity.
Figure 1.

Activity of rare natural Casp6 variants. (a) Protein sequence alignment of 12 human caspases (a) and Casp6 from different species (b) showing A34, E35, A109, and T182 of human Casp6 and respective homologous residues of other caspases in bold. (c) A schematic representation of full-length recombinant pro-Casp6 and its domains, the position of Casp6 processing sites (TETD23, DVVD179, TEVD193), and the epitopes recognised by antibodies. Anti-p20 is a neoepitope antibody against cleaved Casp6 at D179 position. (d) Coomassie stain of SDS-PAGE (top panel) and western-blot analyses (three bottom panels) with Casp6 Pharmingen anti-p10, neoepitope 10630 anti-p20, and Santa Cruz anti-Casp6 (LS, LS-L) antibodies or antiserum of purified recombinant caspases showing large subunit with (LS-L) and without linker (LS), and small subunit (SS) of Casp6. Cropped gels and blots are shown here for clarity, full-length gels and blots are provided in Supplementary Information Fig. S12. (e) The activity of 10–250 nM active site titrated recombinant Casp6. (f) The relationship between initial reaction velocity (v) and substrate concentration of Casp6 catalysed Ac-VEID-AFC cleavage. Data were fitted to Michaelis-Menten equation using nonlinear regression and represent the mean ± standard deviation (SD) from at least three independent experiments.
Table 1.
Comparative catalytic efficiencies of Casp6 variants.
| KM (µM) | kcat (s−1) | kcat/KM (s−1 M−1) | |
|---|---|---|---|
| Casp6-WT | 31.2 ± 6.7 | 2.27 ± 0.30 | 76,008 ± 18,626 |
| Casp6-A34E | 22.7 ± 0.8 | 2.04 ± 0.10 | 89,803 ± 6,453 |
| Casp6-E35K | 37.1 ± 4.9 | 1.71 ± 0.13 | 46,833 ± 7,641 |
| Casp6-A109T | 34.5 ± 1.8 | 2.51 ± 0.16 | 72,683 ± 2,529 |
| Casp6-T182S | 33.3 ± 1.2 | 2.42 ± 0.12 | 72,649 ± 1,192 |
| Casp6-L200A | 315.5 ± 162.0 | 0.05 ± 0.03 | 150 ± 24 |
| Casp6-K284A | 32.6 ± 10.8 | 2.0 ± 1.5 | 58,650 ± 30,269 |
| Casp6-WT-LS-SS | 24.8 ± 4.4 | 2.6 ± 0.4 | 109,148 ± 21,060 |
| Casp6-L200A-LS-SS | 196.2 ± 84.4 | 2.8 ± 0.8 | 15,155 ± 2,351 |
| Casp6-K284A-LS-SS | 27.7 ± 2.7 | 2.8 ± 0.2 | 99,882 ± 9,723 |
ND – not detectable, Data represent mean ± SD of at least three independent experiments.
To assess the effect of mutations on Casp6 structure and function, we solved apo mature crystal structures of Casp6-E35K and Casp6-A109T (Table S2) but crystallisation attempts were not successful for the two other Casp6 variants. Superposition of the Casp6-E35K or Casp6-A109T structure with a previously solved structure of the apo mature Casp6-WT (2WDP)52 did not reveal any significant changes in the active sites of these two variants (Fig. 2a–d), suggesting that the remote mutations, E35K or A109T, do not induce functional changes in the active site catalytic dyad C163-H121 or substrate binding loops of the mature unliganded Casp6. The Casp6-E35K crystal structure revealed some differences in the mutation site compared to the Casp6-WT (2WDP)52 structure. In Casp6-WT the E35 from the large subunit of one protomer forms a weak salt bridge with R254 from the small subunit of the other protomer (and vice versa), stabilising the dimeric Casp6 structure (Fig. 2e). By contrast, in the Casp6-E35K, the two salt bridges are replaced by two repulsive K35-R254 pairs (Fig. 2f). In addition, E35K substitution disrupts a salt bridge between E35 from the large subunit and K284 from the small subunit of the same Casp6-WT protomer (Fig. 2e,f), possibly further destabilising a functional oligomeric state of Casp6. The Casp6-A109T crystal structure did not reveal any significant changes around the mutation site, supporting our findings that A109T substitution does not have a significant impact on Casp6 activity.
Figure 2.

Structures of Casp6 rare variants. Crystal structures of mature apo Casp6-E35K (a,b, light pink) and Casp-A109T (c,d, light orange) superimposed on a mature apo structure of Casp6-WT (PDB: 2WDP, grey). The structures are represented as a cartoon with catalytic residues (H121, C163) and mutated residues (E35/K35, A109/T109) shown as sticks. L1, L2, L3, L4, and L2′ depict active site loops. Right panels (b,d) are the zoom-in images of boxed regions in left panels (a,c). The interaction of K35/E35 with R254 and K284 at the dimer interface of apo Casp6-WT crystal structure (e, dark grey LS, light grey SS) and crystal structure of the apo Casp6-E35K (f, light violet LS, light pink SS). Dotted black lines represent weak electrostatic E35 interaction (3.4–3.6 Å distance) with R254 and K284 in Casp6-WT.
Identification of a putative allosteric pocket
Visual inspection of four amino acid positions affected by the rare single nucleotide polymorphisms on the Ac-VEID-AFC-bound Casp6-WT structure (3OD5)2 revealed that the three amino-acids, A34, E35, and A109, located more than 30 Å away from the Casp6 catalytic site, line a well-defined pocket of low sequence conservation across the caspases (Fig. 3a–c). This pocket is made of residues from the N-terminal end, loops connecting β3 and β4 strands with helix 2 and helix 3, respectively, loop L2′ and C-terminal end of one protomer, and residues from L2 loop and helix 5 of the opposite protomer (Fig. 3a). Because of the size and favourable tertiary architecture of this pocket, we reasoned that it could represent a binding site capable of accommodating small organic compounds. To investigate this, we used an automated active site prediction tool that surveyed the entire surface of the Ac-VEID-AFC-bound Casp6-WT structure. This analysis validated this pocket as a potential druggable site and therefore supported our hypothesis that it could represent a putative allosteric site. Interestingly, the two residues S257 and K36, shown previously as Casp6 allosteric modulation points through phosphorylation and zinc binding43–46, are found inside and at the border of the putative allosteric pocket, respectively (Fig. 3b), further supporting the propensity of allosteric modulation in this region of Casp6.
Figure 3.

Putative Casp6 allosteric pocket. (a) SNP residues A34, E35, and A109 (yellow sticks) are highlighted in the putative allosteric pocket (boxed) from one protomer of the dimeric structure of the Ac-VEID-AFC-bound Casp6-WT (PDB: 3OD5). The protomers of dimeric Casp6 are shown in green and pale green, respectively, and the active-site bound Ac-VEID-AFC molecules are represented by blue spheres. Beta strands (β1–β6), alpha helices (H1–H5), and loops L2 and L2′ are labeled in both protomers. (b) Surface representation of the putative Casp6 allosteric pocket indicating positions of A34, E35, and A109 (yellow), and Casp6 allosteric regulation sites K36 and S257 (orange). (c) Amino acid sequence conservation of the putative allosteric pocket in the human caspase family (left panel) mapped on the 3OD5 crystal structure of Casp6 (right panel).
In silico screen for Casp6 allosteric inhibitors targeting the allosteric pocket
To identify candidate inhibitors of Casp6 activity, we performed a virtual screen by docking of small organic compounds targeting the putative allosteric site of the Ac-VEID-AFC-bound Casp6-WT structure. A total of 57,700 diverse molecules, originating from curated Chembridge and Sigma commercial libraries were screened. Subsequently to docking calculations, the poses of the best ranked molecules based on predicted binding energy were visually inspected to select the candidates that showed both favourable interactions within the putative allosteric site and ligand conformation of low energy. Finally, to ensure chemotype diversity a structural clustering was performed on the best candidates and a selection of 40 compounds was acquired (Tables S3 and S4).
Verification of the inhibitory properties of in silico hits
To assess the potency of in silico hits, a fluorescence-based Ac-VEID-AFC cleavage assay was used to measure recombinant active Casp6 reaction rate in the absence or presence of 100 µM concentration of compounds. A 30% inhibition cut off was arbitrarily chosen for further investigation of putative inhibitors. Eleven of the forty compounds (27.5%) screened showed inhibition of Casp6 (Table 2, Fig. S3). AFC signal interference by compound intrinsic fluorescence was ruled out (Fig. S4). Casp6 inhibition was confirmed for seven compounds (C7, C13, C14, S2, S10, S11 and S13) using a colorimetric Ac-VEID-pNA cleavage assay (Table 2, Fig. S3). The four compounds (C16, C17, C25 and S6) that interfered with the pNA signal (Fig. S4) were tested through Casp6 cleavage of catalytically inactive Casp6-C163A. In the presence of 100 µM of C16, C17, and C25, the rate of Casp6-C163A prodomain cleavage by active Casp6 was reduced by ~30% compared to a DMSO control whereas 100 µM of S6 showed minimal inhibition of active Casp6 (Table 2, Fig. S3). The hits had IC50 values ranging from 13.2 µM to 289 µM (Table 2).
Table 2.
Structure and inhibition data of hit compounds from Sigma (S) and Chembridge (C) libraries.
| Compound | Structure | % Inhibition with 100 µM compound | IC50 (µM)* |
|---|---|---|---|
| C13 |
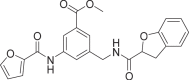
|
57.1 ± 4.7a 86.7 ± 1.8b |
13.2 ± 2.5 |
| S10 |
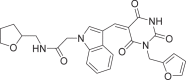
|
59.7 ± 3.8a 42.6 ± 3.4b |
30.2 ± 6.8 |
| C7 |
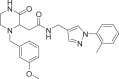
|
38.9 ± 5.5a 92.0 ± 0.3b |
44.1 ± 4.6 |
| C16 |
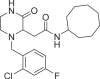
|
33.3 ± 4.8a 28.7 ± 7.4c |
46.4 ± 3.3 |
| S2 |
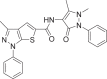
|
31.8 ± 4.8a 52.9 ± 4.1b |
90.0 ± 24.4 |
| C25 |
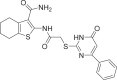
|
34.9 ± 5.1a 27.8 ± 14.0c |
109.5 ± 15.4 |
| S6 |
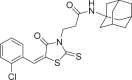
|
32.3 ± 4.3a 6.3 ± 5.1c |
123.0 ± 22.7 |
| C14 |
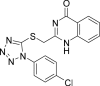
|
34.0 ± 5.0a 60.2 ± 6.5b |
141.8 ± 14.8 |
| S13 |
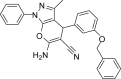
|
33.8 ± 12.1a 41.9 ± 8.2b |
159.0 ± 27.7 |
| C17 |
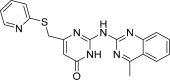
|
30.3 ± 1.7a 23.8 ± 20.0c |
233.4 ± 28.1 |
| S11 |
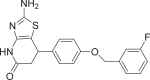
|
30.0 ± 6.0a 56.3 ± 2.3b |
289.0 ± 60.9 |
aFluorescence assay, bcolorimetric assay, cCasp6-C163A cleavage assay, *IC50 values were determined using fluorescence assay.
To probe a preliminary structure-activity relationship (SAR) of the two most active hits, S10 and C13, a few commercially available analogues (Table S5) were tested using the fluorescence-based activity assay. Twelve of the eighteen S10 analogues tested at 100 µM (S10B-S10I, S10K-S10M, and S10P) showed ≥50% inhibition of Casp6 (Table 3, Fig. S3). Compounds S10B–S10F, S10H, S10M, and S10P were verified as Casp6 inhibitors using the colorimetric assay, and compounds S10G, S10I, S10K, and S10L were verified using the Casp6-C163A cleavage assay (Table 3, Fig. S3). These hits had IC50 values ranging from 4.2 μM to 79.9 μM (Table 3). Compounds S10G, S10K, and S10L had IC50 values ~3.8-fold lower than the IC50 of S10 (30.2 μM). Interestingly, compound S10P, which is devoid of the critical pyrimidinetrione scaffold known as being a PAINS53, retained some activity with an IC50 of 80 µM.
Table 3.
Structure and inhibition data of S10 and C13 analogues.
| Compound | Structure | % Inhibition with 100 µM compound | IC50 (µM)* |
|---|---|---|---|
| S10G |
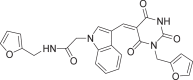
|
98.6 ± 2.4a 99.1 ± 1.5c |
4.2 ± 0.3 |
| S10L |
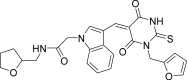
|
89.0 ± 3.0a 90.5 ± 5.2c |
4.4 ± 0.4 |
| S10K |
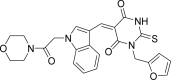
|
81.7 ± 2.0a 81.7 ± 3.4c |
7.6 ± 0.8 |
| S10D |
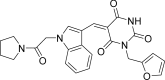
|
96.3 ± 1.6a 53.3 ± 7.8b |
9.8 ± 1.7 |
| S10B |
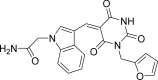
|
92.0 ± 6.7a 64.4 ± 14.7b |
10.5 ± 2.4 |
| S10C |
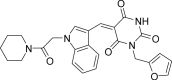
|
96.8 ± 0.7a 62.9 ± 6.8b |
11.0 ± 1.3 |
| S10E |
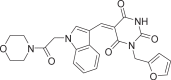
|
94.6 ± 1.1a 46.8 ± 7.4b |
12.8 ± 1.8 |
| S10F |
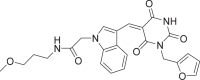
|
93.7 ± 0.8a 52.5 ± 6.5b |
12.9 ± 1.3 |
| S10M |
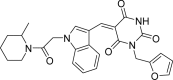
|
81.3 ± 3.3a 43.7 ± 3.1b |
16.4 ± 3.1 |
| S10H |
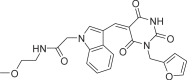
|
92.3 ± 2.7a 53.4 ± 10.4b |
16.5 ± 2.3 |
| S10I |
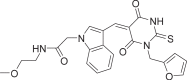
|
74.3 ± 5.8a 81.8 ± 4.6c |
16.8 ± 2.2 |
| S10P |
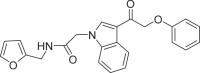
|
59.3 ± 2.5a 44.7 ± 4.7b |
79.9 ± 9.9 |
| C13G |
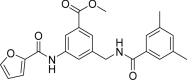
|
55.5 ± 8.8a | 21.5 ± 2.0 |
| C13F |
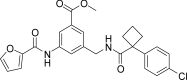
|
43.2 ± 10.9a | 27.0 ± 1.6 |
| C13C |
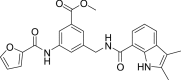
|
24.7 ± 1.7a | 719 ± 76** |
| C13A |
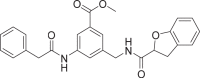
|
21.8 ± 2.5a | 1545 ± 261** |
| C13B |
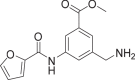
|
2.3 ± 3.1a | 1922 ± 300** |
| C13D |
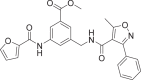
|
5.5 ± 4.1a | 2047 ± 57** |
| C13E |
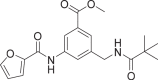
|
−8.3 ± 1.7a | 3370 ± 678** |
aFluorescence assay, bcolorimetric assay, cCasp6-C163A cleavage assay, *IC50 values were determined using fluorescence assay, **approximate IC50 values calculated from not-perfectly fitted dose-response curves due to limited compound solubility (C13A, C13C) or very low compound potency at the highest concentration tested (C13B, C13D, C13E).
In the C13 series, seven analogues were tested, most of them exploring various replacements of the di-hydrobenzofuran right hand side-chain. While two of them (C13F and C13G) retained the activity seen with C13, most of them were found significantly less potent (Table 3).
Compound S10G and C13 inhibit Casp6 through a noncompetitive mechanism
The inhibition of Casp6 activity with S10G and C13 was reversible (Fig. S5). To determine mode of inhibition, kinetic analyses were conducted on Casp6 using S10G and C13 (Fig. 4a). In the Lineweaver-Burk plot, lines of best fit converged at a single point above the x-axis and left from the y-axis, which is characteristic of noncompetitive inhibition54 (Fig. 4b). The dissociation constant (Ki) of S10G was 1.8 µM, which was approximately 3-fold lower than C13 (Ki = 5.3 µM) (Fig. 4c). Moreover, IC50 values determined using six different Ac-VEID-AFC concentrations did not show significant differences, excluding competitive inhibition mode (Fig. 4d).
Figure 4.

Compounds S10G and C13 inhibit Casp6 through a noncompetitive mode of inhibition. (a) Michaelis-Menten kinetic plots of Casp6 enzymatic activity in presence of 1% DMSO or increasing doses S10G (top) and C13 (bottom). Data points represent mean ± SD of three independent experiments. (b) Lineweaver-Burk plots of Casp6 inhibition with S10G (top) and C13 (bottom). Red circle indicates the point where lines converge. (c) The relationship between the slopes of lines in Lineweaver-Burk plot analyses in panel (b) and inhibitor concentration. (d) Plots of IC50 values for S10G (top) and C13 (bottom) with varying concentrations of Ac-VEID-AFC substrate. Data points represent mean ± SD from three independent experiments for each substrate concentration. (e) Binding model of C13 into CASP6 putative allosteric site, derived from molecular docking. Left: C13 (green sticks) occupies the putative allosteric site (white surface and lines; Ala-mutated residues L200 and K284 shown as purple sticks). Right: predicted interactions with Casp6 allosteric pocket residues (pink balls polar, circled blue or red for basic and acidic, respectively. Green balls = hydrophobic. Dotted-arrow: blue and green for backbone and sidechain hydrogen-bond interactions, respectively. Blue shade = solvent exposed area). Dose-response curves of C13 for Casp6-WT and Casp6-K284A (f), and copurified LS and SS of Casp6-WT-LS-SS, Casp6-L200A-LS-SS, and Casp6-K284A-LS-SS (g). Data represent mean ± SD from at least three independent experiments. (h) Global fit of kinetics data to a noncompetitive (mixed) inhibition model for Casp6-WT-LS-SS (left panel, R2 = 0.9635), Casp6-L200A-LS-SS (middle panel, R2 = 0.9699), and Casp6-K284A-LS-SS (right panel, R2 = 0.9794) in the presence of 0–50 µM C13. Data points on the graph represent the mean ± SD, and calculated Ki values represent mean ± standard error (SE) from three independent experiments.
Binding mode analysis of C13
The predicted C13 binding pose, derived from molecular docking, and the two-dimensional ligand interaction diagram (Fig. 4e) revealed several potentially important interactions between the C13 and the Casp6 putative allosteric pocket residues. The K38 backbone amide formed a hydrogen bond with the carbonyl of C13 methyl ester group. Furthermore, the model proposed that the terminal charged amine of K154 sidechain may be involved in a π-cation interaction with the C13 furan moiety. The model also revealed a pretty tight pocket surrounding the furan ring, consistent with the 117-fold reduction in the potency of the benzyl analogue C13A compared to C13 (Fig. S6, Table 3). In addition, the K284 sidechain formed a hydrogen bond with the carbonyl group of the methylamide linker of C13, and the L200 engaged in a hydrophobic interaction with the C13 dihydrobenzofuran end (Fig. 4e). Analogues either lacking the carbonyldihydrobenzofuran moiety (C13B) or wherein the dihydrobenzofuran group has been replaced by a tert-butyl group (C13E) were too short to optimally fill all subpockets of the allosteric site occupied by C13 and showed low potency against Casp6 (Table 3). Replacement of the dihydrobenzofuran moiety in C13 with a 2,3-dimethyl-1H-indole group in C13C and 5-methyl-3-phenyl-isoxazolyl group in C13D resulted in a non-optimal docking poses and a 55-fold and a 155-fold increase in IC50 for Casp6-WT, respectively, compared to C13 (Table 3). On the other hand, the replacement of dihydrobenzofuran group in C13 with a 4-chlorophenyl-cyclobutyl group in C13F, and 3,5-dimethylbenzoyl group in C13G resulted in a rather moderate 2-fold increase in IC50 for Casp6-WT, consistent with the reasonable docking poses obtained with this pair of compounds in the Casp6 allosteric pocket (Fig. S6).
To determine the importance of C13 interactions with L200 and K284 for C13 binding in the Casp6 allosteric pocket, we have generated Casp6-L200A and Casp6-K284A mutants and investigated inhibition with C13. In contrast to Casp6-WT, purified Casp6-L200A and Casp6-K284A underwent poor self-processing when overexpressed in E. coli from a construct encoding a full-length Casp6 (Fig. S5). Casp6-L200A partially self-processed at the TETD23 site generating full-length Casp6 and Casp6 without a prodomain (ΔCasp6). Casp6-K284A underwent complete processing at the TETD23 site but, in contrast to Casp6-WT, it only partially self-cleaved at the TEVD193 site generating ΔCasp6, LS-L and SS fragments (Fig. S5). Consistent with the incomplete self-processing, active site titration of Casp6-L200A did not yield any activity, whereas Casp6-K284A had only ~20% of active sites, in contrast to nearly 100% active sites determined for Casp6-WT samples (Fig. S2). Fully processed forms of Casp6-WT-LS-SS, Casp6-L200A-LS-SS, and Casp6-K284A-LS-SS were obtained by co-expression and purification of LS and SS subunits of WT, L200A or K284A Casp6 from E. coli (Fig. S5). Active site titration with zVAD-FMK inhibitor indicated that both Casp6-K284A-LS-SS and Casp6-WT-LS-SS had more than 80% active sites, and Casp6-L200A-LS-SS had ~178% active sites (Fig. S2), suggesting that zVAD-FMK may not be potent enough to react with Casp6-L200A-LS-SS active sites effectively. The kinetic parameters, KM and kcat, for Ac-VEID-AFC hydrolysis were similar between Casp6-WT and Casp6-K284A as well as Casp6-WT-LS-SS and Casp6-K284A-LS-SS suggesting that K284A mutation does not affect Casp6 catalytic function (Table 1). In contrast, Casp6-L200A and Casp6-L200A-LS-SS displayed 10- and 8-fold higher KM values compared to Casp6-WT and Casp6-WT-LS-SS, respectively. Furthermore, the catalytic function of unprocessed Casp6-L200A was almost completely diminished as reflected by a 45-fold reduction in kcat compared to Casp6-WT. In contrast, fully processed Casp6-L200A-LS-SS had a turnover number comparable to that of Casp6-WT-LS-SS (Table 1). Taken together these results suggest that K284A and L200A mutations negatively affect Casp6 self-processing and L200A impairs peptide substrate binding.
The C13 compound displayed ~2.7-fold higher IC50 value for Casp6-K284A mutant than Casp6-WT (Fig. 4f). Compared to Casp6-WT-LS-SS, the IC50 of C13 was 1.5-fold higher and 1.4-fold lower for the fully processed Casp6-K284A-LS-SS and Casp6-L200A-LS-SS, respectively (Fig. 4g). In the presence of increasing concentrations of C13 compound, an increase in apparent KM and a decrease in apparent Vmax of Ac-VEID-AFC hydrolysis was observed for Casp6-WT-LS-SS, Casp6-L200A-LS-SS, and Casp6-K284A-LS-SS, which indicates a noncompetitive (mixed) model inhibition (Fig. S5). Global fitting of Ac-VEID-AFC hydrolysis velocity data at increasing substrate and C13 concentrations to the velocity equations for different inhibition modalities revealed the best fit for a noncompetitive (mixed) model inhibition for Casp6-WT-LS-SS, Casp6-L200A-LS-SS, and Casp6-K284A-LS-SS (Fig. 4h, Fig. S7). The calculated Ki value of C13 for Casp6-L200A-LS-SS (6.8 µM) was not significantly different from the Ki for Casp6-WT-LS-SS (7.5 µM), whereas Ki for Casp6-K284A-LS-SS (11.8 µM) was 1.6-fold higher compared to the Ki for Casp6-WT-LS-SS (Fig. 4h) suggesting that K284 may play a minor role in binding C13 in the Casp6 allosteric pocket.
For validation purposes, we have synthesised C13 and C13G in our laboratory (Fig. S8). Compared to C13 and C13G obtained from commercial sources, our synthesised compounds displayed lower potency for Casp6.
Evaluation of S10G binding to Casp6 using Hydrogen-Deuterium Exchange Mass Spectrometry (H/DX-MS)
In light of the inconclusive results from site-directed mutagenesis impact on C13 inhibition, a biophysical approach was undertaken using S10G. To evaluate the changes in the Casp6 conformational dynamics affected by S10G binding, we have incubated fully maturated Casp6 with and without S10G in deuterium buffer for between 10 s and 40 min to allow H/D exchange, followed by pepsin digestion and MS for peptide mass analysis. A heat map representing relative differential deuteration profiles of various structural elements in Casp6 over the 40 min H/D exchange time was obtained for Casp6 incubated with and without S10G (Fig. S9). Overall, more than 60% of the backbone amide hydrogens underwent less than 30% H/D exchange within 10 s, indicating that Casp6 is a well-folded and dynamically stable protein. Lower H/D exchange profile was observed for the buried regions of the Casp6 core structure, whereas L2 and L2′ loops displayed highest H/D exchange levels suggesting that these regions are more flexible and solvent exposed in the fully mature Casp6. The peptic peptide coverage was nearly 100% for both Casp6 incubated with and without S10G (Fig. S9), allowing accurate detection of differences in conformational flexibility of Casp6 induced by S10G. Incubation of Casp6 with a 5-fold molar excess of S10G, resulted in a statistically significant decrease in H/D exchange in several peptic peptides (Fig. 5a, Fig. S10), which were mapped on the three-dimensional structure of the active-site liganded Casp6 (Fig. 5b), and cover regions in L2, L2′, L3 and L4 loops, β4 and β5 strands, helix 4, and loop connecting to the helix 5 N-terminal end. Overall, observed changes in deuterium uptake in the presence and absence of S10G for these regions were less than 10%, with the biggest differences detected for the L2′ and L4 loops. In silico docking of S10G onto the Casp6 structure indicated several putative S10G binding sites (Fig. 5c), which partially overlapped with H/DX-MS results, suggesting that S10G may interact with the substrate binding loops and the dimer interface.
Figure 5.

S10G and Casp6 interaction sites. (a) Representative deuterium uptake plots for peptic peptides of Casp6 incubated without S10G (red) and with S10G (black). Data points represent mean ± SD from three independent experiments. (b) Representative peptic peptides from panel (a) are mapped (pink area) on the CASP6 crystal structure (PDB: 3OD5) and compared to potential binding sites of S10G, as predicted by molecular docking (c, orange area). For indication purposes, Ac-VEID-AFC substrate, as found in CASP6-bound structure, is shown as translucent spheres. Same point of view as in Fig. 3.
Discussion
Aiming to identify novel allosteric sites in Casp6, we have investigated how human Casp6 rare non-synonymous missense SNPs resulting in substitutions away from the Casp6 active site, affect Casp6 catalytic efficiency and structure. In contrast to our previous study describing significant loss of activity in Casp6-R65W and Casp6-G66R rare SNP variants51, in the four A34E, E35K, A109T, and T182S remote SNPs investigated here, only the E35K slightly decreased Casp6 catalytic efficiency for a peptide substrate. The crystal structure of apo Casp6-E35K did not reveal hints that could have elucidated a potential mechanism responsible for reduced Casp6 activity. Although E35K substitution may lead to the disruption of few intersubunit salt bridges within Casp6 dimer, we did not observe decreased in vitro stability of Casp6-E35K compared to Casp6-WT or dissociation of enzyme subunits during protein purification (Fig. S11) recently described for Caspase-9 mutant55, which could provide an explanation for the enzyme inactivation.
Located more than 30 Å away from either active site of the Casp6 dimer, the N-terminal loop E35 is found in the neighbourhood of K36, which together with E244 and H287 forms Zn2+-binding exosite43 next to the postulated allosteric pocket. Casp6 inhibition with Zn2+ binding at this exosite, as well as findings that natural substitution E35K and alanine mutants of Zn2+-coordinating residues43 significantly reduce Casp6 catalytic efficiency suggest that this area, remote from Casp6 active sites, is important for Casp6 activity. Additionally, S257, the single reported Casp6 phosphorylation site, whose phosphorylation by ARK5 kinase allosterically inhibits Casp644–46, is located at the postulated Casp6 allosteric pocket, which further supports our hypothesis that small molecules binding in the pocket surrounded by A34E, E35K, and A109T SNPs, could result in inhibition of Casp6 activity. A recent study investigating Casp3 phosphorylation sites, S150 and T152 residues in the C-terminal loop of helix-3 (H3CL), a conserved region also found in the putative allosteric pocket of Casp6, revealed a network of interactions, which couple changes in the allosteric H3CL site to both active sites of the Casp3 dimer56. The localised changes in the H3CL region were shown to propagate through the helix-3 to the connected β-strand containing catalytic histidine as well as affect the active site loop bundle of the opposite protomer56. The authors of the latter study made an excellent insight that the H3CL region in Casp6 is connected to the helix-3, part of which undergoes conformational change into β-strands when Casp6 transits from inactive extended helical to active canonical conformation57,58. This suggests that the putative Casp6 allosteric pocket may indeed have a more central role in regulating Casp6 activity, and perhaps an important biological ligand for this allosteric site remains to be uncovered since Casp6 is not known to be phosphorylated at the S150 site identical to the one in Casp3.
The important structural components of the putative Casp6 allosteric pocket are L2 and L2′ loops. Upon cleavage of the intersubunit linker, the released C-terminal region of the large subunit, and the N-terminal region of the small subunit, become L2, which contains catalytic C163, and L2′ loops, respectively. In a fully mature, active-site liganded conformation of Casp6, a region of the L2 loop of one protomer interacts in the putative allosteric pocket with a segment of the L2′ loop of the opposite protomer, which could represent a putative interprotomer coupling between the postulated allosteric pocket and the active site in the Casp6 dimer. Casp6 allosteric inhibition is often achieved through the conformational changes in the L2′ and L2 loops. Phosphorylation of Casp6 at the S257 results in a steric clash with L2′ loop, which folds over the dimer interface as seen in the apo mature form of Casp6, resulting in misalignment of the substrate binding loops44,45. Small molecule inhibitors bind procaspase-6 allosteric site at the dimer interface and interact with residues in the L2 loop stabilising Casp6 zymogen-like conformation49. Likewise, small molecule covalent inhibitors bind at the analogous allosteric site at the dimer interface of Casp159, 560, 3 and 761, and lock L2′ loop in inactive zymogen-like conformation in Casp7, preventing substrate binding61. Our data indicate that Casp6 L2′ loop L200A mutant almost completely abolishes Casp6 self-processing and catalytic efficiency, consistent with the previous report for the analogous I213A mutation in the L2′ loop of Casp762, which revealed an essential role of L2′ loop in providing a platform for the proper arrangement of the substrate-binding groove in caspases. Interestingly, this was the first study62 to propose that small-molecule binding to the area occupied by I213 of L2′ loop in Casp7, which overlaps with the putative allosteric pocket of Casp6, could provide a novel strategy for modulating caspase activity.
Virtual screening of small molecules against active human Casp6 putative allosteric pocket followed by in vitro evaluation of Casp6 inhibition identified two Casp6 hit inhibitors, S10G and C13. Both compounds display low micromolar Ki and a noncompetitive mode of Casp6 inhibition, suggesting that they bind to an allosteric rather than a catalytic site, which validates our approach. Although our study lacks experimental evidence supporting the direct binding of C13 and S10G to the postulated allosteric pocket, the preliminary SAR of C13, where the core of the C13 chemotype has remained constant, in part supports the predicted C13 binding model in the Casp6 putative allosteric pocket. Modifications at the dihydro-benzofuran end have highlighted the importance of the shape of the pocket delimited by L200. Similarly, a change at the other end of the C13 molecule indicates that the furan ring is likely involved in critical binding interactions as predicted by modelling. It is necessary to assess additional analogues to validate the model, especially in light of the fact that alanine mutagenesis of two Casp6 allosteric pocket residues, L200 and K284, which are predicted to make key interactions with C13 according to our in silico model, did not abolish Casp6 inhibition by C13. Analogues bearing amide and carboxy isosteres, as well as various linkers attaching both ends and the central phenyl ring would be critical to verify the importance of these functionalities for hydrogen bonding or lipophilic interactions. In addition, the fact that Casp6 putative allosteric pocket is mainly composed of flexible loops, complicates accurate predictions of ligand binding using computational tools. Hence, additional C13 in silico binding models have to be experimentally evaluated and methods such as X-ray crystallography could be used to validate C13 binding to the postulated Casp6 allosteric site.
The identification of S10G and Casp6 potential interaction sites using H/DX-MS was inconclusive. S10G contains a pyrimidinetrione PAINS structure53, and may act as a promiscuous compound, hence, one should consider H/DX-MS results with caution. The observed S10G potency for Casp6, however, is not entirely dependent on the presence of pyrimidinetrione structure since the S10P analogue lacking this structure still inhibited Casp6.
This study illustrates challenges faced in the early stage drug discovery and serves as a starting point for the future optimisation of our identified hits towards potent Casp6 allosteric inhibitors.
Methods
Identification of Casp6 variants
Casp6 missense variants were identified from the dbSNP short genetic variation NCBI database (https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?geneId=839).
Compounds
Compounds were purchased from ChemBridge Corporation (San Diego, CA, USA), Sigma-Aldrich (St. Louis, MO, USA) TimTec Inc (Newark, DE, USA), and Vitas-M Laboratory Ltd (Champaign, IL, USA), and solubilised in 100% DMSO at 10 mM stock concentration.
Plasmids
Plasmid pET23b-Casp6-His63 encoding full-length human Casp6 (Addgene plasmid 11823) was distributed by Dr. Guy Salvesen (Sanford Burnham Prebys Medical Discovery Institute, CA, USA). The pET23b-Casp6(WT)p20/p10-His plasmid36 for E. coli codon optimized co-expression of Casp6-WT large and small subunits and pET23b( + ) plasmid encoding Casp6-C163A8 were previously described.
Cloning
Single mutations were introduced into pET23b-Casp6-His plasmid using QuikChange Site Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) with the following primers and corresponding reverse sequence: A34E: 5′-AAA AGA GAA ATG TTT GAT CCG GAA GAA AAG TAC AAA ATG GAC CAC-3′, E35K: 5′-GAG AAA TGT TTG ATC CGG CAA AAA AGT ACA AAA TGG ACC AC-3′, A109T: 5′-TGT CAA CTG TTA GCC ACA CAG ATG CCG ATT GCT TT-3′, T182S: 5′-GGA TGT AGT AGA TAA TCA GTC AGA GAA GTT GGA CAC CAA-3′, L200A: 5′-CCG TTT ACA CGG CCC CTG CTG GAG C-3′, K284A: 5′-AAT GCT AAC TGC CAA GCT GCA TTT C-3′. Mutations were introduced into pET23b-Casp6(WT)p20/p10-His plasmid with the following primers and corresponding reverse sequence: L200A: 5′-GTT TAC ACC GCG CCG GCG GGC-3′, K284A: 5′-CAT GCT GAC CGC GAA ACT GCA CT TC-3′. Plasmids were verified by Sanger sequencing (McGill University and Genome Quebec Innovation Center, Montreal, QC, CA).
Protein purification
Plasmids encoding full-length Casp6-WT and variants, large and small subunits of Casp6-WT and variants, and Casp6-C163A were expressed in E. coli BL21(DE3)pLysS strain (Promega, Fitchburg, WI, USA) and purified51. Purity was evaluated by SDS-PAGE and Coomassie blue staining, and protein concentration was measured using Quick Start Bradford 1x Dye Reagent (Bio-Rad Laboratories).
Casp6 active site titration and activity assays
Casp6 active sites were quantified using zVAD-FMK (N-benzyloxycarbonyl-Val-Ala-Asp-(O-methyl)-fluoromethylketone, MP Biomedicals, Santa Ana, CA, USA)51,64. Casp6 activity was measured in Stennicke’s buffer (SB: 20 mM piperazine-N,N′-bis-(2-ethanesulfonic acid) pH 7.2, 100 mM NaCl, 10 mM DTT, 1 mM EDTA, 0.1% 3-[(3-cholamidopropyl)-dimethylammonio]-2-hydroxy-1-propanesulfonic acid, 10% sucrose)65 using 40 µM Ac-VEID-AFC (Ac-Val-Glu-Ile-Asp-7-Amino-4-trifluoromethylcoumarin, Enzo Life Sciences, Farmingdale, NY, USA)64 and 10-250 nM active site titrated Casp6. Reactions were initiated by Ac-VEID-AFC addition and followed for 20 min at 37 °C in a black clear bottom 96-well microplate (Costar, Corning, NY, USA) in Synergy H4 Hybrid instrument (BioTek, Winooski, VT, USA) with excitation/emission at 380/505 nm. Released AFC was calculated from AFC standard curve (0-25 µM). Casp6 VEIDase activity (released pmol AFC/min) was calculated from the initial linear reaction part. Ac-VEID-AFC hydrolysis initial velocities were measured with 10 nM active-site titrated Casp6 and 1–300 nM Ac-VEID-AFC. Initial velocities v versus substrate concentration [S] were fit to a rectangular hyperbola using Michaelis-Menten equation in GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA).
Western blot analyses
25 ng purified Casp6 were separated by SDS-PAGE on 15% gel, transferred to polyvinylidene fluoride membrane in Trans-Blot Turbo Transfer system (Bio-Rad Laboratories) and probed with 1:10,000 rabbit polyclonal neoepitope antiserum 10630 against active human Casp6 174PLDVVD179 p20 subunit10, 1:250 mouse monoclonal IgG1 clone B93-4 against human Casp6 271GKKQVPCFASMLTKK285 p10 subunit (BD Biosciences, San Jose, CA, USA), and 1:1000 mouse monoclonal IgG1 SC81653 antibody against human pro-Casp6 24–293 aa (Santa Cruz Biotechnology, Dallas, TX, USA). Antibodies were diluted in Tris-buffered saline with 0.1% Tween-20 and 5% non-fat dry milk. Proteins were detected with 1:5000 secondary anti-mouse (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and anti-rabbit (Dako, Glostrup, Denmark) antibodies conjugated to horseradish peroxidase using ECL western blotting detection reagent (GE Healthcare), and Carestream Biomax MR films (Kodak, Rochester, NY, USA).
Crystallisation and structure determination
Fully processed Casp6 mutants E35K and A109T were crystallised in 100 mM sodium acetate pH 4.6 with 2 M sodium chloride. Crystals were grown by hanging drop vapour diffusion at 22 °C and were flash-frozen. X-ray diffraction data was collected at 100 K on beamline F1 with an ADSC Q-270 detector at the Cornell High Energy Synchrotron Source (MacCHESS). Data was processed by HKL200066. The structures were solved by molecular replacement with Phaser67 in Phenix68 using the crystal structure of wild-type Casp6 (PDB: 4FXO) as a search model, and re-built manually in Coot69. Refinement was carried out by phenix.refine70, with non-crystallographic symmetry restraints applied for the A109T mutant structure. Crystallographic data collection and structure refinement statistics are presented in Supplementary Table S2. Structural images were prepared with PyMOL (The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC).
Molecular modelling
The MOE package (Molecular Operating Environment (MOE), 201; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7) was used for structural analysis, protein protonation (Protonate 3D), automated active site prediction (Site Finder module, defaults parameters) and hit compounds binding mode analysis. The ConSurf server was used for analysing sequence conservation of the members of human caspase family, as well as the mapping of this information onto the Casp6 crystal structure71. Molecular graphics were rendered with PyMOL (Version 1.7.1).
Virtual screening by molecular docking
The Sigma Myriascreen II collection (10,000 compounds) and the Chembridge Premium Set (50,000 compounds) were acquired from their commercial provider (www.sigmaaldrich.com and www.chembridge.com, respectively). In-house script was used to filter out compounds with undesired substructures. The retained 57,681 compounds were canonicalized and their three-dimensional coordinates were generated using Chemaxon Standardizer, JChem 5.1.2 (www.chemaxon.com). Protonation at pH 7.4 was performed on Ac-VEID-AFC-bound Casp6-WT structure (PDB: 3OD5) using the Protonate 3D module in MOE with the Amber12:EHT forcefield. The histidine H41 that lines the allosteric site was attributed the HSD protonation state. The AutoDockTools interface72 was used to prepare the Ac-VEID-AFC-bound Casp6-WT protonated structure into PDQT format. The docking grid was defined as a 16 × 16 × 22-Å box around the entire putative allosteric site when docking the compounds library as well as the follow up C13 analogues. Although the C13 compound purchased and tested is a racemic mixture (presence of a chiral centre at the dihydrobenzofuran branching point), the model derived from docking strongly suggests the R-form to be most likely the isomer to optimally bind in the putative site. For blind docking of S10G compound (Fig. 5c) the whole Casp6 structure was considered for the docking search space, also using the PDB entry 3OD5 but in which the peptide substrate was removed. Docking was carried out with Autodock Vina73 using the default 9 output poses per compound docked. Autodock Vina scoring function was used to rank compounds by lowest energy and the top-1% ranked candidates were visualy inspected using PyMOL and the MOE package.
In vitro screening of compounds
In fluorescence-based Casp6 activity assay, 10 µM Ac-VEID-AFC was added to 20 nM Casp6 in SB with 100 μM compound and 1% dimethyl sulfoxide (DMSO), and reaction rate was measured for 20 min at 37 °C as above. Compound potency was calculated by normalising Ac-VEID-AFC cleavage rate to 1% DMSO control. In colorimetric Casp6 activity assay, 200 µM Ac-VEID-pNA (N-acetyl-Val-Glu-Ile-Asp-p-nitroanilide, Enzo Life Sciences) was added to 200 nM Casp6 in SB with 100 μM compound, and Ac-VEID-pNA cleavage rate was measured at 37 °C for 30 min in clear 96-well plate (Sartstedt, Nümbrecht, Germany) using Synergy H4 plate reader at 405 nm. Absorbance was converted to the released pNA amount based on a standard curve of 0–250 µM pNA. Cleavage rates were calculated from the linear assay phase and were normalised to 1% DMSO control. In Casp6-C163A cleavage assay, 0.5 ng/µl Casp6-WT was preincubated with 100 µM inhibitor, 5 µM Q-VD-OPh or 1% DMSO in SB (30 min, RT), followed by transfer on ice, addition of 0.5 µg/µl Casp6-C163A, and incubation (37 °C, 5, 10 and 15 min). Reactions were stopped by adding SDS-PAGE sample buffer (62.5 mM Tris pH 6.8, 2% SDS, 0.1 M DTT, 0.01% Bromphenol Blue, 10% glycerol) and heating (5 min, 95 °C). Samples (2 µl) were analysed on 15% SDS-PAGE gels, proteins were stained with Coomassie blue, and gels were de-stained with 10% acetic acid. The intensities of protein bands were quantified from scanned gels using ImageJ. The band intensity of Casp6-C163A without a prodomain was plotted against the time and the Casp6-C163A cleavage rates were calculated from the slopes of linear plots. The % activity of Casp6-WT in the presence of inhibitors was calculated relative to DMSO control.
IC50 determination
IC50 values were determined against 20 nM Casp6 active sites in fluorescence-based Casp6 activity assay with 10 µM Ac-VEID-AFC and 1 nM-500 µM inhibitor. IC50 was calculated with GraphPad Prism 5 by a nonlinear regression fit for log inhibitor vs relative Casp6 activity (% DMSO control) using a Hill slope of – 1, and top and bottom constraints of 100 and 0, respectively.
Determination of inhibition mechanism
Compounds, S10G (2.5 µM, 4.2 µM, 10 µM), and C13 (5 µM, 14 µM, 50 µM), were added to 20 nM active site titrated Casp6 and enzyme activity was measured with eight different Ac-VEID-AFC substrate concentrations (5.0 μM–200 μM) using fluorescence-based Casp6 activity assay as above. The apparent KM and Vmax values were determined through nonlinear regression analysis of Michaelis-Menten plots (v vs. [S]) using GraphPad Prism 5. The inhibition mode was determined from the pattern of converging Lineweaver-Burk plots (1/v vs. 1/[S])54. The dissociation constant (Ki) for Casp6-inhibitor complex was determined from the x-axis intercept (which is equal to −Ki) in the plot of Lineweaver-Burk slopes vs. inhibitor concentration74. Alternatively, inhibition model and Ki were determined by global fit of kinetic data to the competitive, noncompetitive, noncompetitive (mixed) or uncompetitive inhibition models using respective equations in GraphPad Prism 5. IC50 curves for S10G and C13 against Casp6 were generated using the fluorescence-based activity assay with 5–75 µM Ac-VEID-AFC.
Bioinformatic analyses
Multiple sequence alignments were generated with Clustal Omega tool (http://www.ebi.ac.uk/Tools/msa/clustalo/) using UniProt accession numbers for human caspases: P29466, P42575, P42574, P49662, P51878, P55212, P55210, Q14790, P55211, Q92851, Q6UXS9, P31944, and Casp6 from different species: P55212, K7B3Z2, Q3T0P5, S9XS51, A0A0B8S0B0, O35397, O08738, L5K505, S7N3D7, A0A0Q4ALF8, O93415, A0A0Q3PLE0, Q9IB66, V9L2A1, Q9I8S9, Q52KK6, G0XQH6, Q9NHF9, K1R808.
Hydrogen/deuterium exchange combined with mass spectrometry (HDX-MS)
HDX-MS was carried out as described75. HDX was initiated by diluting 25 µM Casp6 stock solution with or without S10G (125 µM) into the D2O-based buffer (20 mM Tris-HCl, 120 mM NaCl, 2 mM DTT, pD 8.5, based on pD = pHread + 0.40)76 using 1:9 dilution ratio. For Casp6 in the presence of S10G, stock solution of Casp6 with S10G was diluted by D2O-based buffer containing S10G. The HDX incubation period and temperature were set to 10, 60, 900, 2400 s and 25 °C, respectively. HDX was quenched with chilled quenching buffer (300 mM glycine, 8 M urea in H2O, pH 2.4) using 1:2 dilution ratio. Quenched solutions were flash frozen in MeOH containing dry ice, and samples were stored at −80 °C until use. For the undeuterated control, initial dilution was made in H2O buffer. Prior to ultra-high performance liquid chromatography (UHPLC)-MS analysis, the deuterated Casp6 was digested in an on-line immobilised pepsin column prepared in house. Resulting peptides were loaded onto a C18 analytical column (1-mm inner diameter, 50 mm length; Thermo Fisher Scientific, Waltham, MA, USA) equipped to an Agilent 1290 Infinity II UHPLC system. Peptides for each sample were separated using a 5–40% linear gradient of acetonitrile containing 0.1% formic acid for 8 min at a 65 µl/min flow rate. To minimize back-exchange, the columns, solvent delivery lines, injector and other accessories were placed in an ice bath. The C18 column was directly connected to the electrospray ionization source of the LTQ Orbitrap XL (Thermo Fisher Scientific), and mass spectra of peptides were acquired in positive-ion mode for m/z 200–2000. Duplicate measurements were performed for each time point. Identification of peptides was carried out in separate experiments by tandem MS (MS/MS) analysis in data-dependent acquisition mode, using collision-induced dissociation. All MS/MS spectra were analysed using Proteome Discoverer 1.4 (Thermo Fisher Scientific). Peptide searching results were further manually inspected, and only those verifiable were used in HDX analysis. The deuteration (%) as a function of incubation time was determined using HDExaminer 2.3 (Sierra Analytics, Modesto, CA). The first two amino acid residues in peptides were excluded from the analysis.
Supplementary information
Acknowledgements
We thank Maria Putorti for generating expression plasmids of Casp6 A34E, E35K, A109T, and T182S variants. Most computations were made on the supercomputer MsII from Université de Montréal, managed by Calcul Québec and Compute Canada. The operation of this supercomputer is funded by the Canada Foundation for Innovation (CFI), the ministère de l'Économie, de la science et de l’innovation du Québec (MESI) and the Fonds de recherche du Québec - Nature et technologies (FRQ-NT). We thank Dr. Guennadi Kozlov for crystallographic data collection at the Cornell High Energy Synchrotron Source (CHESS). CHESS is supported by the NSF & NIH/NIGMS via NSF award DMR-1332208, and the MacCHESS resource is supported by NIH/NIGMS award GM-103485. This work was supported by funds from the Canadian Institutes of Health Research (CIHR) MOP-243413-BCA-CGAG-45097 to ALB, the Canadian Foundation for Innovation to GLL and ALB, the Cystic Fibrosis Canada and NIH NIDDK RO1DK075302 and CIHR to GLL, and the JGH Foundation to ALB. GLL is a Canada Chair I.
Author Contributions
A.T.-A. performed in vitro screening and inhibition mode characterisation of Chembridge library compounds and Chembridge analogues, generated expression plasmids for L200A, K284A, L200A-LS-SS and K284A-LS-SS Casp6 variants, performed purification and biochemical characterisation of Casp6 variants, generated and analysed data for Figures 1 and 4a–d,f–h, S1–S5, S7, S11–S12, Tables 1–3, wrote the manuscript. A.B. performed in silico docking experiments and identification of putative Casp6 allosteric pocket, generated data for Figures 3, 4e and 5b,c, and S6, contributed to the experimental design, wrote the manuscript. J.L. performed in vitro screening and inhibition mode characterisation of Sigma library compounds and S10 analogues, generated and analysed data for Figures 4a–d, S3-S5, Tables 2 and 3, wrote parts of the manuscript. G.S. performed Casp6 crystallisation, contributed to the experimental design. A.G. solved and analysed X-ray crystal structures of Casp6 variants, generated data for Figures 2 and table S2. L.J.D. synthesised C13 and C13G, performed and analysed LC-MS and NMR of C13 and C13G, prepared Figure S8. N.S. designed and performed HDX-MS experiments, generated and analysed data for Figures S9 and S10. G.L.L. contributed to HDX-MS experimental design and interpretation of results. B.N. contributed to the experimental design and X-ray crystallisation analysis. A.M. contributed to the experimental design and data interpretation, wrote parts of the manuscript. A.C.L. conceptualised experimental design and the manuscript outline, oversaw the study, wrote parts of the manuscript. All authors reviewed the manuscript.
Data Availability
All generated or analysed study data are included in this published article and its Supplementary Information file.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Agne Tubeleviciute-Aydin and Alexandre Beautrait contributed equally.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-41930-7.
References
- 1.Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ. 2015;22:526–539. doi: 10.1038/cdd.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang XJ, et al. Crystal structures of human caspase 6 reveal a new mechanism for intramolecular cleavage self-activation. EMBO Rep. 2010;11:841–847. doi: 10.1038/embor.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talanian RV, et al. Substrate specificities of caspase family proteases. J. Biol. Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- 4.Thornberry NA, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 5.Julien O, et al. Quantitative MS-based enzymology of caspases reveals distinct protein substrate specificities, hierarchies, and cellular roles. Proc. Natl. Acad. Sci. USA. 2016;113:E2001–2010. doi: 10.1073/pnas.1524900113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 7.Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997;16:6914–6925. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klaiman G, Champagne N, LeBlanc AC. Self-activation of Caspase-6 in vitro and in vivo: Caspase-6 activation does not induce cell death in HEK293T cells. Biochim. Biophys. Acta. 2009;1793:592–601. doi: 10.1016/j.bbamcr.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Gray DC, Mahrus S, Wells JA. Activation of specific apoptotic caspases with an engineered small-molecule-activated protease. Cell. 2010;142:637–646. doi: 10.1016/j.cell.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo H, et al. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am. J. Pathol. 2004;165:523–531. doi: 10.1016/S0002-9440(10)63317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albrecht S, et al. Activation of caspase-6 in aging and mild cognitive impairment. Am. J. Pathol. 2007;170:1200–1209. doi: 10.2353/ajpath.2007.060974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albrecht S, Bogdanovic N, Ghetti B, Winblad B, LeBlanc AC. Caspase-6 activation in familial Alzheimer disease brains carrying amyloid precursor protein or presenilin I or presenilin II mutations. J. Neuropathol. Exp. Neurol. 2009;68:1282–1293. doi: 10.1097/NEN.0b013e3181c1da10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LeBlanc A. Increased production of 4 kDa amyloid beta peptide in serum deprived human primary neuron cultures: possible involvement of apoptosis. J. Neurosci. 1995;15:7837–7846. doi: 10.1523/JNEUROSCI.15-12-07837.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gervais FG, et al. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/S0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 15.Tesco G, et al. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007;54:721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LeBlanc A, Liu H, Goodyer C, Bergeron C, Hammond J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J. Biol. Chem. 1999;274:23426–23436. doi: 10.1074/jbc.274.33.23426. [DOI] [PubMed] [Google Scholar]
- 17.Pellegrini L, Passer BJ, Tabaton M, Ganjei JK, D’Adamio L. Alternative, non-secretase processing of Alzheimer’s beta-amyloid precursor protein during apoptosis by caspase-6 and -8. J. Biol. Chem. 1999;274:21011–21016. doi: 10.1074/jbc.274.30.21011. [DOI] [PubMed] [Google Scholar]
- 18.Weidemann A, et al. Proteolytic processing of the Alzheimer’s disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J. Biol. Chem. 1999;274:5823–5829. doi: 10.1074/jbc.274.9.5823. [DOI] [PubMed] [Google Scholar]
- 19.Klaiman G, Petzke TL, Hammond J, LeBlanc AC. Targets of caspase-6 activity in human neurons and Alzheimer disease. Mol. Cell Proteomics. 2008;7:1541–1555. doi: 10.1074/mcp.M800007-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halawani D, et al. Identification of Caspase-6-mediated processing of the valosin containing protein (p97) in Alzheimer’s disease: a novel link to dysfunction in ubiquitin proteasome system-mediated protein degradation. J. Neurosci. 2010;30:6132–6142. doi: 10.1523/JNEUROSCI.5874-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braak, H. & Del Tredici, K. In Neuroanatomy and Pathology of Sporadic Alzheimer’s Disease 57–73 (Springer International Publishing, 2015). [PubMed]
- 22.Ramcharitar J, Afonso VM, Albrecht S, Bennett DA, LeBlanc AC. Caspase-6 activity predicts lower episodic memory ability in aged individuals. Neurobiol. Aging. 2013;34:1815–1824. doi: 10.1016/j.neurobiolaging.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeBlanc AC, et al. Caspase-6 activity in the CA1 region of the hippocampus induces age-dependent memory impairment. Cell Death Differ. 2014;21:696–706. doi: 10.1038/cdd.2013.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Cusack CL, Swahari V, Hampton Henley W, Michael Ramsey J, Deshmukh M. Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat. Commun. 2013;4:1876. doi: 10.1038/ncomms2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simon DJ, et al. A caspase cascade regulating developmental axon degeneration. J. Neurosci. 2012;32:17540–17553. doi: 10.1523/JNEUROSCI.3012-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sivananthan SN, Lee AW, Goodyer CG, LeBlanc AC. Familial amyloid precursor protein mutants cause caspase-6-dependent but amyloid beta-peptide-independent neuronal degeneration in primary human neuron cultures. Cell Death. Dis. 2010;1:e100. doi: 10.1038/cddis.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng TS, Hunot S, Kuida K, Flavell RA. Caspase knockouts: matters of life and death. Cell Death Differ. 1999;6:1043–1053. doi: 10.1038/sj.cdd.4400593. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe C, Shu GL, Zheng TS, Flavell RA, Clark EA. Caspase 6 regulates B cell activation and differentiation into plasma cells. J. Immunol. 2008;181:6810–6819. doi: 10.4049/jimmunol.181.10.6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godefroy N, Foveau B, Albrecht S, Goodyer CG, LeBlanc AC. Expression and Activation of Caspase-6 in Human Fetal and Adult Tissues. PLoS One. 2013;8:e79313. doi: 10.1371/journal.pone.0079313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foveau B, Van Der Kraak L, Beauchemin N, Albrecht S, LeBlanc AC. Inflammation-induced tumorigenesis in mouse colon is caspase-6 independent. PLoS One. 2014;9:e114270. doi: 10.1371/journal.pone.0114270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nyormoi O, Wang Z, Bar-Eli M. Sequence-based discovery of a synthetic peptide inhibitor of caspase 6. Apoptosis. 2003;8:371–376. doi: 10.1023/A:1024173018750. [DOI] [PubMed] [Google Scholar]
- 33.Orth K, Chinnaiyan AM, Garg M, Froelich CJ, Dixit VM. The CED-3/ICE-like protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A. J. Biol. Chem. 1996;271:16443–16446. doi: 10.1074/jbc.271.28.16443. [DOI] [PubMed] [Google Scholar]
- 34.Aharony I, et al. A Huntingtin-based peptide inhibitor of caspase-6 provides protection from mutant Huntingtin-induced motor and behavioral deficits. Hum. Mol. Gen. 2015;24:2604–2614. doi: 10.1093/hmg/ddv023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Calvo M, et al. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- 36.Pakavathkumar P, et al. Caspase vinyl sulfone small molecule inhibitors prevent axonal degeneration in human neurons and reverse cognitive impairment in Caspase-6-overexpressing mice. Mol. Neurodegener. 2017;12:22. doi: 10.1186/s13024-017-0166-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.James KE, et al. Design, synthesis, and evaluation of aza-peptide epoxides as selective and potent inhibitors of caspases-1, -3, -6, and -8. J. Med. Chem. 2004;47:1553–1574. doi: 10.1021/jm0305016. [DOI] [PubMed] [Google Scholar]
- 38.Ekici OD, et al. Design, synthesis, and evaluation of aza-peptide Michael acceptors as selective and potent inhibitors of caspases-2, -3, -6, -7, -8, -9, and -10. J. Med. Chem. 2006;49:5728–5749. doi: 10.1021/jm0601405. [DOI] [PubMed] [Google Scholar]
- 39.Chu W, Rothfuss J, Chu Y, Zhou D, Mach RH. Synthesis and in vitro evaluation of sulfonamide isatin Michael acceptors as small molecule inhibitors of caspase-6. J. Med. Chem. 2009;52:2188–2191. doi: 10.1021/jm900135r. [DOI] [PubMed] [Google Scholar]
- 40.Leyva MJ, et al. Identification and evaluation of small molecule pan-caspase inhibitors in Huntington’s disease models. Chem. Biol. 2010;17:1189–1200. doi: 10.1016/j.chembiol.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pakavathkumar P, Sharma G, Kaushal V, Foveau B, LeBlanc AC. Methylene Blue Inhibits Caspases by Oxidation of the Catalytic Cysteine. Sci. Rep. 2015;5:13730. doi: 10.1038/srep13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dagbay K, et al. A Multipronged Approach for Compiling a Global Map of Allosteric Regulation in the Apoptotic Caspases. Methods Enzymol. 2014;544:215–249. doi: 10.1016/B978-0-12-417158-9.00009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velazquez-Delgado EM, Hardy JA. Zinc-mediated allosteric inhibition of caspase-6. J. Biol. Chem. 2012;287:36000–36011. doi: 10.1074/jbc.M112.397752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Velazquez-Delgado EM, Hardy JA. Phosphorylation regulates assembly of the caspase-6 substrate-binding groove. Structure. 2012;20:742–751. doi: 10.1016/j.str.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao Q, et al. Inhibitory mechanism of caspase-6 phosphorylation revealed by crystal structures, molecular dynamics simulations, and biochemical assays. J. Biol. Chem. 2012;287:15371–15379. doi: 10.1074/jbc.M112.351213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki A, et al. Regulation of caspase-6 and FLIP by the AMPK family member ARK5. Oncogene. 2004;23:7067–7075. doi: 10.1038/sj.onc.1207963. [DOI] [PubMed] [Google Scholar]
- 47.Clark AC. Caspase Allostery and Conformational Selection. Chem. Rev. 2016;116:6666–6706. doi: 10.1021/acs.chemrev.5b00540. [DOI] [PubMed] [Google Scholar]
- 48.Stanger K, et al. Allosteric peptides bind a caspase zymogen and mediate caspase tetramerization. Nat. Chem. Biol. 2012;8:655–660. doi: 10.1038/nchembio.967. [DOI] [PubMed] [Google Scholar]
- 49.Murray J, et al. Tailoring small molecules for an allosteric site on procaspase-6. ChemMedChem. 2014;9(73–77):72. doi: 10.1002/cmdc.201300424. [DOI] [PubMed] [Google Scholar]
- 50.Heise CE, et al. Mechanistic and structural understanding of uncompetitive inhibitors of caspase-6. PLoS One. 2012;7:e50864. doi: 10.1371/journal.pone.0050864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tubeleviciute-Aydin A, et al. Rare human Caspase-6-R65W and Caspase-6-G66R variants identify a novel regulatory region of Caspase-6 activity. Sci. Rep. 2018;8:4428. doi: 10.1038/s41598-018-22283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baumgartner R, et al. The crystal structure of caspase-6, a selective effector of axonal degeneration. Biochem. J. 2009;423:429–439. doi: 10.1042/BJ20090540. [DOI] [PubMed] [Google Scholar]
- 53.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 54.Copeland, R. A. In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists Ch. 3, 48–82 (John Wiley & Sons, Inc., 2005). [PubMed]
- 55.Serrano, B. P. & Hardy, J. A. Phosphorylation by protein kinase A disassembles the caspase-9 core. Cell Death Differ. (2018). [DOI] [PMC free article] [PubMed]
- 56.Thomas ME, 3rd, Grinshpon R, Swartz P, Clark AC. Modifications to a common phosphorylation network provide individualized control in caspases. J. Biol. Chem. 2018;293:5447–5461. doi: 10.1074/jbc.RA117.000728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaidya S, Velazquez-Delgado EM, Abbruzzese G, Hardy JA. Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J. Mol. Biol. 2011;406:75–91. doi: 10.1016/j.jmb.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dagbay KB, Bolik-Coulon N, Savinov SN, Hardy JA. Caspase-6 Undergoes a Distinct Helix-Strand Interconversion upon Substrate Binding. J. Biol. Chem. 2017;292:4885–4897. doi: 10.1074/jbc.M116.773499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheer JM, Romanowski MJ, Wells JA. A common allosteric site and mechanism in caspases. Proc. Natl. Acad. Sci. USA. 2006;103:7595–7600. doi: 10.1073/pnas.0602571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao J, Wells JA. Identification of specific tethered inhibitors for caspase-5. Chem. Biol. Drug Des. 2012;79:209–215. doi: 10.1111/j.1747-0285.2011.01261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hardy JA, Lam J, Nguyen JT, O’Brien T, Wells JA. Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. USA. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Witkowski WA, Hardy JA. L2′ loop is critical for caspase-7 active site formation. Protein Sci. 2009;18:1459–1468. doi: 10.1002/pro.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou Q, et al. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J. Biol. Chem. 1997;272:7797. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]
- 64.Boucher D, Duclos C, Denault JB. General in vitro caspase assay procedures. Methods Mol. Biol. 2014;1133:3–39. doi: 10.1007/978-1-4939-0357-3_1. [DOI] [PubMed] [Google Scholar]
- 65.Stennicke HR, Salvesen GS. Biochemical characteristics of caspases-3, -6, -7, and -8. J. Biol. Chem. 1997;272:25719–25723. doi: 10.1074/jbc.272.41.25719. [DOI] [PubMed] [Google Scholar]
- 66.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 67.McCoy AJ, et al. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adams PD, et al. Acta Crystallogr. D Biol. Crystallogr. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution; pp. 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta. Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Celniker G, et al. ConSurf: Using Evolutionary Data to Raise Testable Hypotheses about Protein Function. Isr. J. Chem. 2013;53:199–206. doi: 10.1002/ijch.201200096. [DOI] [Google Scholar]
- 72.Morris GM, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Copeland, R. A. In Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis 278–280 (Wiley-VCH, Inc, 2000).
- 75.Okiyoneda T, et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 2013;9:444–454. doi: 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Glasoe PK, Long FA. Use of Glass Electrodes to Measure Acidities in Deuterium Oxide. J. Phys. Chem. 1960;64:188–190. doi: 10.1021/j100830a521. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All generated or analysed study data are included in this published article and its Supplementary Information file.