

Abstract
A new chemotype with carbonic anhydrase (CA, EC 4.2.1.1) inhibitory action has been discovered, the homo-sulfocoumarins (3H-1,2-benzoxathiepine 2,2-dioxides) which have been designed considering the (sulfo)coumarins as lead molecules. An original synthetic strategy of a panel of such derivatives led to compounds with a unique inhibitory profile and very high selectivity for the inhibition of the tumour associated (CA IX/XII) over the cytosolic (CA I/II) isoforms. Although the CA inhibition mechanism with these new compounds is unknown for the moment, we hypothesize that it may be similar to that of the sulfocoumarins, i.e. hydrolysis to the corresponding sulfonic acids which thereafter anchor to the zinc-coordinated water molecule within the enzyme active site.
Keywords: Carbonic anhydrase, sulfocoumarin, homo-sulfocoumarins, inhibitor
Introduction
Sulfocoumarins (1,2-benzoxathiine 2,2-dioxides) such as derivatives of type A were discovered by our groups to act as inhibitors of the metalloenzyme carbonic anhydrase (CA, EC 4.2.1.1)1,2. A large series of sulfocoumarins derivatives, among which compounds of type B, were thereafter reported, by using click chemistry or other conventional drug design approaches (Figure 1)3–6.
Figure 1.
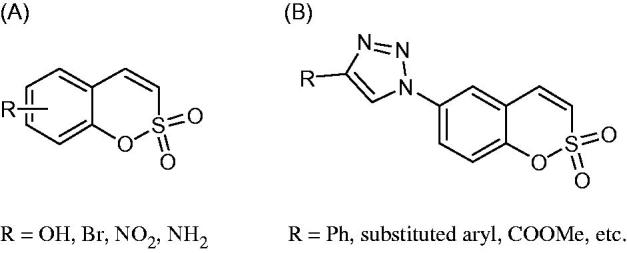
Chemical structure of sulfocoumarins A and B.
A salient feature of this type of CA inhibitor (CAI) was the fact that they showed a very pronounced isoform selectivity for inhibiting tumour-associated CA isoforms (CA IX and XII) over the widespread, cytosolic ones CA I and II1–3. This has been explained when the mechanism of CA inhibition with sulfocoumarins was elucidated, by using kinetic and X-ray crystallographic experiments1. Indeed, in the X-ray crystal structure of the adduct of a CA II/IX mimic complexed with the 6-bromosulfocoumarin A2(A, R = Br) (Figure 1), the 2-dihydroxy-5-bromophenyl-vinyl sulfonic acid D was observed within the enzyme active site, probably due to the CA-mediated hydrolysis of A2 to the cis-sulfonic acid C which was thereafter isomerized to the more stable trans-derivative D (Scheme 1)1.
Scheme 1.
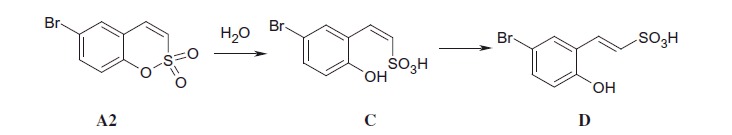
Active site, CA-mediated hydrolysis of A2 to D1.
This inhibition mechanism is similar to the one observed earlier for coumarins7,8 the class of CAIs which constituted the lead compounds for the discovery of sulfocoumarins. Finding isoform-selective CAIs for the 15 different human CA isoforms is a challenging task9,10, but coumarins and sulfocoumarins (and several families of sulfonamides) do show such properties, which make them of great interest for the design of pharmacological agents useful as diuretics, antiglaucoma, anticonvulsant and/or antitumor drugs9–13.
Here, we report the homo-sulfocoumarins or 3H-1,2-benzoxathiepine 2,2-dioxides, which can be considered as homologs of sulfocoumarins or 1,2-benzoxathiine 2,2-dioxides1, where oxathiine ring was expanded by one carbon to form an oxathiepine ring. To the best of our knowledge, there is no reported method for the synthesis of 3H-1,2-benzoxathiepine 2,2-dioxides in the literature. The general strategy for the formation of oxathiepine ring reported in this paper involves a ruthenium-catalysed olefin metathesis as a key step.
Materials and methods
Chemistry
Reagents, starting materials and solvents were obtained from commercial sources and used as received. Thin-layer chromatography was performed on silica gel, spots were visualized with UV light (254 and 365 nm). Melting points were determined on an OptiMelt automated melting point system. IR spectra were measured on Shimadzu FTIR IR Prestige-21 spectrometer. NMR spectra were recorded on Varian Mercury (400 MHz) spectrometer with chemical shifts values (δ) in ppm relative to TMS using the residual DMSO-d6 signal (1H 2.50; 13C 39.52) or CDCl3 signal (1H 7.26; 13C 77.16) as an internal standard. HRMS data were obtained with a Q-TOF micro high resolution mass spectrometer with ESI (ESI+/ESI). Elemental analyses were performed on a CARLO ERBA ELEMENTAL ANALYZER EA 1108.
General procedure for the synthesis of 4-substituted 2-ethenylphenoles (2a–c)14
To a stirred solution of methyltriphenylphosphonium bromide (2.64 eq.) in dry THF (5 ml/1 mmol of corresponding aldehyde), was added tBuOK (2.86–3.12 eq.) in several portions over 20 min. Reaction mixture was stirred for 1 h at RT. Corresponding 2-hydroxy benzaldehyde (1 eq.) was added and stirring continued at room temperature for 24 h. Reaction mixture was diluted with CH2Cl2 (5 ml/1 mmol aldehyde). Organic layer was collected and washed with water (2 × 20 ml) and brine (2 × 20 ml), dried over Na2SO4, solvent was driven off in vacuum. The crude product was purified by column chromatography (silica gel, EtOAc/PhMe1:5).
2-Ethenylphenol (2a)
Compound 2a was prepared according to the general procedure from methyltriphenylphosphonium bromide (18.88 g, 52.9 mmol), tBuOK (6.42 g, 57.2 mmol) and 2-hydroxybenzaldehyde (2.44 g, 20.0 mmol) as yellowish at room temperature melting solid (1.67 g, 70%).1HNMR (400 MHz, CDCl3) δ = 5.37 (dd, 1H, J = 11.3, 1.3 Hz), 5.42 (s, 1H), 5.76 (dd, 1H, J = 17.8, 1.3 Hz), 6.81 (dd, 1H, J = 8.1, 1.1 Hz), 6.90–6.96 (m, 1H), 6.98 (dd, 1H, J = 17.8, 11.3 Hz), 7.12–7.18 (m, 1H), 7.41 (dd, 1H, J = 7.7, 1.7 Hz).
4-Bromo-2-ethenylphenol (2b)
Compound 2b was prepared according to the general procedure from methyltriphenylphosphonium bromide (13.22 g, 37.0 mmol), tBuOK (4.90 g, 43.7 mmol) and 5-bromo-2-hydroxybenzaldehyde (2.81 g, 14.0 mmol) as yellowish at room temperature melting solid (1.64 g, 59%).1H NMR (400 MHz, CDCl3) δ = 4.98 (s, 1H), 5.40 (dd, 1H, J = 11.3, 1.0 Hz), 5.74 (dd, 1H, J = 17.8, 1.0 Hz), 6.68 (d, 1H, J = 8.6 Hz), 6.85 (dd, 1H, J = 17.8, 8.6 Hz), 7.23 (dd, 1H, J = 8.6, 2.4 Hz), 7.49 (d, 1H, J = 2.4 Hz).
2-Ethenyl-4-nitrophenol (2c)
Compound 2c was prepared according to the general procedure from methyltriphenylphosphonium bromide (28.31 g, 79.3 mmol), tBuOK (9.60 g, 85.6 mmol) and 5-nitro-2-hydroxybenzaldehyde (5 g, 30 mmol) as yellow at room temperature melting solid (3.23 g, 65%). 1H NMR (400 MHz, CDCl3) δ = 5.43 (dd, 1H, J = 11.3, 1.1 Hz), 5.87 (dd, 1H, J = 17.8, 1.1 Hz), 6.92–7.00 (m, 2H), 7.96 (dd, 1H, J = 8.9, 2.6 Hz), 8.31(d, 1H, J = 2.6 Hz), 8.82 (s, 1H).
Prop-2-ene-1-sulfonyl chloride (3)15
To a solution of 3-bromoprop-1-ene (24.2 g, 0.20 mol) in water (140 ml) was added Na2SO3 (30 g, 0.24 mol) and the reaction mixture was refluxed overnight. After cooling to room temperature, reaction mixture was washed with Et2O (3 × 35 ml). Aqueous phase was concentrated. Crude white solid was dried under high vacuum at 110 °C for 4 h. To the white solid at 0 °C POCl3 (80 ml) was added, and mixture was refluxed for 4 h. After cooling to room temperature dry THF (60 ml) was added and reaction mixture was vigorously stirred for 10 min and filtered. Filter cake was suspended in dry THF (60 ml), suspension was vigorously stirred for 10 min and filtered. Filtrates were combined and solvent was carefully driven off on rotary evaporator. Residue was distilled in vacuum (10 mbar) and fraction with boiling point 38–42 °C was collected, to give prop-2-ene-1-sulfonil chloride (3) as colourless oil (18.8 g, 67%).
General procedure for the synthesis of 4-substituted 2-ethenyl prop-2-ene-1-sulfonates (4a–c)
To a stirred solution of corresponding 2-ethenylphenol 2 (1 eq.) in CH2Cl2 (10 ml/20 mmol phenol) at 0 °C was added prop-2-ene-1-sulfonyl chloride (3) (1.6 eq.) and Et3N (1.5 eq.). Reaction mixture was stirred overnight (20 h) at room temperature. Water (10 ml/20 mmol phenol) was added, reaction mixture was extracted with EtOAc (3 × 10 ml/20 mmol phenol), combined organic extracts were washed with brine (2 × 10 ml/20 mmol olefin), dried over Na2SO4, filtered and solvent was driven off in vacuum. The crude product was purified by column chromatography (silica gel, CH2Cl2/PhMe 3:2).
2-Ethenylphenyl prop-2-ene-1-sulfonate (4a)
Compound 4a was prepared according to the general procedure from 2-ethenylphenol (2a) (0.50 g, 4.16 mmol), prop-2-ene-1-sulfonyl chloride (3) (0.94 g, 6.69 mmol) and Et3N (0.87 ml, 6.23 mmol) as colourless oil (0.52 g, 56%). IR (film, cm−1) νmax= 1368 (S=O), 1178 (S=O), 1154 (S=O); 1H NMR (400 MHz, CDCl3) δ = 3.96–4.00 (m, 2H), 5.37–5.41 (m, 1H), 5.48–5.54 (m, 2H), 5.79 (dd, 1H, J = 17.6, 0.9 Hz), 5.90–6.01 (m, 1H), 6.99 (dd, 1H, J = 17.6, 11.0 Hz), 7.23–7.34 (m, 2H), 7.57–7.62 (m, 1H); 13C NMR (100 MHz, CDCl3) δ = 55.6, 117.3, 122.8, 123.9, 125.4, 126.9, 127.4, 129.2, 130.3, 131.3, 146.5; HRMS (ESI) m/z [M − 1]− calcd for C11H11O3S: 223.0429, found 223.0435.
4-Bromo-2-ethenylphenyl prop-2-ene-1-sulfonate (4b)
Compound 4b was prepared according to the general procedure from 4-bromo-2-ethenylphenol (2b) (0.50 g, 2.51 mmol), prop-2-ene-1-sulfonyl chloride (3) (0.57 g, 4.05 mmol) and Et3N (0.52 ml, 3.76 mmol) as colourless oil (0.51 g, 67%). IR (film, cm−1) νmax= 1364 (S=O), 1170 (S=O), 1154 (S=O); 1H NMR (400 MHz, CDCl3) δ = 4.00 (dt, 2H, J = 7.4, 0.9 Hz), 5.46 (d, 1H, J = 11.0 Hz), 5.51–5.59 (m, 2H), 5.81 (d, 1H, J = 17.6 Hz), 5.91–6.03 (m, 1H), 6.92 (dd, 1H, J = 17.6, 11.0 Hz), 7.22 (d, 1H, J = 8.6 Hz), 7.41 (dd, 1H, J = 8.6, 2.4 Hz), 7.73 (d, 1H, J = 2.4 Hz); 13C NMR (100 MHz, CDCl3) δ = 55.7, 118.6, 121.0, 123.7, 124.6, 125.7, 129.2, 129.8, 132.0, 133.3, 145.3;HRMS (ESI) m/z [M − 1]− calcd for C11H10BrO3S: 300.9534, found 300.9537.
2-Ethenyl-4-nitrophenyl prop-2-ene-1-sulfonate (4c)
Compound 4c was prepared according to the general procedure from 2-ethenyl-4-nitrophenol (2c) (0.32 g, 1.94 mmol), prop-2-ene-1-sulfonyl chloride (3) (0.44 g, 3.13 mmol) and Et3N (0.41 ml, 2.96 mmol) as yellowish oil (0.30 g, 57%). IR (film, cm−1) νmax= 1350 (S=O), 1159 (S=O); 1H NMR (400 MHz, CDCl3) δ = 4.01 (dt, 2H, J = 7.2, 0.9 Hz), 5.54–5.63 (m, 3H), 5.93–6.05 (m, 2H), 6.99 (dd, 1H, J = 17.6, 11.0 Hz), 7.53 (d, 1H, J = 9.0 Hz), 8.16 (dd, 1H, J = 9.0, 2.8 Hz), 8.48 (d, 1H, J = 2.8 Hz); 13C NMR (100 MHz, CDCl3) δ = 56.3, 120.2, 122.4, 123.4, 123.8, 124.0, 126.2, 128.6, 132.8, 146.5, 150.2; HRMS (ESI) m/z [M − 1]− calcd for C11H10NO5S: 268.0280, found 268.0280.
General procedure for the synthesis of 7-substitued 3H-1,2-benzoxathiepine 2,2-dioxides (6a–c)
To a stirred solution of corresponding 4-substituted 2-ethenyl prop-2-ene-1-sulfonate (1 eq.) in dry toluene (10 ml/0.2 g 4), was added Ru-catalyst 5 (tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene][3-phenyl-1H-inden-1-ylidene]ruthenium(II) dichloride, CAS Nr. 254972–49-1) (0.05 eq.). Reaction mixture was stirred at 70 °C for 4 h. Solvent was driven off in vacuum and the crude product was purified by column chromatography (silica gel, Hex/EtOAc 4:1) with following re-crystallization from EtOAc/Hex. Compound 6c was purified by column chromatography (silica gel, CH2Cl2/Hex 2:1).
3H-1,2-benzoxathiepine 2,2-dioxide (6a)
Compound 5a was prepared according to the general procedure from 2-ethenylphenyl prop-2-ene-1-sulfonate (4a) (100 mg, 0.45 mmol), Ru-catalyst 5 (21 mg, 0.022 mmol) as white solid (76 mg, 87%). Mp 131–132 °C. IR (film, cm−1) νmax= 1369 (S=O), 1176 (S=O); 1H NMR (400 MHz, CDCl3) δ = 4.01 (dd, 2H, J = 6.3, 1.2 Hz), 5.96–6.03 (m, 1H), 6.90 (d, 1H, J = 10.9 Hz), 7.31–7.37 (m, 3H), 7.41–7.46 (m, 1H); 13C NMR (100 MHz, CDCl3) δ = 51.2, 119.5, 123.0, 127.3, 128.4, 130.6, 130.8, 132.9, 147.8;HRMS (ESI) m/z [M − 1]− calcd for C9H7O3S: 195.0116, found 195.0115.
7-Bromo-3H-1,2-benzoxathiepine 2,2-dioxide (6b)
Compound 5b was prepared according to the general procedure from 4-bromo-2-ethenylphenyl prop-2-ene-1-sulfonate (4b) (100 mg, 0.33 mmol), Ru-catalyst 5 (16 mg, 0.017 mmol) as yellowish solid (76 mg, 84%). Mp 129.3–130.3 °C. IR (film, cm−1) νmax= 1360 (S=O), 1170 (S=O), 1154 (S=O); 1H NMR (400 MHz, CDCl3) δ = 4.03 (dd, 2H, J = 6.3, 0.9 Hz), 5.99–6.06 (m, 1H), 6.81 (d, 1H, J = 11.0 Hz), 7.22 (d, 1H, J = 8.6 Hz), 7.47 (d, 1H, J = 2.4 Hz), 7.54 (dd, 1H, J = 8.6, 2.4 Hz); 13C NMR (100 MHz, CDCl3) δ = 51.4, 120.5, 120.9, 124.7, 130.2, 131.6, 133.5, 133.6, 146.7; Anal. Calcd for C9H7BrO3S (275.12): C 39.29, H 2.56, found C 39.19, H 2.59.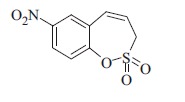
7-Nitro-3H-1,2-benzoxathiepine 2,2-dioxide (6c)
Compound 5c was prepared according to the general procedure from 2-ethenyl-4-nitrophenyl prop-2-ene-1-sulfonate (4c) (100 mg, 0.37 mmol), catalyst 5 (18 mg, 0.019 mmol) as yellowish solid (86 mg, 96%). Mp 130–131 °C. IR (film, cm−1) νmax= 1375 (S=O), 1351 (S=O), 1170 (S=O), 1161 (S=O); 1H NMR (400 MHz, CDCl3) δ = 4.18 (dd, 2H, J = 5.8, 1.2 Hz), 6.05–6.12 (m, 1H), 6.89 (d, 1H, J = 11.3 Hz), 7.48 (d, 1H, J = 8.9 Hz),8.24 (d, 1H, J = 2.6 Hz), 8.28 (dd, 1H, J = 8.9, 2.6 Hz); 13C NMR (100 MHz, CDCl3) δ = 52.4, 121.6, 124.3, 125.6, 126.8, 129.4, 130.8, 151.3; Anal. Calcd for C9H7NO5S (241.22): C 44.81, H 2.92, N 5.81, found C 44.70, H 2.95, N 5.79.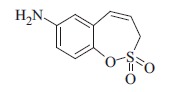
7-Amino-3H-1,2-benzoxathiepine 2,2-dioxide (7)
To a solution of 7-nitro-3H-1,2-benzoxathiepine 2,2-dioxide (6c) (250 mg, 1.04 mmol) in EtOH (4.3 ml) and H2O (2.8 ml) AcOH (0.06 ml, 1.04 mmol) was added following by iron powder (350 mg, 6.27 mmol) at room temperature. Resulting suspension was stirred at 75 °C for 1 h. It was cooled to room temperature, EtOAc (50 ml) was added and washed with sat. aq. NaHCO3 (5 × 30 ml). Organic layer was dried over Na2SO4 and concentrated in vacuum. Re-crystallized of the crude product from EtOAc/Hex afforded 7 (220 mg, 98%) as yellowish solid. Mp 170–171 °C. IR (film, cm−1) νmax=3465 (N–H), 3382 (N–H), 1358 (S=O), 1163 (S=O); 1H NMR (400 MHz, CDCl3) δ = 3.72–3.85 (br s,2H), 3.92 (dd, 2H, J = 6.3, 1.0 Hz), 5.93–6.00 (m, 1H), 6.53 (d, 1H, J = 2.9 Hz), 6.68 (dd, 1H, J = 8.8, 2.6 Hz), 6.80 (d, 1H, J = 10.6 Hz), 7.12 (d, 1H, J = 8.8 Hz); 13C NMR (100 MHz, CDCl3) δ = 50.5, 115.0, 116.8, 119.8, 123.8, 133.4, 140.4, 145.5; HRMS (ESI) m/z [M + H]+ calcd for C9H10NO3S: 212.0381, found 212.0364.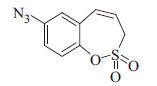
7-Azido-3H-1,2-benzoxathiepine 2,2-dioxide (8)
To a solution of -7-amino-3H-1,2-benzoxathiepine 2,2-dioxide (7) (220 mg, 1.03 mmol) in trifluoroacetic acid (1.3 ml) at 0 °C, slowly was added NaNO2 (80 mg, 1.12 mmol). After 30 min stirring at 0 °C, solution of NaN3 (67 mg, 1.03 mmol) in water (3 ml) was added. Mixture was stirring at 0 °C for1 h. Collection of solid precipitate and drying in vacuum afforded 8 (170 mg, 69%) as brown solid. IR (film, cm−1) νmax= 2116 (N3), 1374 (S=O), 1369 (S=O), 1167 (S=O); 1H NMR (400 MHz, CDCl3) δ = 4.01 (dd, 2H, J = 6.3, 1.2 Hz), 5.99–6.07 (m, 1H), 6.83 (d, 1H, J = 10.9 Hz), 6.94 (d, 1H, J = 2.8 Hz), 7.06 (dd, 1H, J = 8.9, 2.8 Hz), 7.32 (d, 1H, J = 8.9 Hz); 13C NMR (100 MHz, CDCl3) δ = 51.2, 120.5, 120.8, 120.9, 124.5, 129.8, 132.0, 139.2, 144.5.
General procedure for the synthesis of 1,4-disubstitutedtriazolyl compound (9–17)
To a solution of corresponding alkyne (1 eq.) in tBuOH/H2O 1:1 mixture (10 ml)7-azido-3H-1,2-benzoxathiepine 2,2-dioxide (8) (1 eq.), CuSO4·5H2O (2 eq.) and sodium ascorbate (4 eq.) were added and reaction mixture was stirred at room temperature for 10 min. AcOH (19–21 eq.) was added and mixture was stirred for additional 30 min. Solvent was driven off in vacuum and the crude product was purified by reversed phase chromatography (C-18, H2O–MeCN gradient MeCN 10–90%).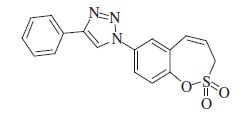
1-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-4-phenyl-1H-1,2,3-triazole (9)
Compound 9 was prepared according to the general procedure from phenylacetylene (13 mg, 0.13 mmol), azide 8 (30 mg, 0.13 mmol), CuSO4·5H2O (65 mg, 0.26 mmol), sodium ascorbate (103 mg, 0.52 mmol), AcOH (0.14 ml, 2.45 mmol) as white solid (41 mg, 95%). Mp 203–204 °C. IR (KBr, cm−1) νmax=1368 (S=O), 1171 (S=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.61 (dd, 2H, J = 5.9, 1.2 Hz), 6.09–6.16 (m, 1H), 7.02 (d, 1H, J = 11.3 Hz), 7.37–7.43 (m, 1H), 7.48–7.54 (m, 2H), 7.63 (d, 1H, J = 8.8 Hz), 7.92–7.97 (m, 2H), 8.04 (dd, 1H, J = 8.8, 2.6 Hz), 8.13 (d, 1H, J = 2.6 Hz), 9.35 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 51.7, 119.9, 121.6, 122.1, 122.7, 124.0, 125.3, 128.4, 129.1, 129.6, 130.0, 130.1, 135.0, 146.3, 147.5; HRMS (ESI) m/z [M + H]+ calcd for C17H14N3O3S: 340.0756, found 340.0755.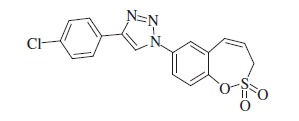
4-(4-Chlorophenyl)-1–(2,2-dioxido-3H-1,2-benzoxathiepin-7-yl)-1H-1,2,3-triazole (10)
Compound 10 was prepared according to the general procedure from 1-chloro-4-ethynylbenzene (17 mg, 0.12 mmol), azide 8 (29 mg, 0.12 mmol), CuSO4·5H2O (61 mg, 0.24 mmol), sodium ascorbate (97 mg, 0.49 mmol), AcOH (0.13 ml, 2.27 mmol) as yellowish solid (34 mg, 74%). Mp 191–192 °C. IR (KBr, cm−1) νmax=1369 (S=O), 1356 (S=O), 1168 (S=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.61 (dd, 2H, J = 5.9, 1.2 Hz), 6.09–6.16 (m, 1H), 7.01 (d, 1H, J = 11.5 Hz), 7.55–7.61 (m, 2H), 7.63 (d, 1H, J = 8.9 Hz), 7.92–7.98 (m, 2H), 8.02 (dd, 1H, J = 8.9, 2.7 Hz), 8.11 (d, 1H, J = 2.7 Hz), 9.38 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 51.7, 120.3, 121.6, 122.1, 122.7, 124.1, 127.0, 129.0, 129.1, 129.6, 130.1, 132.8, 135.0, 146.3, 146.4; HRMS (ESI) m/z [M + H]+ calcd for C17H13ClN3O3S: 374.0366, found 374.0366.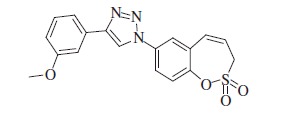
1-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-4-(3-methoxyphenyl)-1H-1,2,3-triazole (11)
Compound 11 was prepared according to the general procedure from 3-ethynylanisole (17 mg, 0.13 mmol), azide 8 (30 mg, 0.13 mmol), CuSO4·5H2O (63 mg, 0.25 mmol), sodium ascorbate (100 mg, 0.50 mmol), AcOH (0.14 ml, 2.45 mmol) as yellowish solid (24 mg, 51%). Mp210–211 °C.IR (KBr, cm−1) νmax=1372 (S=O), 1162 (S=O); 1H NMR (400 MHz, DMSO-d6) δ = 3.84 (s, 3H), 4.61 (dd, 2H, J = 5.8, 1.2 Hz), 6.09–6.16 (m, 1H), 6.94–6.99 (m, 1H), 7.02 (d, 1H, J = 11.5 Hz), 7.39–7.45 (m, 1H), 7.48–7.55 (m, 2H), 7.63 (d, 1H, J = 8.9 Hz), 8.03 (dd, 1H, J = 8.9, 2.7 Hz), 8.12 (d, 1H, J = 2.7 Hz), 9.36 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 51.7, 55.2, 110.6, 114.1, 117.6, 120.1, 121.6, 122.1, 122.6, 124.0, 129.6, 130.1,130.2, 131.4, 135.0, 146.3, 147.4, 159.8; HRMS (ESI) m/z [M + H]+ calcd for C18H16N3O4S: 370.0862, found 370.0876.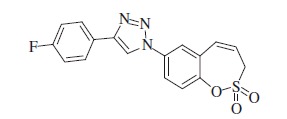
1-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-4–(4-fluorophenyl)-1H-1,2,3-triazole (12)
Compound 12 was prepared according to the general procedure from 1-ethynyl-4-fluorobenzene (30 mg, 0.25 mmol), azide 8 (60 mg, 0.25 mmol), CuSO4·5H2O (126 mg, 0.50 mmol), sodium ascorbate (200 mg, 1.02 mmol), AcOH (0.28 ml, 5.05 mmol) as yellowish solid (60 mg, 66%). Mp 200–201 °C. IR (KBr, cm−1) νmax=1369 (S=O), 1167 (S=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.61 (d, 2H, J = 5.4 Hz), 6.07–6.17 (m, 1H), 7.01 (d, 1H, J = 11.3 Hz), 7.30–7.71 (m, 2H), 7.63 (d, 1H, J = 8.8 Hz), 7.94–8.05 (m, 3H), 8.11 (s, 1H), 9.34 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 51.7, 116.1 (d, J = 21.9 Hz), 119.9, 121.6, 122.1, 122.7, 124.1, 126.6, 127.4 (d, J = 8.3 Hz), 129.7, 130.1, 135.0, 146.3, 146.6, 162.1 (d, J = 245.3 Hz); HRMS (ESI) m/z [M + H]+ calcd for C17H13FN3O3S: 358.0662, found 358.0656.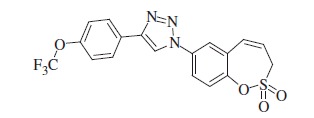
1-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-4-[4-(trifluorometoxy)phenyl]-1H-1,2,3-triazole (13)
Compound 13 was prepared according to the general procedure from 4-(trifluoromethoxy) phenylacetylene (40 mg, 0.21 mmol), azide 8 (50 mg, 0.21 mmol), CuSO4·5H2O (105 mg, 0.42 mmol), sodium ascorbate (167 mg, 0.84 mmol), AcOH (0.23 ml, 4.02 mmol) as yellowish solid (74 mg, 83%). Mp 168–169 °C. IR (film, cm−1) νmax= 1357 (S=O), 1166 (S=O); 1H NMR (400 MHz, CDCl3) δ = 4.13 (dd, 2H, J = 6.0, 1.1Hz), 6.06–6.13 (m, 1H), 6.93 (d, 1H, J = 11.3 Hz), 7.30–7.35 (m, 2H), 7.51 (d, 1H, J = 8.8 Hz), 7.79 (dd, 1H, J = 8.8, 2.5 Hz), 7.85 (d, 1H, J = 2.5 Hz), 7.91–7.98 (m, 2H), 8.25 (s, 1H); 13C NMR (100 MHz, CDCl3) δ = 51.8, 120.6 (q, J = 257.9 Hz), 121.4, 121.7, 122.1, 122.9, 124.7, 127.5, 128.8, 130.0, 131.5, 135.6, 147.3, 149.5, 149.6; HRMS (ESI) m/z [M + H]+ calcd for C18H13F3N3O4S: 424.0579, found 424.0553.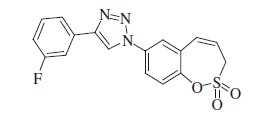
1-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-4–(3-fluorophenyl)-1H-1,2,3-triazole (14)
Compound 14 was prepared according to the general procedure from 1-ethynyl-3-fluorobenzene (25 mg, 0.21 mmol), azide 8 (50 mg, 0.21 mmol), CuSO4·5H2O (105 mg, 0.42 mmol), sodium ascorbate (166 mg, 0.84 mmol), AcOH (0.25 ml, 4.37 mmol) as brownish solid (56 mg, 74%). Mp 188–189 °C. IR (KBr, cm−1) νmax=1354 (S=O), 1175 (S=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.62 (dd, 2H, J = 6.0, 1.3 Hz), 6.09–6.16 (m, 1H), 7.01 (d, 1H, J = 11.6 Hz), 7.20–7.26 (m, 1H), 7.52–7.60 (m, 1H), 7.64 (d, 1H, J = 8.8 Hz), 7.70–7.75 (m, 1H), 7.77–7.81 (m, 1H), 8.02 (dd, 1H, J = 8.9, 2.7 Hz), 8.10 (d, 1H, J = 2.7 Hz), 9.42 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 51.7, 111.9 (d, J = 23.0 Hz), 115.1 (d, J = 20.8 Hz), 120.7, 121.3 (d, J = 2.5 Hz), 121.6, 122.1, 122.7, 124.1, 129.6, 130.0, 131.2 (d, J = 8.7 Hz), 132.4 (d, J = 8.4 Hz), 134.9, 146.3, 146.4, 162.6 (d, J = 243.5 Hz); HRMS (ESI) m/z[M + H]+ calcd for C17H13FN3O3S: 358.0662, found 358.0667.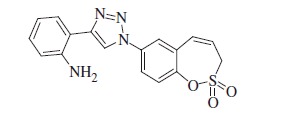
2-[1-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-1H-1,2,3-triazol-4-yl]aniline (15)
Compound 15 was prepared according to the general procedure from 2-ethynylaniline (25 mg, 0.21 mmol), azide 8 (50 mg, 0.21 mmol), CuSO4·5H2O (105 mg, 0.42 mmol), sodium ascorbate (166 mg, 0.84 mmol), AcOH (0.25 ml, 4.37 mmol) as yellowish solid (43 mg, 57%). Mp 190–191 °C. IR (film, cm−1) νmax=3430 (N–H), 3364 (N–H), 1365 (S=O), 1358 (S=O), 1167 (S=O), 1163 (S=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.61 (dd, 2H, J = 6.0, 1.2 Hz), 6.09–6.16 (m, 1H), 6.49–6.85 (m, 2H), 7.01 (d, 1H, J = 11.3 Hz), 7.10–7.18 (m, 1H), 7.59–7.66 (m, 2H), 8.08 (dd, 1H, J = 8.9, 2.4 Hz), 8.16 (d, 1H, J = 2.4 Hz), 9.26 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 51.7, 112.1, 115.9, 116.1, 119.8, 121.8, 122.1, 122.8, 124.0, 127.9, 129.0, 129.6, 130.1, 135.0, 145.8, 146.3, 148.1; HRMS (ESI) m/z[M + H]+ calcd for C17H15N4O3S: 355.0865, found 355.0869.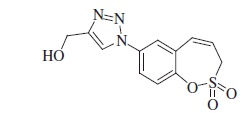
[1-(2,2-dioxido-3H-1,2-benzoxathiepin-7-yl)-1H-1,2,3-triazol-4-yl]methanol (16)
Compound 16 was prepared according to the general procedure from propargyl alcohol (0.012 ml, 0.21 mmol), azide 8 (50 mg, 0.21 mmol), CuSO4·5H2O (105 mg, 0.42 mmol), sodium ascorbate (166 mg, 0.84 mmol), AcOH (0.25 ml, 4.37 mmol) as white solid (50 mg, 81%). Mp 144–145 °C. IR (KBr, cm−1) νmax= 1374 (S=O), 1167 (S=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.59 (d, 2H, J = 5.7 Hz), 4.62 (s, 2H), 6.05–6.13 (m, 1H), 6.98 (d, 1H, J= 11.5 Hz), 7.56 (d, 1H, J = 8.9 Hz), 7.99 (dd, 1H, J = 8.9, 2.6 Hz), 8.09 (d, 1H, J = 2.6 Hz), 8.74 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 51.8, 54.9, 121.3, 121.5, 121.9, 122.6, 123.9, 129.5, 130.1, 135.1, 146.1, 149.4; HRMS (ESI) m/z [M + H]+ calcd for C12H12N3O4S: 294.0549, found 294.0553.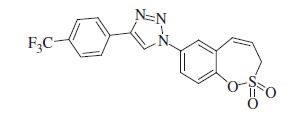
4-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-1-[4-(trifluoromethyl)phenyl]-1H-1,2,3-triazole (17)
Compound 17 was prepared according to the general procedure from 4-(trifluoromethyl)phenylacetylene (36 mg, 0.21 mmol), azide 8 (50 mg, 0.21 mmol), CuSO4·5H2O (105 mg, 0.42 mmol), sodium ascorbate (166 mg; 0.84 mmol), AcOH (0.25 ml, 4.37 mmol) as yellowish solid (73 mg, 85%). Mp 192–193 °C.IR (KBr, cm−1) νmax= 1358 (S=O), 1328 (S=O), 1174 (S=O), 1166 (S=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.62 (dd, 2H, J = 5.9, 1.0 Hz), 6.09–6.16 (m, 1H), 7.01 (d, 1H, J = 11.5 Hz), 7.64 (d, 1H, J = 8.8 Hz), 7.86–7.91 (m, 2H), 8.04 (dd, 1H, J = 8.8, 2.7 Hz), 8.13 (d, 1H, J = 2.7 Hz), 8.13–8.18 (m, 2H), 9.52 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 51.7, 121.2, 121.7, 122.1, 122.8, 124.1, 124.2(q, J = 272.0 Hz), 125.8, 126.1 (q, J = 3.8 Hz), 128.4 (q, J = 32.0 Hz), 129.6, 130.0, 134.0, 134.9, 146.0, 146.4; HRMS (ESI) m/z [M + H]+ calcd for C18H13F3N3O3S: 408.0630, found 408.0626.
CA inhibition assay
An SX.18 MV-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic/inhibition of various CA isozymes16. Phenol Red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.4) as buffer, 0.1 M Na2SO4 or NaClO4 (for maintaining constant the ionic strength; these anions are not inhibitory in the used concentration),17 following the CA-catalysed CO2 hydration reaction for a period of 5–10 s. Saturated CO2 solutions in water at 25 °C were used as substrate. Stock solutions of inhibitors were prepared at a concentration of 10 mM (in DMSO-water 1:1, v/v) and dilutions up to 1 nM done with the assay buffer mentioned above. At least seven different inhibitor concentrations have been used for measuring the inhibition constant. Inhibitor and enzyme solutions were preincubated together for 6 h at 4 °C prior to assay, in order to allow for the formation of the E–I complex. Triplicate experiments were done for each inhibitor concentration, and the values reported throughout the paper are the mean of such results. The inhibition constants were obtained by non-linear least-squares methods using the Cheng–Prusoff equation, as reported earlier17, and represent the mean from at least three different determinations. All CA isozymes used here were recombinant proteins obtained as reported earlier by our group18.
X-ray structure determination
X-Ray diffraction data for compound 6c were collected using a NoniusKappaCCD diffractometre (MoKα radiation, λ = 0.71073 Å), equipped with low temperature Oxford CryosystemsCryostream Plus device (Delft, the Netherlands). Data were collected using KappaCCD Server Software, cell refined by SCALEPACK19, data reduction performed by DENZO20 and SCALEPACK19, structures solved by direct method using SIR2004 and refined by SHELXL9721 as implemented in the program package WinGX22. Software used to prepare CIF file was SHELXL9721 and graphics–ORTEP322.
Crystal data for6c: C9H7NO5S (M = 241.22), monoclinic, P21/a, a = 7.3194(3), b = 14.9000(7) and c = 18.3387(8) Å, β = 101.325(1)°, V = 1961.06(15) Å3, T = 173(2) K, Z = 2, Z' = 1, μ(MoKα) = 0.34 mm−1, 9545 reflections measured, 2150 independent reflections (Rint = 0.083), R1(obs) = 0.058, wR1(obs) = 0.1500, R1(all) = 0.1893, wR1(all) = 0.1096, S = 0.94.
CCDC 1526002 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk.
Results and discussion
Chemistry
The synthesis of homo-sulfocoumarins began with a Wittig reaction in which salicylic aldehydes 1 were converted to the corresponding mono-olefins 2a–c in good yields (Scheme 2). Treatment of compounds 2a–c with allyl sulfonyl chloride (3) provided bis-olefins 4a–c as the key intermediates, again in good yields (see Experimental for details). In the next step, olefin metathesis with the commercially available Ru-catalyst 5 was used, in which bis-olefins 4a–c were converted to 3H-1,2-benzoxathiepine 2,2-dioxides 6a–b in 84–96% yields. To obtain a series of 7-substituted homo-sulfocoumarins, the synthesis of 1,4-triazolyl derivatives 9–17 was thereafter performed. For this purpose, 7-nitro derivative 6c was reduced by elemental iron to the corresponding amine 7 in nearly quantitative yield. Further diazotation of amine 7 followed by in situ treatment with sodium azide afforded the azide 8. Treatment of azide 8 with alkynes under click chemistry condition provides a series of 1,4-triazolyl homo-sulfocoumarins 9–17 in good to excellent yields (see Experimental for details).
Scheme 2.
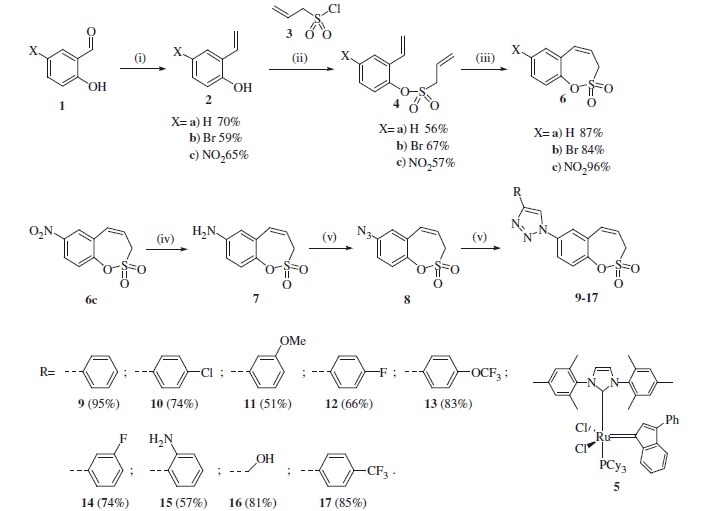
Reagents and conditions: (i) MePPh3Br, tBuOK, THF, RT, 24 h; (ii) NEt3, CH2Cl2, RT, 20 h; (iii) 5, toluene, 70 °C, 4 h; (iv) Fe, AcOH, EtOH, H2O, 70 °C, 1 h, 98%; (v) 1) NaNO2, H2O, TFA, 2) NaN3, H2O, 69%; (vi) alkyne, tBuOH/H2O (1:1), CuSO4, sodium ascorbate, acetic acid, 30 min.
The structures of all synthesized 3H-1,2-benzoxathiepine 2,2-dioxides 6–17 were fully supported by 1H, 13C NMR and IR spectroscopy, MS or elemental analysis. Additionally, the final unequivocal identification of the scaffold of 3H-1,2-benzoxathiepine 2,2-dioxide was established by a single-crystal X-ray structure for compound 6c, shown in Figure 2.
Figure 2.

Single-crystal X-ray structure of 6c (CCDC deposition number 1526002). Thermal ellipsoids are drawn at the 50% probability level (see Experimental for details).
Carbonic anhydrase inhibition
All the synthesized derivatives 6c–17 were evaluated for their efficacy in inhibiting four relevant CA isoforms, i.e. hCA I, II, IX and XII, by using the stopped flow carbon dioxide hydrase assay16, in comparison to the sulphonamide acetazolamide (AAZ, 5-acetamido-1,3,4-thiadiazole-2-sulfonamde) as a standard CAI.
Data of Table 1 show that the cytosolic isoforms hCA I and II (widely distributed enzymes, with important physiological roles in many tissues)9,10 were generally not inhibited by the investigated homo-sulfocoumarins, up to 50 μM concentration of inhibitors in the assay system. Only one derivative, 13, showed a moderate inhibitory profile against hCA II, with an inhibition constant of 5.77 μM.
Table 1.
CA inhibition data against isoforms hCA I, II, IX and XII with homo-sulfocoumarins 6–17 and acetazolamide (AAZ) as standard, by a stopped-flow CO2 hydrase assay14.
| KI (μM)a |
||||
|---|---|---|---|---|
| Compound | hCA I | hCA II | hCA IX | hCA XII |
| 6c | >50 | >50 | 0.027 | 0.64 |
| 7 | >50 | >50 | 3.57 | >50 |
| 9 | >50 | >50 | 1.71 | >50 |
| 10 | >50 | >50 | 3.59 | >50 |
| 11 | >50 | >50 | 2.56 | >50 |
| 12 | >50 | >50 | 1.75 | >50 |
| 13 | >50 | 5.77 | 0.34 | 1.72 |
| 14 | >50 | >50 | 1.15 | >50 |
| 15 | >50 | >50 | 0.46 | 2.32 |
| 16 | >50 | >50 | 0.87 | >50 |
| 17 | >50 | >50 | 0.43 | >50 |
| AAZ | 0.25 | 0.012 | 0.025 | 0.006 |
Errors in the range of ±5% of the reported values, from three different assays.
The tumour associated isoform hCA IX, a validated drug target for antitumor/antimetastatic agents23,24, was on the other hand effectively inhibited by the investigated homo-sulfocoumarins, with KIs ranging between 27 nM and 3.59 μM (Table 1). The structure activity relationship (SAR) was very interesting, as the best inhibitor (6c) incorporated a compact, powerful electron attracting moiety (NO2) whereas the remaining derivatives, incorporating substituted 1,2,3-triazole moieties in position 7 of the homo-sulfocoumarin ring were less effective hCA IX inhibitors. Four submicromolar hCA IX inhibitors were however detected apart 6c, derivatives 13, 15, 16 and 17, which incorporate either the compact hydroxymethyl group at the triazole fragment of the molecule, or substituted phenyls with 4-trifluoromethoxy-, 2-amino-, or 4-trifluoromethyl substituents on the aryl fragment. These derivatives showed KIs ranging between 0.34 and 0.87 μM. The remaining homo-sulfocoumarins were low micromolar hCA IX inhibitors.
The SAR for inhibition of the second tumour-associated isoform, hCA XII, was more complex compared to what discussed above for hCA IX (Table 1). Thus, 8 out of 11 derivatives were inactive (KIs > 50 μM) whereas the remaining ones, 6c, 13 and 15, inhibited hCA XII with KIs in the range of 0.64–2.32 μM.
This inhibition profile is rather similar to the one of sulfocoumarins1–6 and coumarins7,8, which are generally selective inhibitors for the tumour-associated over the cytosolic isoforms. However, some homo-sulfocoumarins showed a very specific, and unique up until now inhibition profile among all classes of CAIs known to date9,10, as they are highly selective for hCA IX over hCA I, II and XII (e.g. 7–12, 14, 16 and 17).
In conclusion, we report here a new chemotype with effective and isoform-selective CAIs, the homo-sulfocoumarins, which show a unique inhibition profile for the tumour-associated CA isoforms hCA IX (and XII) over the cytosolic ones. Although the CA inhibition mechanism with these new compounds is unknown for the moment, we hypothesize that it may be similar to that of the sulfocoumarins, i.e. hydrolysis to the corresponding sulfonic acids which thereafter anchor to the zinc-coordinated water molecule within the enzyme active site.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.a) Grandane A, Belyakov S, Trapencieris P, et al. Facile synthesis of coumarin bioisosteres – 1,2-benzoxathiine 2,2-dioxides. Tetrahedron 2012;68:5541–6. b) Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56: 293–300. [DOI] [PubMed] [Google Scholar]
- 2.Tanc M, Carta F, Bozdag M, et al. 7-Substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg Med Chem 2013;21:4502–10. [DOI] [PubMed] [Google Scholar]
- 3.a) Grandane A, Tanc M, Zalubovskis R, et al. Synthesis of 6-tetrazolyl-substituted sulfocoumarins acting as highly potent and selective inhibitors of the tumor-associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem 2014;22:1522–8. b) Grandane A, Tanc M, Zalubovskis R, et al. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett 2014;24: 1256–60. [DOI] [PubMed] [Google Scholar]
- 4.Grandane A, Tanc M, Di Cesare Mannelli L, et al. 6-Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII Inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem 2015;58:3975–83. [DOI] [PubMed] [Google Scholar]
- 5.Grandane A, Tanc M, Žalubovskis R, et al. Synthesis of 6-aryl-substituted sulfocoumarins and investigation of their carbonic anhydrase inhibitory action. Bioorg Med Chem 2015;23:1430–6. [DOI] [PubMed] [Google Scholar]
- 6.Nocentini A, Ceruso M, Carta F, et al. 7-Aryl-triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrase IX and XII. J Enzyme Inhib Med Chem 2016;31:1226–33. [DOI] [PubMed] [Google Scholar]
- 7.(a) Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62. b) Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53: 335,–44. c) Maresca A, Supuran CT.. Coumarins incorporating hydroxy- and chloro-moieties selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II. Bioorg Med Chem Lett 2010;20: 4511–14. [Google Scholar]
- 8.a) Maresca A, Scozzafava A, Supuran CT.. 7,8-Disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–58 b) Touisni N, Maresca A, McDonald PC, et al. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54: 8271,–7. c) Carta F, Maresca A, Scozzafava A, et al. Novel coumarins and 2-thioxo-coumarins as inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem 2012;20: 2266,–73. d) Davis RA, Vullo D, Maresca A, et al. Natural product coumarins that inhibit human carbonic anhydrases. Bioorg Med Chem 2013;21: 1539,–43. e) Sharma A, Tiwari M, Supuran CT.. Novel coumarins and benzocoumarins acting as isoform-selective inhibitors against the tumor-associated carbonic anhydrase IX. J Enzyme Inhib Med Chem 2014;29: 292,–6. f) Ferraroni M, Carta F, Scozzafava A, Supuran CT.. Thioxocoumarins show an alternative carbonic anhydrase inhibition mechanism compared to coumarins. J Med Chem 2016;59: 462–73. [Google Scholar]
- 9.a) Supuran CT.Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. b) Neri D, Supuran CT.. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10: 767,–77. c) Durdagi S, Vullo D, Pan P, et al. Protein-protein interactions: Inhibition of mammalian carbonic anhydrases I-XV by the murine inhibitor of carbonic anhydrase and other members of the transferrin family. J Med Chem 2012;55: 5529,–35. d) Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2016;31: 894–9. [Google Scholar]
- 10.a) Supuran CT.Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. b) Supuran CT.How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31: 345,–60. c) Supuran CT.Carbonic anhydrases. Bioorg Med Chem 2013;21: 1377,–8. d) Supuran CT.Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12: 61,–88. e) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112: 4421,–68. f) Supuran CT.Legionella pneumophila carbonic anhydrases: underexplored antibacterial drug targets. Pathogens 2016;5: E44. [Google Scholar]
- 11.(a) De Simone G, Alterio V, Supuran CT.. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810. (b) Masini E, Carta F, Scozzafava A, et al. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23: 705,–16. (c) Supuran CT, Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;2: 3467,–74. (d) Supuran CT.Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO2 capture. J Enzyme Inhib Med Chem 2013;28: 229,–30. e) D’Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Commun 2015;51: 302–5. [Google Scholar]
- 12.a) Supuran CT.Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72. b) Supuran CT.Carbonic anhydrase inhibitors: an editorial. Expert Opin Ther Pat 2013;23: 677,–9. c) Winum JY, Supuran CT.. Recent advances in the discovery of zinc-binding motifs for the development of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30: 321,–4. d) De Luca V, Del Prete S, Supuran CT, et al. Protonography, a new technique for the analysis of carbonic anhydrase activity. J Enzyme Inhib Med Chem 2015;30: 277,–82. e) Lomelino CL, Supuran CT, McKenna R, Non-classical inhibition of carbonic anhydrase. Int J Mol Sci 2016;1: E1150. [Google Scholar]
- 13.a) Supuran CT.Acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6. b) Di Fiore A, De Simone G, Alterio V, et al. The anticonvulsant sulfamide JNJ-26990990 and its: S,S-dioxide analog strongly inhibit carbonic anhydrases: solution and X-ray crystallographic studies. Org Biomol Chem 2016;14: 4853,–8. c) Supuran CT.Drug interaction considerations in the therapeutic use of carbonic anhydrase inhibitors. Expert Opin Drug Metab Toxicol 2016;12: 423–31. [Google Scholar]
- 14.Albert S, Horbach R, Deising HB, et al. Synthesis and antimicrobial activity of (E) stilbene derivatives. Bioorg Med Chem 2011;19:5155–66. [DOI] [PubMed] [Google Scholar]
- 15.Dauban P, Dodd RH.. Synthesis of cyclic sulfonamides via intramolecular copper-catalyzed reaction of unsaturated iminoiodinanes. Org Lett 2000;2:2327–9. [DOI] [PubMed] [Google Scholar]
- 16.Khalifah RG.The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73. [PubMed] [Google Scholar]
- 17.Leitans J, Sprudza A, Tanc M, et al. 5-Substituted-(1,2,3-triazol-4-yl)thiophene-2-sulfonamides strongly inhibit human carbonic anhydrases I, II, IX and XII: solution and X-ray crystallographic studies. Bioorg Med Chem 2013;21:5130–8. [DOI] [PubMed] [Google Scholar]
- 18.a) Korkmaz N, Obaidi OA, Senturk M, et al. Synthesis and biological activity of novel thiourea derivatives as carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:75–80. b) Abdel-Aziz AAM, El-Azab AS, Ekinci D, et al. Investigation of arenesulfonyl-2-imidazolidinones as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30: 81,–4. c) Akdemir A, De Monte C, Carradori S, et al. Computational investigation of the selectivity of salen and tetrahydrosalen compounds towards the tumor-associated hCA XII isozyme. J Enzyme Inhib Med Chem 2015;3: 114,–18. d) Leitans J, Kazaks A, Balode A, et al. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem 2015;58: 9004,–9. e) Göçer H, Akincioğlu AS, Gülçin GI, et al. Carbonic anhydrase and acetylcholinesterase inhibitory effects of carbamates and sulfamoylcarbamates. J Enzyme Inhib Med Chem 2015;30: 316,–20. f) Ceruso M, Bragagni M, AlOthman Z, et al. New series of sulfonamides containing amino acid moiety act as effective and selective inhibitors of tumor-associated carbonic anhydrase XII. J Enzyme Inhib Med Chem 2015;3: 430,–4. g) Zolfaghari Emameh R, Syrjänen L, Barker H, et al. Drosophila melanogaster: a model organism for controlling dipteran vectors and pests. J Enzyme Inhib Med Chem 2015;30: 505–13. [Google Scholar]
- 19.Otwinowski Z, Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997;276:307–26. [DOI] [PubMed] [Google Scholar]
- 20.Burla MC, Caliandro R, Camalli M, et al. SIR2004: an improved tool for crystal structure determination and refinement. J Appl Crystallogr 2005;38:381–8. [Google Scholar]
- 21.Sheldrick GM.A short history of SHELX. Acta Crystallogr 2008;64:112–22. [DOI] [PubMed] [Google Scholar]
- 22.Farrugia LJ.WinGX and ORTEP for windows: an update. J Appl Crystallogr 2012;45:849–54. [Google Scholar]
- 23.a) Gieling RG, Babur M, Mamnani L, et al. Antimetastatic effect of sulfamate carbonic anhydrase IX inhibitors in breast carcinoma xenografts. J Med Chem 2012;55:5591–600. b) Winum JY, Maresca A, Carta F, et al. Polypharmacology of sulfonamides: pazopanib, a multitargeted receptor tyrosine kinase inhibitor in clinical use, potently inhibits several mammalian carbonic anhydrases. Chem Commun 2012;48: 8177,–9. c) Lock EF, McDonald PC, Lou Y, et al. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene 2013;32: 5210,–19. d) Ward C, Langdon SP, Mullen P, et al. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat Rev 2013;39: 171–9. [Google Scholar]
- 24.a) Pan J, Lau J, Mesak F, et al. Synthesis and evaluation of 18F-labeled carbonic anhydrase IX inhibitors for imaging with positron emission tomography. J Enzyme Inhib Med Chem 2014;29:249–55. b) Pettersen EO, Ebbesen P, Gieling RG, et al. Targeting tumour hypoxia to prevent cancer metastasis. From biology, biosensing and technology to drug development: The METOXIA consortium. J Enzyme Inhib Med Chem 2015;30: 689–721. [DOI] [PubMed] [Google Scholar]