

Abstract
A diverse set of mono- and bis-sulfonamide was obtained via a direct, chemoselective sulfochlorination of readily available yet hitherto unexplored N-arylpyrazole template. Biochemical profiling of compounds thus obtained against a panel of human carbonic anhydrases (hCA I, hCA II, hCA IV and hCA VII) revealed a number of leads that are promising from the isoform selectivity prospective and exhibit potent inhibition profile (from nanomolar to micromolar range). The observed SAR trends have been rationalized by in silico docking of selected compounds into the active site of all four isoforms. The results reported in this paper clearly attest to the power of direct sulfochlorination as the means to create carbonic anhydrase focused sets in order to identify isoform selective inhibitors of closely related enzymes.
Keywords: Carbonic anhydrases, isoform selectivity, direct sulfochlorination, mono-sulfonamides, bis-sulfonamides, chemoselectivity
Graphical Abstract

Introduction
Spiking a carbo- or heterocyclic compound with a primary sulfonamide group has manifested itself a remarkably efficient strategy to render the molecule somewhat inhibitory toward human carbonic anhydrase (hCA) due to prosthetic Zinc binding by that group (with numerous limitations currently known to that approach) – and, ultimately, determine which of the resulting molecules will have (or at least show tendencies to have) favourable isoform-inhibitory profiles1. These much-needed, early leads can subsequently scrutinized from a structural viewpoint, advanced into clinical status (such as SCL-01112) or gain a more evolutionary look (such as compound 13 developed for treatment of cancer metastases). It’s been a long-standing dogma that the currently available landscape of drugs acting via pan-isoform inhibition mechanism (e.g. acetazolamide, methazolmide, dorzolamide and brinzolamide) are all efficacious but suboptimal in terms of inhibiting several isoforms at the same time (Figure 1)4.
Figure 1.

Advanced and clinically used hCA inhibitors.
They are poor research tools on the one hand (i.e. any attempt to link the biology perturbed by them to reality would be a dicey undertaking). On the other hand, cleaner isoform selectivity of a therapeutic agent has always been a holy grail of pharma companies: such drugs are considered to have fewer off-targets, which usually means less side-effects5. There is one more aspect as to the isoform hCA selectivity worth mentioning here, perhaps even more puzzling. The localization of various hCA isoforms within the cell is uneven and some are more or become more important than others, especially when the disease strikes (Figure 2). Take hCA IX that can is expressed on a cell membrane and became the main defenders of cells in tumors6. Clearly, we need tools to tackle hCA isoform selectivity. One such tool to use would be chemical diversity and, indeed, numerous chemically diverse series of carbonic anhydrase inhibitors (CAIs) have been profiled today4. The power of multicomponent chemistry to deliver CAIs has been relatively underutilized today as was recently reviewed7 and we are currently working to fill this void. Herein, we report a somewhat intermittent approach, namely, a systematic conversion of a set of N-arylpyrazines 2a–t into tractable and SAR-informative set of primary mono- and bis-sulfomamides. The substrates are relevant to, among other pharmacologically sound molecules, known blockbuster antipyretic Celebrex as well as tricyclic congeners 3a–e earlier reported by Marini (Figure 3)8.
Figure 2.
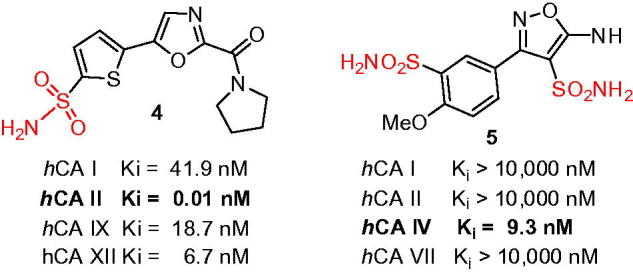
Isoform-selective CAIs derivable by direct sulfochlorination approach.
Figure 3.
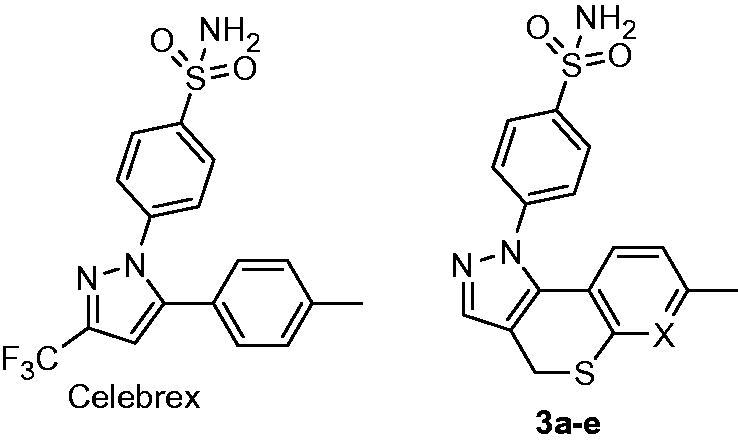
Celebrex and its tricyclic constrained versions 3a–e reported earlier.
Moreover, through some less systematic approach, the direct sulfochlorination of (hetero)aromatics has very recently given rise to: (i) 5-thienyl-1,3-oxazolecarboxamides 4 (where a remarkable potency toward hCA II with Ki = 0.01 nM was achieved)9 and (ii) a series of isoxazole bis-sulfonamides, exemplified by 5 clearly offering an alternative ZBG binging mode and a remarkable Ki of 9.4 nM against hCA IV, an extremely rare hCA to target with such a potency and selectivity (Figure 4)10.
Figure 4.

N-Arylpyrazole substrates 6a-t investigated in direct mono- and bis-sulfochlorination reactions (regiochemistry established for the respective mono- and bis-sulfonamides).
These isolated and nonetheless successful results, prompt us to undertake a direct sulfochlorination approach to produce compounds which would not only provide a wealthy entry into the realm of CAI but also provide the reader with an easy-to-read compendium of methods on direct shofochlorination of Celebrex-like N-arylpyrazoles.
Materials and methods
Chemical syntheses – general
All reactions were carried out in oven-dried glassware in atmosphere of nitrogen. Melting points were measured with a Buchi В-520 melting point apparatus and are uncorrected. Thin-layer chromatography was carried out on Silufol UV-254 silica gel plates using an appropriate mixture of ethyl acetate and hexane. Compounds were visualized with short-wavelength UV light. 1H NMR and 13C NMR spectra were recorded on Bruker MSL-300 spectrometers in DMSO-d6 using TMS as an internal standard. Elemental analyses were obtained at Research Institute for Chemical Crop Protection (Moscow, Russia) using Carlo Erba Strumentazione 1106 analyser. Mass spectra were recorded using Shimadzu LCMS-2020 system with electron impact (EI) ionization. All and reagents and solvents were obtained from commercial sources and used without purification.
General procedure 1 (GP1): regiochemically unambiguous preparation of monosulfonamides 7–8, not requiring chromatographic separation regioisomers of sulfonyl chlorides 10–11
To a well-stirred ice-cold mixture of 6.76 g (58.1 mmol) or of chlorosulfonic acid and 0.76 g (6.4 mmol) thionyl chloride was added, in small portions, an appropriate precursor 6 (5.8 mmol). The mixture was heated at the temperature and for the period of time indicated in Tables 1–3. The reaction mixture was cooled to ambient temperature and poured over ice (250 g). The resulting mixture was extracted with chloroform (100 ml). The organic layer was separated, washed with water (200 ml), 5% aqueous K2CO3, dried over anhydrous CaCl2 and filtered through a short plug of silica. The volatiles were removed in vacuo and the residue dissolved in acetone (15 ml) and the resulting clear solution was treated with 25% aqueous ammonia solution (29.0 mmol). The resulting mixture was heated at 50 °C for 30 min, concentrated in vacuo and the residue was dispersed in water (50 ml) the resulting fine precipitate was separated by filtration, washed with more water (100 ml) and air dried. Crystallization from isopropyl alcohol provided analytically pure mono- (7–8) and bis-sulfamides (9) in yields indicated.
Table 1.
Inhibitory profile of mono-sulfonamides 7a–o against four hCA isoforms.
| Sulfochlorination step |
hCA Ki (μM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Substrate 6 | Structure | T (°C) | Time (h) | hCA I | hCA II | hCA IV | hCA VII |
| 7a | 6a |  |
70 | 6 | 4.78 | 0.072 | >10.0 | 0.80 |
| 7b | 6c | 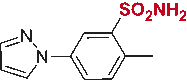 |
70 | 3 | >10.0 | >10.0 | 0.56 | >10.0 |
| 7c | 6f | 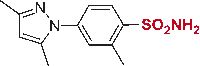 |
20 | 22 | 0.26 | 0.004 | 0.033 | 0.040 |
| 7d | 6e | 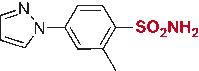 |
70 | 3 | 0.096 | 0.008 | 3.42 | 0.014 |
| 7e | 6h | 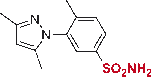 |
20 | 22 | 4.21 | 0.381 | >10.0 | 0.194 |
| 7f | 6g | 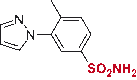 |
70 | 3 | 1.23 | 0.059 | >10.0 | 0.059 |
| 7g | 6j | 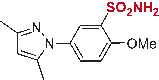 |
20 | 20 | >10.0 | >10.0 | >10.0 | >10.0 |
| 7h | 6i | 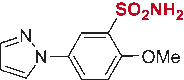 |
20 | 24 | >10.0 | >10.0 | >10.0 | >10.0 |
| 7i | 6l | 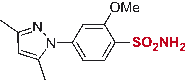 |
20 | 1 | 8.85 | >10.0 | >10.0 | >10.0 |
| 7j | 6k | 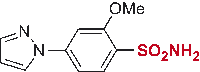 |
20 | 24 | >10.0 | >10.0 | >10.0 | 0.387 |
| 7k | 6n | 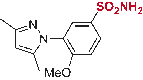 |
10 | 1 | >10.0 | >10.0 | >10.0 | >10.0 |
| 7l | 6m | 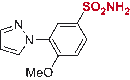 |
20 | 24 | >10.0 | 6.65 | >10.0 | >10.0 |
| 7m | 6q | 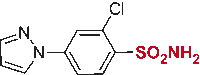 |
80 | 3 | 0.066 | 0.085 | 0.086 | 0.328 |
| 7n | 6s | 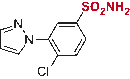 |
90 | 5 | 3.83 | 0.001 | 0.004 | 0.009 |
| 7o | – | 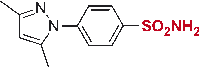 |
– | – | 0.462 | 0.004 | 0.084 | 0.017 |
| Acetazolamide | 0.25 | 0.012 | 0.074 | 0.003 | ||||
Table 2.
Inhibitory profile of mono-sulfonamides 8a–i against four hCA isoforms.
| Sulfochlorination |
hCA Ki (μM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Substrate 6 | Structure | T (°C) | Time (h) | hCA I | hCA II | hCA IV | hCA VII |
| 8a | 6p | 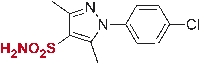 |
70 | 1 | 0.76 | 5.33 | >10.0 | >10.0 |
| 8b | 6b | 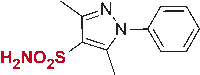 |
70 | 1 | 0.54 | 0.28 | >10.0 | 0.53 |
| 8c | 6d | 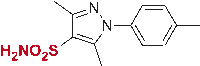 |
20 | 20 | 0.76 | 0.74 | >10.0 | >10.0 |
| 8d | 6f | 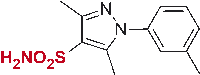 |
20 | 22 | 0.27 | 0.24 | >10.0 | 0.94 |
| 8e | 6h | 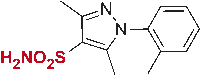 |
20 | 22 | 0.60 | 0.091 | >10.0 | >10.0 |
| 8f | 6o | 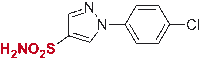 |
90 | 4 | 0.19 | 0.082 | 7.06 | >10.0 |
| 8g | 6r | 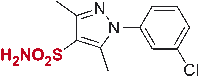 |
70 | 5 | 0.94 | 1.08 | 9.49 | 0.46 |
| 8h | 6q | 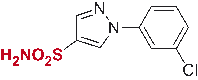 |
80 | 3 | 0.54 | 0.14 | 3.74 | 0.24 |
| 8i | 6t | 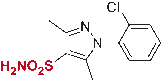 |
70 | 10 | 0.62 | 2.64 | 7.19 | >10.0 |
| Acetazolamide | 0.25 | 0.012 | 0.074 | 0.003 | ||||
Table 3.
Inhibitory profile of mono-sulfonamides 9a–s against four hCA isoforms.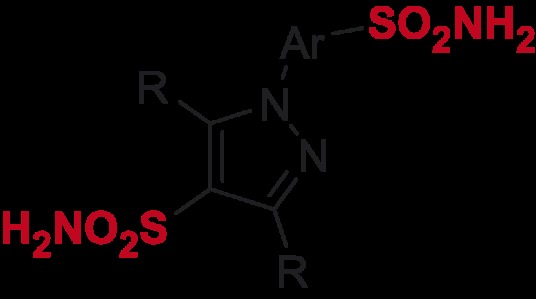
| Sulfochlorination |
hCA Ki (μM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Substrate xx | Structure | T (°C) | Time (h) | hCA I | hCA II | hCA IV | hCA VII |
| 9a | 6b | 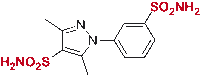 |
70 | 7 | 0.50 | 0.021 | 0.065 | 0.26 |
| 9b | 6b | 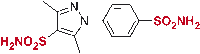 |
70 | 7 | 0.33 | 0.005 | 0.111 | 0.035 |
| 9c | 6a | 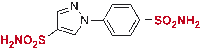 |
100 | 48 | 0.20 | 0.002 | 0.025 | 0.041 |
| 9d | 6d | 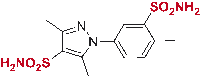 |
70 | 7 | >10.0 | >10.0 | 0.40 | >10.0 |
| 9e | 6c | 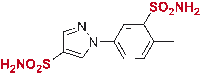 |
100 | 20 | 0.30 | 0.004 | 0.24 | 0.068 |
| 9f | 6f | 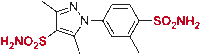 |
70 | 7 | 0.47 | 0.002 | 0.022 | 0.019 |
| 9g | 6e | 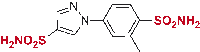 |
100 | 20 | 0.10 | 0.049 | 5.80 | 0.005 |
| 9h | 6h | 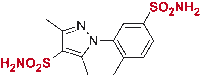 |
70 | 7 | 0.87 | >10.0 | >10.0 | >10.0 |
| 9i | 6g | 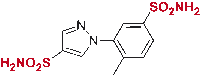 |
100 | 20 | 0.55 | 0.030 | 7.86 | 0.014 |
| 9j | 6j | 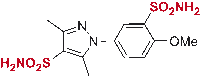 |
70 | 4 | 0.95 | 0.96 | >10.0 | 0.33 |
| 9k | 6l | 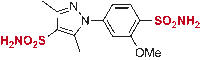 |
70 | 4 | 4.65 | 0.61 | >10.0 | 0.17 |
| 9l | 6n | 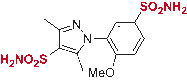 |
70 | 4 | 6.80 | 0.30 | 0.95 | >10.0 |
| 9m | 6p | 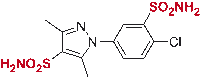 |
100 | 24 | 0.89 | 0.040 | 0.077 | 0.65 |
| 9n | 6o | 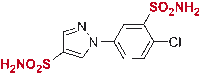 |
120 | 10 | 0.62 | 0.024 | 0.074 | 0.099 |
| 9o | 6r | 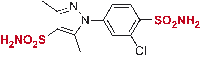 |
100 | 24 | 0.16 | 0.008 | 0.93 | 0.71 |
| 9p | 6q | 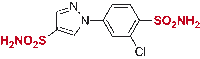 |
120 | 20 | 3.08 | 0.008 | 0.075 | 0.48 |
| 9q | 6t | 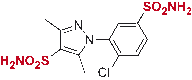 |
100 | 24 | 2.12 | 0.025 | 4.38 | >10.0 |
| 9r | 6s | 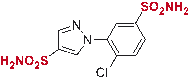 |
120 | 20 | 0.55 | 0.005 | 0.022 | 0.60 |
| Acetazolamide | 0.25 | 0.012 | 0.074 | 0.003 | ||||
4-(1H-pyrazol-1-yl)benzenesulfonamide (7a)
Prepared from 6a according to GP1; white solid, m.p. 323–325°C (i-PrOH), yield 73%; 1H NMR (300 MHz, DMSO-d6) δ ppm 11.44 (br. s., 2H, SO2NH2), 8.50 (d, J = 2.3 Hz, 1H, Hpyrazole), 7.82 (d, J = 8.6 Hz, 2H, HAr), 7.74 (d, J = 1.4 Hz, 1H, Hpyrazole), 7.72 (d, J = 8.6 Hz, 2H, HAr), 6.56 (dd, J1=2.3 Hz, J2=1.4 Hz, 1H, Hpyrazole); 13C NMR (75 MHz, DMSO-d6) δ ppm 146.0, 141.6, 140.1, 128.3, 127.3, 118.0, 108.5; LC/MS (ESI+): m/z [M + H]+ 224.3; Anal. calcd for C9H9N3O2S (223.25): C, 48.42; H, 4.06; N, 18.82; S, 14.36; found: C, 48.39; H, 4.06; N, 18.84; S, 14.37.
2-Methyl-5-(1H-pyrazol-1-yl)benzenesulfonamide (7b)
Prepared from 6c according to GP1; yellow solid, m.p. 147–149°C (i-PrOH), yield 68%. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.38 (d, J = 2.4 Hz, 1H, Hpyrazole), 8.17 (d, J4–6 = 2.4 Hz, 1H, 6-HA), 7.78 (br. s., 2H, SO2NH2), 7.71 (d, J = 1.5 Hz, 1H, Hpyrazole), 7.63 (dd, J3–4 = 8.2, Hz, J4–6 = 2.4 Hz, 1H, 4-HAr), 7.25 (d, J3–4 = 8.2 Hz, 1H, 3-HAr), 6.51 (dd, J1=2.4 Hz, J2=1.5 Hz, 1H, Hpyrazole), 2.54 (s, 3H, Ar-CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.5, 141.1, 137.3, 133.9, 132.2, 127.9, 118.9, 117.5, 108.1, 19.9. LC/MS (ESI+): m/z [M + H]+ 238.3. Anal. calcd for C10H11N3O2S (237.28): C, 50.62; H, 4.67; N, 17.71; S, 13.51; found: C, 50.59; H, 4.68; N, 17.75; S, 13.52.
2-Methyl-4-(1H-pyrazol-1-yl)benzenesulfonamide (7d)
Prepared from 6e according to GP1; white solid, m.p. 161–163 °C (i-PrOH), yield 66%. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.59 (d, J = 2.4 Hz, 1H, Hpyrazole), 7.94 (d, J3–4 = 8.6 Hz, 1H, 3-HAr), 7.89 (d, J3–5 = 2.0 Hz, 1H, 3-HA), 7.82 (dd, J5–6 = 8.6 Hz, J3–5 = 2.0 Hz, 1H, 5-HAr), 7.80 (d, J = 1.6 Hz, 1H, Hpyrazole), 7.42 (br. s., 2H, SO2NH2), 6.59 (dd, J1=2.4 Hz, J2=1.6 Hz, 1H, Hpyrazole), 2.66 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 142.2, 142.0, 139.9, 138.3, 129.2, 128.7, 121.6, 115.6, 108.9, 20.4. LC/MS (ESI+): m/z [M + H]+ 238.3. Anal. calcd for C10H11N3O2S (237.28): C, 50.62; H, 4.67; N, 17.71; S, 13.51; found: C, 50.57; H, 4.67; N, 17.68; S, 13.52.
4-Methyl-3-1H-pyrazol-1-yl)benzenesulfonamide (7f)
Prepared from 6 g according to GP1; light brown solid, m.p. 143–145 °C (i-PrOH), yield 61%. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.12 (d, J = 2.2 Hz, 1H, 5-Hpyrazole), 7.78 (m, 3H, 3-Hpyrazole, 2-HAr, 6-HAr), 7.60 (d, J5–6 = 7.8 Hz, 1H, 5-HAr), 7.42 (s, 2H, SO2NH2), 6.56 (m, 1H, 4-Hpyrazole), 2.29 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ ppm 143.2, 141.1, 140.1, 137.1, 132.4, 131.9, 125.3, 123.5, 107.4, 18.5. LC/MS (ESI+): m/z [M + H]+ 238.3. Anal. calcd for C10H11N3O2S (237.28): C, 50.62; H, 4.67; N, 17.71; S, 13.51; found: C, 50.57; H, 4.67; N, 17.68; S, 13.52.
5-(3,5-Dimethyl-1H-pyrazol-1-yl)-2-methoxybenzenesulfonamide (7g)
Prepared from 6j according to GP1; white solid, m.p. 195–197°C (i-PrOH), yield 78%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.76 (d, J4–6 = 2.7 Hz, 1H, 6-HAr), 7.44 (dd, J3–4 = 8.6 Hz, J4–6 = 2.7 Hz, 1H, 4-HAr), 7.09 (d, J3–4 = 8.6 Hz, 1H, 3-HAr), 6.18 (s, 1H, Hpyrazole), 5.03 (br. s., 2H, SO2NH2, H2O), 3.82 (s, 3H, OCH3), 2.22 (s, 6H, 2xCH3); 13C NMR (75 MHz, DMSO-d6) δ ppm 156.8, 147.1, 142.3, 136.8, 128.9, 127.6, 126.1, 112.9, 107.5, 56.4, 12.8, 12.1. LC/MS (ESI+): m/z [M + H]+ 282.3. Anal. calcd for C12H15N3O3S (281.33): C, 51.23; H, 5.37; N, 14.94; S, 11.40; found: C, 51.17; H, 5.38; N, 14.91; S, 11.41.
2-Methoxy-5-1H-pyrazol-1-yl)benzenesulfonamide (7h)
Prepared from 6i according to GP1; white solid, m.p. 138–140 °C (i-PrOH), yield 63%. 1H NMR (300 MHz, DMSO-d6) δ ppm 8.47 (d, J = 2.0 Hz, 1H, Hpyrazole), 8.18 (d, J4–6 = 2.7 Hz, 1H, 6-HAr), 8.00 (dd, J3–4 = 8.9 Hz, J4–6 = 2.7 Hz, 1H, 4-HAr), 7.74 (s, 1H, Hpyrazole), 7.33 (d, J3–4 = 8.9 Hz, 1H, 3-HAr), 7.23 (s, 2H, SO2NH2), 6.54 (m, 1H, Hpyrazole), 3.95 (s, 3H, OCH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 155.1, 140.8, 136.2, 132.5, 127.9, 121.1, 120.0, 113.5, 107.8, 56.4. LC/MS (ESI+): m/z [M + H]+ 254.3. Anal. calcd for C10H11N3O3S (253.28): C, 47.42; H, 4.38; N, 16.59; S, 12.66; found: C, 47.40; H, 4.38; N, 15.62; S, 12.67.
4-(3,5-Dimethyl-1H-pyrazol-1-yl)-2-methoxybenzenesulfonamide (7i)
Prepared from 6l according to GP1; yellow solid, m.p. 156–158 °C (i-PrOH), yield 72%; 1H NMR (400 MHz, DMSO-d6) δ ppm 11.95 (br. s., 2H, SO2NH2, H2O),7.76 (d, J5–6 = 8.2 Hz, 1H, 6-HAr), 7.08 (d, J3–5 = 1.8 Hz, 1H, 3-HAr), 6.99 (dd, J5–6 = 8.2 Hz, J3–5 = 1.8 Hz, 1H, 5-HAr), 6.15 (s, 1H, Hpyrazole), 3.79 (s, 3H, OCH3), 2.32 (s, 3H, CH3), 2.24 (m, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 157.0, 149.2, 144.3, 140.2, 129.8, 128.8, 114.8, 108.7, 108.7, 56.9, 13.7, 12.9. LC/MS (ESI+): m/z [M + H]+ 282.3. Anal. calcd for C12H15N3O3S (281.33): C, 51.23; H, 5.37; N, 14.94; S, 11.40; found: C, 51.21; H, 5.37; N, 14.95; S, 11.41.
2-Methoxy-4-(1H-pyrazol-1-yl)benzenesulfonamide (7j)
Prepared from 6k according to GP1; white solid, m.p. 221–223°C (i-PrOH), yield 64%. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.66 (d, J = 2.4 Hz, 1H, Hpyrazole), 7.81 (d, J5–6 = 8.4 Hz, 1H, 6-HAr), 7.81 (d, J = 1.0 Hz, 1H, Hpyrazole), 7.63 (d, J3–5 = 1.6 Hz, 1H, 3-HAr), 7.54 (dd, J5–6 = 8.4 Hz, J3–5 = 1.6 Hz, 1H, 5-HAr), 7.12 (s, 2H, SO2NH2), 6.61 (m, 1H, Hpyrazole), 4.00 (s, 3H, OCH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 157.5, 144.0, 142.3, 129.6, 129.3, 129.0, 109.5, 109.0, 103.1, 56.9. LC/MS (ESI+): m/z [M + H]+ 254.3. Anal. calcd for C10H11N3O3S (253.28): C, 47.42; H, 4.38; N, 16.59; S, 12.66; found: C, 47.38; H, 4.38; N, 15.58; S, 12.66.
3-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-methoxybenzenesulfonamide (7k)
Prepared from 6n according to GP1; white solid, m. p. 167–170 °C (i-PrOH); 1H NMR (400 MHz, DMSO-d6) δ ppm 7.64 (dd, J5–6 = 8.6 Hz, J2–6 = 2.2 Hz, 1H, 6-HAr), 7.41 (d, J2–6 = 2.2 Hz, 1H, 2-HAr), 7.14 (d, J5–6 = 8.6 Hz, 1H, 5-HAr), 7.11 (br. s., 2H, SO2NH2), 5.97 (s, 1H, Hpyrazole), 3.78 (s, 3H, OCH3), 2.15 (s, 3H, CH3), 1.99 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 154.4, 148.0, 141.3, 127.6, 127.5, 127.5, 126.7, 112.1, 105.7, 56.4, 13.8, 11.3. LC/MS (ESI+): m/z [M + H]+ 282.3. Anal. calcd for C12H15N3O3S (281.33): C, 51.23; H, 5.37; N, 14.94; S, 11.40; found: C, 51.20; H, 5.37; N, 14.96; S, 11.41.
4-Methoxy-3-(1H-pyrazol-1-yl)benzenesulfonamide (7l)
Prepared from 6m according to GP1; white solid, m.p. 255–257 °C (i-PrOH), yield 61%. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.40 (br. s., 2H, SO2NH2, H2O), 8.17 (d, J = 2.45 Hz, 1H, Hpyrazole), 7.86 (d, J2–6 = 2.2 Hz, 1H, 2-HAr), 7.70 (d, J = 1.7 Hz, 1H, Hpyrazole), 7.56 (dd, J5–6 = 8.6 Hz, J2–6 = 2.2 Hz, 1H, 6-HAr), 7.19 (d, J5–6 = 8.6 Hz, 1H, 5-HAr), 6.47 (m, 1H, Hpyrazole), 3.87 (s, 3H, OCH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 151.42, 141.22, 140.30, 132.30, 128.39, 125.80, 122.75, 112.60, 106.98, 56.73. LC/MS (ESI+): m/z [M + H]+ 254.3. Anal. calcd for C10H11N3O3S (253.28): C, 47.42; H, 4.38; N, 16.59; S, 12.66; found: C, 47.31; H, 4.38; N, 15.64; S, 12.68.
4-Chloro-3-(1H-pyrazol-1-yl)benzenesulfonamide (7n)
Prepared from 6s according to GP1; off-white solid, m.p. 204–206 °C (i-PrOH); 1H NMR (400 MHz, DMSO-d6) δ ppm 8.26 (d, J = 2.2 Hz, 1H, Hpyrazole), 8.00 (d, J2–6 = 1.7 Hz, 1H, 2-HAr), 7.91 (d, J5–6 = 8.3 Hz, 1H, 5-HAr), 7.86 (dd, J5–6 = 8.3 Hz, J2–6 = 1.7 Hz, 1H, 6-HAr), 7.83 (d, J = 1.5 Hz, 1H, Hpyrazole), 7.60 (s, 2H, SO2NH2), 6.59 (m, 1H, Hpyrazole). 13C NMR (75 MHz, DMSO-d6) δ ppm 144.4, 141.8, 138.4, 132.7, 131.9, 131.1, 126.7, 125.4, 107.7. LC/MS (ESI+): m/z [M + H]+ 258.7. Anal. calcd for C9H8ClN3O2S (257.70): C, 41.95; H, 3.13; N, 16.31; S, 12.44; found: C, 41.91; H, 3.13; N, 16.27; S, 12.46.
1-(4-Chlorophenyl)-3,5-dimethyl-1H-pyrazole-4-sulfonamide (8a)
Prepared from 6p according to GP1; white solid, m.p. 164–166 °C (i-PrOH), yield 64%; 1H NMR (400 MHz, DMSO-d6) δ ppm 7.60 (d, J = 9.0 Hz, 2H, HAr), 7.54 (d, J = 9.0 Hz, 2H, HAr), 7.24 (s, 2H, SO2NH2), 2.42 (s, 3H, CH3), 2.36 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.5, 141.0, 137.6, 133.4, 129.8, 127.5, 122.2, 66.8, 13.4, 11.9. LC/MS (ESI+): m/z [M + H]+ 286.7. Anal. calcd for C11H12ClN3O2S (285.75): C, 46.24; H, 4.23; N, 14.71; S, 11.22; found: C, 46.20; H, 4.23; N, 14.75; S, 11.23.
3,5-Dimethyl-1-phenyl-1H-pyrazole-4-sulfonamide (8b)
Prepared from 6b according to GP1; white solid, m.p. 161–163 °C (i-PrOH), yield 67%; 1H NMR (400 MHz, DMSO-d6) δ ppm 7.55 (m, 2H, HAr), 7.49 (m, 3H, HAr), 7.22 (s, 2H, SO2NH2), 2.41 (s, 3H, CH3), 2.36 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ ppm 147.2, 140.8, 138.7, 129.8, 128.9, 125.9, 121.8, 13.5, 12.0. LC/MS (ESI+): m/z [M + H]+ 252.3. Anal. calcd for C11H13N3O2S (251.31): C, 52.57; H, 5.21; N, 16.72; S, 12.76; found: C, 52.54; H, 5.22; N, 16.75; S, 12.78.
3,5-Dimethyl-1-(4-methylphenyl)-1H-pyrazole-4-sulfonamide (8c)
Prepared from 6d according to GP1; white solid, m.p. 171–174 °C (i-PrOH), yield 63%; 1H NMR (400 MHz, DMSO-d6) δ ppm 7.35 (m, 4H, HAr), 7.20 (s, 2H, SO2NH2), 2.39 (s, 3H, CH3), 2.38 (s, 3H, CH3), 2.35 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ ppm 147.0, 140.7, 138.6, 136.4, 130.2, 125.7, 121.7, 21.1, 13.4, 11.9. LC/MS (ESI+): m/z [M + H]+ 266.3. Anal. calcd for C12H15N3O2S (265.34): C, 54.32; H, 5.70; N, 15.84; S, 12.08; found: C, 54.30; H, 5.70; N, 15.82; S, 12.10.
1-(4-Chlorophenyl)-1H-pyrazole-4-sulfonamide (8f)
Prepared from 6o according to GP1; off-white solid, m.p. 147–150 °C (i-PrOH); 1H NMR (300 MHz, DMSO-d6) δ ppm 8.99 (s, 1H, Hpyrazole), 8.04 (s, 1H, Hpyrazole), 7.94 (d, J = 8.9 Hz, 2H, HAr), 7.59 (d, J = 8.9 Hz, 1H, HAr), 7.43 (s, 2H, SO2NH2); 13C NMR (75 MHz, DMSO-d6) δ ppm 139.5, 138.1, 132.1, 130.1, 129.6, 128.9, 121.3. LC/MS (ESI+): m/z [M + H]+ 258.7. Anal. calcd for C9H8ClN3O2S (257.70): C, 41.95; H, 3.13; N, 16.31; S, 12.44; found: C, 41.89; H, 3.13; N, 16.28; S, 12.45.
1-(3-Chlorophenyl)-3,5-dimethyl-1H-pyrazole-4-sulfonamide (8g)
Prepared from 6r according to GP1; white solid, m.p. 182–184 °C (i-PrOH), yield 54%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.56 (m, 4H, HAr), 7.25 (s, 2H, SO2NH2), 2.45 (s, 3H, CH3), 2.37 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.6, 141.1, 139.9, 134.0, 131.3, 128.9, 125.6, 124.5, 122.3, 13.5, 11.9. LC/MS (ESI+): m/z [M + H]+ 286.7. Anal. calcd for C11H12ClN3O2S (285.75): C, 46.24; H, 4.23; N, 14.71; S, 11.22; found: C, 46.18; H, 4.24; N, 14.69; S, 11.24.
1-(2-Chlorophenyl)-3,5-dimethyl-1H-pyrazole-4-sulfonamide (8i)
Prepared from 6t according to GP1; white solid, m.p. 204–206°C (AcOEt), yield 56%. 1H NMR (300 MHz, DMSO-d6) δ ppm 7.73 (d, J = 7.60 Hz, 1H, HAr), 7.59 (m, 3H, HAr), 7.27 (s, 2H, SO2NH2), 2.36 (s, 3H, CH3), 2.21 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.3, 142.2, 136.0, 132.0, 131.5, 130.7, 130.6, 128.9, 121.3, 13.4, 11.2. LC/MS (ESI+): m/z [M + H]+ 286.8. Anal. calcd for C11H12ClN3O2S (285.75): C, 46.24; H, 4.23; N, 14.71; S, 11.22; found: C, 46.21; H, 4.23; N, 14.72; S, 11.22.
General procedure 2 (GP2): regiochemically unambiguous preparation of bis-sulfonamides 9 not requiring chromatographic separation regioisomers of bis-sulfonyl chlorides 12
The procedure is analogous to GP1 except for the double amount of chlorosulfonic acid (13.52 g, 116.1 mmol), thionyl chloride (1.52 g, 12.8 mmol) and 25% aqueous ammonia solution (58.0 mmol) used in the preparation.
1-(4-Sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9c)
Prepared from 6a according to GP2; white solid, m.p. 254–257 °C (i-PrOH), yield 56%. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.07 (s, 1H, Hpyrazole), 8.11 (d, J = 8.9 Hz, 2H, HAr), 8.09 (s, 1H, Hpyrazole), 7.95 (d, J = 8.9 Hz, 2H, HAr), 7.45 (m, 4H, 2 x SO2NH2). 13C NMR (75 MHz, DMSO-d6) δ ppm 143.0, 141.3, 139.9, 129.9, 129.2, 127.8, 119.7. LC/MS (ESI+): m/z [M + H]+ 303.3. Anal. calcd for C9H10N4O4S2 (302.33): C, 35.76; H, 3.33; N, 18.53; S, 21.21; found: C, 35.71; H, 3.34; N, 18.55; S, 21.23.
3,5-Dimethyl-1-(4-methyl-3-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9d)
Prepared from 6d according to GP2; white solid, m.p. 219–221 °C (i-PrOH), yield 52%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.76 (d, J2–6 = 2.2 Hz, 1H, 2-HAr), 7.31 (dd, J5–6 = 8.1, Hz, J2–6 = 2.2 Hz, 1H, 6-HAr), 7.27 (d, J5–6 = 8.1 Hz, 1H, 5-HAr), 6.48 (br. s., 4H, 2 x SO2NH2), 2.57 (s, 3H, Ar-CH3), 2.35 (s, 3H, CH3), 2.28 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.5, 146.2, 138.7, 136.1, 135.7, 131.9, 125.5, 123.9, 107.9, 20.1, 13.1, 11.8. LC/MS (ESI+): m/z [M + H]+ 345.4. Anal. calcd for C12H16N4O4S2 (344.41): C, 41.85; H, 4.68; N, 16.27; S, 18.62; found: C, 41.78; H, 4.69; N, 16.21; S, 18.64.
1-(4-Methyl-3-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9e)
Prepared from 6c according to GP2; white solid, m.p. 217–220°C (i-PrOH), yield 53%. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.97 (s, 1H, Hpyrazole), 8.35 (d, J2–6 = 2.4 Hz, 1H, 2-HA), 8.06 (s, 1H, Hpyrazole), 8.02 (dd, J5–6 = 8.2 Hz, J2–6 = 2.4 Hz, 1H, 6-HAr), 7.55 (s, 2H, SO2NH2), 7.54 (d, J5–6 = 8.2 Hz, 1H, 5-HAr), 7.43 (s, 2H, SO2NH2), 2.62 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 143.8, 139.5, 137.1, 135.4, 134.1, 129.6, 128.8, 122.2, 118.4, 19.7. LC/MS (ESI+): m/z [M + H]+ 317.4. Anal. calcd for C10H12N4O4S2 (316.36): C, 37.97; H, 3.82; N, 17.71; S, 20.27; found: C, 37.94; H, 3.82; N, 17.67; S, 20.30.
3,5-Dimethyl-1-(3-methyl-4-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9f)
Prepared from 6f according to GP2; light grey solid, m.p. 143–146 °C (i-PrOH), yield 41%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.97 (d, J5–6 = 8.6 Hz, 1H, 5-HAr), 7.54 (m, 4H, 2-HAr, 6-HAr, SO2NH2), 7.27 (br. s., 2H, SO2NH2), 2.65 (s, 3H), 2.47 (s, 3H, CH3), 2.37 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.8, 142.2, 141.2, 141.1, 138.2, 128.8, 128.7, 122.9, 122.6, 20.2, 13.4, 12.1. LC/MS (ESI+): m/z [M + H]+ 345.4. Anal. calcd for C12H16N4O4S2 (344.41): C, 41.85; H, 4.68; N, 16.27; S, 18.62; found: C, 41.82; H, 4.69; N, 16.30; S, 18.63.
1-(3-Methyl-4-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9g)
Prepared from 6e according to GP2; white solid, m.p. 205–207 °C (i-PrOH), yield 51%. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.05 (s, 1H, Hpyrazole), 8.07 (s, 1H, Hpyrazole), 7.98 (d, J2–6 = 2.0 Hz, 1H, 2-HAr), 7.96 (d, J5–6 = 8.6 Hz, 1H, 5-HAr), 7.90 (dd, J5–6 = 8.6 Hz, J2–6 = 2.0 Hz, 1H, 6-HAr), 7.47 (s, 2H, SO2NH2), 7.44 (s, 2 H, SO2NH2), 2.66 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 141.2, 141.1, 139.8, 138.6, 129.9, 129.3, 129.1, 122.5, 116.5, 20.4. LC/MS (ESI+): m/z [M + H]+ 317.4. Anal. calcd for C10H11N3O2S (316.36): C, 37.97; H, 3.82; N, 17.71; S, 20.27; found: C, 37.94; H, 3.83; N, 17.74; S, 20.29.
3,5-Dimethyl-1-(2-methyl-5-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9h)
Prepared from 6h according to GP2; white solid, m.p. 308–310 °C (i-PrOH), yield 45%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.63 (dd, J3–4 = 7.8 Hz, J4–6 = 1.3 Hz, 1H, 4-HAr), 7.38 (d, J3–4 = 7.8 Hz, 1H, 3-HAr), 7.37 (d, J4–6 = 1.3 Hz, 1H, 6-HAr), 5.96 (br. s., 2H, SO2NH2), 2.29 (s, 3H, CH3), 2.12 (s, 3H, CH3), 1.96 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ ppm 146.8, 145.7, 139.5, 136.2, 136.2, 130.6, 126.8, 124.9, 124.2, 16.7, 12.6, 10.6. LC/MS (ESI+): m/z [M + H]+ 345.4. Anal. calcd for C12H16N4O4S2 (344.41): C, 41.85; H, 4.68; N, 16.27; S, 18.62; found: C, 41.81; H, 4.68; N, 16.31; S, 18.62.
1-(2-Methyl-5-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9i)
Prepared from 6g according to GP2; white solid, m.p. 185–188 °C (i-PrOH), yield 47%. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.60 (s, 1H, Hpyrazole), 8.05 (s, 1H, Hpyrazole), 7.84 (dd, J3–4 = 7.8 Hz, J4–6 = 1.7 Hz, 1H, 4-HAr), 7.80 (d, J4–6 = 1.3 Hz, 1H, 6-HAr), 7.64 (d, J3–4 = 7.8 Hz, 1H, 3-HAr), 7.44 (s, 2H, SO2NH2), 7.41 (s, 2H, SO2NH2), 2.32 (m, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 143.3, 139.1, 137.6, 132.6, 132.5, 128.7, 128.6, 126.3, 123.8, 18.2. LC/MS (ESI+): m/z [M + H]+ 317.4. Anal. calcd for C10H11N3O2S (316.36): C, 37.97; H, 3.82; N, 17.71; S, 20.27; found: C, 37.92; H, 3.83; N, 17.75; S, 20.30.
1-(4-Methoxy-3-sulfamoylphenyl)-3,5-dimethyl-1H-pyrazole-4-sulfonamide (9j)
Prepared from 6j according to GP2; white solid, m.p. 254–257°C (i-PrOH), yield 79%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.72 (m, 2H, 2-HAr, 6-HAr), 7.35 (d, J5–6 = 8.6 Hz, 1H, 5-HAr), 7.29 (s, 2H, SO2NH2), 7.22 (s, 2H, SO2NH2), 3.97 (s, 3H, OCH3), 2.36 (s, 3H, CH3), 2.40 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 156.3, 147.4, 141.1, 132.4, 130.9, 130.7, 125.4, 121.8, 113.9, 57.1, 13.4, 11.8. LC/MS (ESI+): m/z [M + H]+ 361.4. Anal. calcd for C12H16N4O5S2 (360.41): C, 39.99; H, 4.47; N, 15.55; S, 17.79; found: C, 39.94; H, 4.48; N, 15.52; S, 17.81.
1-(3-Methoxy-4-sulfamoylphenyl)-3,5-dimethyl-1H-pyrazole-4-sulfonamide (9k)
Prepared from 6l according to GP2; white solid, m.p. 256–258 °C (i-PrOH), yield 82%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.85 (d, J5–6 = 8.3 Hz, 1H, 5-HAr), 7.32 (d, J2–6 = 1.8 Hz, 1H, 2-HAr), 7.28 (s, 2H, SO2NH2), 7.22 (s, 1H, SO2NH2), 7.19 (dd, J5–6 = 8.3 Hz, J2–6 = 1.8 Hz, 1H, 6-HAr), 3.95 (s, 3H, OCH3), 2.50 (s, 3H, CH3), 2.36 (m, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 157.02, 147.82, 142.86, 141.29, 131.42, 128.96, 122.60, 116.84, 110.34, 57.11, 13.47, 12.15. LC/MS (ESI+): m/z [M + H]+ 361.4.
1-(2-Methoxy-5-sulfamoylphenyl)-3,5-dimethyl-1H-pyrazole-4-sulfonamide (9l)
Prepared from 6n according to GP2; white solid, m.p. 238–242 °C (i-PrOH), yield 68%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.95 (dd, J3–4 = 8.8 Hz, J4–6 = 2.4 Hz, 1H, 4-HAr), 7.73 (d, J4–6 = 2.4 Hz, 1H, 6-HAr), 7.44 (d, J3–4 = 8.8 Hz, 1H, 3-HAr), 7.36 (s, 2H, SO2NH2), 7.24 (s, 2H, SO2NH2), 3.88 (s, 3H, OCH3), 2.35 (s, 3H, CH3), 2.21 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ ppm 156.8, 147.5, 142.7, 137.0, 129.1, 127.1, 127.0, 121.3, 113.5, 57.0, 13.4, 11.3. LC/MS (ESI+): m/z [M + H]+ 361.4. Anal. calcd for C12H16N4O5S2 (360.41): C, 39.99; H, 4.47; N, 15.55; S, 17.79; found: C, 39.91; H, 4.48; N, 15.60; S, 17.82.
1-(4-Chloro-3-sulfamoylphenyl)-3,5-dimethyl-1H-pyrazole-4-sulfonamide (9m)
Prepared from 6p according to GP2; white solid, m.p. 241–243 °C (AcOEt), yield 39%. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.02 (d, J2–6 = 2.0 Hz, 1H, 2-HAr), 7.80 (m, 4H, 5-HAr, 6-HAr, SO2NH2), 7.29 (s, 2H, SO2NH2), 2.47 (s, 3H, CH3), 2.37 (m, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 148.1, 142.4, 141.4, 137.4, 133.0, 130.4, 129.7, 125.9, 122.7, 13.4, 12.0. LC/MS (ESI+): m/z [M + H]+ 365.8. Anal. calcd for C11H13ClN4O4S2 (264.83): C, 36.21; H, 3.59; N, 15.36; S, 17.58; found: C, 36.18; H, 3.59; N, 15.33; S, 13.59.
1-(4-Chloro-3-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9n)
Prepared from 6o according to GP2; white solid, m.p. 232–235 °C (AcOEt), yield 21%. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.07 (s, 1H, Hpyrazole), 8.48 (d, J2–6 = 2.7 Hz, 1H, 2-HAr), 8.15 (dd, J5–6 = 8.8 Hz, J2–6 = 2.7 Hz, 1H, 6-HAr), 8.09 (s, 1H, Hpyrazole), 7.81 (d, J5–6 = 8.8 Hz, 1H, 5-HAr), 7.79 (s, 2H, SO2NH2), 7.47 (s, 2H, SO2NH2). 13C NMR (75 MHz, DMSO-d6) δ ppm 142.7, 139.9, 137.8, 133.3, 130.1, 129.2, 129.1, 123.4, 120.0. LC/MS (ESI+): m/z [M + H]+ 337.8. Anal. calcd for C9H9ClN4O4S2 (336.78): C, 32.10; H, 2.69; N, 16.64; S, 19.04; found: C, 32.05; H, 2.70; N, 16.69; S, 19.05.
1-(3-Chloro-4-sulfamoylphenyl)-3,5-dimethyl-1H-pyrazole-4-sulfonamide (9o)
Prepared from 6r according to GP2; white solid, m.p. 198–201 °C (AcOEt), yield 43%. 1H NMR (300 MHz, DMSO-d6) δ ppm 7.86 (d, J2–6 = 2.4 Hz, 1H, X-HAr), 7.80 (d, J5–6 = 8.6 Hz, 1H, A-HAr), 7.54 (dd, J5–6 = 8.6, J2–6 = 2.4 Hz, 1H, B-HAr), 7.29 (s, 2H, in exchange, SO2NH2), 2.46 (s, 3H, CH3), 2.36 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.9, 141.3, 138.5, 132.2, 131.6, 131.5, 127.5, 125.9, 122.5, 13.4, 11.9. LC/MS (ESI+): m/z [M + H]+ 365.8. Anal. calcd for C11H13ClN4O4S2 (264.83): C, 36.21; H, 3.59; N, 15.36; S, 17.58; found: C, 36.19; H, 3.59; N, 15.35; S, 13.58.
1-(3-Chloro-4-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9p)
Prepared from 6q according to GP2; white solid, m.p. 195–197 °C (AcOEt), yield 25%. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.07 (s, 1H, Hpyrazole), 8.23 (d, J2–6 = 2.3 Hz, 1H, 2-HAr), 8.06 (s, 1H, Hpyrazole), 7.78 (dd, J5–6 = 8.6 Hz, J2–6 = 2.3 Hz, 1H, 6-HAr),7.94 (d, J5–6 = 8.6 Hz, 1H, 5-HAr), 7.42 (s, 4H, 2 x SO2NH2). 13C NMR (75 MHz, DMSO-d6) δ ppm 139.8, 138.9, 132.6, 132.0, 130.1, 129.9, 129.3, 121.2, 119.5. LC/MS (ESI+): m/z [M + H]+ 337.8. Anal. calcd for C9H9ClN4O4S2 (336.78): C, 32.10; H, 2.69; N, 16.64; S, 19.04; found: C, 32.07; H, 2.69; N, 16.66; S, 19.04.
1-(2-Chloro-5-sulfamoylphenyl)-3,5-dimethyl-1H-pyrazole-4-sulfonamide (9q)
Prepared from 6t according to GP2; white solid, m.p. 230–232 °C (AcOEt), yield 41%. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.00 (dd, J3–4 = 8.3 Hz, J4–6 = 1.7 Hz, 1H, 4-HAr), 7.94 (d, J3–4 = 8.3 Hz, 1H, 3-HAr), 7.91 (d, J4–6 = 1.7 Hz, 1H, 6-HAr), 7.57 (s, 2H, SO2NH2), 7.28 (s, 2H, SO2NH2), 2.38 (s, 3H, CH3), 2.26 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 148.1, 144.6, 142.5, 136.3, 135.0, 131.8, 128.9, 127.7, 121.8, 13.4, 11.3. LC/MS (ESI+): m/z [M + H]+ 365.8. Anal. calcd for C11H13ClN4O4S2 (264.83): C, 36.21; H, 3.59; N, 15.36; S, 17.58; found: C, 36.15; H, 3.60; N, 15.29; S, 13.60.
1-(2-Chloro-5-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9r)
Prepared from 6s according to GP2; white solid, m.p. 235–237 °C (AcOEt), yield 31%. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.71 (s, 1H, Hpyrazole), 8.10 (s, 1H, Hpyrazole), 8.00 (m, 1H, HAr), 7.94 (m, 2H, HAr), 7.39 (s, 4H, 2 x SO2NH2). 13C NMR (75 MHz, DMSO-d6) δ ppm 140.1, 139.4, 132.8, 132.4, 130.1, 130.1, 128.9, 120.9, 118.9. LC/MS (ESI+): m/z [M + H]+ 337.8. Anal. calcd for C9H9ClN4O4S2 (336.78): C, 32.10; H, 2.69; N, 16.64; S, 19.04; found: C, 32.05; H, 2.70; N, 16.69; S, 19.05.
General procedure 3 (GP3): preparation of mono-sulfonamides 7–8 requiring interim chromatographic separation of regioisomeric mono-sulfonyl chlorides 10–11
The procedure is analogous to GP1 except for after evaporation of chloroform, the mixture of regioisomeric mono-sulfonyl chlorides was fractionated on silica gel using an appropriate gradient of ethyl acetate in hexanes as eluent, fractions containing different isomers of mono-sulfonylchlorides were pooled separately, concentrated in vacuo and then, also separately, converted to respective mono-sulfonamides (by treatment with 25% aqueous ammonia) which were characterized.
4-(3,5-Dimethyl-1H-pyrazol-1-yl)-2-methylbenzenesulfonamide (7c)
Prepared (along with 8d) from 6f according to GP3; white solid, m.p. 194–196 °C (AcOEt), yield 35%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.92 (d, J5–6 = 8.2 Hz, 1H, 6-HAr), 7.55 (d, J3–5 = 2.4 Hz, 1H, 3-HA), 7.50 (dd, J5–6 = 8.2 Hz, J3–5 = 2.4 Hz, 1H, 5-HAr), 7.45 (br. s., 2H, SO2NH2), 6.11 (s, 1H, Hpyrazole), 2.64 (s, 3 H, CH3), 2.35 (s, 3H, CH3), 2.18 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 149.1, 142.4, 140.5, 140.0, 137.8, 128.6, 127.0, 120.9, 108.5, 20.2, 13.7, 12.8. LC/MS (ESI+): m/z [M + H]+ 266.3. Anal. calcd for C12H15N3O2S (265.34): C, 54.32; H, 5.70; N, 15.84; S, 12.08; found: C, 54.30; H, 5.70; N, 15.81; S, 12.10.
3-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-methylbenzenesulfonamide (7e)
Prepared (along with 8e) from 6h according to GP3; white solid, m.p. 112–114 °C (i-PrOH), yield 33%; 1H NMR (300 MHz, DMSO-d6) δ ppm 7.89 (dd, J5–6 = 8.0 Hz, J2–6 = 1.5 Hz, 1H, 6-HAr), 7.67 (d, J2–6 = 1.5 Hz, 1H, 2-HAr), 7.65 (d, J5–6 = 7.8 Hz, 1H, 5-HAr), 7.44 (br. s., 2H, SO2NH2), 7.24 (s, 2H, SO2NH2), 2.38 (s, 3H, CH3), 2.23 (s, 3H, CH3), 2.08 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.4, 143.3, 141.8, 140.2, 137.6, 132.3, 127.0, 125.5, 121.3, 17.3, 13.4, 11.3. LC/MS (ESI+): m/z [M + H]+ 266.3. Anal. calcd for C12H15N3O2S (265.34): C, 54.32; H, 5.70; N, 15.84; S, 12.08; found: C, 54.27; H, 5.70; N, 15.86; S, 12.09.
2-Chloro-4-(1H-pyrazol-1-yl)benzenesulfonamide (7m)
Prepared (along with 8h) from 6q according to GP3; white solid, m.p. 181–183 °C, yield 17%. 1H NMR (300 MHz, DMSO-d6) δ ppm 8.68 (d, J = 2.31 Hz, 1H, Hpyrazole), 8.15 (d, J3–5 = 2.0 Hz, 1H, 3-HAr), 8.08 (d, J5–6 = 8.7 Hz, 1H, 6-HAr), 8.00 (dd, J5–6 = 8.7 Hz, J3–5 = 2.0 Hz, 1H, 5-HAr), 7.84 (d, J = 1.32 Hz, 1H, Hpyrazole), 7.66 (s, 2H, SO2NH2), 6.63 (m, 1H, Hpyrazole). 13C NMR (75 MHz, DMSO-d6) δ ppm 142.9, 142.8, 138.5, 132.2, 130.9, 129.2, 120.7, 116.8, 109.6. LC/MS (ESI+): m/z [M + H]+ 258.7. Anal. calcd for C9H8ClN3O2S (257.70): C, 41.95; H, 3.13; N, 16.31; S, 12.44; found: C, 41.88; H, 3.13; N, 16.29; S, 12.46.
3,5-Dimethyl-1-(3-methylphenyl)-1H-pyrazole-4-sulfonamide (8d)
Prepared (along with 7c) from 6f according to GP3; white solid, m.p. 174–176 °C (AcOEt), yield 34%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.42 (t, J = 7.9 Hz, 1H, HAr), 7.28 (m, 3H, HAr), 7.22 (s, 2H, SO2NH2), 2.40 (s, 3H, CH3), 2.38 (s, 3H, CH3), 2.35 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.1, 140.7, 139.5, 138.8, 129.6, 129.5, 126.4, 122.9, 121.8, 21.2, 13.4, 11.9. LC/MS (ESI+): m/z [M + H]+ 266.3. Anal. calcd for C12H15N3O2S (265.34): C, 54.32; H, 5.70; N, 15.84; S, 12.08; found: C, 54.28; H, 5.70; N, 15.79; S, 12.09.
3,5-Dimethyl-1-(2-methylphenyl)-1H-pyrazole-4-sulfonamide (8e)
Prepared (along with 7e) from 6h according to GP3; white solid, m.p. 161–163°C, yield 88%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.45 (m, 2H, HAr), 7.37 (t, J = 7.0 Hz, 1H, HAr), 7.26 (d, J = 7.8 Hz, 1H, HAr), 7.21 (s, 1H, SO2NH2), 2.35 (s, 3H, CH3), 2.17 (s, 3H, CH3), 1.96 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ ppm 146.8, 141.5, 137.6, 135.9, 131.7, 130.2, 128.2, 127.4, 120.7, 17.2, 13.5, 11.3. LC/MS (ESI+): m/z [M + H]+ 266.3. Anal. calcd for C12H15N3O2S (265.34): C, 54.32; H, 5.70; N, 15.84; S, 12.08; found: C, 54.26; H, 5.70; N, 15.81; S, 12.08.
(3-Chlorophenyl)-1H-pyrazole-4-sulfonamide (8h)
Prepared (along with 7m) from 6q according to GP3; white solid, m.p. 82–83 °C, yield 31%. 1H NMR (300 MHz, DMSO-d6) δ ppm 11.63 (br s, 2H, in exchange, SO2NH2), 8.61 (s, 1H, Hpyrazole), 7.96 (s, 1H, HAr), 7.85 (d, J = 8.9 Hz, 1H, HAr), 7.72 (s, 1H, Hpyrazole), 7.49 (t, J = 8.1 Hz, 1H, HAr), 7.34 (d, J = 7.9 Hz, 1H, HAr); 13C NMR (75 MHz, DMSO-d6) δ ppm 141.0, 139.8, 134.4, 133.5, 131.6, 126.8, 126.5, 118.6, 117.3. LC/MS (ESI+): m/z [M + H]+ 258.7. Anal. calcd for C9H8ClN3O2S (257.70): C, 41.95; H, 3.13; N, 16.31; S, 12.44; found: C, 41.91; H, 3.13; N, 16.34; S, 12.44.
General procedure 4 (GP4): preparation of bis-sulfonamides 9 requiring interim chromatographic separation of regioisomeric bis-sulfonyl chlorides 12
The procedure is analogous to GP2 except for after evaporation of chloroform, the mixture of regioisomeric bis-sulfonyl chlorides was fractionated on silica gel using an appropriate gradient of ethyl acetate in hexanes as eluent, fractions containing different isomers of bis-sulfonylchlorides were pooled separately, concentrated in vacuo and then, also separately, converted to respective bis-sulfonamides (by treatment with 25% aqueous ammonia) which were characterized.
3,5-Dimethyl-1-(3-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9a)
Prepared (along with 9b) from 6b according to GP4; white solid, m.p. 223–225 °C (i-PrOH), yield 89%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.90 (m, 2H, HAr), 7.77 (m, 2H, HAr), 7.53 (s, 2H, SO2NH2), 7.28 (s, 2H, SO2NH2), 2.47 (s, 3H, CH3), 2.38 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.9, 145.8, 141.2, 139.0, 130.7, 128.6, 125.7, 122.9, 122.5, 13.4, 12.0. LC/MS (ESI+): m/z [M + H]+ 331.4. Anal. calcd for C11H14N4O4S2 (330.39): C, 39.99; H, 4.27; N, 16.96; S, 19.41; found: C, 39.97; H, 4.27; N, 17.00; S, 19.43.
3,5-Dimethyl-1-(4-sulfamoylphenyl)-1H-pyrazole-4-sulfonamide (9b)
Prepared (along with 9a) from 6b according to GP4; white solid, m.p. 272–274 °C (i-PrOH), yield 92%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.98 (d, J = 8.6 Hz, 2H, HAr), 7.74 (d, J = 8.6 Hz, 2H, HAr), 7.52 (s, 2H, SO2NH2), 7.30 (s, 2H, SO2NH2), 2.51 (s, 3H, CH3), 2.38 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 147.9, 144.0, 141.2, 127.3, 125.9, 122.6, 13.5, 12.1. LC/MS (ESI+): m/z [M + H]+ 331.3. Anal. calcd for C11H14N4O4S2 (330.39): C, 39.99; H, 4.27; N, 16.96; S, 19.41; found: C, 39.93; H, 4.28; N, 17.00; S, 19.42.
4-(3,5-Dimethyl-1H-pyrazol-1-yl)benzenesulfonamide (7o)
A mixture of 4-hydrazinobenzene sulfonamide hydrochloride (10.0 mmol) and acetylacetone (10 mmol) in ethanol (10 ml) was heated at reflux for 90 min and then cooled down to 0 °C. The precipitate formed was collected by filtration and washed with cold ethanol (5 ml). Crystallization from EtOH afforded the title compound in 82% yield.
1H NMR (400 MHz, DMSO-d6) δ ppm 7.91 (d, J = 8.6 Hz, 2H, HAr), 7.72 (d, J = 8.6 Hz, 2H, HAr), 7.43 (br. s., 2H, SO2NH2), 6.13 (s, 1H, Hpyrazole), 2.36 (s, 3H, CH3), 2.19 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ ppm 149.3, 142.3, 140.2, 127.2, 124.0, 108.7, 13.7, 12.8. LC/MS (ESI+): m/z [M + H]+ 252.3. Anal. calcd for C11H13N3O2S (251.31): C, 52.57; H, 5.21; N, 16.72; S, 12.76; found: C, 52.55; H, 5.21; N, 16.70; S, 12.77.
Docking studies
The crystal structure of hCA I (pdb code 1AZM11), hCA II (pdb code 2AW112), hCA IV (pdb code 1ZNC13), and hCA VII (pdb code 3ML514) was taken from the Protein Data Bank15. After adding hydrogen atoms and removing complexed ligands, the four proteins were minimized using Amber 14 software16 and parm03 force field at 300 K. The four proteins were placed in a rectangular parallelepiped water box, an explicit solvent model for water, TIP3P, was used and the complexes were solvated with a 20 Å water cap. Sodium ions were added as counter ions to neutralize the system. Two steps of minimization were then carried out; in the first stage, we kept the protein fixed with a position restraint of 500 kcal/mol Å2 and we solely minimized the positions of the water molecules. In the second stage, we minimized the entire system through 5000 steps of steepest descent followed by conjugate gradient (CG) until a convergence of 0.05 kcal/Å•mol. Automated docking was carried out by means of the AUTODOCK 4.2 program17 using the improved force field18. Autodock Tools was used in order to identify the torsion angles in the ligand, add the solvent model and assign the Kollman atomic charges to the protein. The ligand charge was calculated using the Gasteiger method. The sulfonamide group involved in the interaction with the Zinc ion was considered as deprotonated, as reported in literature19,20. A grid spacing of 0.375 Å and a distance-dependent function of the dielectric constant were used for the energetic map calculations. Using the Lamarckian Genetic Algorithm, the docked compounds were subjected to 100 runs of the Autodock search, using 500,000 steps of energy evaluation and the default values of the other parameters. Cluster analysis was performed on the results using an RMS tolerance of 2.0 Å and the best docked conformations were taken into account.
Carbonic anhydrase inhibition assay
An applied photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity21. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Tris (pH 8.3) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.005 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlier, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house22–25.
Results and discussion
Compound synthesis
In order to create and investigate a set of the required compounds for this study, we began by performing synthesis of mono-sulfonamides 7a–n or 8a–i as well as of bis-sulfonamides 9a–s by direct sulfochlorination of a large set of N-arylpyrazoles 6a–t (all of which are known compounds and/or can be prepared according to straightforward technique26), followed by conversion of the respective mono-and bis-sulfochlorites 9, 10 and 11 on treatment with aqueous ammonia (Scheme 1).
Scheme 1.
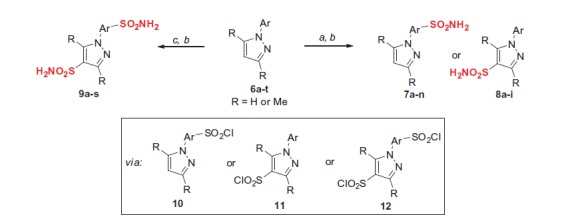
Mono- and bis-sulfonamide synthesis via direct sulfochlorination of 6a–t. Reagents and conditions: (a) ClSO3H (10 equiv.), SOCl2 (1.1 equiv.), 10–70 °C, 1–24 h; (b) aq. NH3 (20 equiv.), acetone, 50 °C, 1 h; (c) ClSO3H (20 equiv.), SOCl2 (2.2 equiv.), 70–120 °C, 7–48 h.
The regiospecificity of the sulfochlorination was unequivocally established for every substrate 6a–t by means for correlational NOESY spectroscopy (ESI) and is depicted in Figure 4 in a straightforward fashion.
On close observation, it is the relative stereoelectronic character of the phenyl vs. the pyrazole unit that governed the direction of the first sulfochlorination. With some exceptions, dimethylpyrazole unit was the first affected with electron-neutral or moderately electron-rich aryls. There are a few mixed situations (6f and 6h). Clearly, introduction of an anisyl group swayed the sulfochlorination completely to that group and made it impossible to produce bis-sulfonamides from those compounds (leading to only tar formation when attempting (6i–k, 6m). Some compounds (6f, 6h, 6q) allowed making mono-sulfonamides at both the phenyl and the pyrazoles portions of the molecule, thereby contributing even more to the diversity of this hCA-probing set compounds. Altogether, to the best of our knowledge, the one presented in Figure 4 is the most comprehensive mono- and bis-sulfochlorination match presented to-date. Of course, some “arrows” require rather mild conditions to realize; others – a lot more forcing conditions, particularly when it comes to achieving the second sulfochlorination. For clarity of the presentation in Figure 4, the reader is referred to Tables 1–3 and the ESI for specific reaction times and temperatures.
Compound 7o which constitutes an important SAR point could not be prepared as described in Scheme 1, was prepared by a direct route26 shown in Scheme 2.
Scheme 2.
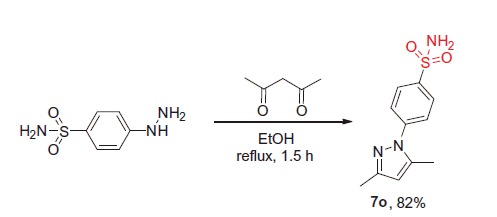
Preparation of compound 7o.
Biological activity
The inhibitory profile obtained for mono-sulfonamides 7a–o in a stopped-flow kinetics assay against human CA I, II, IV and VII is shown in Table 1.
Several observations emerge from the data in Table 1. Clearly, some respectable hCA II levels are achievable. Compounds 7c and 7o are distinctly acetazolamide-like. The difficult-to-inhibit hCA IV is not giving high inhibition results throughout, considering the “detrimental methoxy” phenomenon present in 7g–7l (previously noted by us10 and tentatively justified). When it comes to hCA IV isoform, some striking restoration of potency is observed in 7b (on top of a high selectivity) and, particularly, in 7n (where overall selectivity is not that good). The hCA VII selectivity of compound 7j is also quite notable (and was not ablated, in this case, by the “detrimental methoxy” phenomenon). Altogether, from this set alone, compounds 7b and 7j can be developed as selective probes for hCA IV and VII, respectively.
The set of compounds presented in Table 2 is marked by a virtual absence of hCA IV activity. The compounds can be regarded as weaker, nonselective analogues of acetazolamide and are primarily of interest as a reference set to compare with the bis-sufonamide set discussed below.
Among bis-sulfonamides 9a–r, several instances of restoring specific inhibitory potencies (compared to the respective mono-sulfonamide parts 7 or 8) can be noted, which is suggestive of a possibility of alternative binding mode compared to either 7 or 8. Most notable example is provided by compound 9l whose analogue 7k was inactive throughout the panel (most likely, due to the “detrimental methoxy” effect noted earlier). However, the potency is restored against three isoforms in 9l, which is indicative of the inhibitor’s binding to the target at the pyrazole sulfonamide portion.
Notable examples of isoform selectivity identified within 9a–r set include: 9d (selective hCA IV inhibitor); 9h (selective inhibitor of hCA I) which, cf.7f, demonstrates the power of an additional sulfonamide in ablating activity against all other isoforms; 9q (selective hCA II inhibitor).
In silico modelling
In order to identify the possible binding mode of the new mono- and bis-sulfonamides disclosed herein and also rationalize the SAR trends observed, representative compounds were docked into the hCA I, II, IV and VII X-ray structures. Figure 5(A) shows the docking of compound 7b into the active site of hCA IV. The sulfonamide group acts as a zinc binding group (ZBG) and forms hydrogen bonds with the protein backbone and the hydroxy group of T225; the phenyl ring does not show important lipophilic interactions whereas the methyl substituent in inserted into a lipophilic cleft mainly delineated by V142, I163, L224 and V233. With regards to the pyrazole ring, it points towards H88 and shows an H-bond with T226. The docking analysis of this compound into hCA I, II and VII highlights a completely different binding mode for these three enzymes. As shown in Figure 6, in all three cases the pyrazole ring points towards the entrance of the binding site and the phenyl ring shows lipophilic interactions with V121, V142 and L197 (hCA II numbering). The sulfonamide group acts as a ZBG with an uncommon coordination, with one of the two oxygens that coordinates the zinc ion and the nitrogen that forms an H-bond T198. This different binding disposition is in agreement with the selectivity profile of this compound (potent inhibition of hCA IV with virtually no activity against the other three isoforms) and could be due to small differences in the lipophilic cleft in which the methyl group interacts in hCA IV, as this enzyme exhibits the non-conserved I163 that is substituted in the other three CA subtypes by a Leucine residue. The substitution of the methyl with a methoxy substituent in the benzene ring (as in compound 7h) triggers the loss of hCA IV inhibition activity. As shown in Figure 5(B), the methoxy group is not able to interact into the lipophilic cleft mainly delineated by V142, I163, L224 and V233 and for this reason this compound shows a binding disposition very similar to that observed for compound 7b into hCA I, II and VII (Figure 6). The pyrazole ring points towards the entrance of the binding site, the phenyl ring shows lipophilic interactions with V142, I163, V165 and L224 whereas one of the two oxygens of the sulfonamide group coordinates the zinc ion and the amide nitrogen forms an H-bond T225.
Figure 5.

Docking of compound 7b (A) and 7h (B) into hCA IV.
Figure 6.

Docking of compound 7b into hCA I (A), hCA II (B) and hCA VII (C).
Compound 8h highlights a good hCA I, II and VII inhibition activity with selectivity against hCA IV. Docking studies suggests that this compound interacts with a similar binding disposition into the four different CA subtypes. The sulfonamide group acts as the ZBG and forms hydrogen bonds with the protein backbone and the hydroxy group of T198 (hCA II numbering), the pyrazole ring shows a lipophilic interaction with the conserved L197 whereas the chlorophenyl group shows lipophilic interactions with the conserved V121, L140 and L197. Furthermore, this aromatic ring shows a lipophilic interaction with F130 (L132 for hCA I and F133 for hCA VII) that partially occludes the binding site cavity (see Figure 7). In hCA IV this residue is substituted by an asparagine residue and corresponds to a region that in hCA IV is far away from the binding site, thus leaving the chlorophenyl ring more exposed to the solvent (Figure 7).
Figure 7.

Docking of compound 8h into hCA I (A), hCA II (B), hCA IV (C) and hCA VII (D).
Finally, the analysis of the docking results for compound 9r suggests that in the four CA subtypes the sulfonamide group attached to the o-chlorophenyl fragment act as the ZBG. The phenylpyrazole portion shows lipophilic interactions with F130, L140, L197, P201 (hCA II numbering), and the sulfonamide group attached to the pyrazole ring forms an H-bond with the oxygen backbone of P200 (Figure 8).
Figure 8.

Docking of compound 9r into hCA II.
Conclusions
In this work, we systematically harnessed the power of direct sulfochlorination of a series of known, diversely substituted N-arylpyrazole to arrive at three distinct series of compounds. In each series, SAR generalizations have been made and a number of selective compounds (working against only one target in the panel of four or having high selectivity indices) have been identified. The observed selectivity patterns have been rationalized by modelling. The compounds thus identified can serve as isoform-selective tool inhibitors to probe for cellular processes and their linkage to particular hCA isoforms.
Funding Statement
This research was supported by the Russian Scientific Foundation (project grant 14-50-00069).
Acknowledgement
This research was supported by the Russian Scientific Foundation (project grant 14-50-00069).
Disclosure ststement
No potential conflict of interest was reported by the authors.
References
- 1.Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. [DOI] [PubMed] [Google Scholar]
- 2.http://www.clinicaltrials.gov [published 11 August 2014; last accessed 20 May 2017]. [Google Scholar]
- 3.Zhang Z.PCT Int. Appl. 2017004543. Chem Abstr 2017;166:134991. [Google Scholar]
- 4.Supuran CT.Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. [DOI] [PubMed] [Google Scholar]
- 5.Bowes J, Brown AJ, Hamon J, et al. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov 2012;11:909–22. [DOI] [PubMed] [Google Scholar]
- 6.McDonald PC, Winum JY, Supuran CT, Dedhar S.. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012;3:84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalinin S, Supuran CT, Krasavin M.. Multicomponent chemistry in the synthesis of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2016;31:185–219. [DOI] [PubMed] [Google Scholar]
- 8.Marini AM, Maresca A, Aggarwal M, et al. Tricyclic sulfonamides incorporating benzothiopyrano[4,3-c]pyrazole and pyridothiopyrano[4,3-c]pyrazole effectively inhibit α- and β-carbonic anhydrase: X-ray crystallography and solution investigations on 15 isoforms. J Med Chem 2012;55:9619–29. [DOI] [PubMed] [Google Scholar]
- 9.Krasavin M, Korsakov M, Dorogov M, et al. Probing the ‘bipolar’ nature of the carbonic anhydrase active site: aromatic sulfonamides containing 1,3-oxazol-5-yl moiety as picomolar inhibitors of cytosolic CA I and CA II isoforms. Eur J Med Chem 2015;101:334–47. [DOI] [PubMed] [Google Scholar]
- 10.Krasavin M, Korsakov M, Zvonaryova Z, et al. Human carbonic anhydrase inhibitory profile of mono- and bis-sulfonamides synthesized via a direct sulfochlorination of 3- and 4-(hetero)arylisoxazol-5-amine scaffolds. Bioorg Med Chem 2017;25:1914–25. [DOI] [PubMed] [Google Scholar]
- 11.Chakravarty S, Kannan KK.. Drug-protein interactions. Refined structures of three sulfonamide drug complexes of human carbonic anhydrase I enzyme. J Mol Biol 1994;243:298–309. [DOI] [PubMed] [Google Scholar]
- 12.Di Fiore A, Pedone C, D'Ambrosio K, et al. Carbonic anhydrase inhibitors: valdecoxib binds to a different active site region of the human isoform II as compared to the structurally related cyclooxygenase II “selective” inhibitor celecoxib. Bioorg Med Chem Lett 2006;16:437–42. [DOI] [PubMed] [Google Scholar]
- 13.Stams T, Nair SK, Okuyama T, et al. Crystal structure of the secretory form of membrane-associated human carbonic anhydrase IV at 2.8-A resolution. Proc Natl Acad Sci U S A 1996;93:13589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Fiore A, Truppo E, Supuran CT, et al. Crystal structure of the C183S/C217S mutant of human CA VII in complex with acetazolamide. Bioorg Med Chem Lett 2010;20:5023–6. [DOI] [PubMed] [Google Scholar]
- 15.Berman HM, Battistuz T, Bhat TN, et al. The protein data bank. Acta Crystallogr D Biol Crystallogr 2002;58:899–907. [DOI] [PubMed] [Google Scholar]
- 16.Case DA, Berryman JT, Betz RM, et al. AMBER, version 14. San Francisco, CA: University of California; 2015. [Google Scholar]
- 17.Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santos-Martins D, Forli S, Ramos MJ, Olson AJ.. AutoDock4Zn: an improved AutoDock force field for small-molecule docking to zinc metalloproteins. J Chem Inf Model 2014;54:2371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cappalonga AM, Alexander RS, Christianson DW.. Structural comparison of sulfodiimine and sulfonamide inhibitors in their complexes with zinc enzymes. J Biol Chem 1992;267:19192–7. [DOI] [PubMed] [Google Scholar]
- 20.Tuccinardi T, Nuti E, Ortore G, et al. Analysis of human carbonic anhydrase II: docking reliability and receptor-based 3D-QSAR study. J Chem Inf Model 2007;47:515–25. [DOI] [PubMed] [Google Scholar]
- 21.Khalifah RG.The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73. [PubMed] [Google Scholar]
- 22.Maresca A, Carta F, Vullo D, Supuran CT.. Dithiocarbamates strongly inhibit the β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:407–11. [DOI] [PubMed] [Google Scholar]
- 23.Ekinci D, Kurbanoglu NI, Salamci E, et al. Carbonic anhydrase inhibitors: inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J Enzyme Inhib Med Chem 2012;27:845–8. [DOI] [PubMed] [Google Scholar]
- 24.Ekinci D, Karagoz L, Ekinci D, et al. Carbonic anhydrase inhibitors: in vitro inhibition of α isoforms (hCA I, hCA II, bCA III, hCA IV) by flavonoids. J Enzyme Inhib Med Chem 2013;28:283–8. [DOI] [PubMed] [Google Scholar]
- 25.Alp C, Maresca A, Alp NA, et al. Secondary/tertiary benzenesulfonamides with inhibitory action against the cytosolic human carbonic anhydrase isoforms I and II. J Enzyme Inhib Med Chem 2013;28:294–8. [DOI] [PubMed] [Google Scholar]
- 26.Stanovni B, Svete J.. Pyrazoles. Sci Synth 2002;121:15–225. [Google Scholar]