

Abstract
The endocannabinoid (eCB) signaling system modulates neurotransmission and inflammation, amongst other physiological functions. Its newest member, α/β-hydrolase domain containing 6 (ABHD6), has emerged as a promising therapeutic target to treat several devastating diseases, including epilepsy. We review the molecular mechanisms that mediate and control eCB signaling and within it, the specific role of ABHD6. We also discuss how ABHD6 controls the abundance of additional lipids and the trafficking of ionotropic receptors to plasma membranes. We finish with several unexplored questions regarding this novel enzyme. Our current understanding of the molecular mechanism and biological function of ABHD6 provides a strong foundation for the development of small molecule therapeutics to treat devastating diseases.
Keywords: ABHD6, endocannabinoids, enzymes, neurons, neuroinflammation, epilepsy
The canonical endocannabinoid signaling system
The existence of an endocannabinoid (eCB) signaling system was first inferred from studies on the bioactivity of Δ9-tetrahydrocannabinol (THC), the psychoactive compound produced by the cannabis plant [1]. This signaling system consists of two cannabinoid (CB) receptors (CB1R and CB2R), two main signaling lipids (anandamide, AEA, and 2-arachidonoylglycerol, 2-AG) that activate these receptors, and the enzymes that produce and inactivate eCBs. The lipid nature of eCBs provides both autocrine and paracrine modalities to regulate the physiology and function of cells in the brain and periphery. Accordingly, eCBs are 1) produced in an activity-dependent manner by independent lipases located at the plasma membrane, 2) exhibit distinct pharmacology at CB receptors and 3) are inactivated by independent enzymes [1]. Thus, the eCB signaling system encompasses parallel signaling lipids that independently regulate membrane receptors and fundamental physiological functions throughout the body.
The CB1R is a G protein-coupled receptor expressed by many cell types and most abundantly expressed in neuron terminals [2]. In neurons, through the activation of G proteins, CB1R regulates the activity of second messengers and effector proteins, including adenylyl cyclase and extracellular signal–regulated kinases (ERK) kinases activities, the opening probability of L-, N- and P/Q-type calcium channels and of potassium hyperpolarization-activated cyclic nucleotide–gated (HCN) channels, the combination of which controls action potential-dependent vesicular neurotransmitter release [3–7]. Under certain conditions, CB1Rs activate phospholipase C and PI3K pathways through G protein β/γ subunits also reduce presynaptic neurotransmitter release [8–11]. The prolonged, repeated activation of CB1Rs regulates gene expression and synaptic arborization [12–14]. Thus, activation of CB1Rs expressed by neurons regulates neurotransmitter release and is involved in eCB-dependent synaptic plasticity [15].
The CB2R also couples to G proteins, has 44% protein homology to CB1R and is primarily expressed by hematopoietic cells [16, 17]. eCBs also modulate CB2R activity, but with a different pharmacological action than at CB1R, which is likely to explain a portion of the select modulatory control of immune cell function by CB2R [2]. Activation of CB2Rs reduces inflammation by lowering the production of immune mediators, reducing macrophage’s ability to process antigens and prime helper T-cells, and by eliminating immune cells by apoptosis [18]. Small molecules that target CB2Rs also regulate immune cell proliferation and migration [19]. Accumulating evidence show that CB2Rs are also expressed in neurons and recent work uncovered cell-type specific roles of CB2Rs on contextual fear, spatial memory and anxiety, illustrating an increasing appreciation for CB2Rs in neurons [20]. There are several additional receptors that are also modulated by phytocannabinoids (phytoCB), synthetic CB ligands and eCBs, including GPR55, several transient receptor potential (TRP) channels and glycine receptors [1]. These are all likely to play roles in the bioactivity of phytoCBs that are independent of what is often referred to as the canonical eCB signaling pathway that encompasses CB1R, CB2R, AEA, and 2-AG [21–23].
This first eCB, AEA, is a partial agonist of CB1R [24]. The production and release of AEA is tonically low and is increased through the stimulation of a multi-step enzymatic pathway that starts by generating its precursor, N-arachidonoylphosphatidylethanolamide (NAPE), which is converted to NAPE-specific phospholipase D (PLD) into AEA and phosphatidic acid [25–29]. The second eCB, 2-AG, is 200 times more abundant than AEA in mouse brain, and its production is transiently increased by rises in intracellular calcium that activates diacylglycerol lipase (DGL) to cleave DAG and produce 2-AG [30–35] (Figure 1, Key Figure). 2-AG then acts at several targets, including as a paracrine full agonist at both CB1R and CB2R and an autocrine positive allosteric modulator of gamma-aminobutyric acid receptors A (GABAA receptors).
Figure 1, Key Figure. Molecular components involved in the regulation of 2-AG signaling by neurons.
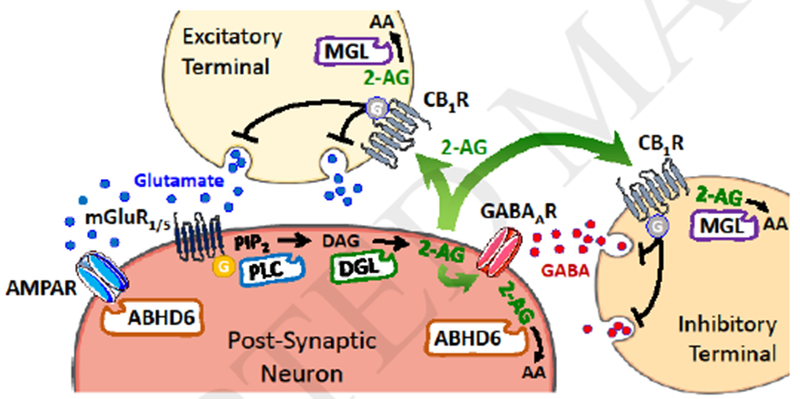
Stimulation of excitatory neurons releases glutamate (blue spheres), activating metabotropic glutamate receptors 1/5 (mGluR1/5) on the postsynaptic neuron, which couple to G proteins (orange sphere), activate phospholipase C (PLC) and lead to the production of diacylglycerol (DAG) from phosphoinositol bisphosphate (PIP2). DAG is cleaved by DAG lipase (DGL), which produces 2-AG that acts as 1) paracrine agonist at CB1 receptors (CB1R) expressed by excitatory terminals, 2) paracrine agonist at CB1R expressed by inhibitory terminals that release GABA (red spheres) to activate GABAA receptors on the post-synaptic neurons and 3) autocrine positive allosteric modulator of GABAA receptors on the post-synaptic neurons. Excess 2-AG is hydrolyzed to arachidonic acid (AA) and glycerol (not shown) by monoacylglycerol lipase (MGL) in presynaptic neurons or by ABHD6 in postsynaptic neurons. ABHD6 interacts with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and controls their trafficking to the membrane of post-synaptic compartments.
The potency and efficacy of eCBs to activate CB receptors is regulated by at least six mechanisms: 1) the amount of eCB produced, 2) the assisted travelling of eCBs from their site of synthesis to CB receptors (presynaptic, postsynaptic and on neighboring cells), which is most likely mediated by fatty acid binding proteins [36], 3) the number of CB receptors expressed and their functional coupling, which are controlled by agonist-induced receptor recycling and desensitization, 4) CB receptor biased signaling to different second messengers, 5) clearance of eCBs by uptake through the plasma membranes, and 6) eCB inactivation by intracellular enzymatic hydrolysis, which primarily involves fatty acid amide hydrolase (FAAH) for AEA and monoacyglycerol lipase (MGL) and α/β-hydrolase domain containing 6 (ABHD6) for 2-AG (Figure 1).
Significant effort dedicated to understanding the molecular mechanism by which eCBs are hydrolyzed has revealed clear similarities and cardinal differences in the enzymatic mechanisms that hydrolyze eCBs. While the majority of AEA hydrolysis in the brain is carried by FAAH, 2-AG hydrolysis involves multiple enzymes, including 85% by MGL, 4% by ABHD6 and 9% by α/β-serine hydrolase domain 12 (ABHD12), at least when measured in mouse brain homogenates [37]. Importantly, recent evidence shows that the overall activity of 2-AG hydrolyzing enzymes in brain homogenates do not fully explain the relative ability of each enzyme to control the levels and efficacy of 2-AG at CB receptors, and that their subcellular expression pattern and the proximity of 2-AG production to its specific target better reflects their contribution to the regulation of signaling. Here we will review the exciting new evidence that established ABHD6’s place in eCB signaling, as well as several emerging evidences of its role in controlling other eCB-independent signaling pathways, and how better understanding these molecular interactions will help develop novel therapeutics.
Initial evidence for new 2-AG-hydrolyzing activities and identification of ABHD6
The identification of a novel eCB-hydrolyzing activity and the demonstration of its ability to control eCB signaling requires addressing several fundamental criteria. This often starts with the pharmacological and genetic demonstration that blocking the candidate enzyme halts eCB hydrolysis in both tissue homogenates and intact cells, leads to an increase in the levels of the eCB, and that this activity controls the ability of eCB to activate its target receptors. The first evidence that suggested the existence of another enzyme besides FAAH and MGL that hydrolyzes 2-AG came from Muccioli et al, which found that the mouse microglial cell line, BV-2, does not express MGL yet actively hydrolyzes 2-AG [38]. Concomitantly, three novel enzyme candidates that hydrolyze 2-AG (ABHD6, ABHD12 and neuropathy target esterase) were identified by a multi-dimensional protein identification platform, known as activity-based protein profiling (ABPP) (see Glossary) [37, 39]. The demonstration that ABHD6 is responsible for hydrolyzing 2-AG in BV-2 cells that do not express MGL was established by using the first generation ABHD6 inhibitor, WWL70, and by short interfering RNA (siRNA) knockdown, both of which reduced 2-AG hydrolysis in BV-2 homogenates and intact BV-2 cells [40]. ABHD6 expression was detected in several cells, including neurons, and its pharmacological and genetic inhibition enhanced the magnitude of 2-AG activation of CB1Rs and CB2R [40]. Together, these studies established that ABHD6 regulates 2-AG signaling through CB1Rs in neurons and CB2Rs in immune cells and provided initial evidence that ABHD6 might represent a new member of the eCB signaling system.
ABHD6: A member of the serine hydrolase family
ABHD6 is a 30kDa integral membrane protein that belongs to the serine hydrolase family, one of the largest family of enzymes. Although all members of this family contain the canonical hydrolase fold of an 8-stranded parallel α/β structure with a nucleophile-his-acid catalytic triad, each member is often different in sequence, substrate specificity and catalytic activity [41]. For example, ABHD6 and ABHD12 both hydrolyze 2-AG to similar extents when their activities are measured in mouse brain homogenates despite only sharing less than 20% sequence homology [37]. ABHD6 includes a catalytic triad determined by amino acids S148-D278-H306 that is responsible for 2-AG hydrolysis [41,42]
Expression profile of ABHD6
The expression profile of ABHD6 RNA in tissue and cells suggests high expression in the immune system, particularly the spleen, and the small intestine, then followed by the brain (particularly the cerebral cortex, pituitary gland and hippocampus)i. Single cell RNA-seq analyses of mouse brain shows that ABHD6 mRNA expression is different in young and adult brain. In young brains, ABHD6 mRNA is found in progenitor cells and astrocytes, while in adults, it is abundant in select types of neurons (such as GABAergic) and astrocytes [43]ii. ABHD6 mRNA expression is regulated by hormones such as estrogen, suggesting sex-dependent differences in its expression [44]. Together, these results suggest that ABHD6 expression in select cell types is likely to change as a function of development and sex.
Further, the expression profile of ABHD6 protein in brain tissue appears to be differentially regulated in distinct brain areas and neural cell subtypes. ABHD6 enzymatic activity measured using ABPP in mouse brain has shown that the highest enzyme activity is in the frontal cortex, hippocampus, striatum, and cerebellum, where it is higher than ABHD12 and even MGL [45]. ABHD6 is primarily expressed on the cytoplasmic side by principal glutamatergic neurons, with some expression on GABA interneurons and astrocytes [40]. In murine neurons in culture, ABHD6 expression is prominently localized to dendrites and colocalized with the post-synaptic protein microtubule associated protein 2 (MAP2) and not with presynaptic CB1R, suggesting a post-synaptic expression pattern that complements the presynaptic expression profile of MGL (Figure 1) [40, 46]. It is likely that the distinct subcellular localization of ABHD6 and MGL (post- and presynaptic, respectively) allows for differential spatial and temporal control of 2-AG levels.
Activity of ABHD6:
Studies in mouse brain homogenates have shown that there are several lipid substrates that can be hydrolyzed by ABHD6 including monoacylglycerols (MAG) with saturated acyl chains varying from 8-18 carbons [42]. Thus, ABHD6 hydrolyzes select lipids with distinct activities 1-arachidonoyl-glycerol (1-AG) > 2-AG > 2-lauroyl-glycerol (2-LG) [42]. Independent of eCB signaling, ABHD6 hydrolyzes 90% of multichain lipid constituents bis(monoacylglycero)phosphate (BMP) and lysobisphosphatidic acid in late endosomes and lysosomes in the liver [47]. Together, these studies suggest that ABHD6 will regulate the levels of several classes of lipids, including signaling, metabolic and structural lipids.
ABHD6 inhibitors
Several ABHD6 inhibitors with distinct chemical scaffolds have been developed, including carbamate-based inhibitors (WWL70, WWL123, KT-182, KT-195 and MJN193) that block its hydrolyzing activity through a covalent modification of the catalytic S148 [37, 48, 49] and fatty acid-based inhibitors (UCM710, CP7B, JAC88, JAC93) that block substrate hydrolysis by competition [50] (Figure 2). It is postulated that with carbamate-based inhibitors, the hydrogen bond between the inhibitor’s carbonyl group and the main chain of P80 of ABHD6 is weakened and a new hydrogen bond is formed with the side chain of S148, facilitating a covalent bond [51]. However, the selectivity of early generation carbamate-based and fatty acid-based ABHD6 inhibitors has been questioned by studies reporting their ability to inhibit other enzymes. For example, WWL70 also modulates cyclooxygenase 2 (COX-2) and prostaglandin E1/2 (PGE-1/2) activities in microglia [52]. The carbamate-based inhibitor MJN110 inhibits MGL and ABHD6 activities, although ABHD6 to a lesser extent [53]. Many fatty acid-based inhibitors reduce ABHD6, MGL and FAAH activities with similar IC50s within the micromolar (μM) range and thus cannot pharmacologically differentiate these activities. For example, UCM710 inhibits ABHD6 and FAAH with IC50s of 2.4 and 4.0 μM, respectively, and MGL of 30 μM[50].
Figure 2. ABHD6 activity and inhibition.
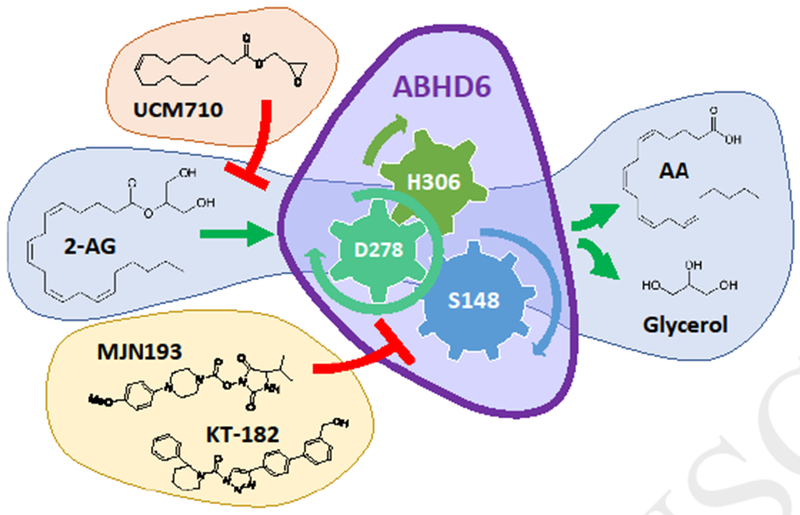
The lipid substrate (e.g. 2-AG) is hydrolyzed by ABHD6 (purple shape) containing a catalytic triad made by amino acids S148, D278 and H306 and leads to the release of arachidonic acid (AA) and glycerol. Fatty-acid based inhibitors (e.g. UCM710) compete with the lipid substrate whereas irreversible carbamates inhibitors (KT-182) and reversible inhibitors (MJN193) interact with catalytic triad.
However, the next generation of carbamate-based ABHD6 inhibitors have reached greater potency, selectivity and in vivo activity, including the brain-penetrant compound KT-182, the peripherally-restricted compound KT-203, and the orally-available compound KT-185 [49]. In addition, triazole urea featuring chiral hydroxylated 2-benzylpiperidines have recently been synthesized to act as dual inhibitors of both DGL and ABHD6, citing the chirality of the carbon at the C2 substituent and position of the C5 hydroxyl to dictate inhibitory activity on the enzymes in mouse brain extracts [54]. Finally, the recent chemical biology tools based on fluorescence activity-based probes that are being developed to try and differentiate the enzymatic activity of these enzymes will certainly help develop more select inhibitors [55]. Thus, much effort has been dedicated to developing selective inhibitors of ABHD6 to unravel the role of this enzyme in regulating cell functions and explore the possibility of targeting this enzyme for therapeutic purposes.
eCB-dependent and eCB-independent roles of ABHD6 in modulating neuronal functions
Evidence suggests that ABHD6 activity controls eCB-dependent neuronal functions through remarkably distinct mechanisms that impact long-term, but not short-term, synaptic plasticity. An involvement of ABHD6 in long-term synaptic depression (LTD) was first suggested when it was observed that addition of ABHD6 inhibitor, WWL70, reduced the threshold to LTD at glutamatergic synapses which persisted for at least 40 min after induction [40]. By sharp contrast, WWL70 did not affect short-term plasticity, which is exemplified by depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE) [56–58]. Specifically, these two forms of short-term CB1R-mediated synaptic plasticity that regulate GABAergic and glutamatergic transmission, respectively, and are mediated by retrograde 2-AG signaling [59]. Since the durations of DSE and DSI are determined largely by enzymatic degradation of 2-AG, one might predict that blocking 2-AG hydrolysis would extend the time course of DSE/DSI [46, 60]. However, in vitro recordings of neurons revealed that it is mediated by MGL and another enzyme known to modify 2-AG, COX-2 and remains unaffected by ABHD6 inhibition [46, 60, 61]. These studies provide an important differentiator between the biological roles plays by ABHD6, MGL and COX-2 by emphasizing their involvement in short-term versus long-term synaptic plasticity mechanisms.
The initial clue for the existence of an eCB-independent function of ABHD6 in regulating neuronal function came from unbiased high-resolution proteomics on brain tissue unveiling that ABHD6 is part of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor macromolecular complex and reducing its expression results in decreasing AMPA receptor surface expression and ensuing glutamatergic transmission independently of cannabinoid receptors [62, 63] (Figure 1). The reduction in surface expression of the AMPA receptor happens via reduction in surface expression of one of the subunits of AMPA, GluA1. It was found that the C-terminal tail of GluA1 is required for binding between ABHD6 and AMPA subunits GluA1, GluA2 and GluA3 [64]. Importantly, mutation of the S148 site on ABHD6, which is critical to its serine hydrolase activity, did not affect the ability of ABHD6 to regulate AMPA expression, showing that this function of ABHD6 is independent of the enzyme’s ability to hydrolyze eCBs [62]. It is interesting to note that this reduction in glutamatergic signaling by ABHD6’s regulation of functional AMPA receptor expression contrasts with its eCB-dependent role of enhancing the sensitivity of LTD induction [62]. Therefore, ABHD6’s impact on long-term synaptic plasticity at excitatory synapses results from the net effect of two competing mechanisms involving eCB-dependent and eCB-independent mechanisms.
It is important to point out that the findings with ABHD6 inhibitors or genetic deletion of ABHD6 in mice on obesity and diabetes are also independent of eCB signaling as indicated by the results showing the liver, adipose, pancreas eCB signaling were unaltered when ABHD6 is blocked down [65–67]. Thus, ABHD6 is likely to play both eCB-dependent and eCB-independent roles in both central and peripheral tissues.
Pathological consequences of impaired ABHD6 activity
Sever reports show that ABHD6 expression and activity changes in select tissues and as a function of specific pathological processes. ABHD6 expression is increased in select cancer subtypes such as bone, prostate, leukemia and Burkitt’s lymphoma, as well as in systemic lupus erythematosus [41, 68, 69]. The gene is also known to be targeted by the Epstein-Barr virus (EBV), a DNA tumor virus associated with Hodgkin’s lymphoma and post-transplant lymphoma [70]. Decreased ABHD6 expression may also be involved in select pathological processes, as shown in inflammatory-associated pathologies where infection of macrophages by Salmonella typhimurium downregulates hydrolysis activity of ABHD6 and FAAH, which increases 2-AG and AEA levels and enhances phagocytic function in host cells [71]. Thus, changes in ABHD6 expression and functionality may be involved in select pathological processes, including cancer, which provides a framework to test the therapeutic value of targeting ABHD6 for the treatment of select maladies.
Targeting ABHD6 for therapeutic benefit
An initial clue to maladies that might benefit from ABHD6 inhibitors came from discovering that ABHD6 blockade regulates activity-dependent production of 2-AG and ensuing activation of CB1R, a characteristic of certain epilepsies [40]. Accordingly, ABHD6 inhibition by WWL123 significantly decreases seizure frequency in mice models of chemically-induced and genetically-induced seizures [72]. However, it is interesting to note that ABHD6 inhibition reduced these seizures by increasing GABAA receptor activity and not by activating CB1Rs or CB2Rs [72, 73] (Figure 1). This suggests that ABHD6 inhibitors dampen excessive excitatory transmission in seizures by two mechanisms that provide therapeutic advantages: 1) increasing the level of an endogenous ligand, 2-AG, instead of directly targeting the receptor (here GABAA receptors) and 2) allosterically increasing GABAA receptor signaling instead of targeting the orthostatic site of the receptors, a combination of assets that will likely reduce desensitization and therapeutic tolerance [74].
Additional diseases that might benefit for ABHD6 inhibitors are fragile × mental retardation, and traumatic brain injury (TBI). Specifically, in the mouse model of fragile × mental retardation, ABHD6 inhibitors rescue the impaired eCB-dependent LTD [75]. In this model, the transgenic fragile × mental retardation 1 protein directly disrupts the expression of DGL mRNA, reduces 2-AG signaling and increased mGluR5 function, the combination of which impairs neurotransmission in this pathology [75]. In TBI, the ensuing secondary injury mediated by excitotoxicity, neuroinflammation and oxidative stress propagates because of insufficient increase in 2-AG levels to counteract these pathological processes [76]. In a TBI mouse model, post-injury treatment with the ABHD6 inhibitor WWL70 improved motor coordination and working memory performance but did not affect spatial learning or memory impairments [77]. Remarkably, WWL70 attenuated several TBI-induced neurodegenerative processes through both CB1R and CB2R, including reduced lesion volume and neuronal loss, suppressed expression of intercellular adhesion molecule 1 (ICAM-1), a marker of inflammation and blood-brain-barrier (BBB) dysfunction, and attenuated nitric oxide and prostaglandin E2 production, two mediators of proinflammatory responses [77]. It should be mentioned that the effect of ABHD6 blockade in fragile × mental retardation and in TBI were carried out using WWL70 and thus will require further validation using genetic tools and more selective ABHD6 inhibitors.
ABHD6 inhibitors have also been tested in experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis (MS) [78]. Here chronic treatment with WWL70 ameliorated clinical symptoms and accordingly reduced both mRNA levels of proinflammatory mediators tumor necrotic factor (TNF-α), inducible nitric oxide synthase (iNOS) and interleukin (IL)-iβ, and the number of CD4+ T-cell infiltrates and TNF-α+ cells [78]. This therapeutic effect was blocked by the CB2R antagonist, AM630, and lost in the CB2R-knockout (KO) mice, confirming a CB2R-dependent mechanism [78]. As mentioned above, the lack of selectivity of WWL70 required further validations [52]. Accordingly, recent work revisited the therapeutic value of ABHD6 inhibitors in MS with the next generation ABHD6 inhibitor, KT-182, and showed that treatment did ameliorate neurological symptoms of EAE but had a small overall effect in disease progression [79]. In another model of MS, the cuprizone model of non-immune dependent demyelination, which allows to study a different aspect of MS pathogenesis, KT-182 only partially attenuated myelin damage, astrogliosis and microglia/macrophage reactivity without affecting oligodendrocytes [80]. Taken together, these results indicate limited efficacy in therapeutic efficacy of ABHD6 inhibitors in autoimmune-mediated diseases.
When considering systemic diseases, ABHD6 inhibitors might help for the treatment of obesity as indicated by its role in the control of lipid metabolism [42]. Specifically, downregulation of ABHD6 expression in the liver protects against body weight and fat mass gain, increases energy expenditure, and improved glucose and insulin tolerance in mice fed a high-fat diet [65]. β cell-specific ABHD6-KO mice show similar results suggesting that ABHD6 negatively regulates glucose-stimulated insulin secretion [66, 67]. Both functions of ABHD6 are independent of eCB signaling [65–67]. Remarkably, mice lacking ABHD6 in the ventromedial hypothalamus exhibit CB1R-mediated blunting effects on fasting-induced feeding and consume less food overall, expend less energy and have reduced adaptive thermogenesis in response to a metabolic challenge, suggesting a role for ABHD6 in the counter-regulatory responses associated with metabolic shifts [81]. Together, these studies identify central and peripheral maladies that might benefit from blocking ABHD6 activity.
Conclusion and future perspectives.
Our understanding of the molecular mechanism and biological function of ABHD6, as well as the therapeutic potential of ABHD6 inhibitors, has rapidly increased during the last decade. We’ve learned that ABHD6 finetunes multiple signaling pathways and that it functions as a molecular hub that controls several biological functions. The studies published on the molecular mechanisms and biological function of ABHD6 during the last decade provided the foundation to establish this enzyme both as a bona fide member of the eCB signaling system and revealed a versatile molecular machinery that regulates additional signaling systems, including GABAA and AMPA receptors through distinct mechanisms (Figure 3A). However, important questions remain unanswered regarding ABHD6’s multifunctionality (see Outstanding Questions). While ABHD6 is highly expressed in the brain, it is only responsible for a very small portion of 2-AG hydrolysis when its activity is measured in brain homogenates. What are the major functional lipids metabolized by ABHD6? While it is known that the serine-hydrolase activity of ABHD6 cleaves several lipid substrates, an unbiased global metabolomics analysis of the changes trigged by the acute and prolonged blockade of ABHD6 activity has yet to be reported and will likely identify novel lipid substrates and role in lipid membrane turn-over (Figure 3B). Furthermore, the remarkable ability of ABHD6 to directly interact with AMPA receptors independently of its serine-hydrolase activity and chaperone their trafficking to the plasma membrane raises fundamental questions about the specific ABHD6-AMPA receptors amino acid interaction sites, the type of molecular motor that controls this trafficking and whether additional receptors use this platform to traffic to the plasma membrane (Figure 3C). The range of signaling lipids and interacting receptors that are modulated by ABHD6 suggests a multifunctional role that positions this protein as a critical molecular hub to modulate multiple signaling systems
Figure 3. Multifunctional activity of ABHD6.
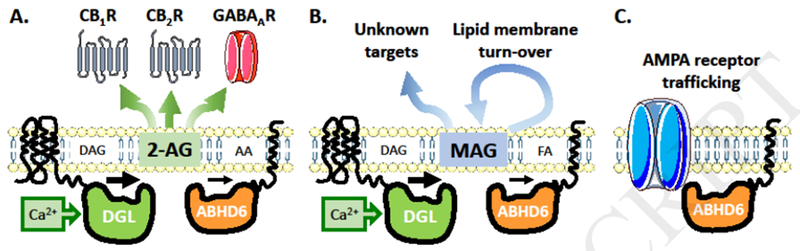
A. Rise in intracellular calcium concentration activates diacylglycerol lipase (DGL) that cleaves diacylglycerol (DAG) into 2-AG. ABHD6 hydrolyzes 2-AG into arachidonic acid (AA) and glycerol (not shown). Thus, ABHD6 controls the activity-dependent 2-AG accumulation and eventual activation of cannabinoid receptors CB1R and CB2R and GABAA receptors. B. ABHD6 controls the hydrolysis of monoacylglycerols (MAG) into fatty acids (FA). MAG accumulation might module the activity of unknown receptors targets and/or modify lipid membrane composition. C. ABHD6 acts as a chaperone to traffic AMPA receptors to the plasma membrane.
Outstanding Questions.
What are the global metabolomic and phenotypic changes caused by either acute or prolonged blockade of ABHD6 activity?
What are the specific amino acids sites on ABHD6 involved in its multifunctional activity?
What is the cell-specific expression pattern of ABHD6, and does this change during development, aging and select disease processes?
Are there different therapeutic benefits of ABHD6 inhibition in rapid versus slow-progressing neuronal dysfunctions and diseases?
We also have a limited understanding of the cell-specific expression pattern of ABHD6 and how it changes during development and aging. ABHD6 expression is upregulated by estrogen, indicating that its expression will be tightly controlled by hormone levels [44]. The remarkably high expression of ABHD6 in astrocytes tallies well with the recent studies establishing a dominant role for eCB signaling in this glial subtype and raises the question whether other glial cell types might express this enzyme and whether ABHD6 expression changes as a function of reactive gliosis associated with select neurological diseases [82]. The tools that were recently developed to follow ABHD6 expression and activity, which include specific antibodies that were validated using genetic controls and fluorescence activity-based probes that differentiate serine hydrolase enzymatic activities, will certainly help answer these questions [55, 83].
Further, what are the next therapeutic indications that might benefit from targeting ABHD6? The activity-dependent regulation of 2-AG levels by ABHD6 suggest that its inhibitors might show benefit in rapid onset neuronal dysfunctions and diseases, such as stroke and pain, and more limited efficacy in slow-progressing neuronal dysfunctions and diseases. Is there a role for ABHD6 in brain development? Probably “yes,” as suggested by the changes in ABHD6 expression in select cell types as a function of brain development and sex, and that this enzyme regulates eCB signaling and AMPA receptor expression at the plasma membrane, two key molecular mechanisms involved in neuronal differentiation.
Selective manipulation of ABHD6 activities represents an exciting opportunity to develop novel therapeutic modalities. However, the utility of targeting of ABHD6 for therapeutic benefit should be systematically validated using genetic approaches, such as knockout and knockdown strategies. Furthermore, there remains a clear need for developing highly selective inhibitors that target the different activities carried by ABHD6 and lack off-target activity before moving towards the clinic. The escalating interest to study ABHD6’s role in eCB signaling and beyond promises to continue on this steep trajectory, a venture will help promptly move ABHD6 inhibitors towards the clinic for the treatment of several devastating diseases.
Highlights.
ABHD6 is a bona fide member of the endocannabinoid signaling system, enzymatically controlling levels of the most abundant endocannabinoid 2-AG, controlling activation of CB1 and CB2 receptors, and thereby modulating cell function.
ABHD6 modulates neuronal activity in an endocannabinoid-independent manner, through AMPA receptor trafficking and allosteric modulation of GABA receptor signaling. This illustrates a valuable multifunctionality of this enzyme with multiple therapeutic possibilities.
Novel inhibitors targeting ABHD6 have been developed with increasing selectivity and sensitivity that led to elucidation of its biological function.
Inhibition of ABHD6 has emerged as a promising therapeutic treatment of select neurological diseases.
Resources:
Acknowledgments:
Supported by NIH grants DA026430 and NS098777.
Glossary
- ABPP:
activity-based protein profiling; used in functional proteomic technology utilizing specific chemical probes to target and identify mechanistically related classes of enzymes
- DSE:
depolarization-induced suppression of excitation; following neuronal depolarization, glutamate-mediated neurotransmission is reduced
- DSI:
depolarization-induced suppression of inhibition; following neuronal depolarization, GABA-mediated neurotransmission is reduced
- LTD:
long-term synaptic depression; long-lasting decreases in synaptic strength following prolonged activation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: Nephi Stella is the founder of Stella Therapeutic Inc.
References
- 1.Mechoulam R et al. (2014) Early phytocannabinoid chemistry to endocannabinoids and beyond. Nature Reviews Neuroscience 15 (11), 757–764. [DOI] [PubMed] [Google Scholar]
- 2.Elphick MR (2012) The evolution and comparative neurobiology of endocannabinoid signalling. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 367 (1607), 3201–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caulfield MP and Brown DA (1992) Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. British Journal of Pharmacology 106 (2), 231–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hampson AJ and Grimaldi M, Cannabidiol and (−) Δ9-tetrahydrocannabinol are neuroprotective antioxidants, Proceedings of the …, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackie K and Hille B (1992) Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proceedings of the National Academy of Sciences of the United States of America 89 (9), 3825–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Q et al. (2012) Effects of anandamide on potassium channels in rat ventricular myocytes: a suppression of I(to) and augmentation of K(ATP) channels. American journal of physiology. Cell physiology 302 (6), C924–30. [DOI] [PubMed] [Google Scholar]
- 7.Maroso M et al. (2016) Cannabinoid Control of Learning and Memory through HCN Channels. Neuron 89 (5), 1059–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho BY et al. (1999) Coupling of the expressed cannabinoid CB1 and CB2 receptors to phospholipase C and G protein-coupled inwardly rectifying K+ channels. Receptors & channels 6 (5), 363–374. [PubMed] [Google Scholar]
- 9.Kreitzer AC and Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29 (3), 717–727. [DOI] [PubMed] [Google Scholar]
- 10.Ohno-Shosaku T et al. (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29 (3), 729–738. [DOI] [PubMed] [Google Scholar]
- 11.Wilson RI and Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410 (6828), 588–592. [DOI] [PubMed] [Google Scholar]
- 12.Roland AB et al. (2014) Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. eLife 3, 1489–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marsicano G et al. (2003) CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302 (5642), 84–88. [DOI] [PubMed] [Google Scholar]
- 14.Maccarrone M et al. (2014) Programming of neural cells by (endo) cannabinoids: from physiological rules to emerging therapies. Nature Reviews Neuroscience 15 (12), 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castillo PE et al. (2012) Endocannabinoid Signaling and Synaptic Function. Neuron 76 (1), 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munro S et al. (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365 (6441), 61–65. [DOI] [PubMed] [Google Scholar]
- 17.Shire D et al. (1996) Molecular cloning, expression and function of the murine CB2 peripheral cannabinoid receptor. Elsevier 1307 (2), 132–136. [DOI] [PubMed] [Google Scholar]
- 18.Guindon J and Hohmann A (2008) Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. British journal of pharmacology 153 (2), 319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stella N (2004) Cannabinoid signaling in glial cells. Glia 48 (4), 267–277. [DOI] [PubMed] [Google Scholar]
- 20.Li Y and Kim J (2017) Distinct roles of neuronal and microglial CB2 cannabinoid receptors in the mouse hippocampus. NSC 363, 11–25. [DOI] [PubMed] [Google Scholar]
- 21.Pertwee RG (2006) Cannabinoid pharmacology: the first 66 years. British Journal of Pharmacology 147 Suppl 1, S163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Petrocellis L et al. (2011) Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. British Journal of Pharmacology 163 (7), 1479–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiong W et al. (2011) Cannabinoid potentiation of glycine receptors contributes to cannabis-induced analgesia. Nature Chemical Biology 7 (5), 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogel Z et al. (1993) Anandamide, a brain endogenous compound, interacts specifically with the cannabinoid receptors and inhibits adenylate cyclase. The Journal of Neurochemistry 61, 352–355. [DOI] [PubMed] [Google Scholar]
- 25.Devane WA et al. (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258 (5090), 1946–1949. [DOI] [PubMed] [Google Scholar]
- 26.Felder CC et al. (1993) Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proceedings of the National Academy of Sciences of the United States of America 90 (16), 7656–7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogel Z et al. (1993) Anandamide, a Brain Endogenous Compound, Interacts Specifically with Cannabinoid Receptors and Inhibits Adenylate Cyclase. Journal of Neurochemistry 61 (1), 352–355. [DOI] [PubMed] [Google Scholar]
- 28.Di Marzo V et al. (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372 (6507), 686–691. [DOI] [PubMed] [Google Scholar]
- 29.Sugiura T et al. (1996) Enzymatic synthesis of anandamide, an endogenous cannabinoid receptor ligand, through N-acylphosphatidylethanolamine pathway in testis: involvement of Ca 2+ …. Elsevier 218 (1), 113–117. [DOI] [PubMed] [Google Scholar]
- 30.Mechoulam R et al. (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochemical pharmacology 50 (1), 83–90. [DOI] [PubMed] [Google Scholar]
- 31.Stella N et al. (1997) A second endogenous cannabinoid that modulates long-term potentiation. Nature 388 (6644), 773–778. [DOI] [PubMed] [Google Scholar]
- 32.Sugiura T et al. (1995) 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochemical and biophysical research communications 215 (1), 89–97. [DOI] [PubMed] [Google Scholar]
- 33.Beltramo M and Piomelli D (2000) Carrier-mediated transport and enzymatic hydrolysis of the endogenous cannabinoid 2-arachidonylglycerol. Neuroreport 11 (6), 1231–1235. [DOI] [PubMed] [Google Scholar]
- 34.Dinh TP et al. (2002) A role for monoglyceride lipase in 2-arachidonoylglycerol inactivation. Chemistry and physics of lipids 121 (1-2), 149–158. [DOI] [PubMed] [Google Scholar]
- 35.Stella N and Piomelli D (2001) Receptor-dependent formation of endogenous cannabinoids in cortical neurons. European journal of pharmacology 425 (3), 189–196. [DOI] [PubMed] [Google Scholar]
- 36.Kaczocha M et al. (2009) Identification of intracellular carriers for the endocannabinoid anandamide. Proceedings of the National Academy of Sciences 106 (15), 6375–6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blankman JL et al. (2007) A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chemistry & Biology 14 (12), 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muccioli GG et al. (2007) Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. The Journal of neuroscience : the official journal of the Society for Neuroscience 27 (11), 2883–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Tienhoven M et al. (2002) Human neuropathy target esterase catalyzes hydrolysis of membrane lipids. Journal of Biological Chemistry 277 (23), 20942–20948. [DOI] [PubMed] [Google Scholar]
- 40.Marrs WR et al. (2010) The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci 13 (8), 951–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li F et al. (2008) An unannotated α/β hydrolase superfamily member, ABHD6 differentially expressed among cancer cell lines. Molecular Biology Reports 36 (4), 691–696. [DOI] [PubMed] [Google Scholar]
- 42.Navia-Paldanius D et al. (2012) Biochemical and pharmacological characterization of human α/β-hydrolase domain containing 6 (ABHD6) and 12 (ABHD12). Journal of lipid research 53 (11), 2413–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gokce O et al. (2016) Cellular Taxonomy of the Mouse Striatum as Revealed by Single-Cell RNA-Seq. Cell Rep 16 (4), 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drehmer MN et al. (2018) Gene Expression of ABHD6, a Key Factor in the Endocannabinoid System, Can Be Modulated by Female Hormones in Human Immune Cells. Biochemical genetics, 1–11. [DOI] [PubMed] [Google Scholar]
- 45.Baggelaar MP et al. (2017) Chemical Proteomics Maps Brain Region Specific Activity of Endocannabinoid Hydrolases. ACS chemical biology 12 (3), 852–861. [DOI] [PubMed] [Google Scholar]
- 46.Straiker A et al. (2009) Monoacyl glycerol lipase (MGL) limits the duration of endocannabinoid-mediated depolarization-induced suppression of excitation (DSE) in autaptic hippocampal neurons. Molecular pharmacology, mol. 109.059030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pribasnig MA et al. (2015) α/β Hydrolase Domain-containing 6 (ABHD6) Degrades the Late Endosomal/Lysosomal Lipid Bis(monoacylglycero)phosphate. The Journal of biological chemistry 290 (50), 29869–29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bowman AL and Makriyannis A (2012) Highly Predictive Ligand-based Pharmacophore and Homology Models of ABHD6. Chemical Biology & Drug Design 81 (3), 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsu K-L et al. (2013) Discovery and Optimization of Piperidyl-1,2,3-Triazole Ureas as Potent, Selective, and in Vivo-Active Inhibitors of α/β-Hydrolase Domain Containing 6 (ABHD6). Journal of Medicinal Chemistry 56 (21), 8270–8279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marrs WR et al. (2011) Dual inhibition of alpha/beta-hydrolase domain 6 and fatty acid amide hydrolase increases endocannabinoid levels in neurons. J Biol Chem 286 (33), 28723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaczor AA et al. (2015) Comparative molecular field analysis and molecular dynamics studies of α/β hydrolase domain containing 6 (ABHD6) inhibitors. Journal of molecular modeling 21 (10), 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanaka M et al. (2017) WWL70 attenuates PGE 2 production derived from 2-arachidonoylglycerol in microglia by ABHD6-independent mechanism. Journal of Neuroinflammation 14 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Owens RA et al. (2017) Inhibition of the endocannabinoid-regulating enzyme monoacylglycerol lipase elicits a CB1 receptor-mediated discriminative stimulus in mice. Neuropharmacology 125, 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng H et al. (2017) Chiral disubstituted piperidinyl ureas: a class of dual diacylglycerol lipase-α and ABHD6 inhibitors. MedChemComm 8, 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Rooden EJ et al. (2018) Design and Synthesis of Quenched Activity-based Probes for Diacylglycerol Lipase and α,β-Hydrolase Domain Containing Protein 6. Chemistry, an Asian journal. [DOI] [PubMed] [Google Scholar]
- 56.Di S et al. (2005) Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. The Journal of physiology 569 (Pt 3), 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diana MA and Marty A (2004) Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). British Journal of Pharmacology 142 (1), 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Telleria-Diaz A et al. (2010) Spinal antinociceptive effects of cyclooxygenase inhibition during inflammation: Involvement of prostaglandins and endocannabinoids. Pain 148 (1), 26–35. [DOI] [PubMed] [Google Scholar]
- 59.Chevaleyre V et al. (2006) Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 29, 37–76. [DOI] [PubMed] [Google Scholar]
- 60.Straiker A and Mackie K (2009) Cannabinoid signaling in inhibitory autaptic hippocampal neurons. Neurobiology of Disease 163 (1), 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Straiker A et al. (2011) COX-2 and fatty acid amide hydrolase can regulate the time course of depolarization-induced suppression of excitation. British Journal of Pharmacology 164 (6), 1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wei M et al. (2016) α/β-Hydrolase domain-containing 6 (ABHD6) negatively regulates the surface delivery and synaptic function of AMPA receptors. Proceedings of the National Academy of Sciences 113 (19), E2695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwenk J et al. (2012) High-Resolution Proteomics Unravel Architecture and Molecular Diversity of Native AMPA Receptor Complexes. Neuron 74 (4), 621–633. [DOI] [PubMed] [Google Scholar]
- 64.Wei M et al. (2017) The Inhibitory Effect of α/β-Hydrolase Domain-Containing 6 (ABHD6) on the Surface Targeting of GluA2- and GluA3-Containing AMPA Receptors. Frontiers in molecular neuroscience 10, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thomas G et al. (2013) The Serine Hydrolase ABHD6 Is a Critical Regulator of the Metabolic Syndrome. CellReports 5 (2), 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao S et al. (2015) α/β-Hydrolase domain-6 and saturated long chain monoacylglycerol regulate insulin secretion promoted by both fuel and non-fuel stimuli. Molecular metabolism 4 (12), 940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao S et al. (2016) α/β-Hydrolase Domain 6 Deletion Induces Adipose Browning and Prevents Obesity and Type 2 Diabetes. CellReports 14 (12), 2872–2888. [DOI] [PubMed] [Google Scholar]
- 68.Max D et al. (2009) High expression of the evolutionarily conserved alpha/beta hydrolase domain containing 6 (ABHD6) in Ewing tumors. Cancer science 100 (12), 2383–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oparina NY et al. (2014) PXK locus in systemic lupus erythematosus: fine mapping and functional analysis reveals novel susceptibility gene ABHD6. Annals of the Rheumatic Diseases. [DOI] [PubMed] [Google Scholar]
- 70.Maier S et al. (2006) Cellular target genes of Epstein-Barr virus nuclear antigen 2. Journal of Virology 80 (19), 9761–9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee JH (2018) Endocannabinoid hydrolases in avian HD11 macrophages identified by chemoproteomics: inactivation by small-molecule inhibitors and pathogen-induced downregulation of their activity. Molecular and Cellular Biochemistry 444 (1), 125–141. [DOI] [PubMed] [Google Scholar]
- 72.Naydenov AV et al. (2014) ABHD6 Blockade Exerts Antiepileptic Activity in PTZ-Induced Seizures and in Spontaneous Seizures in R6/2 Mice. Neuron 83 (2), 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sigel E et al. (2011) The major central endocannabinoid directly acts at GABA(A) receptors. Proceedings of the National Academy of Sciences 108 (44), 18150–18155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McKinney DL et al. (2008) Dose-related differences in the regional pattern of cannabinoid receptor adaptation and in vivo tolerance development to Δ9-tetrahydrocannabinol. Journal of Pharmacology and Experimental Therapeutics 324 (2), 664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jung K-M et al. (2012) Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile × syndrome. Nature communications 3 (1), 1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Panikashvili D et al. (2001) An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413, 527–31. [DOI] [PubMed] [Google Scholar]
- 77.Tchantchou F and Zhang Y (2013) Selective inhibition of alpha/beta-hydrolase domain 6 attenuates neurodegeneration, alleviates blood brain barrier breakdown, and improves functional recovery in a mouse model of traumatic brain injury. Journal of neurotrauma 30 (7), 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wen J et al. (2015) Activation of CB2 receptor is required for the therapeutic effect of ABHD6 inhibition in experimental autoimmune encephalomyelitis. Neuropharmacology 99, 196–209. [DOI] [PubMed] [Google Scholar]
- 79.Manterola A et al. (2018) Deregulation of the endocannabinoid system and therapeutic potential of ABHD6 blockade in the cuprizone model of demyelination. Biochemical pharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manterola A et al. (2018) Re-examining the potential of targeting ABHD6 in multiple sclerosis: Efficacy of systemic and peripherally restricted inhibitors in experimental autoimmune encephalomyelitis. Neuropharmacology, 1–33. [DOI] [PubMed] [Google Scholar]
- 81.Fisette A et al. (2016) α/β-Hydrolase Domain 6 in the Ventromedial Hypothalamus Controls Energy Metabolism Flexibility. CellReports 17 (5), 1217–1226. [DOI] [PubMed] [Google Scholar]
- 82.Metna-Laurent M and Marsicano G (2015) Rising stars: modulation of brain functions by astroglial type-1 cannabinoid receptors. Glia 63 (3), 353–364. [DOI] [PubMed] [Google Scholar]
- 83.Zhao S et al. (2014) α/β-Hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell metabolism 19 (6), 993–1007. [DOI] [PubMed] [Google Scholar]