

Abstract
The development of adenosine A2A receptor antagonists has received much interest in recent years for the treatment of neurodegenerative diseases. Based on docking studies, a new series of 2-arylbenzoxazoles has been identified as potential A2AR antagonists. Structure-affinity relationship was investigated in position 2, 5 and 6 of the benzoxazole heterocycle leading to compounds with a micromolar affinity towards the A2A receptor. Compound F1, with an affinity of 1 μm, presented good absorption, distribution, metabolism and excretion properties with an excellent aqueous solubility (184 μm) without being cytotoxic at 100 μm. This compound, along with low-molecular weight compound D1 (Ki = 10 μm), can be easily modulated and thus considered as relevant starting points for further hit-to-lead optimisation.
Keywords: Benzoxazole, A2A receptor, solubility, neurodegenerative disease
Introduction
Alzheimer’s (AD) and Parkinson’s diseases (PD), currently the most important neurodegenerative pathologies, are characterised by a progressive neuronal death1. Current therapies are restricted to symptomatic interventions and do not prevent progressive neuronal loss. Therefore, novel therapeutic solutions are needed and one of the promising strategies consists of targeting the adenosine A2A receptor.
Epidemiological studies have shown that people consuming regularly over a lifetime caffeine-based beverages are substantially less likely to develop PD and AD2,3. Caffeine exerts its biological effects primarily by antagonising adenosine receptors (GPCRs). The adenosine A2A receptor subtype, one of the four adenosine receptors, was shown in multiple studies to be responsible for the neuroprotective effects of caffeine in experimental models of AD and PD4–6. Besides, many A2A antagonists have been synthesised over the past few years and some showed promising results in managing cognitive dysfunction in both diseases7. For example, antagonists (Figure 1) such as Istradefylline (KW-6002), Preladenant (SCH 42088) and Tozadenant (SYN 115)9 have been investigated clinically in PD with promising results, especially Istradefylline which has been approved in Japan as an adjunct to L-DOPA therapy10,11. Less research work has been undertaken regarding AD but it is now well established that A2A antagonists lead to the improvement of spatial memory accompanied by the decrease of Aβ amyloid burden, Tau hyperphosphorylation and neurotoxicity6,12.
Figure 1.
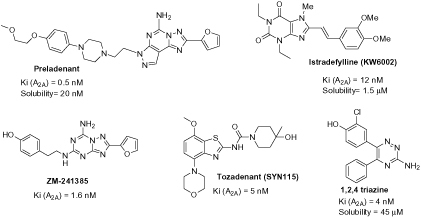
Selective adenosine A2A receptor antagonists.
Therefore, developing A2A antagonists constitutes a promising therapeutic strategy for the treatment of both AD and PD. However, although many antagonists have been developed so far, constant drawbacks remain such as high toxicity and poor solubility7,8,13,14. These important limitations have obstructed the development of drugs targeting this receptor. Therefore, one of the main challenges regarding A2A antagonist development is to improve solubility and lower toxicity while keeping good affinity at A2A receptor. Selectivity parameters are now more debated since studies have highlighted the therapeutic potential of dual A1/A2A antagonists15 as well as a non-selective ligand proven by caffeine, for neurodegenerative disease.
With the aim of developing novel A2A antagonists with good water solubility, we designed a series of benzoxazoles bearing a protonable amine function (Figure 2(A)). We first focused on a diffuse hydrophobic zone located at the top of the active site that is generally occupied by an aryl group in well-known antagonists (Figure 2(B)). Because of the presence of an acidic cluster (Glu169, Asp170) in this pocket, a tertiary amine which can allow for an ionic interaction could be a good alternative to an aryl group (Figure 2(C)). The present work describes the medicinal chemistry approach leading to a series of 2-arylbenzoxazoles which best compounds display micromolar affinity towards the A2A receptor and good water solubility.
Figure 2.

Molecular modelling-guided design. (A) Representation of various modulations around the benzoxazole scaffold. (B) Predicted binding mode of ZM-241385 in the apoA2AR-T4E pocket (dark) compared with the X-ray binding mode (gray). (C) Putative binding mode of compound F1 in the apoA2AR-T4E pocket.
Methods
Chemistry
All reagents and solvents were purchased and used without further purification. Reactions were monitored by TLC performed on MachereyeNagel Alugram® Sil 60/UV254 sheets (thickness 0.2 mm). Some purification of products was carried out by column chromatography using MachereyeNagel silica gel (230e400 mesh). Melting points were determined on a BÜCHI B-540 apparatus and are uncorrected. NMR spectra were recorded on a Bruker Avance 300 spectrometer operating at 300 MHz (1H) or 75 MHz (13 C). Chemical shifts are in parts per million (ppm) and were referenced to the residual proton peaks in deuterated solvents. Mass spectra were recorded with an LCMS (Waters Alliance Micromass ZQ 2000). LCMS analysis was performed using a Waters XBridge C18 column (5 μm particle size column, dimensions 50 × 4.6 mm). A gradient starting from 98% H2O/formate buffer 5 mm (pH 3.8) and reaching 100% CH3CN/formate buffer 5 mm (pH 3.8) within 4 min at a flow rate of 2 ml/min was used followed by a return to the starting conditions within 1 min. FT-IR spectra were recorded on a Thermo Nicolet Avatar 320 FT-IR spectrometer. The purity of final compounds was verified by high-pressure liquid chromatography (HPLC) columns: C4 Interchrom UPTISPHERE. Analytical HPLC was performed on a Shimadzu LC-2010AHT system equipped with a UV detector set at 254 and 215 nm. Compounds were dissolved in 50 ml acetonitrile and 950 ml buffer A and injected into the system. The following eluent systems were used: buffer A (H2O/TFA, 100:0.1) and buffer B (CH3CN/H2O/TFA, 80:20:0.1). HPLC retention times (HPLC tR) were obtained at a flow rate of 0.2 ml/min for 35 min using the following conditions: a gradient run from 100% of buffer A over 1 min, then to 100% of buffer B over the next 30 min.
General procedure for the synthesis of amide (2a–2d)
To a solution of acid (17.8 mmol) in DCM (100 ml) at 0 °C were added thionyl chloride (71.4 mmol) and 5 drops of DMF. This solution was stirred for 3 h at reflux, cooled to room temperature and concentrated in vacuo. The residue was diluted in 50 ml of EtOAc and added to a solution of aminophenol (16.6 mmol) and Et3N (35.6 mmol) in 35 ml of EtOAc at 0 °C. The reaction mixture was stirred at room temperature overnight, hydrolysed with water and extracted twice with EtOAc. An acid–base workup with saturated NaHCO3 and 1 M HCl solution was performed and the organic layer was concentrated in vacuo. The solid was then suspended in a mixture of EtOH/H2O (250 ml/10 ml) and NaOH was added (54 mmol). The reaction mixture was stirred at reflux for 4 h, cooled to room temperature, acidified slowly with 6 M HCl up to acidic pH. Resulting solid was filtered, washed with water and recrystallised in an appropriate solvent.
N-(2-Hydroxy-5-methylphenyl)furan-2-carboxamide (2a): The title compound was prepared from 2-furoic acid and 4-methyl-2-aminophenol to afford 2a as a beige solid (80%): mp 186 °C (acetonitrile). 1H NMR (300 MHz, [D6]DMSO): 9.76 (br s, 1H, OH), 9.09 (br s, 1H, NH), 7.92 (m, 1H, H5'), 7.72 (m, 1H, H6), 7.28 (dd, 1H, H4, J = 2.2 Hz and J = 8.4 Hz), 6.79 (m, 2H, H3 and H3'), 6.70 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 2.21 (s, 3H, CH3). IR (ν, cm−1): 3380 (NH), 2750–3100 (OH), 1640 (C = O). LC-MS (ESI) m/z found: 218 [M + H]+.
N-(2-Hydroxy-5-methylphenyl)-3,4-dimethoxybenzamide (2b): The title compound was prepared from 3,4-dimethoxybenzoic acid and 4-methyl-2-aminophenol to afford 2b as a white solid (82%): mp 164 °C (acetonitrile). 1H NMR (300 MHz, CDCl3): 9.47 (br s, 1H, OH), 9.41 (br s, 1H, NH), 7.61 (dd, 1H, H4, J = 2.0 Hz and J = 8.4 Hz), 7.55 (d, 1H, H6, J = 2.0 Hz), 7.42 (m, 1H, H2'), 7.07 (d, 1H, H3, J = 8.4 Hz), 6.83 (dd, 1H, H6', J = 1.7 Hz and J = 8.3 Hz), 6.79 (d, 1H, H5', J = 8.3 Hz), 3.84 (s, 3H, OMe), 3.82 (s, 3H, OMe), 2.22 (s, 3H, CH3). IR (ν, cm−1): 3368 (NH), 3182–2854 (OH), 1653 (C = O). LC-MS (ESI) m/z found: 288 [M + H]+.
N-(2-Hydroxy-4-methylphenyl)furan-2-carboxamide (2c): The title compound was prepared from furoic acid and 5-methyl-2-aminophenol to afford 2c as a brown solid (72%): mp 170 °C (acetonitrile). 1H NMR (300 MHz, CDCl3): 8.85 (br s, 1H, OH), 8.34 (br s, 1H, NH), 7.50 (m, 1H, H5'), 7.28 (m, 1H, H3'), 7.10 (d, 1H, H6, J = 8.1 Hz), 6.87 (m, 1H, H3), 6.72 (m, 1H, H5), 6.58 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 2.30 (s, 3H, CH3). IR (ν, cm−1): 3388 (NH), 3182–2854 (OH), 1649 (C = O).
N-(2-Hydroxy-5-nitro-phenyl) furan-2-carboxamide (2d): The title compound was prepared from furoic acid and 4-nitro-2-aminophenol to afford 2d as a yellow solid (84%): mp 165 °C (methanol). 1H NMR (300 MHz, CDCl3): 9.77 (br s, 1H, OH), 9.13 (br s, 1H, NH), 8.79 (d, 1H, H6, J = 1.9 Hz), 8.25 (dd, 1H, H4, J = 1.9 Hz and J = 8.6 Hz), 8.07 (m, 1H, H5'), 7.81 (d, 1H, H3', J = 3.5 Hz), 7.32 (d, 1H, H3, J = 8.6 Hz), 6.78 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz). IR (ν, cm−1): 3376 (NH), 3000–2500 (OH), 1640 (C = O).
General procedure for the synthesis of benzoxazole (3a–3d)
A suspension of amide 2 (2.21 mmol) and TsOH (5.53 mmol) was refluxed in toluene (150 ml) equipped with a Dean–Stark apparatus until complete dissolution for 17 h. The solution was then cooled to room temperature, hydrolysed with water and basified with a 6 M solution of NaOH up to basic pH (10–12). The organic layer was separated, dried over K2CO3 and concentrated in vacuo. Solid was suspended from the appropriate solvent and filtered.
2-(Furan-2-yl)-5-methyl-1,3-benzoxazole (3a): The title compound was prepared from amide 2a to afford 3a as a beige solid (82%): mp 64 °C (petroleum ether). 1H NMR (300 MHz, CDCl3): 7.67 (m, 1H, H5'), 7.54 (m, 1H, H3'), 7.44 (d, 1H, H7, J = 8.3 Hz), 7.25 (m, 1H, H4), 7.17 (m, 1H, H6), 6.70 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 2.49 (s, 3H, CH3). LC-MS (ESI) m/z found: 200 [M + H]+.
2-(3,4-Dimethoxyphenyl)-5-methyl-1,3-benzoxazole (3b): The title compound was prepared from amide 2b to afford 3b as a beige solid (80%): mp 136 °C (diethyl ether). 1H NMR (300 MHz, CDCl3): 7.83 (dd, 1H, H6', J = 1.9 Hz and J = 8.4 Hz), 7.75 (d, 1H, H2', J = 1.9 Hz), 7.53 (m, 1H, H6), 7.43 (d, 1H, H5', J = 8.4 Hz), 7.13 (m, 1H, H4), 6.98 (d, 1H, H7, J = 8.4 Hz), 4.02 (s, 3H, OMe), 3.97 (s, 3H, OMe), 2.48 (s, 3H, CH3). LC-MS (ESI) m/z found: 270 [M + H]+.
2-(Furan-2-yl)-6-methyl-1,3-benzoxazole (3c): The title compound was prepared from amide 2c to afford 3c as a beige solid (68%): mp 54 °C (diethyl ether). 1H NMR (300 MHz, CDCl3): 8.04 (m, 1H, H5'), 7.61 (d, 1H, H4, J = 8.1 Hz), 7.40 (d, 1H, H3', J = 3.5 Hz), 7.38 (m, 1H, H7), 7.27–7.18 (m, 1H, H5), 6.79 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 2.44 (s, 3H, CH3). LC-MS (ESI) m/z found: 200 [M + H]+.
2-(Furan-2-yl)-5-nitro-1,3-benzoxazole (3d): The title compound was prepared from amide 2b to afford 3d as a yellow solid (82%): mp 182 °C (diethyl ether). 1H NMR (300 MHz, CDCl3): 8.62 (d, 1H, H4, J = 2.1 Hz), 8.34 (dd, 1H, H6, J = 2.1 Hz and J = 8.4 Hz), 7.75 (d, 1H, H7, J = 8.4 Hz), 7.68 (m, 1H, H5'), 7.39 (d, 1H, H3', J = 3.5 Hz), 6.68 (dd, 1H, H4',J = 1.7 Hz and J = 3.5 Hz). IR (ν, cm−1): 1519 (Ar-NO2). LC-MS (ESI) m/z found: 231 [M + H]+.
Methyl 2-[2-(3,4-dimethoxyphenyl)-1,3-benzoxazol-5-yl] acetate (3e): To a suspension of methyl 2-(3-amino-4-hydroxyphenyl) acetate (1.6 g, 8.83 mmol) in T3P (solution in EtOAc) (4.2 g, 13.3 mmol), were added 3,4-dimethoxybenzoic acid (1.61 g, 8.83 mmol) and DIPEA (1.46 ml, 8.83 mmol). The reaction mixture was heated overnight at 120 °C, cooled to room temperature, suspended in water and extracted three times with EtOAc. Combined organic layers were washed 1 M NaOH solution, dried over MgSO4 and concentrated in vacuo. Solid was recrystallised from EtOH to afford compound (3e) as a beige solid (1.59 g, 55%): mp 110 °C. 1H NMR (300 MHz, CDCl3): 7.82 (dd, 1H, H6', J = 8.4 Hz and J = 1.8 Hz), 7.74 (d, 1H, H4, J = 1.8 Hz), 7.64 (m, 1H, H2'), 7.49 (d, 1H, H7, J = 8.4 Hz), 7.24 (dd, 1H, H6, J = 8.4 Hz and J = 1.8 Hz), 6.98 (d, 1H, H5', J = 8.4 Hz), 4.01 (s, 3H, OMe), 3.95 (s, 3H, OMe), 3.75 (s, 2H, CH2), 3.70 (s, 3H, CO2Me). IR (ν, cm−1): 1730 (ester C = O). LC-MS (ESI) m/z found: 328 [M + H]+.
General procedure for the synthesis of compound (4a–4c)
To a solution of compound (3a–3c) (20.1 mmol) in CCl4 (150 ml) was added N-bromosuccinimide (NBS) (24.1 mmol) and benzoyl peroxide (1.41 mmol) and the reaction mixture was refluxed under a halogen lamp (230 W). After 3 h and 30 min stirring, the mixture was cooled to room temperature and the succinimide was filtered off. Then, the solution was concentrated in vacuo, and the solid was suspended in diethyl ether and filtered.
5-(Bromomethyl)-2-(furan-2-yl)-1,3-benzoxazole (4a): The title compound was prepared from benzoxazole 3a to afford 4a as a beige solid (70%): mp 130 °C. 1H NMR (300 MHz, CDCl3): 7.77 (m, 1H, H5'), 7.67 (m, 1H, H3'), 7.54 (d, 1H, H7, J = 8.4 Hz), 7.42 (dd, 1H, H6, J = 1.8 Hz and J = 8.4 Hz), 7.30 (m, 1H, H4), 6.64 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 4.64 (s, 2H, CH2). LC-MS (ESI) m/z found: 278 [M + H]+, 280 [M + H]+.
6-(Bromomethyl)-2-(furan-2-yl)-1,3-benzoxazole (4b): The title compound was prepared from benzoxazole 3b to afford 4b as a brown solid (65%): mp 122 °C, 1H NMR (300 MHz, CDCl3): 7.72–7.68 (m, 2H, H5' and H4), 7.60 (m, 1H, H7'), 7.40 (dd, 1H, H5, J = 8.2 Hz and J = 1.6 Hz), 7.30 (dd, 1H, H3', J = 0.7 Hz and J = 3.5 Hz), 6.63 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 4.64 (s, 2H, CH2). IR (ν, cm−1): 750 (C-Br).
5-(Bromomethyl)-2-(3,4-dimethoxyphenyl)-1,3-benzoxazole (4c): The title compound was prepared from benzoxazole 3c to afford 4c as a beige solid (75%): mp 140 °C. 1H NMR (300 MHz, CDCl3): 7.86 (dd, 1H, H6', J = 1.9 Hz and J = 8.4 Hz), 7.76 (m, 2H, H4 and H2'), 7.53 (d, 1H, H7 or H5', J = 8.5 Hz), 7.39 (dd, 1H, H6, J = 1.7 Hz and J = 8.4 Hz), 7.00 (d, 1H, H7 or H5', J = 8.4 Hz), 4.65 (s, 2H, CH2), 4.02 (s, 3H, OMe), 3.99 (s, 3H, OMe). IR (ν, cm−1): 750 (C-Br).
General procedure for the synthesis of compound (A1–A3 and A5–A8)
To a solution of amine (2.05 mmol) in acetone (15 ml) were added compound (4a–4c) (1.87 mmol) and Et3N (2.05 mmol). The reaction mixture was refluxed for 1 h, cooled to room temperature and concentrated in vacuo. Solid was suspended in water and extracted three times with EtOAc. Combined organic layers were dried over MgSO4 and concentrated in vacuo to give a solid which was then purified.
2-(Furan-2-yl)-5-(piperidin-1-ylmethyl)-1,3-benzoxazole hydrochloride (A1): The title compound was prepared from compound 4a and piperidine. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from acetonitrile to afford A1 as a white solid (74%): mp >300 °C. 1H NMR (300 MHz, [D6]DMSO): 12.43 (br s, 1H, NH+), 8.02 (m, 1H, H6), 7.75 (m, 1H, H5'), 7.70 (m, 1H, H4), 7.66 (d, 1H, H7, J = 8.1 Hz), 7.32 (d, 1H, H3', J = 3.5 Hz), 6.65 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 4.26 (d, 2H, CH2,J = 5.0 Hz), 3.47 (m, 2H, Hpiperidine), 2.61 (m, 2H, Hpiperidine), 2.33 (m, 2H, Hpiperidine), 1.92–1.80 (m, 3H, Hpiperidine), 1.35 (m, 1H, Hpiperidine). 13C NMR (75 MHz, [D6]DMSO): 156.5 (C), 151.0 (C), 146.3 (CH), 142.1 (C), 142.0 (C), 129.1 (CH), 125.1 (C), 122.9 (CH), 115.3 (CH), 112.5 (CH), 111.8 (CH), 60.9 (CH2), 52.3 (2 CH2), 22.5 (2 CH2), 22.1 (CH2). LC-MS (ESI) m/z found: 283 [M + H]+. HPLC: C4 column: tR = 16.2 min, purity 98%.
2-(Furan-2-yl)-5-[(4-phenylepiperazin-1-yl)methyl]-1,3-benzoxazole hydrochloride (A2): The title compound was prepared from compound 4a and phenylpiperazine. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from ethanol to afford A2 as a white solid (65%): mp >300 °C. 1H NMR (300 MHz, [D6]DMSO): 13.12 (br s, 1H, NH+), 8.04 (m, 1H, H5'), 7.83 (m, 1H, H6), 7.68 (m, 2H), 7.33 (d, 1H, H3',J = 3.5 Hz), 7.30–7.25 (m, 2H), 6.98–6.89 (m, 3H), 6.66 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 4.37 (s, 2H, CH2), 3.75–3.52 (m, 6H, Hpiperazine), 3.06 (m, 2H, Hpiperazine). 13C NMR (75 MHz, [D6]DMSO): 156.0 (C), 150.6 (C), 150.0 (C), 148.0 (CH), 141.9 (C), 141.7 (C), 129.6 (2 CH), 129.4 (CH), 127.5 (C), 123.5 (CH), 120.4 (CH), 116.4 (2 CH), 116.2 (CH), 113.4 (CH), 111.6 (CH), 58.7 (CH2), 50.6 (2 CH2), 45.7 (2 CH2). LC-MS (ESI) m/z Found: 360 [M + H]+. HPLC: C4 column: tR = 15.5 min, purity >99%.tert-Butyl4-{[2-(Furan-2-yl)-1,3-benzoxazol-5-yl]methyl}piperazine-1-carboxylate (A3): The title compound was prepared from compound 4a and boc-piperazine. Solid was recrystallised from methanol to afford A3 as a white solid (83%): mp 128 °C (methanol). 1H NMR (300 MHz, CDCl3): 7.69 (m, 1H, H5'), 7.67 (m, 1H, H4), 7.49 (d, 1H, H7, J = 8.3 Hz), 7.34 (dd, 1H, H6, J = 1.5 Hz and J = 8.3 Hz), 7.26 (m, 1H, H3'), 6.62 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.63 (s, 2H, CH2), 3.44 (t, 4H, Hpiperazine, J = 4.9 Hz), 2.42 (t, 4H, Hpiperazine, J = 3.5 Hz), 1.45 (s, 9H, (3 CH3). NMR 13C (CDCl3, δ ppm): 163.0 (C), 155.6 (C), 154.0 (C), 149.8 (C), 147.7 (C), 141.9 (C), 141.8 (CH), 132.2 (CH), 127.9 (C), 121.5 (CH), 115.8 (CH), 113.3 (CH), 111.2 (CH), 79.6 (2 CH2), 60.8 (CH2), 51.9 (2 CH2), 26.5 (3 CH3). IR (ν, cm−1): 1679 (C = O). LC-MS (ESI) m/z found: 328 [M-tBu + H]+, 284 [M + H]+. HPLC: C4 column: tR = 14.7 min, purity >99%.
4-(4-{[2-(Furan-2-yl)-1,3-benzoxazol-5-yl]methyl}piperazin-1-yl)phenol (A5): The title compound was prepared from compound 4a and 4-piperazinephenol. Solid was purified by flash chromatography using DCM/EtOAc (9/1) to afford A5 as a white solid (83%): mp 212 °C. 1H NMR (300 MHz, [D6]DMSO): 8.79 (br s, 1H, OH), 8.07 (m, 1H, H5'), 7.71 (m, 2H, H7 and H4), 7.46 (d, 1H, H3', J = 3.5 Hz), 7.41 (dd, 1H, H6, J = 1.2 Hz and J = 8.4 Hz), 6.82 (dd, 1H, H4', J = 1.8 Hz and J = 3.5 Hz), 6.78 (d, 2H, Hphenyl, J = 8.7 Hz), 6.63 (d, 2H, Hphenyl, J = 8.7 Hz), 3.64 (s, 2H, CH2), 2.96 (m, 4H, Hpiperazine), 2.55 (m, 4H, Hpiperazine). 13C NMR (75 MHz, [D6]DMSO): 155.4 (C), 151.3 (C), 149.2 (C), 147.6 (CH), 144.7 (C), 142.1 (C), 141.7 (C), 135.9 (C), 126.9 (CH), 120.2 (CH), 118.2 (2 CH), 115.9 (2 CH), 115.5 (CH), 113.3 (CH), 110.8 (CH), 62.2 (CH2), 53.2 (2 CH2), 50.5 (2 CH2). IR (ν, cm−1): 3195–2780 (OH). LC-MS (ESI) m/z found: 376 [M + H]+. HPLC: C4 column: tR = 10.2 min, purity 99% C18 column: tR = 14.3 min, purity >99%.
2-(Furan-2-yl)-5-({4-[4-(2-methoxyethoxy)phenyl]piperazin-1-yl}methyl)-1,3-benzoxazole (A6): The title compound was prepared from compound 4a and 1-[4-(2-methoxyethoxy)phenyl]piperazine. Solid was recrystallised from ethanol to afford A6 as a white solid (33%): mp 123 °C. RMN 1H (300 MHz, [D6]DMSO): 8.08 (m, 1H, H5'), 7.72 (m, 2H, H7 and H4), 7.46 (dd, 1H, H3', J = 0.7 Hz and J = 3.5 Hz), 7.40 (dd, 1H, H6, J = 1.4 Hz and J = 8.4 Hz), 6.88–6.79 (m, 5H, H4', Hphenyl), 3.99 (m, 2H, OCH2), 3.64 (s, 2H, CH2), 3.61 (m, 2H, CH2O), 3.29 (s, 3H, OCH3), 3.01 (m, 4H, Hpiperazine), 2.54 (m, 4H, Hpiperazine). RMN 13C (75 MHz, [D6]DMSO): 155.4 (C), 152.5 (C), 149.2 (CH), 147.5 (C), 145.9 (C), 142.1 (C), 141.7 (C), 135.9 (C), 126.1 (C), 119.9 (CH), 118.1 (2 CH), 115.4 (2 CH), 114.1 (CH), 112.3 (CH), 110.2 (CH), 71.2 (CH2), 67.7 (CH2), 60.8 (CH2), 53.4 (2 CH2), 50.6 (2 CH2), 33.7 (CH3). LC-MS (ESI) m/z found: 434 [M + H]+. HPLC: C4 column: tR = 16.1 min, purity >99%.
2-(Furan-2-yl)-6-(piperidin-1-ylmethyl)-1,3-benzoxazole hydrochloride (A7): The title compound was prepared from compound 4c and piperidine. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from acetonitrile to afford A7 (276 mg, 68%): mp >300 °C. 1H NMR (300 MHz, [D6]DMSO): 11.08 (br s, 1H, NH+), 8.16–8.10 (m, 2H, H7 and H5'), 7.82 (d, 1H, H4, J = 8.1 Hz), 7.63 (m, 1H, H5), 7.51 (d, 1H, H3', J = 3.2 Hz), 6.83 (m, 1H, H4'), 4.38 (m, 2H, CH2), 3.28 (m, 2H, Hpiperidine), 2.83 (m, 2H, Hpiperidine), 1.76–1.66 (m, 5H, Hpiperidine), 1.35 (m, 1H, Hpiperidine). 13C NMR (75 MHz, [D6]DMSO): 156.1 (C), 149.9 (C), 148.0 (CH), 142.4 (C), 141.7 (C), 129.0 (C), 127.8 (CH), 120.1 (CH), 116.3 (CH), 114.4 (CH), 113.4 (CH), 59.1 (CH2), 52.0 (2 CH2), 22.6 (2 CH2), 21.9 (CH2). LC-MS (ESI) m/z found: 283 [M + H]+. HPLC: C4 column: tR = 15.7 min, purity 96%.
2-(3,4-Dimethoxyphenyl)-5-(piperidin-1-ylmethyl)-1,3-benzoxazole hydrochloride (A8): The title compound was prepared from compound 4b and piperidine. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from acetonitrile to afford A8 as a white solid (200 mg, 60%): mp >300 °C, 1H NMR (300 MHz, [D6]DMSO): 12.31 (br s, 1H, NH+), 7.90 (dd, 1H, H6', J = 1.6 Hz and J = 8.4 Hz), 7.82 (dd, 1H, H6, J = 1.9 Hz and J = 8.4 Hz), 7.78 (d, 1H, H2', J = 1.6 Hz), 7.71 (d, 1H, H4, J = 1.9 Hz), 7.62 (d, 1H, H5', J = 8.4 Hz), 6.97 (d, 1H, H7, J = 8.4 Hz), 4.27 (d, 2H, CH2, J = 5.1 Hz), 3.99 (s, 3H, OMe), 3.96 (s, 3H, OMe), 3.45 (m, 2H, Hpiperidine), 2.64 (m, 2H, Hpiperidine), 2.34 (m, 2H, Hpiperidine), 1.86 (m, 3H, Hpiperidine), 1.35 (m, 1H, Hpiperidine). 13C NMR (75 MHz, [D6]DMSO): δ 164.4 (C), 152.4 (C), 151.5 (C), 149.3 (C), 142.6 (C), 128.4 (CH), 124.7 (C), 122.5 (CH), 121.5 (C), 119.0 (CH), 111.5 (CH), 111.1 (CH), 110.1 (CH), 60.9 (CH2), 56.1 (CH3), 56.1 (CH3), 52.6 (2 CH2), 22.5 (2 CH2), 22.0 (CH2). LC-MS (ESI) m/z found: 353 [M + H]+. HPLC: C4 column: tR = 14.3 min, purity 99%.
Synthesis of 2-(Furan-2-yl)-5-(piperazin-1-ylmethyl)-1,3-benzoxazole (A4): A solution of compound (A3) (100 mg, 0.35 mmol) diluted in 5 ml of DCM with TFA (2 ml, 26 mmol) was stirred for 3 h at room temperature, hydrolysed with water and basified with 1 M solution of NaOH up to basic pH and extracted three times with DCM. Combined organic layers were dried over MgSO4 and concentrated in vacuo. Solid was suspended in diethyl ether with a drop of ethanol and filtered to afford compound (A4) as a beige solid (60%): mp 192 °C. 1H NMR (300 MHz, [D6]DMSO): 8.08 (m, 1H, H5'), 7.74–7.71 (m, 2H, H4 and H7), 7.46 (dd, 1H, H3', J = 0.7 Hz and J = 3.5 Hz), 7.38 (dd, 1H, H6, J = 1.6 Hz and J = 8.3 Hz), 6.83 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.65 (s, 2H, CH2), 3.45–3.39 (m, 4H, Hpiperazine), 2.43 (m, 4H, Hpiperazine). 13C NMR (75 MHz, [D6]DMSO): 155.4 (C), 149.3 (C), 147.6 (CH), 142.0 (C), 141.7 (C), 135.2 (C), 127.0 (CH), 120.3 (CH), 115.6 (CH), 113.3 (CH), 110.9 (CH), 61.9 (CH2), 50.6 (2 CH2), 44.1 (2 CH2). LC-MS (ESI) m/z found: 284 [M + H]+. HPLC: C4 column: tR = 15.8 min, purity 99%.
2-[2-(Furan-2-yl)-1,3-benzoxazol-5-yl]acetonitrile (5): To a solution of 5-(bromomethyl)-2-(3,4-dimethoxyphenyl)-1,3-benzoxazole (4a) (1.23 g, 3.53 mmol) in a mixture of EtOH (56 ml) and H2O (15 ml) was added KCN (1.15 g, 17.7 mmol). After one night stirring at reflux, the reaction mixture was cooled to room temperature and concentrated in vacuo. Solid was suspended in water and extracted three times with EtOAc. Combined organic layer were dried over MgSO4 and concentrated in vacuo. Solid was then recrystallised in methanol to afford (5) as a beige solid (582 mg, 56%): mp 140 °C. 1H NMR (300 MHz, CDCl3): 7.71–7.70 (m, 2H, H5' and H3'), 7.58 (d, 1H, H7, J = 8.3 Hz), 7.36–7.31 (m, 2H, H4 and H6), 6.64 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.90 (s, 2H, CH2). IR (ν, cm−1): 2240 (CN). LC-MS (ESI) m/z found: 225 [M + H]+.
2-[2-(Furan-2-yl)-1,3-benzoxazol-5-yl] acetic acid (6): A solution of compound (5) (1.1 g, 4.91 mmol) in a mixture of AcOH (5.5 ml), H2SO4 (5.5 ml) and H2O (5.5 ml) was refluxed for 17 h. After cooling to room temperature, cold water was added and the solution was extracted with EtOAc. The organic layer was extracted twice with a saturated NaHCO3 solution, and then the aqueous layer was acidified with 1 M HCl solution and extracted twice with EtOAc. Combined organic layer were dried over MgSO4 and concentrated under vacuum. Solid was suspended in diethyl ether and filtered to afford compound (6) as a white solid (702 mg, 60%): mp 203 °C. 1H NMR (300 MHz, CDCl3): 12.38 (br s, 1H, OH), 8.07 (m, 1H, H5'), 7.68 (d, 1H, H7, J = 8.4 Hz), 7.66 (m, 1H, H3'), 7.45 (m, 1H, H4), 7.32 (dd, 1H, H6, J = 1.8 Hz and J = 8.4 Hz), 6.80 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.72 (s, 2H, CH2). IR (ν, cm−1): 1710 (acid C = O). LC-MS (ESI) m/z found: 244 [M + H]+.
General procedure for the synthesis of amide (7a–7d)
To a solution of acid (6) (1.5 mmol) in toluene (4 ml) was added at 0 °C SOCl2 (5.97 mmol). The mixture was refluxed during 1 h, cooled to room temperature and concentrated in vacuo. The oil was then diluted with EtOAc (26 ml) and added dropwise to a solution of amine (1.6 mmol) and Et3N (2.25 mmol) in EtOAc (30 ml) while being stirred and cooled in an ice bath. After 1 h and 30 min stirring at room temperature, the mixture was hydrolysed with water, extracted twice with EtOAc and combined organic layers were washed with a saturated NaHCO3 solution, brine, dried over MgSO4 and concentrated in vacuo to give a solid which was suspended in diethyl ether and filtered.
2-[2-(Furan-2-yl)-1,3-benzoxazol-5-yl]-1-(piperidin-1-yl)ethan-1-one (7a): The title compound was prepared from compound 6 and piperidine to afford 7a as a white solid (76%): mp 112 °C. 1H NMR (300 MHz, CDCl3): 8.16 (d, 1H, H5', J = 1.7 Hz), 7.71 (m, 2H, H4 and H7), 7.68 (m, 1H, H6), 7.34 (d, 1H, H3', J = 3.5 Hz), 6.81 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.78 (s, 2H, CH2), 3.39 (m, 4H, Hpiperidine), 1.68 (m, 4H, Hpiperidine), 1.47 (m, 2H, Hpiperidine). IR (ν, cm−1): 1640 (C = O).
2-[2-(Furan-2-yl)-1,3-benzoxazol-5-yl]-1-(4-phenylpiperazin-1-yl)ethan-1-one (7b): The title compound was prepared from compound 6 and phenylpiperazine to afford 7b as a white solid (78%): mp 128 °C. 1H NMR (300 MHz, CDCl3): 7.68 (m, 1H, H5'), 7.63 (m, 1H, H3'), 7.52 (d, 1H, H7, J = 8.4 Hz), 7.32–7.23 (m, 4H, Hphenyl), 6.89–6.87 (m, 3H, H4, H6 and Hphenyl), 6.63 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.91 (s, 2H, CH2), 3.83 (t, 2H, Hpiperazine, J = 4.6 Hz), 3.66 (t, 2H, Hpiperazine, J = 4.6 Hz), 3.16 (t, 2H, Hpiperazine, J = 4.6 Hz), 3.02 (t, 2H, Hpiperazine, J = 4.6 Hz). IR (ν, cm−1): 1640 (C = O).
2-[2-(Furan-2-yl)-1,3-benzoxazole-5-yl]-1-(morpholine-4-yl) ethan-1-one (7c): The title compound was prepared from compound 6 and morpholine to afford 7c as a white solid (57%): mp 118 °C. 1H NMR (300 MHz, CDCl3): 7.67 (m, 1H, H5'), 7.59 (m, 1H, H3'), 7.52 (d, 1H, H7, J = 8.4 Hz), 7.27 (m, 2H, H4 and H6), 6.63 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.85 (s, 2H, CH2), 3.66 (m, 4H, Hmorpholine), 3.50 (m, 4H, Hmorpholine). IR (ν, cm−1): 1630 (C = O).
2-[2-(Furan-2-yl)-1,3-benzoxazol-5-yl]-1-{4-[4-(2-methoxyethoxy)phenyl]piperazin-1-yl}ethan-1-one (7d): The title compound was prepared from compound 6 and 1-[4-(2-methoxyethoxy)phenyl]piperazine to afford 7d as a beige solid (73%): mp 114 °C. 1H NMR (300 MHz, [D6]DMSO): 7.67 (m, 1H, H5'), 7.62 (m, 1H, H3'), 7.51 (d, 1H, H7, J = 8.4 Hz), 7.29 (m, 2H, H6 and H4), 6.84 (m, 4H, Hphenyl and Hphenyl), 6.63 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 4.05 (t, 2H, CH2O,J = 4.6 Hz), 3.89 (s, 2H, CH2), 3.81 (t, 2H, Hpiperazine, J = 4.7 Hz), 3.71 (t, 2H, CH2O, J = 4.6 Hz), 3.63 (t, 2H, Hpiperazine, J = 4.7 Hz), 3.43 (s, 3H, OCH3), 3.02 (t, 2H, Hpiperazine, J = 4.7 Hz), 2.88 (t, 2H, Hpiperazine, J = 4.7 Hz). IR (ν, cm−1): 1630 (C = O).
General procedure for the synthesis of compound (B1–B4)
To a solution of amide (1.06 mmol) (7a–7d) in THF (5 ml) was added LiAlH4 (2.65 mmol). After 1 h stirring at room temperature, water (0.1 ml), 1 M NaOH solution (0.1 ml) and water (0.3 ml) were added successively to get a white mineral solid which was filtered off and washed with EtOAc (50 ml). Organic layer was then washed with water, brine solution, dried over MgSO4 and concentrated in vacuo to afford a solid which was then purified.
2-(Furan-2-yl)-5-[2-(piperidine-1-yl) ethyl]-1,3-benzoxazole hydrochloride (B1): The title compound was prepared from amide 7a. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from acetonitrile to afford B1 as a white solid (10%): mp >300 °C. 1H NMR (300 MHz, [D6]DMSO): 10.53 (br s, 1H, NH+), 8.0 (m, 1H, H5'), 7.74 (d, 1H, H7, J = 8.4 Hz), 7.69 (m, 1H, H4), 7.46 (d, 1H, H3', J = 3.5 Hz), 7.33 (dd, 1H, H6, J = 3.4 and J = 8.4 Hz), 6.82 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.52–3.48 (m, 2H, CH2CH2), 3.33–3.19 (m, 4H, Hpiperidine), 2.93–2.90 (m, 2H, CH2CH2), 1.80–1.70 (m, 5H, Hpiperidine), 1.43–1.39 (m, 1H, Hpiperidine). 13C NMR (75 MHz, [D6]DMSO): 155.5 (C), 149.0 (C), 147.7 (CH), 142.0 (C), 141.9 (C), 134.9 (C), 126.8 (CH), 120.1 (CH), 115.7 (CH), 113.3 (CH), 111.3 (CH), 57.2 (CH2), 52.5 (2 CH2), 29.7 (CH2), 22.8 (2 CH2), 21.9 (CH2). LC-MS (ESI) m/z found: 297 [M + H]+. HPLC: C4 column: tR = 16.3 min, purity >99%.
2-(Furan-2-yl)-5-[2-(4-phenylpiperazin-1-yl) ethyl]-1,3-benzoxazole (B2): The title compound was prepared from amide 7b. Solid was recrystallised from ethanol to afford B2 as a white solid (12%): mp 160 °C. 1H NMR (300 MHz, [D6]DMSO): 8.06 (m, 1H, H5'), 7.65 (m, 1H, H4), 7.56 (1H, d, H7, J = 8.2 Hz), 7.43 (d, 1H, H3', J = 3.5 Hz), 7.31 (dd, 1H, H6, J = 1.3 Hz and J = 8.2 Hz), 7.22–7.17 (m, 2H, Hphenyl), 6.92 (d, 2H, Hphenyl,J = 7.9 Hz), 6.80 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 6.78–6.73 (m, 1H, Hphenyl), 3.12 (m, 4H, Hpiperazine), 2.88 (t, 2H, CH2CH2, J = 7.3 Hz), 2.64–2.57 (m, 6H, CH2CH2 and Hpiperazine). 13C NMR (75 MHz, [D6]DMSO): 155.2 (C), 151.5 (C), 148.5 (C), 147.5 (CH), 141.7 (C), 142.1 (C), 138.2 (C), 129.4 (CH), 126.9 (CH), 120.0 (CH), 119.2 (CH), 115.8 (CH), 115.4 (CH), 113.2 (CH), 110.8 (CH), 60.4 (CH2), 53.1 (2 CH2), 48.7 (2 CH2), 33.0 (CH2). LC-MS (ESI) m/z found: 374 [M + H]+. HPLC: C4 column: tR = 15.8 min, purity >99%.
2-(Furan-2-yl)-5-[2-(morpholin-4-yl) ethyl]-1,3-benzoxazole hydrochloride (B3): The title compound was prepared from amide 7c. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from acetonitrile to afford B3 as a white solid (80%): mp >300 °C. 1H NMR (300 MHz, [D6]DMSO): 11.36 (br s, 1H, NH+), 8.06 (m, 1H, H5'), 7.73–7.67 (m, 2H, H7 and H4), 7.43 (m, 1H, H3'), 7.31 (m, 1H, H6), 7.80 (m, 1H, H4'), 3.92–3.82 (m, 4H, CH2CH2), 3.31–3.18 (m, 8H, Hmorpholine). LC-MS (ESI) m/z found: 299 [M + H]+. HPLC: C4 column: tR = 14.8 min, purity 99%.
2-(Furan-2-yl)-5-(2-{4-[4-(2-methoxyethoxy)phenyl]piperazin-1-yl}ethyl)-1,3-benzoxazole (B4): The title compound was prepared from amide 7d. Solid was suspended recrystallised from acetonitrile to afford B4 as a beige solid (73%): mp >300 °C. 1H NMR (300 MHz, CDCl3): 7.67 (m, 1H, H5'), 7.62 (m, 1H, H3'), 7.48 (d, 1H, H7, J = 8.3 Hz), 7.27 (m, 2H, H6 and H4), 7.22 (m, 1H, Hphenyl), 6.90 (m, 4H, Hphenyl), 6.63 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 4.09 (t, 2H, OCH2,J = 4.6 Hz), 3.73 (t, 2H, MeOCH2,J = 4.6 Hz), 3.45 (s, 3H, OMe), 3.15 (m, 4H, Hpiperazine), 2.98 (m, 2H, CH2CH2), 2.73 (m, 6H, Hpiperazine and CH2CH2). 13C NMR (75 MHz, CDCl3): 155.5 (C), 152.9 (C), 148.8 (C), 145.9 (C), 145.7 (CH), 142.7 (C), 141.9 (C), 137.3 (C), 126.1 (CH), 119.9 (CH), 118.1 (2 CH), 115.4 (2 CH), 114.1 (CH), 112.3 (CH), 110.2 (CH), 71.2 (CH2), 67.7 (CH2), 60.8 (CH2), 59.2 (CH2), 53.4 (2 CH2), 50.6 (2 CH2), 33.7 (CH3). LC-MS (ESI) m/z found: 448 [M + H]+. HPLC: C4 column: tR = 15.2 min, purity 99%.
General procedure for the synthesis of acid (8a–8b)
A solution of dimethyl malonate (9.06 mmol) in acetone (33 ml) with K2CO3 (13.6 mmol) was stirred for 20 min at room temperature. Then compound (4a, 4c) (4.53 mmol) was added and the mixture was stirred at reflux for 2 h, cooled to room temperature and concentrated in vacuo. Solid was suspended in water and extracted twice with EtOAc. Combined organic layer were dried over MgSO4 and then concentrated in vacuo. The solid was suspended in H2O (7 ml) and NaOH (18.1 mmol) was added. The mixture was stirred overnight at 40 °C and then washed with EtOAc. The aqueous layer was acidified with 6 M HCl solution up to acid pH (1–3) and the formed solid was filtered. Crude was heated in DMF (5 ml) at 80 °C for 3 h, cooled to room temperature, hydrolysed with water and acidified with 1 M HCl solution and extracted three times with EtOAc. Combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo. Solid was suspended in diethyl ether and filtered.
3-[2-(Furan-2-yl)-1,3-benzoxazol-5-yl] propanoic acid (8a): The title compound was prepared from compound 4a to afford 8a as a white solid (43%): mp 207 °C. 1H NMR (300 MHz, CDCl3): 12.19 (br s, 1H, OH), 8.05 (m, 1H, H5'), 7.66–7.62 (m, 2H, H6 and H4), 7.42 (d, 1H, H3', J = 3.5 Hz), 7.28 (d, 1H, H7, J = 8.2 Hz), 6.79 (m, 1H, H4'), 2.94 (t, 2H, CH2CH2, J = 7.5 Hz), 2.59 (t, 2H, CH2CH2, J = 7.5 Hz). IR (ν, cm−1): 1686 (C = O). LC-MS (ESI) m/z found: 258 [M + H]+.
3-[2-(3,4-Dimethoxyphenyl)-1,3-benzoxazol-5-yl] propanoic acid (8b): The title compound was prepared from compound 4c to afford 8b as a white solid (47%): mp 182 °C. 1H NMR (300 MHz, CDCl3): 12.14 (br s, 1H, OH), 7.77 (dd, 1H, H6', J = 2.0 Hz and J = 8.4 Hz), 7.67–7.60 (m, 3H, H4, H2' and H5'), 7.25 (dd, 1H, H6, J = 1.6 Hz and J = 8.3 Hz), 7.17 (d, 1H, H7, J = 8.3 Hz), 3.88 (s, 3H, OMe), 3.86 (s, 3H, OMe), 2.94 (t, 2H, CH2CH2, J = 7.7 Hz), 2.59 (t, 2H, CH2CH2, J = 7.7 Hz). IR (ν, cm−1): 1710 (C = O).
Synthesis of compounds (9a–9c)
Same procedure as described for compounds (7a–7d) has been used.
3-(2-(Furan-2-yl)-1,3-benzoxazol-5-yl)-1-(4-phenylpiperazin-1-yl)propan-1-one (9a): The title compound was prepared from compound 8a and phenylpiperazine to afford 9a as a white solid (55%): mp 164 °C. 1H NMR (300 MHz, CDCl3): 7.66 (m, 1H, H5'), 7.58 (m, 1H, H3'), 7.48 (d, 1H, H7, J = 8.3 Hz), 7.29–7.23 (m, 4H, Hphenyl), 6.91–6.87 (m, 3H, H4, H6 and Hphenyl), 6.62 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.57 (m, 2H, Hpiperazine, J = 5.7 Hz), 3.55 (m, 2H, Hpiperazine), 3.17–3.11 (m, 4H, CH2CH2 and Hpiperazine), 3.04 (m, 2H, Hpiperazine), 2.73 (t, 2H, CH2CH2, J = 7.7 Hz). IR (ν, cm−1): 1658 (C = O).
3-[2-(3,4-Dimethoxyphenyl)-1,3-benzoxazole-5-yl]-1-(piperidin-1-yl)propan-1-one (9b): The title compound was prepared from compound 8b and piperidine to afford 9b as a white solid (66%): mp 130 °C. 1H NMR (300 MHz, CDCl3): 7.85 (dd, 1H, H6', J = 8.4 Hz and J = 1.9 Hz), 7.75 (d, 1H, H2', J = 1.9 Hz), 7.56 (m, 1H, H4), 7.58 (d, 1H, H7', J = 8.4 Hz), 7.21 (dd, 1H, H6, J = 1.5 Hz and J = 8.1 Hz), 7.17 (d, 1H, H7, J = 8.4 Hz), 4.02 (s, 3H, OMe), 3.97 (s, 3H, OMe), 3.57 (t, 1H, CH2CH2, J = 5.6 Hz), 3.35 (t, 2H, Hpiperidine, J = 5.6 Hz), 3.10 (t, 2H, CH2CH2, J = 7.7 Hz), 2.67 (t, 2H, CH2CH2, J = 7.7 Hz), 1.68–1.47 (m, 6H, Hpiperidine). IR (ν, cm−1): 1625 (C = O).
3-(2-(3,4-Dimethoxyphenyl)-1,3-benzoxazol-5-yl)-1-(4-phenylpiperazin-1-yl)propan-1-one (9c): The title compound was prepared from compound 8b and phenylpiperazine to afford 9c as a white solid (54%): mp 160 °C. 1H NMR (300 MHz, CDCl3): 7.85 (dd, 1H, H6', J = 2.0 Hz and J = 8.5 Hz), 7.75 (d, 1H, H2', J = 2.0 Hz), 7.60 (d, 1H, H4, J = 1.2 Hz), 7.47 (d, 1H, H7, J = 8.3 Hz), 7.28–7.20 (m, 3H, H6 and Hphenyl), 6.99 (d, 1H, H5', J = 8.5 Hz), 6.87–6.82 (m, 3H, Hphenyl), 4.02 (s, 3H, OMe), 3.98 (s, 3H, OMe), 3.79 (m, 2H, Hpiperazine), 3.55 (m, 2H, Hpiperazine), 3.16–3.11 (m, 4H, CH2CH2 and Hpiperazine), 3.03 (m, 2H, Hpiperazine), 2.74 (t, 2H, CH2CH2, J = 7.4 Hz). IR (ν, cm−1): 1635 (C = O).
Synthesis of compounds (C1–C3)
Same procedure as described for compounds (B1–B4) has been used.
2-(Furan-2-yl)-5-(3-(4-phenylpiperazin-1-yl)propyl)-1,3-benzoxazole hydrochloride (C1): The title compound was prepared from compound 9a. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from acetonitrile to afford C1 as a white solid (12%): mp >300 °C. 1H NMR (300 MHz, CDCl3): 11.00 (br s, 1H, NH+), 8.07 (m, 1H, H5'), 7.71–7.67 (m, 2H, H4 and H3'), 7.44 (m, 1H, H6), 7.32–7.24 (m, 3H, H7 and Hphenyl), 6.98 (m, 2H, Hphenyl), 6.81 (m, 2H, Hphenyl and H4'), 3.76 (m, 2H), 3.57 (m, 2H), 3.12 (m, 6H), 2.80 (m, 2H, CH2CH2CH2), 2.14 (m, 2H, CH2CH2CH2). 13C NMR (75 MHz, [D6]DMSO): 155.3 (C), 150.1 (C), 148.7 (C), 147.6 (CH), 142.0 (C), 141.9 (C), 138.3 (C), 129.6 (2 CH), 126.9 (CH), 120.4 (CH), 119.6 (CH), 116.4 (2 CH), 115.5 (CH), 113.3 (CH), 111.0 (CH), 55.5 (CH2), 51.1 (2 CH2), 45.9 (2 CH2), 32.4 (CH2), 25.6 (CH2). LC-MS (ESI) m/z found: 388 [M + H]+. HPLC: C4 column: tR = 15.8 min, purity 98%.
2-(3,4-Dimethoxyphenyl)-5-[3-(piperidin-1-yl)propyl]-1,3-benzoxazole hydrochloride (C2): The title compound was prepared from compound 9b. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from ethyl acetate to afford C2 as a white solid (72%): mp 214 °C. 1H NMR (300 MHz, [D6]DMSO): 10.20 (br s, 1H, NH+), 7.77 (dd, 1H, H6', J = 2.0 Hz and J = 8.4 Hz), 7.68 (d, 1H, H5', J = 8.4 Hz), 7.66–7.63 (m, 2H, H2' and H4), 7.21 (dd, 1H, H6, J = 1.5 Hz and J = 8.3 Hz), 7.17 (d, 1H, H7, J = 8.3 Hz), 3.88 (s, 3H, OMe), 3.86 (s, 3H, OMe), 3.38 (m, 2H, CH2CH2CH2), 2.99 (m, 2H, CH2CH2CH2), 2.76 (m, 2H, CH2CH2CH2), 2.08 (m, 2H, Hpiperidine), 1.75–1.65 (m, 5H, Hpiperidine), 1.35 (m, 1H, Hpiperidine). 13C NMR (75 MHz, [D6]DMSO): 163.2 (C), 152.5 (C), 149.5 (C), 149.3 (C), 142.4 (C), 137.8 (C), 125.9 (C), 121.3 (CH), 119.2 (CH), 119.2 (CH), 112.4 (CH), 110.9 (CH), 110.2 (CH), 56.2 (CH3), 56.1 (CH3), 55.9 (CH2), 52.4 (2 CH2), 32.5 (CH2), 25.6 (CH2), 22.9 (2 CH2), 21.9 (CH2). LC-MS (ESI) m/z found: 381 [M + H]+. HPLC: C4 column: tR = 17.3 min, purity 98%.
2-(3,4-Dimethoxyphenyl)-5-[3-(4-phenylpiperazin-1-yl)propyl]-1,3-benzoxazole hydrochloride (C3): The title compound was prepared from compound 9c. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from ethanol to afford C3 as a white solid (73%): mp 194 °C. 1H NMR (300 MHz, [D6]DMSO): 10.81 (br s, 1H, NH+), 7.77 (dd, 1H, H6', J = 1.8 Hz and J = 8.4 Hz), 7.70–7.65 (m, 3H, H4, H6 and H7), 7.29–7.22 (m, 3H, H2' and Hphenyl), 7.17 (d, 1H, H5', J = 8.4 Hz), 6.98 (m, 2H, Hphenyl), 6.87–6.82 (m, 1H, Hphenyl), 3.88 (s, 3H, OMe), 3.86 (s, 3H, OMe), 3.77 (m, 2H, CH2CH2CH2), 3.56 (m, 2H, Hpiperazine), 3.11 (m, 6H, Hpiperazine), 2.79 (t, 2H, CH2CH2CH2, J = 7.4 Hz), 2.13 (m, 2H, CH2CH2CH2). 13C NMR (75 MHz, [D6]DMSO): 163.2 (C), 152.5 (C), 150.1 (C), 149.5 (C), 149.3 (C), 142.4 (C), 137.8 (C), 129.6 (2 CH), 125.9 (CH), 121.3 (CH), 120.4 (C), 119.3 (CH), 119.2 (CH), 116.4 (2 CH), 112.4 (CH), 110.9 (CH), 110.2 (CH), 56.2 (CH2), 56.1 (CH3), 55.5 (CH3), 51.1 (2 CH2), 45.9 (2 CH2), 32.5 (CH2), 25.7 (CH2). LC-MS (ESI) m/z found: 458 [M + H]+. HPLC: C4 column: tR = 16.6 min, purity >99%.
2-(2-(3,4-dimethoxyphenyl)-1,3-benzoxazol-5-yl)ethanol (10)
To a solution of compound (3e) (1 g, 3.06 mmol) in THF (10 ml) was added LiAlH4 (340 mg, 9.2 mmol). After 1 h stirring at room temperature, water (0.34 ml), 1 M NaOH solution (0.34 ml) and water (1.02 ml) were added successively until get a white solid which was filtered off and washed with EtOAc (60 ml). Organic layer was washed with water, dried over MgSO4 and concentrated in vacuo. Solid was recrystallised in acetonitrile to afford compound (10) as a beige solid (603 mg, 86%): mp 210 °C. 1H NMR (300 MHz, CDCl3): 9.13 (br s, 1H, OH) 7.86 (dd, 1H, H6', J = 1.8 Hz and J = 8.4 Hz), 7.76 (d, 1H, H2', J = 1.8 Hz), 7.61 (m, 1H, H4), 7.50 (d, 1H, H7, J = 8.4 Hz), 7.24 (dd, 1H, H6, J = 1.2 Hz and J = 8.4 Hz), 7.00 (d, 1H, H5', J = 8.4 Hz), 4.03 (s, 3H, OMe), 3.98 (s, 3H, OMe), 3.92 (m, 2H, CH2CH2), 3.64 (t, 2H, CH2CH2, J = 6.6 Hz). IR (ν, cm−1): 3400 (OH). LC-MS (ESI) m/z found: 300 [M + H]+.
2-(2-(3,4-Dimethoxyphenyl)-1,3-benzoxazol-5-yl)ethyl methanesulfonate (11)
To a solution of compound (10) (800 mg, 2.37 mmol) in DCM (20 ml) with Et3N (0.49 ml, 3.55 mmol) at 0 °C, was added dropwise mesyl chloride (0.28 ml, 3.55 mmol). After 4 h stirring at room temperature, mixture was hydrolysed with water and extracted twice with DCM. Combined organic layers were dried over MgSO4 and concentrated in vacuo. Solid was suspended in diethyl ether and filtered to afford compound (11) as a beige solid (895 mg, 100%): mp 112 °C. 1H NMR (300 MHz, CDCl3): 7.85 (dd, 1H, H6', J = 8.4 Hz and J = 2.0 Hz), 7.76 (d, 1H, H4', J = 2.0 Hz), 7.61 (d, 1H, H4, J = 1.6 Hz), 7.51 (d, 1H, H7, J = 8.3 Hz), 7.21 (dd, 1H, H6, J = 8.3 Hz and J = 1.6 Hz), 7.00 (d, 1H, H5', J = 8.4 Hz), 4.47 (t, 2H, CH2CH2, J = 6.8 Hz), 4.02 (s, 3H, OMe), 3.97 (s, 3H, OMe), 3.18 (t, 2H, CH2CH2, J = 6.9 Hz), 2.88 (s, 3H, CH3). LC-MS (ESI) m/z found: 378 [M + H]+.
General procedure for the synthesis of compound (B5–B6)
To a solution of compound 11 (0.79 mmol) in DMF (8 ml), were added K2CO3 (1.46 mmol) and amine (1.03 mmol). After overnight stirring at 80 °C, the reaction mixture was cooled, hydrolysed with water and extracted three times with EtOAc. Combined organic layers were dried over MgSO4 and concentrated in vacuo to afford a solid which was then purified.
2-(3,4-Dimethoxyphenyl)-5-(2-(piperidin-1-yl)ethyl)-1,3-benzoxazole hydrochloride (B5): The title compound was prepared from compound 11 and piperidine. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from ethanol to afford B5 as a white solid (25%): mp 260 °C. 1H NMR (300 MHz, [D6]DMSO): 10.63 (br m, 1H, NH+), 7.79 (dd, 1H, H6', J = 8.4 Hz and J = 2.0 Hz), 7.73 (d, 1H, H7, J = 8.3 Hz), 7.68 (d, 1H, H2', J = 1.3 Hz), 7.66 (d, 1H, H4, J = 2.0 Hz), 7.31 (dd, 1H, H6, J = 8.4 Hz and J = 1.7 Hz), 7.18 (d, 1H, H5', J = 8.6 Hz), 3.89 (s, 3H, OMe), 3.87 (s, 3H, OMe), 3.50 (m, 2H, CH2CH2), 3.27–3.20 (m, 4H, CH2CH2 and Hpiperidine), 2.92 (m, 2H, Hpiperidine), 1.82–1.70 (m, 5H, Hpiperidine), 1.42 (m, 1H, Hpiperidine). 13C NMR (75 MHz, [D6]DMSO): 163.4 (C), 152.5 (C), 149.5 (C), 149.3 (C), 142.4 (C), 137.8 (C), 125.9 (C), 121.3 (CH), 119.2 (CH), 119.2 (CH), 112.4 (CH), 110.9 (CH), 110.2 (CH), 57.3 (CH2), 56.2 (CH3), 56.1 (CH3), 52.4 (2 CH2), 29.7 (CH2), 22.8 (2 CH2), 21.9 (CH2). LC-MS (ESI) m/z found: 367 [M + H]+. HPLC: C4 column: tR = 16.1 min, purity 99%.
2-(3,4-Dimethoxyphenyl)-5-[2-(4-phenylpiperazin-1-yl)ethyl]-1,3-benzoxazole (B6): The title compound was prepared from compound 11 and phenylpiperazine. Solid was recrystallised from ethanol to afford B6 as a white solid (28%): mp 140 °C. 1H NMR (300 MHz, CDCl3): 7.85 (dd, 1H, H6', J = 2.0 Hz and J = 8.4 Hz), 7.76 (d, 1H, H2', J = 2.0 Hz), 7.61 (m, 1H, H4), 7.48 (d, 1H, H5', J = 8.4 Hz), 7.31–7.26 (m, 2H, H7 and Hphenyl), 7.20 (dd, 1H, H6, J = 1.5 Hz and J = 8.3 Hz), 7.01–6.85 (m, 4H, Hphenyl), 4.03 (s, 3H, OMe), 3.98 (s, 3H, OMe), 3.27–3.24 (t, 4H, Hpiperazine,J = 4.8 Hz), 3.01–2.96 (m, 2H, CH2CH2), 2.75–2.71 (m, 6H, CH2CH2 and Hpiperazine). 13C NMR (300 MHz, CDCl3): 163.4 (C), 152.0 (C), 151.3 (C), 149.4 (C), 149.2 (C), 142.5 (C), 136.9 (C), 129.1 (2 CH), 125.5 (CH), 121.1 (CH), 119.9 (C), 119.7 (CH), 119.4 (CH), 116.1 (2 CH), 111.0 (CH), 110.0 (2 CH), 60.9 (CH2), 56.1 (CH3), 56.0 (CH3), 53.3 (2 CH2), 49.2 (2 CH2), 33.7 (CH2). LC-MS (ESI) m/z found: 444 [M + H]+. HPLC: C4 column: tR = 15.7 min, purity 99%.
2-(Furan-2-yl)-1,3-benzoxazol-5-amine (D1)
To a solution of 2-(furan-2-yl)-5-nitro-1,3-benzoxazole (3.07 g, 13.3 mmol) (3d) in EtOH (130 ml) were added Pd/C (10%, 100 mg) and hydrazine monohydrate (0.78 ml, 16 mmol). The mixture was heated at 70 °C for 3 h, cooled to room temperature, the Pd/C was filtered off and the filtrate was concentrated in vacuo. Crude was suspended in water and extracted three times with EtOAc. Combined organic layer were washed with brine, dried over MgSO4 and concentrated in vacuo. Solid was recrystallised from hexane to afford compound (D1) as grey solid (2.16 g, 81%): mp 112 °C. 1H NMR (300 MHz, [D6]DMSO): 8.01 (m, 1H, H5'), 7.40 (d, 1H, H7, J = 8.7 Hz), 7.34 (d, 1H, H3', J = 3.5 Hz), 6.84 (d, 1H, H4, J = 2.1 Hz), 6.77 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 6.66 (dd, 1H, H6, J = 2.1 Hz and J = 8.7 Hz), 5.16 (br s, 2H, NH2). 13C NMR (75 MHz, [D6]DMSO): 154.9 (C), 147.3 (C), 147.0 (CH), 142.6 (C), 142.5 (C), 142.4 (C), 114.4 (CH), 113.6 (CH), 113.1 (CH), 110.9 (CH), 103.0 (CH). IR (ν, cm−1): 3424 (NH2). LC-MS (ESI) m/z found: 201 [M + H]+. HPLC: C4 column: tR = 14.2 min, purity 98%.
N-(4-(Benzyloxy)phenethyl)-2-(furan-2-yl)-1,3benzoxazol-5-amine (12)
To a solution of compound D1 (800 mg, 4 mmol) in DMF (15 ml) were added 1-(benzyloxy)-4-(2-bromoethyl)benzene (1.4 g, 4.8 mmol)16 and K2CO3 (828 mg, 5.99 mmol). The reaction mixture was stirred at 80 °C overnight, cooled to room temperature, hydrolysed with water, acidified with 1 M HCl solution, and extracted with EtOAc three times. Combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo to give a yellow oil. Purification by flash chromatography was realised with EtOAc/Cyclohexane as solvent (1/9 up to 3/7) to give the product as a yellow oil which was suspended in diethyl ether and filtered to afford compound 12 as a pale brown solid (430 mg, 26%): mp 184 °C. 1H NMR (75 MHz, [D6]DMSO, J Hz): 8.10 (m, 1H, H5'), 7.75 (m, 1H), 7.58 (m, 1H), 7.46–7.26 (m, 7H), 7.21 (d, 2H, Hphenyl, J = 8.7 Hz), 6.96 (d, 2H, Hphenyl, J = 8.7 Hz), 6.83 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 5.06 (s, 2H, CH2), 3.52 (t, 2H, CH2CH2, J = 7.5 Hz), 2.99 (t, 2H, CH2CH2, J = 7.5 Hz). LC-MS (ESI) m/z found: 411 [M + H]+.
4-(2-{[2-(Furan-2-yl)-1,3-benzoxazol-5-yl]amino}ethyl)phenol (E2)
A solution of compound 12 (740 mg, 1.66 mmol) in MeOH (15 ml) with Pd/C (50 mg) under H2 atmosphere was stirred at 25 °C for overnight. Pd/C was filtered off and the filtrate was concentrated in vacuo. Solid was recrystallised in CH3CN to afford compound E2 as a yellow crystal (104 mg, 18%): mp 174 °C. 1H NMR (300 MHz, [D6]DMSO): 9.16 (br s, 1H, OH), 8.01 (m, 1H, H5'), 7.43 (d, 1H, H7, J = 8.7 Hz), 7.33 (d, 1H, H3', J = 3.5 Hz), 7.07 (d, 2H, Hphenyl, J = 8.4 Hz), 6.80 (m, 1H, H4), 6.77 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 6.69 (d, 2H, Hphenyl, J = 8.4 Hz), 6.71 (m, 1H, H6), 5.73 (br t, 1H, J = 5.6 Hz, NH), 3.24–3.17 (m, 2H, J = 6.9 Hz, CH2CH2), 2.75 (t, 2H, J = 7.6 Hz, CH2CH2). 13C NMR (75 MHz, [D6]DMSO): 156.1 (C), 154.9 (C), 147.7 (C), 147.0 (CH), 142.8 (C), 142.5 (C), 142.2 (C); 130.3 (C), 130.0 (2 CH), 115.5 (2 CH), 114.4 (CH), 113.1 (CH), 112.8 (CH), 111.1 (CH), 100.2 (CH); 46.1 (CH2), 34.4 (CH2). IR (ν, cm−1): 3340 (OH). LC-MS (ESI) m/z found: 321 [M + H]+. HPLC: C4 column: tR = 14.8 min, purity 97%.
2-(Furan-2-yl)-N-(2-(piperidin-1-yl)ethyl)-1,3 benzoxazol-5-amine hydrochloride (E1)
To a solution of compound D1 (800 mg, 4 mmol) in DMF (16 ml) were added K2CO3 (1.7 g, 12 mmol) and N-2-chloroethyl piperidine hydrochloride (1471 mg, 7.99 mmol). The reaction mixture was stirred at 70 °C overnight, cooled to room temperature, hydrolysed with water and extracted three times with EtOAc. Combined organic layer were dried over MgSO4 and concentrated in vacuo. Purification by flash chromatography was performed with DCM/EtOH/NH3 (90/10/1) as a solvent. The obtained yellow oil was suspended in diethyl ether with HCl gas to formed a solid which was filtered to afford compound E1 as a beige solid (11 mg, 8%): mp 169 °C. 1H NMR (300 MHz, [D6]DMSO): 10.69 (br m, 1H, NH+), 8.04 (m, 1H, H5'), 7.55 (d, 1H, H7, J = 8.8 Hz), 7.38 (m, 1H, H3'), 7.09 (m, 1H, H4), 6.88 (dd, 1H, H6, J = 1.9 Hz and J = 8.8 Hz), 6.79 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 5.37 (br m, 1H, NH), 3.57 (t, 2H, CH2CH2, J = 6.3 Hz), 3.49–3.46 (m, 2H, Hpiperidine), 3.24 (t, 2H, CH2CH2, J = 6.2 Hz), 2.90 (m, 2H, Hpiperidine), 1.79 (m, 5H, Hpiperidine), 1.39 (m, 1H, Hpiperidine). 13C NMR (75 MHz, [D6]DMSO): 155.3 (C), 147.2 (CH), 145.0 (C), 143.6 (C), 142.7 (C), 124.3 (C), 114.9 (CH), 114.1 (CH), 113.2 (CH), 111.5 (CH), 102.6 (CH), 54.5 (CH2), 52.7 (2 CH2), 39.2 (CH2), 22.8 (2 CH2), 21.8 (CH2). LC-MS (ESI) m/z found: 312 [M + H]+. HPLC: C4 column: tR = 15.9 min, purity 99%.
2-Bromo-N-(2-(furan-2-yl)-1,3-benzoxazol-5-yl)acetamide (13): To a solution of compound D1 (210 mg, 1.05 mmol) and Et3N (0.18 ml, 1.26 mmol) in DCM (10 ml) at 0 °C was added dropwise a solution of bromoacetyl bromide (0.11 ml, 1.26 mmol) diluted in DCM (5 ml). The reaction mixture was stirred at room temperature for 2 h, hydrolysed with water and extracted twice with DCM. Combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo. The solid was suspended in diethyl ether and filtered to afford compound 13 as a white solid (249 mg, 74%): mp 203 °C. 1H NMR (300 MHz, CDCl3): 8.27 (br s, 1H, NH), 7.96 (m,1H, H5'), 7.70–7.69 (m, 1H, H4), 7.56–7.50 (m, 2H, H7 and H6), 7.30 (dd, 1H, H3', J = 0.7 Hz and J = 3.5 Hz), 6.39 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 4.08 (s, 2H, CH2). IR (ν, cm−1): 3246 (NH), 1643 (C = O).
General procedure for the synthesis of compounds (F1–F3)
To a solution of compound 13 (0.59 mmol) in acetone (5 ml) were added K2CO3 (0.88 mmol) and the amine (0.65 mmol). The reaction mixture was refluxed for 2 h, cooled to room temperature and concentrated in vacuo. The crude was suspended in water and extracted three times with EtOAc. Combined organic layer were washed with brine, dried over MgSO4 and concentrated in vacuo to afford a solid which was then purified.
N-[2-(Furan-2-yl)-1,3-benzoxazol-5-yl]-2-(piperidin-1-yl)acetamide hydrochloride (F1): The title compound was prepared from compound 13 and piperidine. Solid was suspended in diethyl ether with HCl gas, concentrated in vacuo and recrystallised from ethanol to afford F1 as a white solid (57%): mp >300 °C. 1H NMR (300 MHz, [D6]DMSO): 11.26 (br s, 1H, NH+), 9.99 (br s, 1H, NH), 8.17 (m, 1H, H5'), 8.08 (m, 1H, H4), 7.76 (d, 1H, H7, J = 8.8 Hz), 7.58 (dd, 1H, H6, J = 1.9 Hz and J = 8.8 Hz), 7.46 (d, 1H, H3', J = 3.5 Hz), 6.82 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 4.18 (s, 2H, CH2), 3.50–3.47 (m, 2H, Hpiperidine), 3.10 (m, 2H, Hpiperidine), 1.80–1.67 (m, 5H, Hpiperidine), 1.40 (m, 1H, Hpiperidine). 13C NMR (75 MHz, [D6]DMSO): 163.4 (C), 156.0 (C), 147.7 (CH), 146.6 (C), 141.9 (C), 141.8 (C), 135.9 (C), 118.2 (CH), 115.8 (CH), 113.3 (CH), 111.4 (CH), 110.8 (CH), 57.6 (CH2), 53.4 (2 CH2), 22.7 (2 CH2), 21.6 (CH2). LC-MS (ESI) m/z found: 326 [M + H]+. HPLC: C4 column: tR = 15.1 min, purity 98%.
N-[2-(Furan-2-yl)-1,3-benzoxazol-5-yl]-2-(4-phenylpiperazin-1-yl)acetamide (F2): The title compound was prepared from compound 13 and phenylpiperazine. Solid was recrystallised from ethanol to afford F2 as a white solid (48%): mp 174 °C. 1H NMR (300 MHz, CDCl3): 9.26 (br s, 1H, NH+), 8.02 (m, 1H, H4), 7.68–7.67 (m, 1H, H5'), 7.56 (dd, 1H, H6, J = 2.0 Hz and J = 8.8 Hz), 7.51 (d, 1H, H7, J = 8.6 Hz), 7.3–7.29 (m, 3H, Hphenyl and H3'), 6.98–6.88 (m, 3H, Hphenyl and Hphenyl), 6.63 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.30 (t, 4H, Hpiperazine, J = 4.9 Hz), 3.26 (s, 2H, CH2), 2.83 (t, 4H, Hpiperazine,J = 4.9 Hz). 13C NMR (75 MHz, CDCl3): 168.2 (C), 156.2 (C), 152.9 (C), 146.9 (C), 145.8 (CH), 142.5 (C), 142.2 (C), 134.9 (C), 129.2 (2 CH), 120.2 (CH), 117.8 (CH), 116.3 (2 CH), 114.5 (CH), 112.3 (CH), 111.2 (CH), 110.5 (CH), 62.0 (CH2), 50.6 (2 CH2), 49.5 (2 CH2). LC-MS (ESI) m/z found: 403 [M + H]+. HPLC: C4 column: tR = 14.7 min, purity >99%.tert-Butyl-4-({[2-(furan-2-yl)-1,3-benzoxazol-5-yl]carbamoyl}methyl)piperazine-1-carboxylate (F3): The title compound was prepared from compound 13 and tert-butyl piperazine-1-carboxylate. Solid was recrystallised from ethanol to afford F3 as a white solid (90%): mp 186 °C. 1H NMR (300 MHz, CDCl3): 9.15 (br s, 1H, NH), 7.98 (d, 1H, H4, J = 2.0 Hz), 7.68–7.67 (m, 1H, H5'), 7.55 (dd, 1H, H6, J = 2.0 Hz and J = 8.6 Hz), 7.50 (d, 1H, H7, J = 8.6 Hz), 7.28 (m, 1H, H3'), 6.62 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 3.54 (t, 4H, Hpiperazine, J = 4.9 Hz), 3.19 (s, 2H, CH2), 2.60 (t, 4H, Hpiperazine, J = 4.9 Hz), 1.48 (s, 9H, 3 CH3). 13C NMR (75 MHz, CDCl3): 167.9 (C), 156.2 (C), 154.6 (C), 147.0 (C), 145.9 (CH), 142.5 (C), 142.2 (C), 134.8 (C), 117.8 (CH), 114.5 (CH), 112.3 (CH), 111.2 (CH), 110.5 (CH), 80.1 (CH2), 62.1 (2 CH2), 53.3 (2 CH2), 28.4 (3 CH3). IR (μ, cm−1): 1684 (C = O), 1673 (C = O). LC-MS (ESI) m/z found: 371 [M-tBu + H]+, 427 [M + H]+. HPLC: C4 column: tR = 15.1 min, purity 99%.
N-(2-(Furan-2-yl)-1,3-benzoxazol-5-yl)-2-(piperazin-1-yl)acetamide dihydrochloride (F4): To a solution of (F3) (180 mg, 0.422 mmol) in MeOH (10 ml) was added 6 M HCl (1.41 ml, 8.44 mmol) and the reaction mixture was stirred at 40 °C for 15 h. The precipitated product was filtered and washed with diethyl ether to afford compound (F4) as a white solid (121 mg, 72%): mp 192 °C. 1H NMR (300 MHz, [D6]DMSO): 11.16 (br s, 1H, NH), 9.88 (br s, 2H, NH2+), 8.16 (d, 1H, H4, J = 1.8 Hz), 8.07 (m, 1H, H5'), 7.75 (d, 1H, H7, J = 8.8 Hz), 7.59 (dd, 1H, H6, J = 1.8 Hz and J = 8.8 Hz), 7.45–7.44 (m, 1H, H3'), 6.81 (dd, 1H, H4', J = 1.7 Hz and J = 3.5 Hz), 4.22 (s, 2H, CH2), 3.57 (s, 4H, Hpiperazine, J = 4.9 Hz), 3.43 (s, 4H, Hpiperazine, J = 4.9 Hz). 13C NMR (75 MHz, [D6]DMSO): 163.7 (C), 155.9 (C), 147.7 (CH), 146.5 (C), 141.9 (C), 141.8 (C), 135.9 (C), 118.2 (CH), 115.7 (CH), 113.3 (CH), 111.4 (CH), 110.8 (CH), 57.6 (CH2), 49.1 (4 CH2). LC-MS (ESI) m/z found: 327 [M + H]+. HPLC: C4 column: tR = 14.9 min, purity 99%.
Molecular docking
Molecular docking was performed using Gold suite v5.217 within the Hermes v1.6 GUI (CCDC©). Thus after adding hydrogens, water molecules were deleted and docking was performed in a 10 Å around co-crystallised ligands then ligands deleted. Early termination of three docking solutions within 1.5 Å was set up in order to highlight ligands converging towards a few binding modes.
Pharmacological assays
Displacement binding assays
Radioligands were obtained from the following source: [3H]-ZM241385 from IsoBio (10–50 Ci/ml), [3H]-DPCPX from PerkinElmer (Waltham, MA) (120 Ci/mmol), [3H]-CPX from Perkin Elmer (120 Ci/mmol), [125I]-AB-MECA from Chelatec (2200 Ci/mmol) (Saint-Herblain, France).
For A2A receptor binding evaluation, the stock solution of compounds was prepared in DMSO. The final concentration of DMSO was no more than 3% for radioligand binding assay.
Competition binding curves of the A2A receptor antagonist [3H]-ZM241385 by the designed A2A antagonists described above, were performed as before18 in Human HEK293 A2AR membranes (PerkinElmer). 0.5 ml of membranes (0.5 U of A2AR) were incubated with [3H]-ZM241385 (2 nm) and increasing concentrations of the designed A2AR antagonists (0–600 nm) in a final volume of 300 μl in the presence of 1 U/ml of adenosine deaminase (Roche, Basel, Switzerland). All samples were assayed in duplicate. Non-specific binding was determined for each assay in the presence of the antagonist ZM-24135 (8.3 nm). Microplates were incubated for 1 h at room temperature and the reaction was stopped by vacuum filtration with a Skatron semi-automatic cell harvester with chilled incubation solution (pH 7.4, Tris 50 mm MgCl 10 mm) to filter mats 1.5 mm (Molecular Devices, Sunnyvale, CA). Three millilitres of scintillation cocktail (OptiPhase “HiSafe” 2, PerkinElmer) were added and radioactivity bound to the filters was determined after 12 h. Molecules inhibited binding by ≥30% at 10 μm were submitted to Ki evaluation. This percentage was calculated using Excel 2013 as a ratio of data with ligands to data without ligands. Data were analysed using Graph Pad Prism, version 5.01 (San Diego, CA). Inhibition constants (Ki) were calculated from the IC50 values by non-linear regression analysis, the Cheng and Prusoff equation and KD value of 1.0 nm were used. Displacement reference curves were performed with ZM-24135 (0–6 nm in 6%, 40% or 60% of DMSO). No difference was observed between each concentration.
Affinity towards A1R (human recombinant CHO cells, [3H]-DPCPX (1 nm), Cerep catalogue reference 0002, as described by Townsend-Nicholson19), A2BR (human recombinant HEK-293 cells, [3H]-CPX (5 nm), Cerep catalogue reference 0005, as described by Stehle20) and A3R (human recombinant HEK-293 cells, [125I]-AB-MECA (0.15 nm), Cerep catalogue reference 0006, as described by Salvatore21) was determined by CEREP laboratories. KDvalues used were: 1.7 nm for [3H]-DPCPX, 65 nm for [3H]-CPX and 0.15 nm for [125I]-AB-MECA. For these three receptors, the stock solution of compounds was prepared in DMSO. The final concentration of DMSO was no more than 1%. Data were analysed using SigmaPlot® version 4.0 for Windows® (© 1997 by SPSS Inc., Chicago, IL).
Cell culture and cytotoxicity assay
The human neuroblastoma cell line (SY5Y) was cultured in Dulbecco’s modified Eagle medium (DMEM) (Gibco, Waltham, MA) supplemented with 2 mm L-glutamine, 100 mg/ml streptomycin, 100 IU/ml penicillin, 1 mm non-essential amino acids and 10% (v/v) heat-inactivated foetal bovine serum (Sigma-Aldrich, Saint-Louis, MO), and grown at 37 °C in a humidified incubator with 5% CO2. Cells were seeded at 2000 cells per well onto 96-well plates in DMEM medium. Cells were starved for 24 h to obtain synchronous cultures and were then incubated in a culture medium that contained various concentrations of test compounds, each dissolved in less than 0.1% DMSO. After 72 h of incubation, cell growth was estimated by the colorimetric MTT (thiazolyl blue tetrazolium bromide) assay.
Absorption, distribution, metabolism and excretion (ADME) assessment
Aqueous solubility (in phosphate-buffered saline, PBS, pH 7.4; incubation room temperature for 24 h as described by Lipinski22 Eurofins Cerep SA catalogue reference G235), partition coefficient (log D, n-octanol/PBS, pH 7.4, room temperature for 60 min as described by Sangster23; Eurofins Cerep SA catalogue reference 0417), human plasma protein binding evaluated at 10 μm concentration for 4 h at 37 °C as described by Banker24 (Eurofins Cerep SA catalogue reference 2194), A-B and B-A permeability coefficient evaluated at 10 μm for 40 min as described by Hidalgo25 (Papp, Caco-2 cells, pH 6.5/7.4; Eurofins Cerep SA catalogue reference G228), metabolic stability in human liver microsomes evaluated at 0.1 μm concentration for 0, 15, 30, 45, 60 min at 37 °C as described by Obach26 (Eurofins Cerep SA catalogue reference 0607) were determined in standard assays by Eurofins Cerep SA, France www.cerep.fr).
Results and discussion
Structure-based insights
Our design was guided by molecular modelling studies which took into consideration the two structures of adenosine A2A receptors co-crystallised with high-affinity A2A antagonists ZM-241385 and T4E (1,2,4-triazines) in respective 3EML27,28 and 3UZC29 PDB entries. Although several molecular dynamics studies have shown the importance of water molecules for ligand recognition30,31, these molecules represent a bias in molecular docking with many options like their rigid pre-docking displacement, a tolerance for moving them or simply delete them. Since T4E-bound structures show that the key water molecules in ZM-241385-bound crystal structures could be displaced by aryl substituents, we decided to avoid this experimental bias in order to benefit with the whole cavity.
In contrast with ZM-241385 bound structure, the T4E-bound one was identified as the most suitable target to predict correct docking poses of both T4E (no data show) and ZM-241385 within a 2.0 Å structural deviation in comparison with experimental co-crystallised poses (Figure 2(B)). This is due to the greater volume of T4E-bound pocket that allows to accommodate bulky di-aryl substituted triazine as well as linear ZM-241385 ligands. Consequently, apoA2AR-T4E pocket appears to be more relevant to accommodate a wide diversity of chemical structures and was therefore used to perform our docking calculations.
A set of benzoxazole-based molecules have been docked, using Gold suite v5.2 within the Hermes v1.6 GUI (CCDC©), into the apoA2AR-T4E pocket and the ones that satisfied interactions with essential amino acids32 Phe168, Glu169, Trp246 and Asn253 were selected. ZM-241385 was shown as an example (Figure 2(B)) rather than the triazine because it is structurally closer to our benzoxazole ligands (Figure 2(C)). As illustrated in Figure 2(B), the central heterocycle of ZM-241385 interacts through a hydrogen bond with Asn253 and π-stacking with Phe168. Benzoxazole ring seems to recapitulate these interactions and could, therefore, constitute a good alternative as a central core. The furan, found in many A2A antagonists, was selected to interact with Trp246 by aromatic interaction and thus made the antagonist character of our ligands. Indeed, interaction with this key amino acid is well known to lock the A2A receptor33 and more generally class-A GPCR34 in their inactive conformation. The 3,4 dimethoxyphenyl, found on Istradefylline, was also used to create this interaction. Furthermore, different nature and size of linkers was used to bring selected amine function. Indeed, these basic functions in designed ligands (Figure 2(C)) occupy the same pocket as the phenol part of the ZM-241385 and allow not only to interact with Glu169 through an ionic interaction but also to improve solubility.
Chemistry
Synthetic routes used to prepare benzoxazole derivatives are depicted in Schemes 1–3. The first set of benzoxazoles (3a–d) was prepared from commercially available aminophenols using two different synthetic routes (Scheme 1). The first one led to molecules 3a–d, in two steps. Indeed, aminophenol derivatives 1a–e reacted with appropriate acyl chlorides to give an equimolar mixture of mono and diacyl compounds which, when treated under basic conditions, gave amides 2a–d. Subsequent cyclisation under acidic conditions afforded benzoxazoles 3a–d in good yield35. The second synthetic route allowed to transform aminophenol 1e into compound 3e in one step using T3P®36.
Scheme 1.
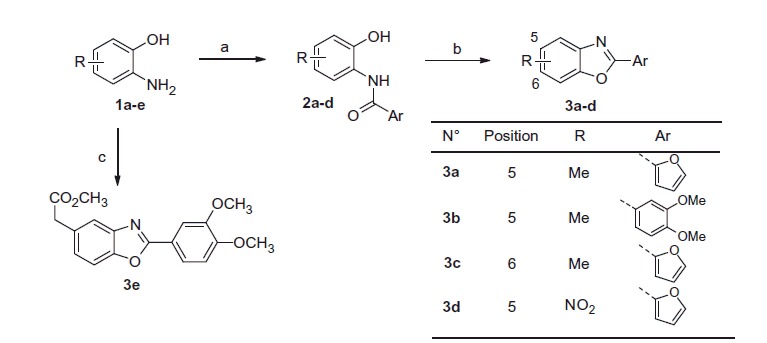
Reagents and conditions: (a) i) ArCO2H, SOCl2, DMF, DCM, ii) Et3N, EtOAc, aminophenol (1a–d); iii) NaOH, EtOH/H2O, then 6 M HCl, 60–80% over 2 steps; (b) APTS, toluene, reflux, 70–80%, (c) T3P® (50% in EtOAc), DIPEA, 3,4-dimethoxybenzoic acid, 35%.
Scheme 2.
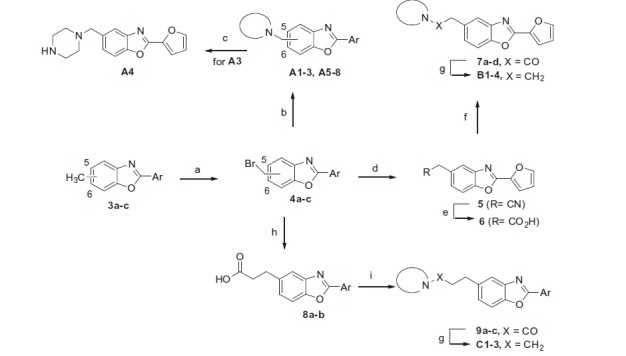
Reagents and conditions: (a) NBS, benzoyl peroxide, CCl4, reflux/hυ (230 W), 60–75%; (b) R2R1NH, Et3N, acetone, 50–85%; (c) TFA, DCM, 60%; (d) KCN, EtOH/H2O, 50–60%; (e) H2O/H2SO4/AcOH, reflux, 60–70%; (f) i) SOCl2, toluene; ii) secondary amine, Et3N, EtOAc, 50–60%; (g) LiAlH4, THF, 60–75%; (h) i) dimethyl malonate, K2CO3, acetone; ii) NaOH, H2O then 6 M HCl; iii) DMF, reflux, 40–43%.
Scheme 3.
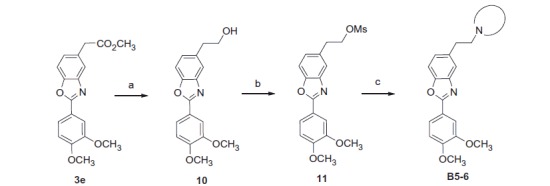
Reagents and conditions: (a) LiAlH4, THF, 86%; (b) MsCl, Et3N, DCM, quant.; (c) secondary amine, K2CO3, DMF, 60 °C, 25–28%.
A radical bromination of compounds 3a–c was performed to generate the key brominated intermediates 4a–c (Scheme 2) which allowed the introduction of the tertiary amine at different distances from the central heterocycle. Molecules A1–8 displaying a one-methylene linker were obtained by reacting 4a–c with various amines. Compound A4 was synthesised from A3 deprotection using TFA. Based on binding results (see Table 1), we focused the further synthetic effort on C-5 substituted compounds. Treatment of 4a with potassium cyanide followed by an acidic hydrolysis gave carboxylic acid 6 which was subjected to amidification and reduction to afford target compounds B1–4 (two-methylene linker).
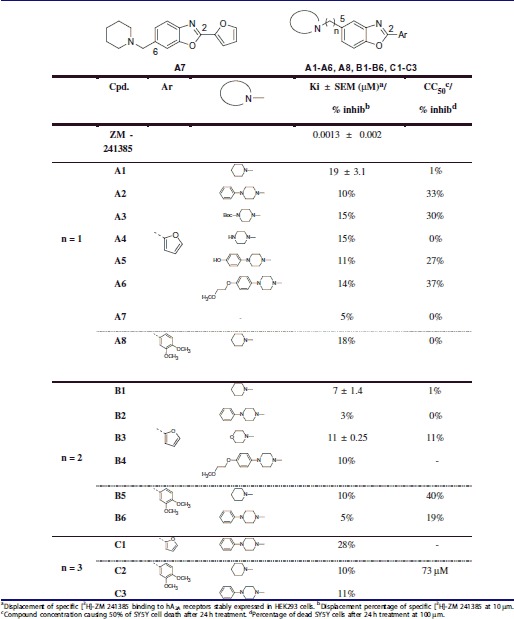
Table 1.
A2A receptor affinity and cytotoxicity data of compounds A1–8, B1–6 and C1–3.
To get molecules C1–3 (three-methylene linker), the same procedure was used starting from carboxylic acids 8a–b, obtained by malonic substitution performed on compounds 4a–b followed by a basic hydrolysis and then a decarboxylation reaction by heating in DMF.
To obtain molecules B5–6 (Scheme 3), ester function of compound 3e was first reduced to alcohol 10 using LiAlH4. The latter was then activated by the action of mesyl chloride to afford molecule 11. The classical nucleophilic substitution was then performed to give compounds B5–6.
To get target molecules with an amide (F1–4) or ethylamine linker (E1–2), 5-nitro benzoxazole derivative (3d) was reduced with hydrazine hydrate in the presence of Pd/C to afford compound D1 (Scheme 4). Nucleophilic substitution gave compounds E1 and 12. The latter was deprotected under H2 atmosphere afforded E2. To get molecules F1–3, treatment of D1 with bromoacetyl bromide gave compound 13 that was substituted with various amines. Compound F4 was obtained from F3 deprotection using 6 M HCl in MeOH.
Scheme 4.
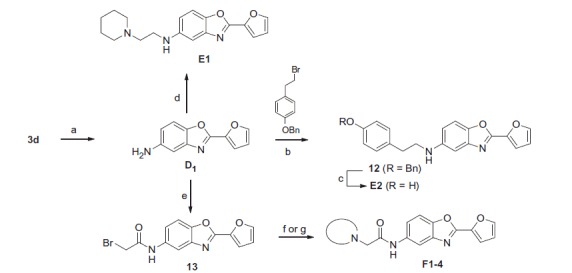
Reagents and conditions: (a) hydrazine hydrate, Pd/C (10%), EtOH, 81%; (b) 1-(benzyloxy)-4-(2-bromoethyl)benzene, K2CO3, DMF, 70 °C, 15%; (c) H2, Pd/C (10%), MeOH, 48%; (d) N-chloroethylpiperidine, K2CO3, DMF, 70 °C, 8%; (e) bromoacetylbromide, NEt3, DCM, 75%; (f) for F1–3, secondary amine, K2CO3, acetone, 48–90%; (g) for F4, i) boc-piperazine, K2CO3, acetone, ii) 6 M HCl, MeOH, 72%.
Structure-affinity relationship and early ADME studies
Affinities of benzoxazole derivatives for the human adenosine receptor were determined by a competitive radioligand displacement assay using [3H]-ZM241385 as radioligand18. All compounds were first screened at 10 μm concentration, and Ki values were determined for those exhibiting a specific displacement superior to 35%.
The first set of derivatives A1–8, allowed drawing some early SARs (Table 1). First, comparing molecules A1–6, piperidine emerged as the preferred amine since a dramatic decrease in affinity was observed with all other selected ones. Surprisingly, the phenylpiperazine group featured in many A2A antagonists was not tolerated in our series.
Regarding the position of the protonable amine function, compounds with a chain at the C-5 position appeared to exhibit a higher affinity than a compound with a chain at the C-6 position (molecules A1 versus A7). Therefore, the protonable amine was placed in the C-5 position for following molecules. Concerning the aryl in position 2 of the benzoxazole, replacing the furan of A1 by a 3,4-dimethoxyphenyl (A8) totally abolished affinity. This result is in agreement with literature since the preference of the adenosine A2A receptor for furan substituent is well documented even if the 3,4-dimethoxyphenyl is found on the Istradefylline molecule7,13.
Throughout Table 1, a similar tendency was observed for the other analogues suggesting that the piperidine chain at C-5 and the furan at C-2 of the benzoxazole ring are the best moieties for A2A receptor affinity.
Concerning the linker, the comparison between B1 and A1 revealed that increasing the length to two carbons is beneficial for affinity. Moreover, as can be seen from affinity data of E1 (11 μm) and F1 (1 μm) (Table 2), a three-atom linker is also well tolerated. When comparing the latter two compounds, rigidifying the linker by incorporating an amide in place of an amine allows an increase in affinity. Besides, F1 was found to be the most active compound in this series with a Ki value of 1 μm. As can be seen from Figure 2(C) the three-atom linker seems necessary to allow the interaction between the piperidine and Glu169. This ligand also exhibited a slight selectivity (see Table 4 of the Supplementary Material) versus A1 receptor (2.5-fold). Concerning the two others adenosine receptors, preliminary studies showed a probably good interaction with hA2B (73% inhibition at 10 μm) but a highly selectivity over adenosine A3 receptor (11% inhibition at 10 μm). These informations do not constitute a brake for the development of F1 as a potential drug candidate at this “hit” identification stage.
Table 2.
A2A receptor affinity and cytotoxicity data of compounds D1, E1–2 and F1–4.
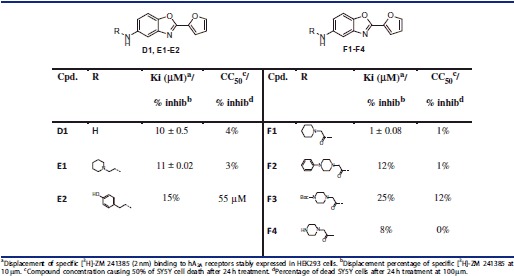
Interestingly, replacing the protonable amine of E1 by the aminoethyl phenol chain present in ZM-241385 (E2) led to a drastic drop in affinity, highlighting the importance of the tertiary amine in this series.
Finally, compound D1 (Ki = 10 μm), despite the lack of the protonable amine, could be considered a promising hit for future A2A antagonist development. Indeed, given its low molecular weight (MW = 200 g.mol−1), it offers multiple possibilities for further modifications.
The most interesting compounds of this series, F1 and D1, were subjected to preliminary pharmacokinetics studies (Table 3). As expected, molecule F1 displaying a protonable amine exhibits a higher solubility (184 μm) than molecule D1 (28 μm) in PBS solution at pH 7.4. When compared to reference A2A antagonists, these two hits show a higher solubility37–40. Indeed, Istradefylline (KW-6002) and Preladenant (SCH 42088), the two reference antagonists exhibit solubility values of 1.5 μm and 20 nm, respectively.
Table 3.
Preliminary ADME studies of F1.
| Permeability Caco-2-(10−6 cm/s)c |
|||||||
|---|---|---|---|---|---|---|---|
| Cpd. | Solubility (μm) PBS, pH 7.4a | PPB (%)b | A/B | B/A | LogD7.4d | HLMet1/2 (min) | Clintf (μl/min/mg) |
| F1 | 184 | 83 | 50 | 24 | 2.33 | 11 | 630 |
aEvaluated after 24 h stirring. bPPB = plasma protein binding. Compound was tested at 10 μm concentration. cPermeability = Compound was tested at 10 μm concentration at pH 6.5/7.4. dDetermined between a mixture PBS7.4/octanol. eHLM = human liver microsome. fClint = Compound was tested at 0.1 μm concentration.
Moreover, F1 displayed a good partition coefficient (log D7.4) of 2.33 which is within the same range as reference A2A antagonists currently in clinical studies41. These results confirm the importance of having a tertiary amine which allows a sharp increase in solubility while keeping a good log D7.4 value. At 10 μm concentration, a correct value of permeability coefficient (Caco-2 cells, pH 6.5/7.4) is observed (50 × 10−6 cm/s). The efflux ratio (B/AA/B) of 0.5 also suggested that F1 was probably distributed through P-glycoprotein (P-gp), an important transporter protein found in cell throughout the body. A good human plasma protein binding (mean of 83%) was also observed compared to reference A2A antagonist which expressed high PPB around 98%. Nevertheless, this compound also exhibited a high clearance and thus a short half time in human liver microsome at 0.1 μm.
Finally, no toxicity was observed for active compounds at 100 μm when tested on neuroblastoma cell lines (SY5Y, Tables 1,2).
Conclusions
Reported results showed a set of benzoxazole derivatives, diversely substituted at the C-2 and C-5 positions, as new “hits” molecules for adenosine A2A receptor. Among the synthesised compounds, those featured by a furan at the C-2 position combined with a piperidine and an amide-based linker at C-5 resulted in the most active compound (F1) toward the hA2AR (Ki = 1 μm). Furthermore, the latter presented good preliminary ADME properties with a very interesting solubility (184 μm) as well as good log D7.4 (2.33) without cytotoxicity at 100 μm. Thus, both F1 and D1, which can be easily modulated in position 7, appear to be promising starting points for further optimisation.
Supplementary Material
Acknowledgements
We express our thanks to the 300 MHz NMR facilities funded by the Region Nord-Pas de Calais (France), the Ministere de la Jeunesse, de l’Education Nationale et de la Recherche (MJENR) and the Fonds Europeens de Developpement Regional (FEDER). This work was supported by Lille 2 University, ANR “Adoratau”, Comue Univ Lille Nord de France. Romain Duroux is the recipient of a fellowship from Lille 2 University.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Fargo K, Bleiler L.. Alzheimer’s disease facts and figures. Alzheimers Dement 2014;10:47–92. [Google Scholar]
- 2.Panza F, Solfrizzi V, Barulli MR, et al. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: a systematic review. J Nutr Health Aging 2015;19:313–28. [DOI] [PubMed] [Google Scholar]
- 3.Solfrizzi V, Panza F, Imbimbo BP, et al. Coffee consumption habits and the risk of mild cognitive impairment: the Italian longitudinal study on aging. J Alzheimers Dis 2015;47:889–99. [DOI] [PubMed] [Google Scholar]
- 4.Flaten V, Laurent C, Coelho JE, et al. From epidemiology to pathophysiology: what about caffeine in Alzheimer’s disease? Biochem Soc Trans 2014;42:587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu K, Di Luca DG, Orrú M, et al. Neuroprotection by caffeine in the MPTP model of Parkinson’s disease and its dependence on adenosine A2A receptors. Neuroscience 2016;322:129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dall’Igna OP, Fett P, Gomes MW, et al. Caffeine and adenosine A(2a) receptor antagonists prevent beta-amyloid (25-35)-induced cognitive deficits in mice. Exp Neurol 2007;203:241–5. [DOI] [PubMed] [Google Scholar]
- 7.Preti D, Baraldi PG, Moorman AR, et al. History and perspectives of A2A adenosine receptor antagonists as potential therapeutic agents. Med Res Rev 2015;35:790–848. [DOI] [PubMed] [Google Scholar]
- 8.Shook BC, Jackson PF.. Adenosine A(2A) receptor antagonists and Parkinson’s disease. ACS Chem Neurosci 2011;2:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hauser RA, Olanow CW, Kieburtz KD, et al. Tozadenant (SYN115) in patients with Parkinson’s disease who have motor fluctuations on levodopa: a phase 2b, double-blind, randomised trial. Lancet Neurol 2014;13:767–76. [DOI] [PubMed] [Google Scholar]
- 10.Pinna A.Adenosine A2A receptor antagonists in Parkinson’s disease: progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 2014;28:455–74. [DOI] [PubMed] [Google Scholar]
- 11.Kondo T, Mizuno Y, Japanese Istradefylline Study Group . A long-term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin Neuropharmacol 2015;38:41–6. [DOI] [PubMed] [Google Scholar]
- 12.Laurent C, Burnouf S, Ferry B, et al. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol Psychiatry 2016;21:97–107. [DOI] [PubMed] [Google Scholar]
- 13.de Lera Ruiz M, Lim YH, Zheng J.. Adenosine A2A receptor as a drug discovery target. J Med Chem 2014;57:3623–50. [DOI] [PubMed] [Google Scholar]
- 14.Müller CE, Ferré S.. Blocking striatal adenosine A2A receptors: a new strategy for basal ganglia disorders. Recent Pat CNS Drug Discov 2007;2:1–21. [DOI] [PubMed] [Google Scholar]
- 15.Robinson SJ, Petzer JP, Terre’Blanche G, et al. 2-Aminopyrimidines as dual adenosine A1/A2A antagonists. Eur J Med Chem 2015;104:177–88. [DOI] [PubMed] [Google Scholar]
- 16.Tokuyama H, Okano K, Fujiwara H, et al. Total synthesis of dictyodendrins A-E. Chem Asian J 2011;6:560–72. [DOI] [PubMed] [Google Scholar]
- 17.Jones G, Willett P, Glen RC.. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol 1995;245:43–53. [DOI] [PubMed] [Google Scholar]
- 18.Lopes LV, Cunha RA, Ribeiro JA.. Cross talk between A1 and A2A adenosine receptors in the hippocampus and cortex of young adult and old rats. J Neurophysiol 1999;82:3196–203. [DOI] [PubMed] [Google Scholar]
- 19.Townsend-Nicholson A, Schofield PR.. A threonine residue in the seventh transmembrane domain of the human A1 adenosine receptor mediates specific agonist binding. J Biol Chem 1994;269:2373–6. [PubMed] [Google Scholar]
- 20.Stehle JH, Rivkees SC, Lee JJ, et al. Molecular cloning and expression of the cDNA for a novel A2-adenosine receptor subtype. Mol Endocrinol 1992;6:384–93. [DOI] [PubMed] [Google Scholar]
- 21.Salvatore CA, Jacobson MA, Taylor HE, et al. Molecular cloning and characterization of the human A3 adenosine receptor. Proc Natl Acad Sci U S A 1993;90:10365–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;46:3–26. [DOI] [PubMed] [Google Scholar]
- 23.Sangster J. Wiley series in solution chemistry. Volume 2. Chichester: John Wiley and Sons; 1997. [Google Scholar]
- 24.Banker MJ, Clark TH, Williams JA.. Development and validation of a 96-well equilibrium dialysis apparatus for measuring plasma protein binding. J Pharm Sci 2003;92:967. [DOI] [PubMed] [Google Scholar]
- 25.Hidalgo IJ, Raub TJ, Borchardt RT.. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989;96:736–49. [PubMed] [Google Scholar]
- 26.Obach RS, Baxter JG, Liston TE, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther 1997;283:46–58. [PubMed] [Google Scholar]
- 27.Jaakola VP, Griffith MT, Hanson MA, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008;322:1211–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu W, Chun E, Thompson AA, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012;337:232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Congreve M, Andrews SP, Doré AS, et al. Discovery of 1,2,4-triazine derivatives as adenosine A(2A) antagonists using structure based drug design. J Med Chem 2012;55:1898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabbadin D, Ciancetta A, Moro S.. Perturbation of fluid dynamics properties of water molecules during G protein-coupled receptor-ligand recognition: the human A2A adenosine receptor as a key study. J Chem Inf Model 2014;54:2846–55. [DOI] [PubMed] [Google Scholar]
- 31.Lenselink EB, Beuming T, Sherman W, et al. Selecting an optimal number of binding site waters to improve virtual screening enrichments against the adenosine A2A receptor. J Chem Inf Model 2014;54:1737–46. [DOI] [PubMed] [Google Scholar]
- 32.Keränen H, Gutiérrez-de-Terán H, Åqvist J.. Structural and energetic effects of A2A adenosine receptor mutations on agonist and antagonist binding. PLoS One 2014;9:e108492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan S, Hu Z, Filipek S, et al. W246(6.48) opens a gate for a continuous intrinsic water pathway during activation of the adenosine A2A receptor. Angew Chem Int Ed Engl 2015;54:556–9. [DOI] [PubMed] [Google Scholar]
- 34.Venkatakrishnan AJ, Deupi X, Lebon G, et al. Molecular signatures of G-protein-coupled receptors. Nature 2013;494:185–94. [DOI] [PubMed] [Google Scholar]
- 35.Wisastra R, Ghizzoni M, Boltjes A, et al. Anacardic acid derived salicylates are inhibitors or activators of lipoxygenases. Bioorg Med Chem 2012;20:5027–32. [DOI] [PubMed] [Google Scholar]
- 36.Wen X, Bakali JE, Deprez-Poulain R, et al. Efficient propylphosphonic anhydride (®T3P) mediated synthesis of benzothiazoles, benzoxazoles and benzimidazoles. Tetrahedron Lett 2012;53:2440–3. [Google Scholar]
- 37.Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau Ministry of Health Labour and Welfare [Online] 2015. Available from: https://www.pmda.go.jp/files/000153870.pdf/
- 38.Müller CE.Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem Biodivers 2009;6:2071–83. [DOI] [PubMed] [Google Scholar]
- 39.Slee DH, Zhang X, Moorjani M, et al. Identification of novel, water-soluble, 2-amino-N-pyrimidin-4-yl acetamides as A2A receptor antagonists with in vivo efficacy. J Med Chem 2008;51:400–6. [DOI] [PubMed] [Google Scholar]
- 40.Mikkelsen GK, Langgård M, Schroder TJ, et al. Synthesis and SAR studies of analogues of 4-(3,3-dimethyl-butyrylamino)-3,5-difluoro-N-thiazol-2-yl-benzamide (Lu AA41063) as adenosine A2A receptor ligands with improved aqueous solubility. Bioorg Med Chem Lett 2015;25:1212–16. [DOI] [PubMed] [Google Scholar]
- 41.Zheng J, Yang Z, Li X, et al. Optimization of 6-heterocyclic-2-(1H-pyrazol-1-yl)-N-(pyridin-2-yl)pyrimidin-4-amine as potent adenosine A2A receptor antagonists for the treatment of Parkinson’s disease. ACS Chem Neurosci 2014;5:674–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.