

Skeletal muscle oxidative capacity and fatigue resistance can be improved with endurance training, but the mechanism is not fully understood. Ivarsson et al. find that the signaling pathway that increases fatigue resistance in muscle is triggered by a mild Ca2+ leak from the sarcoplasmic reticulum.
Abstract
Effective practices to improve skeletal muscle fatigue resistance are crucial for athletes as well as patients with dysfunctional muscles. To this end, it is important to identify the cellular signaling pathway that triggers mitochondrial biogenesis and thereby increases oxidative capacity and fatigue resistance in skeletal muscle fibers. Here, we test the hypothesis that the stress induced in skeletal muscle fibers by endurance exercise causes a reduction in the association of FK506-binding protein 12 (FKBP12) with ryanodine receptor 1 (RYR1). This will result in a mild Ca2+ leak from the sarcoplasmic reticulum (SR), which could trigger mitochondrial biogenesis and improved fatigue resistance. After giving mice access to an in-cage running wheel for three weeks, we observed decreased FKBP12 association to RYR1, increased baseline [Ca2+]i, and signaling associated with greater mitochondrial biogenesis in muscle, including PGC1α1. After six weeks of voluntary running, FKBP12 association is normalized, baseline [Ca2+]i returned to values below that of nonrunning controls, and signaling for increased mitochondrial biogenesis was no longer present. The adaptations toward improved endurance exercise performance that were observed with training could be mimicked by pharmacological agents that destabilize RYR1 and thereby induce a modest Ca2+ leak. We conclude that a mild RYR1 SR Ca2+ leak is a key trigger for the signaling pathway that increases muscle fatigue resistance.
Introduction
Ca2+ plays a central role in intracellular signaling in most cell types. Ca2+ plays an essential role in initiating contraction in skeletal muscle. The neural activation triggers action potentials that propagate along the surface membrane of muscle fibers and into the transverse tubular system, where they activate voltage sensors, the dihydropyridine receptors (DHPRs). Activated DHPRs open the SR Ca2+ release channel, ryanodine receptor 1 (RYR1), resulting in an increase in the free cytosolic [Ca2+] ([Ca2+]i). Increasing [Ca2+]i exposes the myosin-binding site on the actin filament, leading to repetitive cycles of myosin head (cross-bridge) binding and muscle contraction (Gordon et al., 2000; Allen et al., 2008). Cross-bridge cycles and the continuous pumping of Ca2+ back into the SR during contractions are highly energy-demanding processes. Thus, oxidative capacity is a key determinant of muscle endurance, i.e., the ability to maintain contractile function during repeated cycles of SR Ca2+ releases and contractions (Holloszy et al., 1970; Holloszy and Coyle, 1984; Allen et al., 2008; Ekblom-Bak et al., 2014; Cheng et al., 2018).
The large RYR1 tetramer (∼2 megadalton) is the core of a protein complex, which includes the FK506 -binding protein 12 (FKBP12, also known as calstabin1; Ahern et al., 1994; Zalk et al., 2015). If FKBP12 is dissociated from RYR1, the channel has an increased open probability, i.e., it becomes “leaky” (Ahern et al., 1997). Prolonged periods of stress may lead to hyperphosphorylation of RYR1, causing a severe SR Ca2+ leak due to disassociation of FKBP12 (Marx et al., 2000; Aydin et al., 2008). Modifications of RYR1 induced by reactive oxygen/nitrogen species also causes severe disassociation of FKBP12 and subsequent SR Ca2+ leakage (Bellinger et al., 2008). Severe FKBP12 disassociation from RYR1 is linked to muscle weakness in overtraining (Bellinger et al., 2008), as well as various diseases, such as muscle dystrophy (Bellinger et al., 2009), breast cancer–related cachexia (Waning et al., 2015), and ventilator-induced diaphragmatic dysfunction (Matecki et al., 2016).
However, increased SR Ca2+ leak leading to increased [Ca2+]i at rest may not always be deleterious. A minor elevation of baseline [Ca2+]i, which in itself is not sufficient to initiate contraction, may act as a signal for increased mitochondrial biogenesis by activating Ca2+-sensing proteins, such as Ca2+-calmodulin–dependent protein kinase II, calcineurin, or cAMP response element–binding protein, resulting in transcription of proteins important for oxidative capacity (Ojuka et al., 2002; Wu et al., 2002; Tavi and Westerblad, 2011; Bruno et al., 2014). Furthermore, a prolonged increase in [Ca2+]i can increase the expression of peroxisome proliferator–activated receptor γ coactivator-1α (PGC1α; Wu et al., 2002; Ojuka et al., 2003; Wright et al., 2007), which is tightly associated with mitochondrial numbers, volume and protein content (Wu et al., 1999; Lehman et al., 2000; Baar et al., 2002), and improved exercise endurance in humans (Pilegaard et al., 2003; Norrbom et al., 2004).
Endurance exercise is the most effective method to increase oxidative capacity and mitochondrial content in skeletal muscle (Holloszy and Coyle, 1984; Booth and Thomason, 1991). Nevertheless, the immediate cellular triggers promoting increased oxidative capacity in response to endurance training are not fully understood. The present study is based on the hypothesis that the stress induced in skeletal muscle fibers by endurance exercise causes decreased FKBP12 association to RYR1 and SR Ca2+ leak, which trigger mitochondrial biogenesis, leading to improved fatigue resistance. The hypothesis was tested by investigating RYR1 modifications, cellular Ca2+ handling, mitochondrial biogenesis, and fatigue resistance in muscles from mice either engaged in voluntary running or exposed to pharmacological agents that induce SR Ca2+ leak.
Materials and methods
Ethical approval
Animal experiments complied with the Swedish Animal Welfare Act, the Swedish Welfare Ordinance, and applicable regulations and recommendations from Swedish authorities. The study was approved by the Stockholm North Ethical Committee on Animal Experiments (no. N120/13). We used a total of 81 male C57BL/6N (Harlan) mice with an age of 8 wk at the start of interventions. Animals were housed in a temperature-controlled environment with a 12-h light–dark cycle and were provided with standard rodent chow and water ad libitum. Mice were euthanized by rapid cervical dislocation before tissue extraction.
Mouse experiments
Mice were individually housed in cages equipped with a wireless low profile running wheel (ENV-044; Med Associates), and running distance was continuously measured for up to 6 wk. Sedentary controls were similarly housed with locked running wheels. Pharmacologically treated mice were housed in standard cages and injected intraperitoneally every 48 h for 3 wk with 0.7% sterile NaCl solution containing 0.1% DMSO with or without 10 ng/g body weight rapamycin (Sigma-Aldrich) or synthetic ligand of FKBP12 (SLF; Cayman Chemicals). These drugs destabilize RYR1, and the concentration of rapamycin and SLF used here has previously been shown not to have any measurable mTOR (mammalian target of rapamycin)-mediated effects (Lee et al., 2014).
In vivo endurance test
An exhaustion test was conducted after 3 wk on sedentary and voluntary running mice and mice injected with rapamycin or SLF. Endurance was assessed using a mouse treadmill (Exer 3/6; Columbus Instruments). Mice were prepared for the exhaustion test by running on the treadmill for 10 min/d for 4 d before the test. During the test, mice ran at 25° uphill, and an initial 10-min warm-up at a speed of 10 m/min was followed by gradual speed increases of 2 m/min every 2 min. Endurance was scored as the speed during the last interval completed before exhaustion. Exhaustion was determined as the time when the mouse withstood three mild electric shocks (0.1 mA, 2 Hz) without attempts to continue running. This exhaustion test provides an indirect measure of mouse maximal oxygen uptake (VO2max) (Kemi et al., 2002).
Force and [Ca2+]i measurements in single muscle fibers
Intact, single muscle fibers were dissected from flexor digitorum brevis (FDB) muscles as described elsewhere (Cheng and Westerblad, 2017). The isolated fiber was mounted in a stimulation chamber at optimum length and superfused with Tyrode’s solution (in mM): NaCl, 121; KCl, 5.0; CaCl2, 1.8; MgCl2, 0.5; NaH2PO4, 0.4; NaHCO3, 24.0; EDTA, 0.1; glucose, 5.5. Fetal calf serum (0.2%) was added to the solution to improve muscle fiber survival. The solution was bubbled with 5% CO2–95% O2, which gives an extracellular pH of 7.4. Experiments were performed at room temperature (∼25°C). Tetanic stimulation was achieved by supramaximal current pulses (duration, 0.5 ms) delivered via platinum plate electrodes lying parallel to the muscle fiber.
The fluorescent Ca2+ indicator indo-1 (Invitrogen/Molecular Probes) was microinjected into the isolated fiber. The fiber was allowed to rest for at least 30 min after being injected with indo-1. It was then stimulated by individual 350-ms stimulation trains at 10 to 150 Hz given at 1-min intervals. Indo-1 was excited at 360 nm, and emitted fluorescence was measured at 405 nm and 495 nm. The fluorescence ratio of indo-1 was converted to [Ca2+]i using an intracellularly established calibration curve (Andrade et al., 1998). Tetanic [Ca2+]i was measured as the mean indo-1 fluorescence during tetanic stimulation trains and basal [Ca2+]i as the mean over ∼200 ms before stimulation. Tetanic force was measured as the mean over 100 ms where force was maximal. Fatigue was induced by repeated tetanic stimulations (70 Hz, 350-ms duration) given at 2-s intervals for 100 contractions.
Measurements of cellular Ca2+ entry
Mn2+ quenching of fura-2 fluorescence (Hopf et al., 1996) was used to assess cellular Ca2+ entry in FDB fibers of sedentary control mice and of mice performing 3 or 6 wk voluntary running. Mouse FDB muscles were isolated and incubated in DMEM (Sigma-Aldrich) with 10% fetal calf serum and 0.3% type I collagenase (Sigma-Aldrich) for 2 h at 37°C. Dissociated single muscle fibers were plated on laminin coated glass bottom dishes (Mattek) in DMEM with 10% fetal calf serum and incubated for at least 1 h at 37°C. Fibers were loaded with 4 ng/µl fura-2 AM (Invitrogen/Molecular Probes) for 30 min and then washed with Tyrode’s for 30 min. Fura-2 was excited at 360 nm (i.e., the isosbestic point where fluorescence is independent of Ca2+), and emitted fluorescence was measured at 495 nm. Baseline fluorescence was measured for 50 s, after which 1 mM MnCl2 was added and the signal was followed for 50 s. Fibers were then stimulated with 50 repeated 70-Hz, 350-ms tetani given at 2-s intervals, and the fluorescent signal was followed for 500 s subsequent to stimulation, after which MnCl2 was washed out. The slope of fluorescence decay was measured for 50 s before and between 100 and 400 s after stimulation and expressed relative to the fluorescent signal at the start of the respective measurement period.
Western blotting and immunoprecipitation (IP)
Mouse tibialis anterior muscles were homogenized with a ground glass homogenizer in ice-cold homogenization buffer (20 µl per mg wet weight, pH 7.6) consisting of (in mM): HEPES, 20; NaCl, 150; EDTA, 5; KF, 25; Na3VO4, 1; and 20% glycerol, 0.5% Triton X-100, and protease inhibitor cocktail (Roche; 1 tablet/50 ml). The homogenate was centrifuged at 700 g for 10 min at 4°C. Protein content of the supernatant was determined using the Bradford assay (#500-0006; Bio-Rad) and frozen at −80°C until protein analysis. On the day of gel electrophoresis, samples were diluted 1:1 in Laemmli buffer (Bio-Rad) with 5% 2-mercaptoethanol and heated to 95°C for 5 min. For IP, 1 µg anti-RYR1 (ab2868, Abcam) was bound to 12 µl G-protein Dynal magnetic beads (Invitrogen) per the manufacturer’s instructions. Samples were diluted to 0.5 µg/µl in 400 µl ice-cold RIPA buffer, pH 7.5, consisting of (in mM): Trizma-HCl, 10; NaCl, 150; NaF, 5; Na3VO4, 1; 1% Triton X-100, and protease inhibitor cocktail (Roche; 1 tablet/50 ml). The samples were then mixed with antibody-bead complex and incubated under rotation at 4°C overnight. The samples were gently washed four times with RIPA buffer. Peptides were then eluded from the beads with 50 µl Laemmli buffer (Bio-Rad) with 5% 2-mercaptoethanol and heated to 95°C for 5 min. For the gel electrophoresis, 10 µg protein or 15 µl IP eluate was run on a 4–12% precast Bis–Tris gel (NP0336PK2, NuPAGE; Invitrogen) and transferred onto polyvinylidine fluoride membranes (Immobilon FL; Millipore, Billerica). Membranes were then blocked with Li-Cor blocking buffer (LI-COR Biosciences) followed by incubation overnight at 4°C with the following antibodies diluted in blocking buffer; mouse anti-RYR1 (ab2868, Abcam), rabbit anti-RyR-pho (phospho S2843; ab592205; Abcam), mouse anti-oxphos complex IV (cytochrome c oxidase) subunit 1 (COX1; #459600, Invitrogen), rabbit anti-PGC1α (ab54481; Abcam), rabbit anti-actin (ab1801; Abcam), rabbit anti-FKBP12 (ab2918; Abcam), mouse anti-DHPR(α 2 subunit; ab2864; Abcam), mouse anti-SERCA1 (ab2819; Abcam), and rabbit anti-STIM1 (ab108994; Abcam). Membranes were washed and incubated with secondary antibody IRDye 680–conjugated donkey anti-mouse IgG and IRDye 800–conjugated donkey anti-rabbit IgG (926–68072, 926–32213, LI-COR). Immunoreactive bands were visualized using infrared fluorescence (IR-Odyssey scanner; LI-COR Biosciences). Band density was established using Image Studio v 2.0.38 (LI-COR Biosciences), and the band density of actin was used as loading control. Actin and RYR1 were used as loading control for the Western blot and IP experiments, respectively. Equal protein loading to each lane was also verified with protein staining of membranes (#161-0436; Bio-Rad). For S6K1 signaling, membrane was first stained with rabbit anti-S6K1 (phospho T412; ab78413; Abcam), blotted with secondary IRDye 800–conjugated donkey anti-rabbit IgG (926–68072, 926–32213; LI-COR) and subsequently scanned. Membrane was then stripped using Restore Fluorescent Western Blot Stripping Buffer (ThermoFisher), per the manufacturer’s instructions. Membrane was relocked and blotted with rabbit anti-S6K1 antibody [E343] (ab32529; Abcam) and reblotted with IRDye 800–conjugated donkey anti-rabbit IgG (926–68072, 926–32213; LI-COR). Signal is calculated as relative S6K1 (phospho-T412) to total S6K1.
Enzymatic activity
Mouse tibialis anterior muscles were homogenized in ice-cold buffer (pH 7.4; 50 µl/mg wet weight) consisting of (in mM): Tris, 50; sodium citrate, 5; MnCl2, 0.6; cysteine, 1; and 0.05% (vol/vol) Triton X-100, pH 7.4. The homogenate was centrifuged for 1 min at 1,400 g (4°C) and aliquots of the supernatant were frozen at −80°C until citrate synthase activity assay. Citrate synthase activities were analyzed with standard spectrophotometric method based on generation of free coenzyme A reacting with 5,5′-dithio-bis-[2-nitrobenzoic acid] (DTNB; Bass et al., 1969). The activities were measured at room temperature under conditions that yielded linearity with respect to extract volume and time. The supernatant protein content was determined using the Bradford assay (BioRad), and activities were adjusted for protein content.
Messenger RNA (mRNA) expression
Total RNA was obtained from mouse extensor digitorum longus by mechanical homogenization using 20× RNeasy lysis buffer (QIAGEN) with 1% β-mercaptoethanol. Homogenates were diluted in 1 ml Trizol (Invitrogen) and mRNA was purified via centrifugation. The extracted mRNA was treated with DNase (Ambion; Invitrogen), and 500 ng complementary DNA (cDNA) was prepared using reverse transcript-PCR (SuperScript III First-Strand Synthetis RT-PCR; Invitrogen). The cDNA was frozen at −80°C until the day of quantitative PCR. Final cDNA was diluted to 2.5 ng/µl, and real-time determination of transcript abundance was performed with SYBR-Green–based real-time quantitative PCR (BioRad) using 1.5 µl cDNA solution per reaction (i.e., ∼3.75 ng cDNA) in 5 µl final volume. Table 1 provides the sequences of primers used. Each gene is calculated as relative to the transcript abundance of the housekeeping gene hypoxanthine guanine phosphoribosyl transferase (HPRT), which did not differ between groups. Data are presented as relative expression compared with average control, which was set to 1.0.
Table 1. Forward and reverse qPCR primer sequence for gene expression analysis.
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|
| HPRT | ACAGGCCAGACTTTGTTGGA | ACTTGCGCTCATCTTAGGCT |
| PGC1α1 | GGACATGTGCAGCCAAGACTCT | CACTTCAATCCACCCAGAAAGCT |
| TFAM | CACCCAGATGCAAAACTTTCAG | CTGCTCTTTATACTTGCTCACAG |
| COX5B | ACCCTAATCTAGTCCCGTCC | CAGCCAAAACCAGATGACAG |
| MEF2c | GCTGTTCCAGTACGCCAGC | AGTGCGTGGGGTGAGTGCA |
Statistical analyses
Data are presented as mean ± SEM or as boxplots showing median, quartiles, and min/max. Statistical significant changes were assessed using a two-tailed paired or unpaired Student’s t test or one-way ANOVA with Holm–Sidak post hoc test when comparing more than two groups. Significant differences were set as P < 0.05. All statistical analyses were performed with the SigmaPlot 13.0 software for Windows (Systat Software).
Results
Increasing voluntary running causes RYR1 destabilization and increased baseline [Ca2+]i and triggers signaling for mitochondrial biogenesis
Mice given access to an in-cage running wheel performed prolonged low-intensity exercise ∼12 h a day and gradually increased their voluntary running distance up to ∼15 km per day during the first 3 wk and thereafter the daily running distance remained stable (Fig. 1 A). After 3 wk of running, there was an increased SR Ca2+ leak as judged from a modest dissociation of FKBP12 from RYR1 (Fig. 1 B), accompanied by a ∼25% increased baseline [Ca2+]i in isolated FDB fibers (Fig. 1 C). Conversely, after 6 wk, when the running distance had reached a steady state, FKBP12’s association with RYR1 had returned to the control level (Fig. 1 B), and baseline [Ca2+]i was actually ∼50% lower than in FDB fibers of nonrunning controls (Fig. 1 C). Previous studies have linked exercise and cold exposure–induced leaky RYR1 to hyperphosphorylation likely due to adrenergic activation of protein kinase A, which phosphorylates S2843 on RYR1 (Aydin et al., 2008; Bellinger et al., 2008). We observed an increase in average RYR1 phosphorylation after 3 wk of running, but this increase did not reach statistical significance (P = 0.22; Fig. 1 D). Thus, further experiments are required to reveal the mechanisms underlying the FKBP12 dissociation from RYR1 and the resulting increase in baseline [Ca2+]i after 3 wk of increasing running, but this is outside the scope of the present study.
Figure 1.
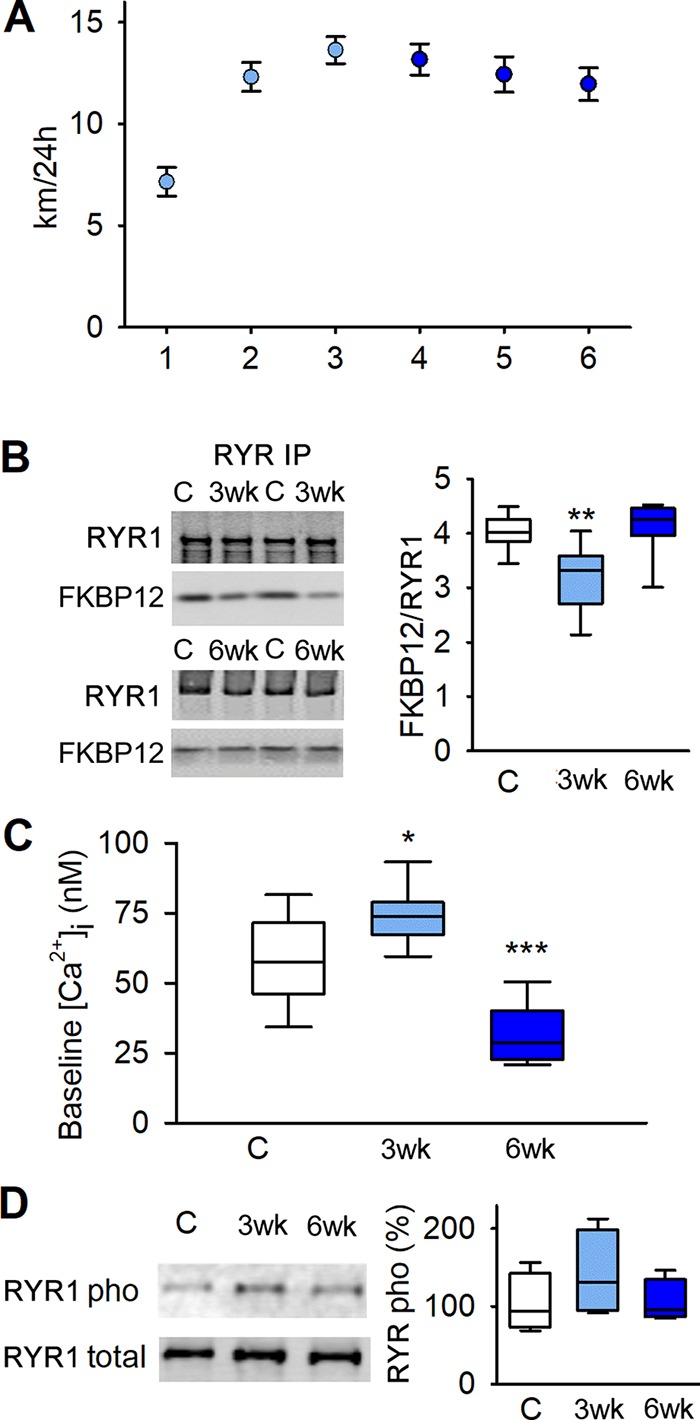
Voluntary running causes leaky RYR1 and elevated baseline [Ca2+]i. (A) Mean (± SEM) of voluntary 24-h running distance of mice with up to 6-wk access to an in-cage running wheel (n = 20). (B) Original blots (left) and boxplot data (right) of immunoprecipitated RYR1 in tibialis anterior from mice with locked wheels (controls), 3-wk runners, and 6-wk runners blotted for coprecipitated FKBP12; data are presented relative to the mean value in controls (n = 17, 10, and 7, respectively). (C) Boxplot of baseline [Ca2+]i in dissected single FDB fibers from controls, and 3- and 6-wk runners (n = 14, 9, and 6 respectively). (D) Original blots (left) and boxplot of relative S2834 phosphorylation per total RYR1; data presented relative to the mean pixel density in controls, which was set to 100%. *, P < 0.05; **, P < 0.01; ***, P < 0.001, 3-wk or 6-wk vs. controls with one-way ANOVA. 3wk, 3-wk runners; 6wk, 6-wk runners; C, controls. Box plots show median, quartiles, and min/max.
After 3 wk of running, there was a marked increase in skeletal muscle mRNA expression of the mitochondrial biogenesis promoting genes of PGC1α1, myocyte enhancer factor-2c (MEF2c), and mitochondrial transcription factor A (TFAM; Fig. 2 A), as well as the protein level of PGC1α1 (Fig. 2 B). Intriguingly, all of these mitochondria-promoting factors had returned to the control level after 6 wk of running, i.e., they followed the same temporal pattern as the FKBP12 dissociation from RYR1. However, the mRNA expression of cytochrome c oxidase subunit 5B (COX5B) and the protein content of the mitochondrial DNA (mtDNA)-encoded cytochrome c oxidase subunit 1 (COX1), which reflects the mitochondrial volume in muscle, were increased after 3 wk of running and remained high at 6 wk of running (Fig. 2, A and B). Thus, with continued running, the mitochondrial content was maintained at a higher level than in control despite no concomitant up-regulation of mitochondria-promoting factors, which indicates temporal and quantitative differences in the signaling for the initial increase in mitochondrial biogenesis and subsequent maintenance at a higher level.
Figure 2.
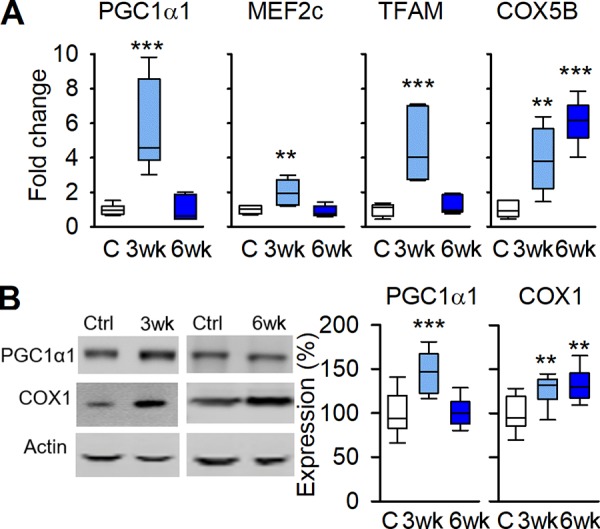
Signaling-promoting mitochondrial biogenesis follows the temporal pattern of leaky RYR1. (A) Boxplots showing mRNA expression of PGC1α1, MEF2c, TFAM, and COX5B relative to HPRT in controls and 3- and 6-wk runners; values were normalized to mean expression of sedentary controls, which was set to 1.0 (n = 6). (B) Original blots (left) and boxplots of relative expression (right) of PGC1α1 and COX1 protein with actin as loading control; data are presented relative to the mean pixel density in controls, which was set to 100% (n = 13, 6, and 7 respectively). **, P < 0.01; ***, P < 0.001, 3-wk or 6-wk vs. controls with one-way ANOVA. Box plots show median, quartiles, and min/max.
The decreased [Ca2+]i at rest after 6 wk of running was unexpected and experiments were performed to reveal underlying mechanisms. In addition to RYR1 resting conductance, the rates of active reuptake of Ca2+ into the SR by the SR Ca2+-ATPase (SERCA1) and Ca2+ entering the cell via store-operated Ca2+ entry (SOCE) can affect [Ca2+]i at rest. Compared with control muscles, the protein expression of SERCA1 was increased by ∼70% and ∼120% after 3 and 6 wk of running, whereas the SR Ca2+ sensor that activates SOCE, the stromal-interacting molecule 1 (STIM1; Roos et al., 2005; Wei-LaPierre et al., 2013), showed a progressive decrease by ∼20% and ∼40% (Fig. 3 A). We also used Mn2+ quenching of fura-2 to assess the rate of SOCE (Wei-LaPierre et al., 2013). The rate of Mn2+ quenching was low in fully rested FDB fibers, and there was no difference between fibers from control mice and mice running for 3 or 6 wk. The rate of Mn2+ quenching was markedly higher after a series of tetanic contractions, and in this case, the rate was significantly lower in fibers of 6-wk runners than of controls (Fig. 3 B), as expected from the down-regulation of STIM1 with running.
Figure 3.
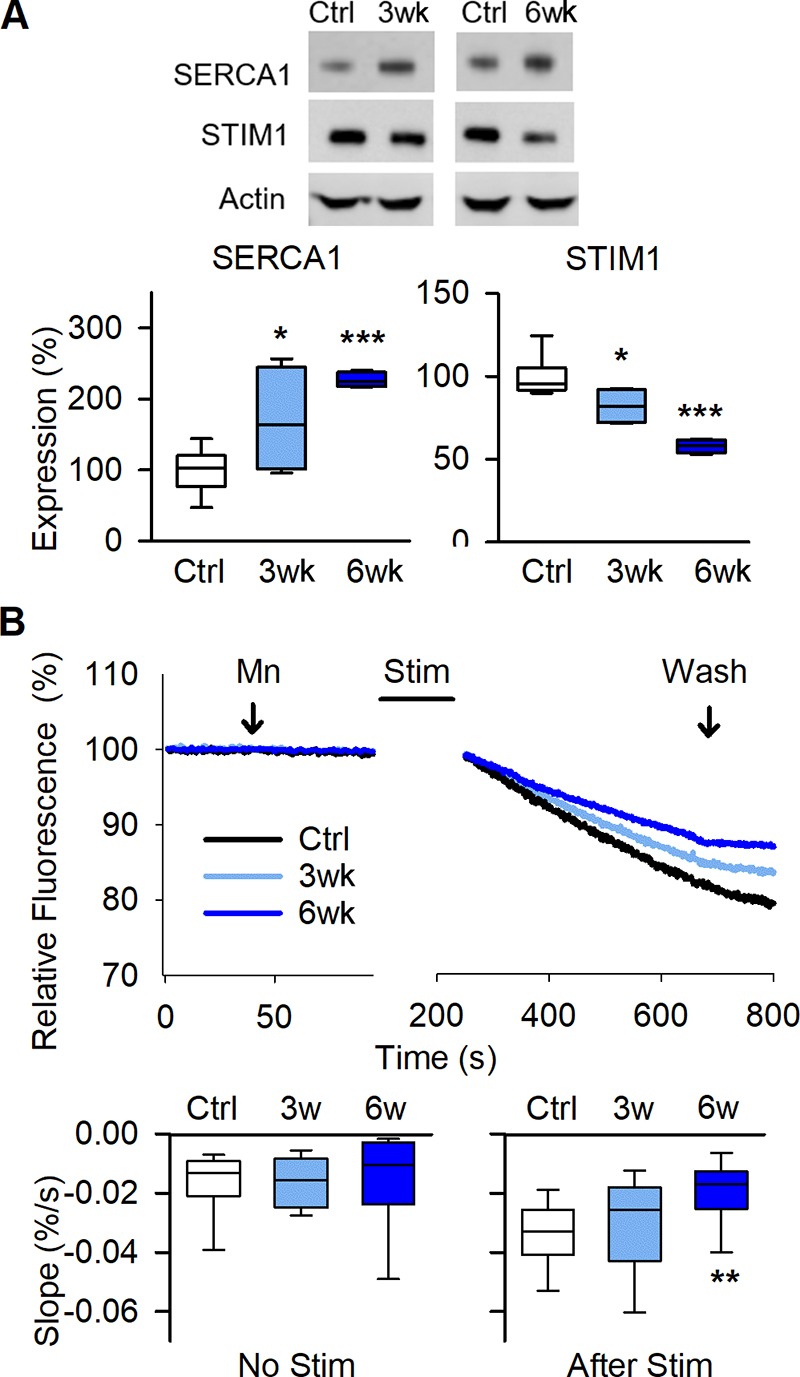
Voluntary running enhances SR Ca2+ pumping and decreases store-operated Ca2+ entry. (A) Original Western blots (top) and boxplots of relative expression (below) show increased SERCA1 and decreased STIM1 protein expression in tibialis anterior muscles of 3-wk and 6-wk running mice with actin as loading control; data are presented relative to the mean pixel density in controls, which was set to 100% (n = 8, 4, and 4 for control, 3 wk, and 6 wk, respectively). (B) Mn2+ quenching of fura-2 fluorescence shows decreased rate of Ca2+ entry after repeated contractions in FDB fibers from 6-wk running mice. Representative fluorescent signal traces (above) over time obtained before and after stimulating the isolated fibers to produce repeated tetanic contractions. Boxplots comparisons of the slope (below) of fura-2 fluorescence decay before (left) and after (right) repeated tetanic contractions (n = 13–14 fibers from eight, four, and four mice for control, 3 wk, and 6 wk, respectively). *, P < 0.05; **, P < 0.01; ***, P < 0.001, 3 wk or 6 wk vs. sedentary controls (Ctrl) with one-way ANOVA. Box plots show median, quartiles, and min/max.
In the unfatigued state, single FDB fibers dissected from control and running mice showed similar force-frequency and [Ca2+]i-frequency relationships (Fig. 4 A). FDB fibers from 3- and 6-wk runners maintained tetanic [Ca2+]i and force better than nonrunning controls during the induction of fatigue by repeated tetanic contractions (70 Hz, 350 ms given at 2-s intervals; Fig. 4, B and C).
Figure 4.
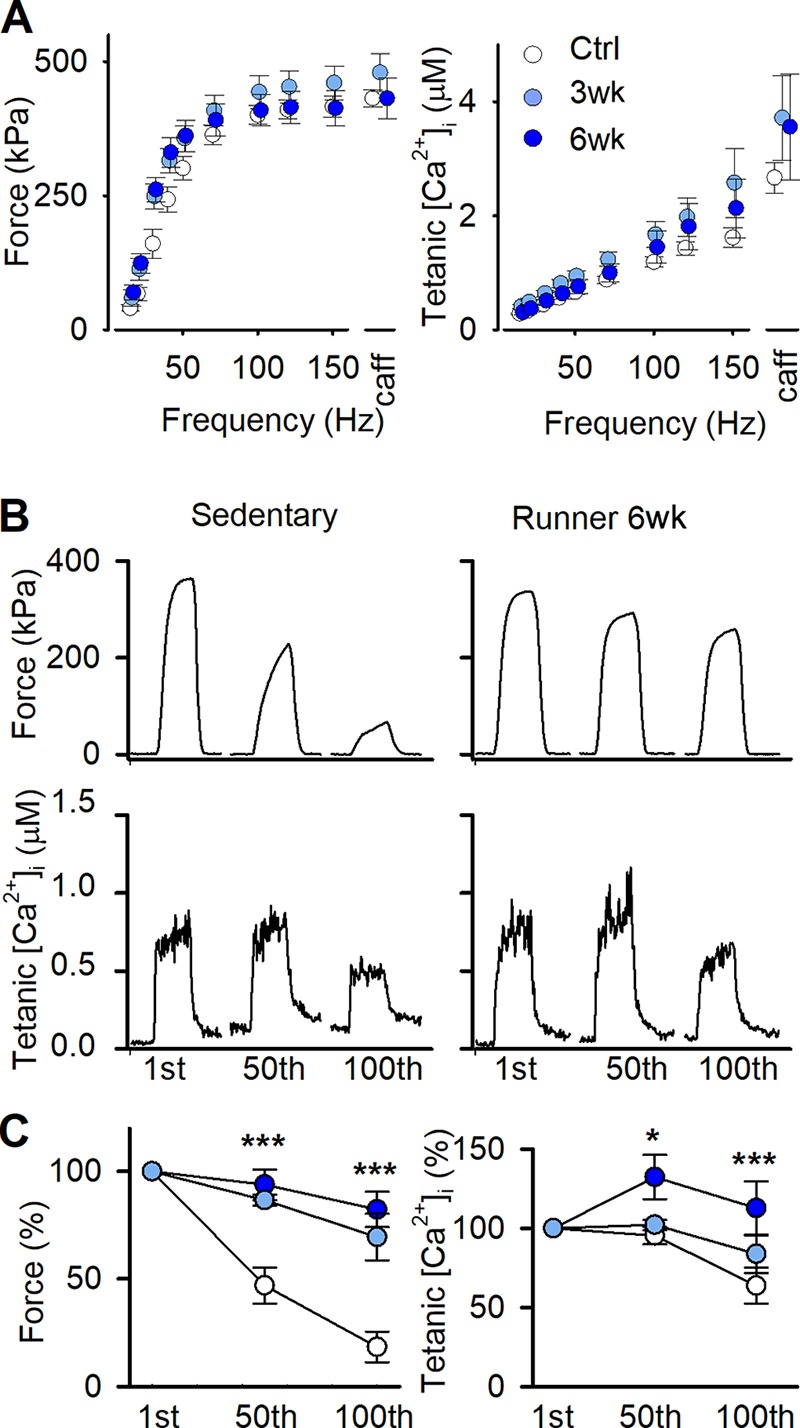
Dissected single FDB fibers are more fatigue resistant after 3 and 6 wk of voluntary running. (A) Mean (± SEM) force (left) and [Ca2+]i (right) against stimulation frequency in individual FDB fibers of control mice with locked wheels (Ctrl; open circles), and mice running for 3 wk (3wk; light blue circles) and 6 wk (6wk; dark blue circles). Tetani at 150 Hz were also produced in the presence of 5 mM caffeine (caff) to assess maximum force generating capacity and total SR Ca2+ content (n = 17, 9, and 6 fibers from 10, 5, and 5 mice, respectively). (B) Representative force and [Ca2+]i records of the 1st, 50th, and 100th fatiguing tetanic contractions (70-Hz, 350-ms tetani given every 2 s) of individual FDB fibers from control (left) and 6-wk running (right) mice. (C) Mean (± SEM) relative tetanic force (left) and [Ca2+]i (right) during fatiguing 70-Hz, 350-ms tetani given every 2 s (n = 17, 9, and 6 fibers from 10, 5, and 5 mice for control 3 wk, and 6 wk, respectively). *, P < 0.05; ***, P < 0.001 50th or 100th contraction vs. first contraction with one-way ANOVA.
The decay of [Ca2+]i after tetanic stimulation can be used to broadly distinguish between effects due to alterations in SR Ca2+ pumping or passive SR Ca2+ leak. Averaged decays of [Ca2+]i after a 150-Hz tetanus showed no notable difference between muscle fibers from controls and runners in the early fast phase (Fig. 5 A). However, after the initial fast phase, [Ca2+]i leveled off at higher and lower [Ca2+]i in fibers of 3- and 6-wk runners, respectively (Fig. 5 B). The late slow phase of [Ca2+]i can be used to assess changes in SR Ca2+ pumping and SR Ca2+ leak (Klein et al., 1991; Westerblad and Allen, 1993). For this assessment, we first fitted the slow [Ca2+]i decay of each individual fiber to the sum of two exponential functions (Fig. 5 C). From these fits, the relation between the rate of [Ca2+]i decline (d[Ca2+]i /dt) and [Ca2+]i was obtained (Fig. 5 D), and data points from this relation were fitted to the following equation:
where A is a factor reflecting the rate of SR Ca2+ pumping, L represents the SR Ca2+ leak, and N is a power function related to the Ca2+ binding per SR pump unit (Klein et al., 1991). N = 4.03 ± 0.24 (n = 39 curve fits) when allowed to fit freely, which is similar to what others have reported (Klein et al., 1991; Westerblad and Allen, 1993; Head et al., 2015). To facilitate the comparison of L and A between groups, N was set to four for all fibers, and values for L and A in each fiber were obtained when d[Ca2+]i/dt − A([Ca2+]i)4 + L = 0. This assessment gave about twice as large SR Ca2+ leak (L) in 3-wk runners than in sedentary controls, whereas the leak in 6-wk runners was similar to controls (Fig. 5 E). Furthermore, the assessment gave a significantly higher SR Ca2+ uptake rate (A) in 6-wk runners than in controls (Fig. 5 F), which fits with the marked increase in SERCA1 protein expression in the 6-wk group (see Fig. 3 A). Thus, [Ca2+]i decay assessment indicates that the higher resting [Ca2+]i in 3-wk runners is mainly due to increased leak, whereas the decreased resting [Ca2+]i in 6-wk runners can be explained by increased pump rate.
Figure 5.
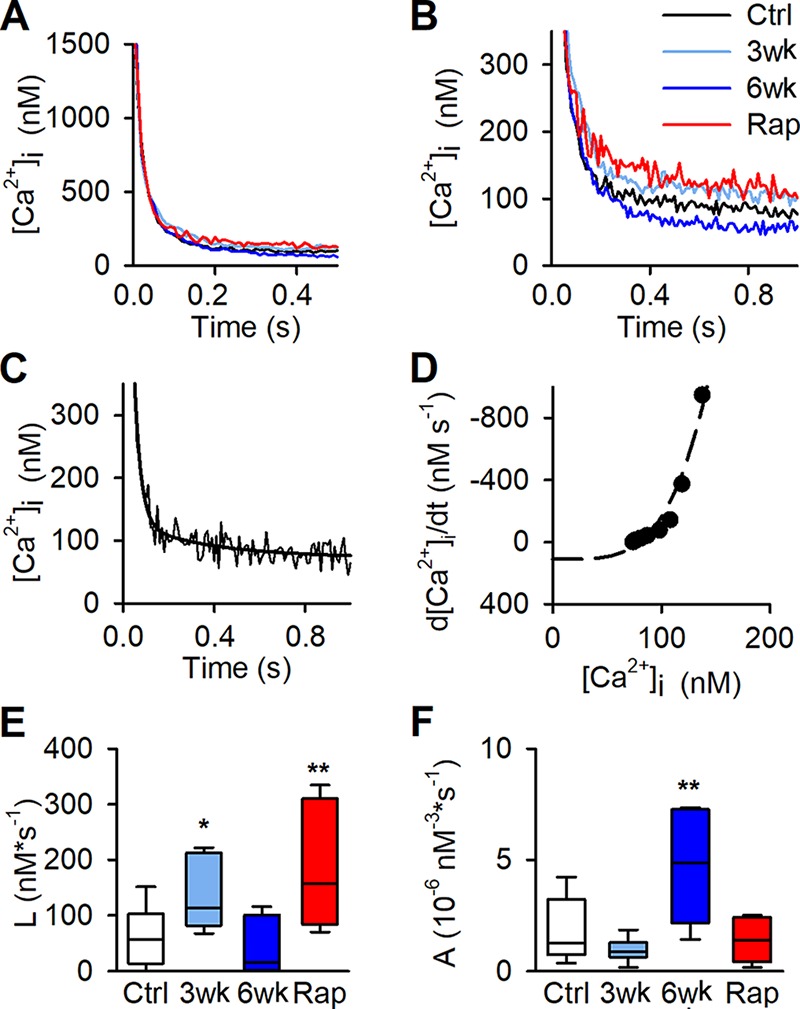
Decay of [Ca2+]i following a tetanic contraction in dissected single fibers. (A) Average decays of [Ca2+]i following a 150-Hz contractions in FDB fibers from control mice (Ctrl; black), mice running for 3 wk (3wk; light blue) and 6 wk (6wk; dark blue), and rapamycin-injected (Rap; red) mice (n = 17, 9, 6, and 5 fibers from 10, 5, 5, and 5 mice for control, 3 wk, 6 wk, and Rap, respectively). There was no notable difference between groups in the fast-decay phase at high [Ca2+]i. (B) The slow phase of average decays levels off at higher [Ca2+]i in fibers of 3-wk runners and rapamycin-injected mice and at lower [Ca2+]i in 6-wk runners. (C) Typical example of [Ca2+]i decay in Ctrl fiber with least-square curve fit (black line). (D) Data points of the relation between the rate of decline (d[Ca2+]i/dt) and [Ca2+]i were fitted to the following equation: d[Ca2+]i/dt = A[Ca2+]iN − L, where N was set to 4, for each individual fiber. Values for L and A in each fiber was extrapolated when d[Ca2+]i/dt − A([Ca2+]i)4 + L = 0. (E and F) Boxplots of calculated SR Ca2+ leak (L; E) and the factor reflecting the rate of SR Ca2+ pumping (A; F). *, P < 0.05; **, P < 0.01, 3 wk, 6 wk, or Rap vs. controls with one-way ANOVA. Box plots show median, quartiles, and min/max.
SR Ca2+ leak–induced mitochondrial biogenesis can be achieved by RYR1-destabilizing pharmacological agents
To investigate a causative role of increases in SR Ca2+ leak and baseline [Ca2+]i in the triggering of mitochondrial biogenesis and improved performance during endurance exercise, we pharmacologically induced a FKBP12 dissociation from RYR1 by injecting mice with a low dose (10 ng/g) of rapamycin or SLF every 48 h for 3 wk. Injection of either of these compounds resulted in FKBP12 dissociation from RYR1 to a degree similar to that seen after 3 wk of voluntary running (Fig. 6 A). Measurements of [Ca2+]i and force performed in FDB fibers from rapamycin-injected mice show a higher baseline [Ca2+]i than in fibers from control mice (Fig. 6 B), whereas tetanic force and [Ca2+]i did not differ from controls (Fig. 6 C). Assessments made on the decay of [Ca2+]i after tetanic stimulation of fibers from rapamycin-injected mice gave a picture similar to that in 3-wk runners; i.e., a calculated leak that was approximately three times higher than in sedentary controls combined with no significant difference in SR Ca2+ uptake rate (Fig. 5).
Figure 6.
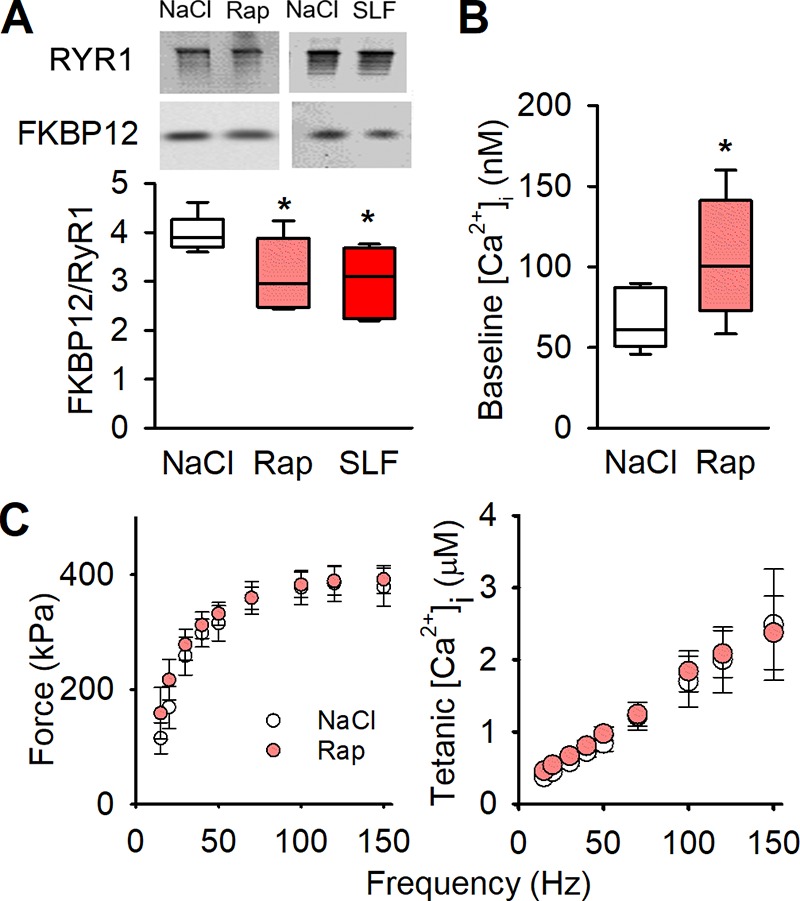
Pharmacologically destabilizing RYR1 causes increased baseline [Ca2+]i, with no negative effects on tetanic [Ca2+]i or contractile force. (A) Original blots (top) and boxplot data (below) of immunoprecipitated RYR1 blotted for coprecipitated FKBP12 in tibialis anterior muscles from sedentary mice injected with NaCl, rapamycin (Rap), or SLF every 48 h for 3 wk (n = 11, 5, and 6, respectively). (B) Boxplots of baseline [Ca2+]i in dissected single FDB fibers from NaCl- and rapamycin-injected mice (n = 7 vs. 5, from four mice). (C) Dissected single fiber from mice injected for 3 wk with rapamycin (light red circles) show no difference in tetanic force or tetanic [Ca2+]i compared with NaCl-injected controls (open circles). Mean (± SEM) n = 7 vs. 5, from four mice). *, P < 0.05 Rap or SLF vs. controls (NaCl) with one-way ANOVA. Box plots show median, quartiles, and min/max.
The mRNA expression of PGC1α1, TFAM, and COX5B were increased in muscle of rapamycin- and SLF-injected mice (Fig. 7 A). Furthermore, PGC1α1 protein expression was also higher in both the rapamycin- and SLF-treated groups than in NaCl-treated controls (Fig. 7 B). Conversely, the mRNA expression of MEF2c did not increase with the pharmacologically induced SR Ca2+ leak (Fig. 7 A), which contrasts with the results obtained in 3-wk runners (see Fig. 2 A). Rapamycin at higher dosage is a known inhibitor of mTOR (Ballou and Lin, 2008). However, neither rapamycin nor SLF affected the phosphorylation of S6K1, which is a downstream target of mTOR (Fig. 7 C).
Figure 7.
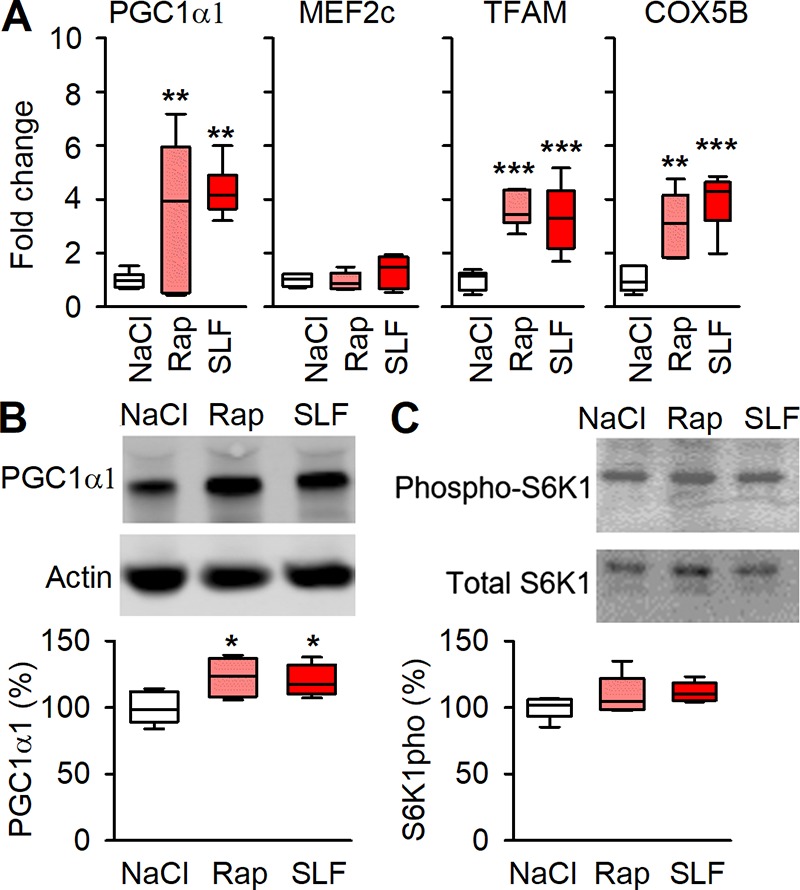
Pharmacologically destabilizing RYR1 causes signaling promoting mitochondrial biogenesis. (A) Boxplots of relative mRNA expression of PGC1α1, MEF2c, TFAM, and COX5B shown relative to HPRT expression in NaCl-, rapamycin- and SLF-injected mice, values were normalized to mean expression of NaCl, which was set to 1.0 (n = 6). (B) Original Western blots (top) and boxplot of relative expression (below) for PGC1α1 with actin as loading control; data presented as relative to the average NaCl, which was set to 100% (n = 6). (C) Original Western blots (top) and boxplot of relative T412 phosphorylation per total S6K1 (below) show no significant effects of Rapamycin or SLF on S6K1; data presented relative to average NaCl which was set to 100% . *, P < 0.05; **, P < 0.01; ***, P < 0.001 Rap or SLF vs. controls (NaCl) with one-way ANOVA. Box plots show median, quartiles, and min/max.
Citrate synthase activity was similarly increased in muscles of rapamycin- and SLF-injected mice, and 3-wk runners (Fig. 8 A). Accordingly, a treadmill running test, during which the mice were forced to run at incrementally increased speed every 2 min, showed ∼25% higher running speed at exhaustion both in rapamycin- and SLF-treated mice, and their performance was similar to that of mice running for 3 wk (median running speed during the last completed interval: NaCl, 18 m/min; Rap, 22 m/min; SLF, 23 m/min; 3-wk runners, 24 m/min; Fig. 8 B). Thus, the pharmacologically induced SR Ca2+ leak leads to improved running endurance.
Figure 8.
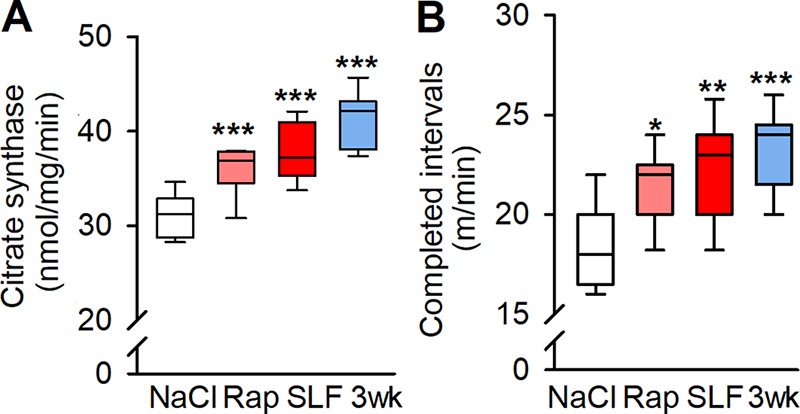
Prolonged pharmacological destabilization of RYR1 increases mouse oxidative capacity and endurance exercise performance. (A) Citrate synthase activity in tibialis anterior muscles of NaCl-, rapamycin-, and SLF-injected mice and 3-wk runners (n = 13, 9, 6, and 7, respectively). (B) Boxplot of mouse endurances in a treadmill test expressed as the speed of the last completed 2-min interval of sequentially increased speed in four groups of animals: NaCl, rapamycin, and SLF injected and 3 wk runners (3 wk; n = 20 control vs. 10 in each of the other groups). *, P < 0.05; **, P < 0.01; ***, P < 0.001 Rap, SLF, or 3 wk vs. controls (NaCl) with one-way ANOVA. Box plots show median, quartiles, and min/max.
Discussion
Depletion of FKBP12 from RYR1 resulting in severe SR Ca2+ leak has been linked to muscle weakness in several diseases, as well as in aging and overtraining (Aydin et al., 2008; Bellinger et al., 2008, 2009; Andersson et al., 2011; Lanner et al., 2012; Waning et al., 2015; Matecki et al., 2016). Conversely, here we show that moderate dissociation of FKBP12 from RYR1 results in a mild form of SR Ca2+ leak accompanied by cell signaling that promotes mitochondrial biogenesis, resulting in increased muscle fatigue resistance and improved performance during a treadmill running endurance exercise test.
Modifications of RYR1 resulting in leaky SR seems to be a common feature following endurance exercise, where the severity of RYR1 modification relates to the intensity of the exercise performed. In a recent study, we observed a severe reduction of the full-size protein and appearance of fragments of RYR1 in Western blots performed on vastus lateralis muscles biopsies obtained from recreationally active human subjects after a session of high-intensity interval training (HIIT; Place et al., 2015). Such RYR1 fragmentation was not observed with the low-intensity exercise used in the present study. Moreover, RYR1 fragmentation did not occur in vastus lateralis muscles of recreationally active subjects running a marathon, where instead FKBP12 dissociation from RYR1 was observed (Place et al., 2015). A likely scenario underlying the difference is that the HIIT-induced RYR1 fragmentation is linked to a very rapid production of reactive oxygen/nitrogen species and the rate of production of these highly reactive molecules is much lower during exercises at lower intensities (Sakellariou et al., 2013). Interestingly, RYR1 fragmentation was not observed after a HIIT session in vastus lateralis muscles of elite endurance athletes (Place et al., 2015), and we here show that FKBP12 association to RYR1 returned to preexercise levels and baseline [Ca2+]i was lowered in muscles of 6-wk running mice despite continued running at almost constant distance (see Fig. 1). Thus, RYR1 is generally less susceptible to modifications in the endurance-trained state, both regarding fragmentation after HIIT exercise and FKBP12 dissociation from RYR1 with low-intensity exercise.
In the short term, baseline [Ca2+]i in skeletal muscle fibers reflects the balance between Ca2+ fluxes in and out of the SR, whereas in the longer term Ca2+ fluxes over the sarcolemma become more important (Ríos, 2010). There were no major differences between muscle fibers of control mice and 3- and 6-wk runners in the total SR Ca2+ content as judged from similar [Ca2+]i during tetanic contractions at 150 Hz in the presence of caffeine (see Fig. 4 A), during which most Ca2+ in the SR is released to the cytosol (Allen and Westerblad, 1995). In muscles of 3-wk runners, we observed FKBP12 dissociation from RYR1 accompanied by SR Ca2+ leak, promoting an increase resting [Ca2+]i. At the same instance, SERCA1 was up-regulated and STIM1 down-regulated and both these protein changes act toward decreased resting [Ca2+]i. The net effect of all these changes was a slight (∼25%) but significant increase in resting [Ca2+]i in FDB fibers of 3-wk runners (see Fig. 1 C). Conversely, in muscles of 6-wk runners, FKBP12’s association with RYR1 had returned to pretraining levels, the protein expression of SERCA1 and STIM1 was further increased and decreased, respectively, and measured SR Ca2+ uptake rate was higher than sedentary controls. Accordingly, resting [Ca2+]i was lower than the pretraining level in FDB fibers of 6-wk runners. Furthermore, the increased Ca2+ entry seen after repeated stimulation became less pronounced with endurance exercise (see Fig. 3 B). This complex adaptive pattern underscores the fundamental importance of tightly controlled cellular Ca2+ handling. Thus, a modest increase in resting [Ca2+]i emerges as a key trigger of beneficial adaptations such as more tightly controlled Ca2+ homeostasis and increased fatigue resistance, whereas large and prolonged increases are associated with deleterious changes and muscle pathologies (Aydin et al., 2008; Bellinger et al., 2008, 2009; Andersson et al., 2011; Lanner, 2012; Waning et al., 2015; Matecki et al., 2016). In line with this, increased SOCE has been shown to promote skeletal muscle growth and endurance (Wei-LaPierre et al., 2013), but it is also linked to various myopathies (Pan et al., 2014).
Increased baseline [Ca2+]i can increase mitochondrial biogenesis in vitro (Ojuka et al., 2002, 2003; Wright et al., 2007). Furthermore, Lee et al. (2014) showed that pharmacologically destabilizing RYR1 can induce protein synthesis in skeletal muscle and boost endurance training adaptation in mice. A link between mild SR Ca2+ leak and improved muscle function has also been observed in mice exposed to a cold environment, where FDB muscles showed increased oxidative capacity and improved endurance (Aydin et al., 2008; Bruton et al., 2010). Similarly, mutations in the ATCN3 gene resulting in α-actinin-3 protein deficiency, prevalent among elite endurance athletes, are coupled to increased SR Ca2+ leak (Yang et al., 2003; Head et al., 2015). However, there is fine balance between beneficial and deleterious effects of SR Ca2+ leak. Deleterious effects are, for instance, observed in soleus muscle partaking in the thermogenic response in cold-exposed mice, which display severe FKBP12 dissociation from RYR1 and markedly reduced force production; i.e., a hallmark of muscle “overtraining” (Aydin et al., 2008).
[Ca2+]i is generally regarded as a broad second messenger, being initiated by a host of different receptors and which functions as an activator for several different downstream signaling cascades (Clapham, 2007). There are a number of potential pathways through which Ca2+ could affect transcription of mRNAs related to oxidative capacity, including signaling dependent on Ca2+-calmodulin kinase II, calcineurin, and cAMP response element–binding protein (Wu et al., 2002; Tavi and Westerblad, 2011; Ito et al., 2013; Bruno et al., 2014). These are relatively slow Ca2+ sensors and are unlikely to respond to the transient increase in [Ca2+]i during individual contractions (Tavi and Westerblad, 2011). However, these Ca2+ sensors might be activated by the repeated contractions produced during endurance exercise. Intriguingly, we here show similar increases in markers of mitochondrial biogenesis and fatigue resistance in muscles of mice performing wheel running and in sedentary mice exposed to the RYR1 destabilizing drugs rapamycin and SLF, which suggests a greater importance of a prolonged but modest increase in resting [Ca2+]i than the transient increases in [Ca2+]i during the repeated contractions of running. Moreover, wheel running requires a large increase in energy consumption, whereas the increase in energy metabolism with RYR1-destabilizing drugs would be small, although some increase would be expected due to increased SR Ca2+ pumping to balance the increased SR Ca2+ leak. Thus, these data indicate that the Ca2+-related signaling was more important for the observed increases mitochondrial biogenesis and fatigue resistance than signaling induced by energy metabolic stress (e.g., activation of AMP-kinase; Kahn et al., 2005). Accordingly, the running distance remained constant after 3 wk and at 6 wk of training, resting [Ca2+]i was lower than the preexercise level, and the energy requirement would be expected to be similar to that at 3 wk. At this point, the muscles had apparently entered a new stable state with increased fatigue resistance, but without gene activation toward further adaptations (see Fig. 1). This fits with a general pattern where the accumulating effect of running-induced bursts of, for instance, mRNA for PGC1α leads to an increase in PGC-1α protein, which after some delay results in an increased concentration of mitochondrial proteins. When the running exercise proceeds at a constant level, the amplitude of the mRNA bursts gradually declines while the content of mitochondrial proteins remains elevated (Perry et al., 2010). Moreover, the cellular mitochondrial content depends on the balance between the rates of synthesis (mitochondrial biogenesis) and removal of dysfunctional or damaged mitochondria via a selective degradation process known as mitophagy (Hood et al., 2018). In untrained muscle, acute endurance exercise has been shown to increase the rate of mitophagy (Vainshtein et al., 2015; Chen et al., 2018). In endurance-trained muscles, on the other hand, recent studies indicate a reduced basal rate of mitophagy as well as a blunted mitophagic response to acute endurance exercise, which possibly reflects an improved health of the mitochondrial pool in trained muscle (Schwalm et al., 2017; Carter et al., 2018; Chen et al., 2018; Kim et al., 2018). Thus, a decreased rate of mitochondrial degradation via mitophagy might explain how an increased cellular mitochondrial content can be maintained during constant running exercise despite signaling promoting mitochondrial biogenesis being returned to the untrained level.
Intriguingly, mice exposed to the RYR-destabilizing drugs rapamycin and SLF, as well as mice having access to a running wheel, performed better than controls in the treadmill running exhaustion test (see Fig. 8 B), which is considered to provide an indirect measure of mouse VO2max (Kemi et al., 2002). The ability of the heart to deliver O2 to the working muscles is generally believed to be the limiting factor for VO2max (Levine, 2008). Running wheel exercise is likely to increase cardiac pumping capacity, but it is unclear as to how exposure to RYR-destabilizing drugs without concomitant endurance exercise would improve cardiac function. One possible mechanism would be that exposure to these drugs improve cardiomyocyte function via Ca2+-dependent signaling similar to that in skeletal muscle fibers, although an increased SR Ca2+ leak induced by dissociation of FKBP 12.6 from RYR2 in cardiomyocytes is generally associated with impaired cardiac function and heart disease (Lehnart et al., 2004; MacMillan, 2013). Alternatively, the improved performance in the treadmill running exhaustion test reflects the increased mitochondrial capacity and fatigue resistance in skeletal muscles.
Mice exposed to the RYR1-destabilizing drugs rapamycin and SLF showed increased baseline [Ca2+]i, together with increases in mRNA expression of the mitochondrial biogenesis–promoting genes PGC1α1 and TFAM. This was followed by increased citrate synthase activity and improved performance in an exercise endurance test, i.e., adaptations similar to those seen in mice performing voluntary wheel running. However, the mRNA expression of MEF2c increased with running, but not with exposure to the RYR1-destabilizing drugs. MEF2c acts as a transcriptional regulator for normal glucose metabolism, glycogen utilization, and energy homeostasis in skeletal muscle (Anderson et al., 2015). Thus, not all changes in gene transcription induced by endurance training can be triggered by increased resting [Ca2+]i and the expression of MEF2c is apparently controlled by other types of endurance exercise–induced metabolic stress.
Conclusion
In conclusion, endurance exercise induces RYR1 modifications and a modest SR Ca2+ leak in skeletal muscle fibers. Leaky RYR1 and the subsequent increase in baseline [Ca2+]i trigger expression of mitochondrial biogenesis–promoting genes, resulting in increased oxidative capacity, increased fatigue resistance, and improved performance during endurance exercises.
Acknowledgments
This work was supported by the Swedish Research Council (grants K2014-52X-10842-21-5 and 2016-02457), the Swedish Research Council for Sport Science (grant FO2016-0033), the Lars Hierta Memorial Foundation, and the Swedish Military Forces’ Research Authority.
The authors declare no competing financial interests.
Author contributions: N. Ivarsson, J.T. Lanner, C.M. Mattsson, B. Ekblom, and H. Westerblad conceptualized the study. N. Ivarsson, J.T. Lanner, and H. Westerblad drafted the manuscript. N. Ivarsson performed in vivo mouse experiments. N. Ivarsson, A.J. Cheng, and J.D. Bruton performed mouse single fiber and biochemical experiments. All authors participated in the final draft of the manuscript.
Eduardo Ríos served as editor.
References
- Ahern G.P., Junankar P.R., and Dulhunty A.F.. 1994. Single channel activity of the ryanodine receptor calcium release channel is modulated by FK-506. FEBS Lett. 352:369–374. 10.1016/0014-5793(94)01001-3 [DOI] [PubMed] [Google Scholar]
- Ahern G.P., Junankar P.R., and Dulhunty A.F.. 1997. Subconductance states in single-channel activity of skeletal muscle ryanodine receptors after removal of FKBP12. Biophys. J. 72:146–162. 10.1016/S0006-3495(97)78654-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen D.G., and Westerblad H.. 1995. The effects of caffeine on intracellular calcium, force and the rate of relaxation of mouse skeletal muscle. J. Physiol. 487:331–342. 10.1113/jphysiol.1995.sp020883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen D.G., Lamb G.D., and Westerblad H.. 2008. Skeletal muscle fatigue: cellular mechanisms. Physiol. Rev. 88:287–332. 10.1152/physrev.00015.2007 [DOI] [PubMed] [Google Scholar]
- Anderson C.M., Hu J., Barnes R.M., Heidt A.B., Cornelissen I., and Black B.L.. 2015. Myocyte enhancer factor 2C function in skeletal muscle is required for normal growth and glucose metabolism in mice. Skelet. Muscle. 5:7 10.1186/s13395-015-0031-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson D.C., Betzenhauser M.J., Reiken S., Meli A.C., Umanskaya A., Xie W., Shiomi T., Zalk R., Lacampagne A., and Marks A.R.. 2011. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 14:196–207. 10.1016/j.cmet.2011.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade F.H., Reid M.B., Allen D.G., and Westerblad H.. 1998. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 509:565–575. 10.1111/j.1469-7793.1998.565bn.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin J., Shabalina I.G., Place N., Reiken S., Zhang S.J., Bellinger A.M., Nedergaard J., Cannon B., Marks A.R., Bruton J.D., and Westerblad H.. 2008. Nonshivering thermogenesis protects against defective calcium handling in muscle. FASEB J. 22:3919–3924. 10.1096/fj.08-113712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baar K., Wende A.R., Jones T.E., Marison M., Nolte L.A., Chen M., Kelly D.P., and Holloszy J.O.. 2002. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J. 16:1879–1886. 10.1096/fj.02-0367com [DOI] [PubMed] [Google Scholar]
- Ballou L.M., and Lin R.Z.. 2008. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 1:27–36. 10.1007/s12154-008-0003-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass A., Brdiczka D., Eyer P., Hofer S., and Pette D.. 1969. Metabolic differentiation of distinct muscle types at the level of enzymatic organization. Eur. J. Biochem. 10:198–206. 10.1111/j.1432-1033.1969.tb00674.x [DOI] [PubMed] [Google Scholar]
- Bellinger A.M., Reiken S., Dura M., Murphy P.W., Deng S.X., Landry D.W., Nieman D., Lehnart S.E., Samaru M., LaCampagne A., and Marks A.R.. 2008. Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc. Natl. Acad. Sci. USA. 105:2198–2202. 10.1073/pnas.0711074105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger A.M., Reiken S., Carlson C., Mongillo M., Liu X., Rothman L., Matecki S., Lacampagne A., and Marks A.R.. 2009. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 15:325–330. 10.1038/nm.1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth F.W., and Thomason D.B.. 1991. Molecular and cellular adaptation of muscle in response to exercise: perspectives of various models. Physiol. Rev. 71:541–585. 10.1152/physrev.1991.71.2.541 [DOI] [PubMed] [Google Scholar]
- Bruno N.E., Kelly K.A., Hawkins R., Bramah-Lawani M., Amelio A.L., Nwachukwu J.C., Nettles K.W., and Conkright M.D.. 2014. Creb coactivators direct anabolic responses and enhance performance of skeletal muscle. EMBO J. 33:1027–1043. 10.1002/embj.201386145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton J.D., Aydin J., Yamada T., Shabalina I.G., Ivarsson N., Zhang S.J., Wada M., Tavi P., Nedergaard J., Katz A., and Westerblad H.. 2010. Increased fatigue resistance linked to Ca2+-stimulated mitochondrial biogenesis in muscle fibres of cold-acclimated mice. J. Physiol. 588:4275–4288. 10.1113/jphysiol.2010.198598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H.N., Kim Y., Erlich A.T., Zarrin-Khat D., and Hood D.A.. 2018. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 596:3567–3584. 10.1113/JP275998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.C.W., Erlich A.T., and Hood D.A.. 2018. Role of Parkin and endurance training on mitochondrial turnover in skeletal muscle. Skelet. Muscle. 8:10 10.1186/s13395-018-0157-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A.J., and Westerblad H.. 2017. Mechanical isolation, and measurement of force and myoplasmic free [Ca2+] in fully intact single skeletal muscle fibers. Nat. Protoc. 12:1763–1776. 10.1038/nprot.2017.056 [DOI] [PubMed] [Google Scholar]
- Cheng A.J., Place N., and Westerblad H.. 2018. Molecular basis for exercise-induced fatigue: the importance of strictly controlled cellular Ca2+ handling. Cold Spring Harb. Perspect. Med. 8:a029710 10.1101/cshperspect.a029710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham D.E. 2007. Calcium signaling. Cell. 131:1047–1058. 10.1016/j.cell.2007.11.028 [DOI] [PubMed] [Google Scholar]
- Ekblom-Bak E., Björkman F., Hellenius M.L., and Ekblom B.. 2014. A new submaximal cycle ergometer test for prediction of VO2max. Scand. J. Med. Sci. Sports. 24:319–326. 10.1111/sms.12014 [DOI] [PubMed] [Google Scholar]
- Gordon A.M., Homsher E., and Regnier M.. 2000. Regulation of contraction in striated muscle. Physiol. Rev. 80:853–924. 10.1152/physrev.2000.80.2.853 [DOI] [PubMed] [Google Scholar]
- Head S.I., Chan S., Houweling P.J., Quinlan K.G.R., Murphy R., Wagner S., Friedrich O., and North K.N.. 2015. Altered Ca2+ kinetics associated with α-actinin-3 deficiency may explain positive selection for ACTN3 null allele in human evolution. PLoS Genet. 11:e1004862 10.1371/journal.pgen.1004862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloszy J.O., and Coyle E.F.. 1984. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J. Appl. Physiol. 56:831–838. 10.1152/jappl.1984.56.4.831 [DOI] [PubMed] [Google Scholar]
- Holloszy J.O., Oscai L.B., Don I.J., and Molé P.A.. 1970. Mitochondrial citric acid cycle and related enzymes: adaptive response to exercise. Biochem. Biophys. Res. Commun. 40:1368–1373. 10.1016/0006-291X(70)90017-3 [DOI] [PubMed] [Google Scholar]
- Hood D.A., Memme J.M., Oliveira A.N., and Triolo M.. 2018. Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu. Rev. Physiol. 10.1146/annurev-physiol-020518-114310 [DOI] [PubMed] [Google Scholar]
- Hopf F.W., Reddy P., Hong J., and Steinhardt R.A.. 1996. A capacitative calcium current in cultured skeletal muscle cells is mediated by the calcium-specific leak channel and inhibited by dihydropyridine compounds. J. Biol. Chem. 271:22358–22367. 10.1074/jbc.271.37.22358 [DOI] [PubMed] [Google Scholar]
- Ito N., Ruegg U.T., Kudo A., Miyagoe-Suzuki Y., and Takeda S.. 2013. Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat. Med. 19:101–106. 10.1038/nm.3019 [DOI] [PubMed] [Google Scholar]
- Kahn B.B., Alquier T., Carling D., and Hardie D.G.. 2005. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1:15–25. 10.1016/j.cmet.2004.12.003 [DOI] [PubMed] [Google Scholar]
- Kemi O.J., Loennechen J.P., Wisløff U., and Ellingsen Ø.. 2002. Intensity-controlled treadmill running in mice: cardiac and skeletal muscle hypertrophy. J. Appl. Physiol. 93:1301–1309. 10.1152/japplphysiol.00231.2002 [DOI] [PubMed] [Google Scholar]
- Kim Y., Triolo M., Erlich A.T., and Hood D.A.. 2018. Regulation of autophagic and mitophagic flux during chronic contractile activity-induced muscle adaptations. Pflugers Arch. 10.1007/s00424-018-2225-x [DOI] [PubMed] [Google Scholar]
- Klein M.G., Kovacs L., Simon B.J., and Schneider M.F.. 1991. Decline of myoplasmic Ca2+, recovery of calcium release and sarcoplasmic Ca2+ pump properties in frog skeletal muscle. J. Physiol. 441:639–671. 10.1113/jphysiol.1991.sp018771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner J.T. 2012. Ryanodine receptor physiology and its role in disease. Adv. Exp. Med. Biol. 740:217–234. 10.1007/978-94-007-2888-2_9 [DOI] [PubMed] [Google Scholar]
- Lanner J.T., Georgiou D.K., Dagnino-Acosta A., Ainbinder A., Cheng Q., Joshi A.D., Chen Z., Yarotskyy V., Oakes J.M., Lee C.S., et al. . 2012. AICAR prevents heat-induced sudden death in RyR1 mutant mice independent of AMPK activation. Nat. Med. 18:244–251. 10.1038/nm.2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.S., Georgiou D.K., Dagnino-Acosta A., Xu J., Ismailov I.I. II, Knoblauch M., Monroe T.O., Ji R., Hanna A.D., Joshi A.D., et al. . 2014. Ligands for FKBP12 increase Ca2+ influx and protein synthesis to improve skeletal muscle function. J. Biol. Chem. 289:25556–25570. 10.1074/jbc.M114.586289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman J.J., Barger P.M., Kovacs A., Saffitz J.E., Medeiros D.M., and Kelly D.P.. 2000. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 106:847–856. 10.1172/JCI10268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart S.E., Wehrens X.H., Kushnir A., and Marks A.R.. 2004. Cardiac ryanodine receptor function and regulation in heart disease. Ann. N. Y. Acad. Sci. 1015:144–159. 10.1196/annals.1302.012 [DOI] [PubMed] [Google Scholar]
- Levine B.D. 2008. VO2max: what do we know, and what do we still need to know? J. Physiol. 586:25–34. 10.1113/jphysiol.2007.147629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan D. 2013. FK506 binding proteins: cellular regulators of intracellular Ca2+ signalling. Eur. J. Pharmacol. 700:181–193. 10.1016/j.ejphar.2012.12.029 [DOI] [PubMed] [Google Scholar]
- Marx S.O., Reiken S., Hisamatsu Y., Jayaraman T., Burkhoff D., Rosemblit N., and Marks A.R.. 2000. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 101:365–376. 10.1016/S0092-8674(00)80847-8 [DOI] [PubMed] [Google Scholar]
- Matecki S., Dridi H., Jung B., Saint N., Reiken S.R., Scheuermann V., Mrozek S., Santulli G., Umanskaya A., Petrof B.J., et al. . 2016. Leaky ryanodine receptors contribute to diaphragmatic weakness during mechanical ventilation. Proc. Natl. Acad. Sci. USA. 113:9069–9074. 10.1073/pnas.1609707113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrbom J., Sundberg C.J., Ameln H., Kraus W.E., Jansson E., and Gustafsson T.. 2004. PGC-1α mRNA expression is influenced by metabolic perturbation in exercising human skeletal muscle. J. Appl. Physiol. 96:189–194. 10.1152/japplphysiol.00765.2003 [DOI] [PubMed] [Google Scholar]
- Ojuka E.O., Jones T.E., Han D.H., Chen M., Wamhoff B.R., Sturek M., and Holloszy J.O.. 2002. Intermittent increases in cytosolic Ca2+ stimulate mitochondrial biogenesis in muscle cells. Am. J. Physiol. Endocrinol. Metab. 283:E1040–E1045. 10.1152/ajpendo.00242.2002 [DOI] [PubMed] [Google Scholar]
- Ojuka E.O., Jones T.E., Han D.H., Chen M., and Holloszy J.O.. 2003. Raising Ca2+ in L6 myotubes mimics effects of exercise on mitochondrial biogenesis in muscle. FASEB J. 17:675–681. 10.1096/fj.02-0951com [DOI] [PubMed] [Google Scholar]
- Pan Z., Brotto M., and Ma J.. 2014. Store-operated Ca2+ entry in muscle physiology and diseases. BMB Rep. 47:69–79. 10.5483/BMBRep.2014.47.2.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry C.G., Lally J., Holloway G.P., Heigenhauser G.J., Bonen A., and Spriet L.L.. 2010. Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J. Physiol. 588:4795–4810. 10.1113/jphysiol.2010.199448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilegaard H., Saltin B., and Neufer P.D.. 2003. Exercise induces transient transcriptional activation of the PGC-1alpha gene in human skeletal muscle. J. Physiol. 546:851–858. 10.1113/jphysiol.2002.034850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Place N., Ivarsson N., Venckunas T., Neyroud D., Brazaitis M., Cheng A.J., Ochala J., Kamandulis S., Girard S., Volungevičius G., et al. . 2015. Ryanodine receptor fragmentation and sarcoplasmic reticulum Ca2+ leak after one session of high-intensity interval exercise. Proc. Natl. Acad. Sci. USA. 112:15492–15497. 10.1073/pnas.1507176112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos E. 2010. The cell boundary theorem: a simple law of the control of cytosolic calcium concentration. J. Physiol. Sci. 60:81–84. 10.1007/s12576-009-0069-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J., DiGregorio P.J., Yeromin A.V., Ohlsen K., Lioudyno M., Zhang S., Safrina O., Kozak J.A., Wagner S.L., Cahalan M.D., et al. . 2005. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169:435–445. 10.1083/jcb.200502019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellariou G.K., Vasilaki A., Palomero J., Kayani A., Zibrik L., McArdle A., and Jackson M.J.. 2013. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 18:603–621. 10.1089/ars.2012.4623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalm C., Deldicque L., and Francaux M.. 2017. Lack of activation of mitophagy during endurance exercise in human. Med. Sci. Sports Exerc. 49:1552–1561. 10.1249/MSS.0000000000001256 [DOI] [PubMed] [Google Scholar]
- Tavi P., and Westerblad H.. 2011. The role of in vivo Ca2+ signals acting on Ca2+-calmodulin-dependent proteins for skeletal muscle plasticity. J. Physiol. 589:5021–5031. 10.1113/jphysiol.2011.212860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainshtein A., Tryon L.D., Pauly M., and Hood D.A.. 2015. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiol. Cell Physiol. 308:C710–C719. 10.1152/ajpcell.00380.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waning D.L., Mohammad K.S., Reiken S., Xie W., Andersson D.C., John S., Chiechi A., Wright L.E., Umanskaya A., Niewolna M., et al. . 2015. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 21:1262–1271. 10.1038/nm.3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei-LaPierre L., Carrell E.M., Boncompagni S., Protasi F., and Dirksen R.T.. 2013. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 4:2805 10.1038/ncomms3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerblad H., and Allen D.G.. 1993. The influence of intracellular pH on contraction, relaxation and [Ca2+]i in intact single fibres from mouse muscle. J. Physiol. 466:611–628. [PMC free article] [PubMed] [Google Scholar]
- Wright D.C., Geiger P.C., Han D.H., Jones T.E., and Holloszy J.O.. 2007. Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1α and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J. Biol. Chem. 282:18793–18799. 10.1074/jbc.M611252200 [DOI] [PubMed] [Google Scholar]
- Wu H., Kanatous S.B., Thurmond F.A., Gallardo T., Isotani E., Bassel-Duby R., and Williams R.S.. 2002. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 296:349–352. 10.1126/science.1071163 [DOI] [PubMed] [Google Scholar]
- Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R.C., and Spiegelman B.M.. 1999. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 98:115–124. 10.1016/S0092-8674(00)80611-X [DOI] [PubMed] [Google Scholar]
- Yang N., MacArthur D.G., Gulbin J.P., Hahn A.G., Beggs A.H., Easteal S., and North K.. 2003. ACTN3 genotype is associated with human elite athletic performance. Am. J. Hum. Genet. 73:627–631. 10.1086/377590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalk R., Clarke O.B., des Georges A., Grassucci R.A., Reiken S., Mancia F., Hendrickson W.A., Frank J., and Marks A.R.. 2015. Structure of a mammalian ryanodine receptor. Nature. 517:44–49. 10.1038/nature13950 [DOI] [PMC free article] [PubMed] [Google Scholar]