

Abstract
Background:
Sialic acid–binding immunoglobulin-like lectin (Siglec)-8 is a cell-surface protein expressed selectively on human eosinophils, mast cells, and basophils, making it an ideal target for the treatment of diseases involving these cell types. However, the effective delivery of therapeutic agents to these cells requires an understanding of the dynamics of Siglec-8 surface expression.
Objectives:
We sought to determine whether Siglec-8 is endocytosed in human eosinophils and malignant mast cells, identify mechanisms underlying its endocytosis, and demonstrate whether a toxin can be targeted to Siglec-8–bearing cells to kill these cells.
Methods:
Siglec-8 surface dynamics were examined by flow cytometry using peripheral blood eosinophils, mast cell lines, and Siglec-8–transduced cells in the presence of inhibitors targeting components of endocytic pathways. Siglec-8 intracellular trafficking was followed by confocal microscopy. The ribosome-inhibiting protein saporin was conjugated to a Siglec-8–specific antibody to examine the targeting of an agent to these cells through Siglec-8 endocytosis.
Results:
Siglec-8 endocytosis required actin rearrangement, tyrosine kinase and protein kinase C activities, and both clathrin and lipid rafts. Internalized Siglec-8 localized to the lysosomal compartment. Maximal endocytosis in Siglec-8–transduced HEK293T cells required an intact immunoreceptor tyrosine-based inhibitory motif. Siglec-8 was also shuttled to the surface via a distinct pathway. Sialidase treatment of eosinophils revealed that Siglec-8 is partially masked by sialylated cis ligands. Targeting saporin to Siglec-8 consistently caused extensive cell death in eosinophils and the human mast cell leukemia cell line HMC-1.2.
Conclusions:
Therapeutic payloads can be targeted selectively to eosinophils and malignant mast cells by exploiting this Siglec-8 endocytic pathway. (J Allergy Clin Immunol 2018;141:1774–85.)
Keywords: Siglec-8, endocytosis, eosinophil, mast cell, allergic diseases, chronic eosinophilic leukemia, systemic mastocytosis, immunotoxin, targeting
Eosinophils, basophils, and mast cells contribute to host defense responses against parasitic infections, but are also involved in allergic disorders including asthma and certain myeloproliferative disorders.1,2 Currently, there are no therapies that selectively and directly target these cells. Sialic acid–binding immunoglobulin-type lectin (Siglec)-8 is a human glycan-binding receptor expressed selectively on eosinophils, mast cells, and basophils.3 Notably, Siglec-8 is expressed at normal levels on eosinophils and basophils from patients diagnosed with hypereosinophilic syndrome, chronic eosinophilic leukemia, or chronic myeloid leukemia as well as on bone marrow mast cells from patients diagnosed with indolent systemic mastocytosis.4 Consistent with the presence of a cytoplasmic immunoreceptor tyrosine-based inhibitory motif (ITIM) in the protein,5 Siglec-8 may act as an inhibitory receptor. Ligation of Siglec-8 on eosinophils using a primary antibody and a secondary cross-linking antibody leads to cell death.6 Priming eosinophils with IL-5, GM-CSF, or IL-33 potentiates this cell death function of Siglec-8 and eliminates the requirement for a secondary cross-linking antibody to achieve consistently high levels of cell death.6,7 In contrast, Siglec-8 ligation on mast cells does not cause cell death but rather inhibits FcεRI-mediated Ca2+ flux and prostaglandin D2 and histamine release.8 There are no published data regarding the function of Siglec-8 on basophils. No specific endogenous ligand for Siglec-8 has yet been defined, but high-molecular-weight O-linked Siglec-8 sialoglycoprotein ligands have been detected in extracts of human trachea and cultured tracheal gland cells.9
In addition to its ligand-induced inhibitory functions, Siglec-8 may represent a target via which to deliver toxins, chemotherapeutic agents, corticosteroids, or other drugs selectively into Siglec-8–expressing cells to treat various diseases involving eosinophils, mast cells, and basophils and achieve the selectivity that is currently lacking. However, the delivery of therapeutic payloads to these cell types requires knowledge of the endocytosis of Siglec-8, which has not been elucidated.
In this report, we examine the endocytosis of Siglec-8, identify the pathway and proteins involved, and determine whether this process can be leveraged to deliver a toxic payload to kill human eosinophils and malignant mast cells. Our findings reveal unique ways in which Siglec-8 endocytosis may be modulated and leveraged to selectively target eosinophils and mast cells. Finally, we demonstrate that the dynamics of Siglec-8 cell-surface expression are well suited for the targeting of therapeutic payloads to Siglec-8–expressing cells.
METHODS
Human eosinophil isolation and culture
Written informed consent for blood donation was obtained using an institutional review board–approved protocol. Eosinophils from blood donors were purified from peripheral blood using density gradient centrifugation, erythrocyte hypotonic lysis, and immunomagnetic negative selection with CD16 beads (Miltenyi Biotec, Bergisch Gladbach, Germany) as described.10 Purity and viability were consistently greater than 95% as determined by Siglec-8 staining and 4′−6-diamidino-2-phenylindole (DAPI) exclusion. Cells were cultured in RPMI 1640 medium supplemented with 10% FCS and 1% penicillin/streptomycin (all from Gibco, Grand Island, NY) as well as up to 30 ng/mL rhIL-5 (R&D Systems, Minneapolis, Minn) as previously described.6 Each experiment was performed with eosinophils only from a single donor; samples were not pooled for any analysis.
Mast cell lines and culture conditions
The human mast cell leukemia cell line HMC-1.2 was generously provided by P. Valent of the Medical University of Vienna (Vienna, Austria).11 HMC-1.2 cells were cultured in Iscove’s modified Dulbecco’s medium supplemented with 25 mM HEPES, 3.024 g/L sodium bicarbonate, 10% FCS, 1% L-glutamine, 1% penicillin-streptomycin, and 0.1% β-mercaptoethanol (all from Gibco). The LUVA human mast cell line was generously provided by J. Steinke and L. Borish of the University of Virginia School of Medicine (Charlottesville, Va).12 LUVA cells were cultured in StemPro-34 serum-free medium supplemented with the StemPro-34 nutrient supplement, 1% L-glutamine, and 1% penicillin/streptomycin.
Flow cytometric analysis of Siglec-8 surface expression
The expression and internalization of Siglec-8 was assessed with a delayed secondary staining method, whereby the Siglec-8 initially present on the cell surface was bound by an unlabeled mouse IgG1 anti–Siglec-8 mAb (clone 2C4 or 1H10, the latter generously provided by Dr Nenad Tomasevic, Allakos, Inc, San Carlos, Calif) or isotype-matched control mAb (clone MOPC-21; Tonbo Biosciences, San Diego, Calif) at 2.5 μg/mL at 4°C, the excess unbound antibody was washed off, and the cells were incubated at 37°C for various durations to permit internalization. The antibody remaining at the surface was detected by addition of a fluorescein isothiocyanate–labeled secondary antimouse IgG1 antibody (BioLegend, San Diego, Calif). Siglec-8 that had been shuttled or recycled to the surface during the incubation at 37°C was detected with a directly Alexa Fluor 647–conjugated 2C4 mAb after staining with the secondary antibody and extensive washing. To address the possibility of Siglec-8 shedding, surface Siglec-8 was bound by Alexa Fluor 647–conjugated 2C4 mAb, and fluorescence was detected after the cells were incubated at 37°C for various durations to permit internalization. This method was designed to measure both surface and intracellular Siglec-8; loss of fluorescence would indicate shedding of Siglec-8. Cells were analyzed using a BD LSR II flow cytometer (BD Biosciences, San Jose, Calif) and FlowJo software version 10 (Tree Star, Ashland, Ore). Mean fluorescence intensity of isotype control samples was subtracted from experimental mean fluorescence intensities to obtain adjusted mean fluorescence intensities, which were used in further calculations. The calculation of loss of Siglec-8 from the cell surface was based on the loss of fluorescence relative to fresh cells kept on ice that had not been incubated at 37°C (t = 0). The same formula was used to assess shedding of Siglec-8 from the cell surface.
Assessing the effects of Siglec-8 intracellular motifs on endocytosis in Siglec-8–transduced HEK293T cells
Tyrosine residues in the cytoplasmic signaling motifs (Y447 of the ITIM, Y470 of the ITSM) of Siglec-8 were mutated to phenylalanine residues using a QuikChange II XL Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, Calif). Full-length Siglec-8 and the mutant versions were separately cloned into the multiple cloning site of a pCDH-CMV-EF1-GFP-PURO lentiviral vector and lentiviral particles were produced by the DNA/RNA Delivery Core at Northwestern University. The lentiviral particles were used to transduce HEK293T cells generously provided by N. Lu of Northwestern University (Chicago, Ill). Total loss of Siglec-8 from the cell surface and any potential shedding of Siglec-8 at 120 minutes were measured by flow cytometry following delayed secondary staining and, in parallel, detection of Siglec-8 with an Alexa Fluor 647–conjugated anti–Siglec-8 mAb (2C4) in the transduced (GFP+) population as described above. Endocytosis calculations accounted for the loss of Siglec-8 from the cell surface due to shedding after normalization to initial levels of surface Siglec-8:
Confocal microscopy
The cells were incubated in the presence or absence of DQ Green BSA, a self-quenched dye conjugate of BSA that fluoresces when the BSA is cleaved by proteases and the quenching is relieved (10 μg/mL; Thermo Fisher Scientific, Carlsbad, Calif) for 120 minutes at 37°C to mark lysosomes either before labeling of Siglec-8 on the cell surface or following Siglec-8 labeling that would lead to the visualization of internalized Siglec-8. Eosinophils were incubated with Alexa Fluor 647–conjugated anti–Siglec-8 mAb (2C4) or Alexa Fluor 647– conjugated isotype-matched control mAb (MOPC-21; both at 2.5 μg/mL) during a 20-minute incubation at 4°C. The eosinophils were cytospun onto glass slides and fixed with 4% paraformaldehyde (Affymetrix, Santa Clara, Calif) for 15 minutes at room temperature. Slides were mounted using Fluoromount-G mounting medium (Electron Microscopy Services, Hatfield, Pa) and imaged using a Nikon A1R+ confocal laser microscope (Tokyo, Japan) using a 100×/1.45 NA oil-immersion objective lens and 488-and 640-nm lasers with a 1.5-AU pinhole. Images were acquired and analyzed using Nikon Elements software.
Siglec-8 targeting and assessment of cell death
Eosinophil cell death was assessed after 18 to 24 hours of incubation with 2C4 mAb or isotype control (MOPC-21; both at 2.5 μg/mL) or equimolar concentrations of saporin-conjugated 2C4 or MOPC-21 (produced for us by Advanced Targeting Systems, San Diego, Calif) in the presence or absence of 15 ng/mL rhIL-5. Cell death was assessed with annexin V (BD Biosciences)/DAPI (Thermo Fisher Scientific) staining. Cell death induction in the HMC-1.2 human mast cell line was assessed after 7 to 13 days (unless otherwise indicated) of culture in medium containing 0.78 μg/mL of the nonionic surfactant saponin (Sigma-Aldrich, St Louis, Mo) following a 10-minute incubation at 4°C with 1.25 μg/mL of 2C4 or MOPC-21 mAbs or equimolar concentrations of their saporin-conjugated counterparts and extensive washing.
Statistical analysis
Data are presented as means ± SDs unless otherwise indicated. Statistical significance was determined by ANOVA and either a Dunnett or Tukey multiple comparisons test, as appropriate, using GraphPad Prism version 6.0e for Mac OS X (La Jolla, Calif). When appropriate, single outliers were identified and removed from the data using a Grubbs’ outlier test. Statistical differences were considered significant at P < .05.
RESULTS
Siglec-8 is internalized in human eosinophils and mast cell lines
Because Siglec-8 endocytosis has not been studied previously, we first sought to confirm that Siglec-8, like other siglecs, is indeed internalized following its engagement. To measure Siglec-8 endocytosis, cell-surface Siglec-8 was bound by an unlabeled anti–Siglec-8 mAb and, following incubation at 37°C to permit endocytosis, any mAb remaining at the cell surface was detected using a labeled secondary antibody. Upon antibody engagement of the receptor on primary human eosinophils, Siglec-8 was slowly internalized—about half of the initial pool of Siglec-8 surface molecules was internalized by about 90 minutes—and about 20% of this pool remained even after extended incubations (Fig 1, A). Because soluble forms of Siglec-8 have been detected in patient sera,13 the possibility that Siglec-8 shedding from the cell surface contributed to these measures of Siglec-8 endocytosis was examined. To assess Siglec-8 shedding, we detected Siglec-8 directly with a fluorescently labeled mAb and incubated the cells at 37°C as before. This staining protocol measures total cell-associated Siglec-8 (ie, both cell-surface and internalized). No loss of signal was detected throughout this time course, demonstrating that the loss of signal in the previous experiment was indeed due to receptor endocytosis rather than shedding from the cell surface (Fig 1, B). Internalization of surface Siglec-8 was also measured on the human mast cell leukemia cell line HMC-1.2 in a similar fashion (Fig 1, C). As with the eosinophils, no Siglec-8 shedding from the HMC-1.2 cells was detected (Fig 1, D), indicating that the loss from the cell surface was the result of endocytosis in these cells as well. To extend these findings, the internalization of Siglec-8 was also confirmed in the LUVA human mast cell line (see Fig E1 in this article’s Online Repository at www.jacionline.org). To explore the possibility that the clone of anti–Siglec-8 mAb used in these experiments (2C4) is unique in its ability to induce endocytosis, we examined the ability of another clone (1H10) recognizing a distinct epitope on a different extracellular domain (unpublished observations) to induce Siglec-8 endocytosis. As shown, both clones cause Siglec-8 endocytosis on eosinophils (see Fig E2 in this article’s Online Repository at www.jacionline.org).
FIG 1.
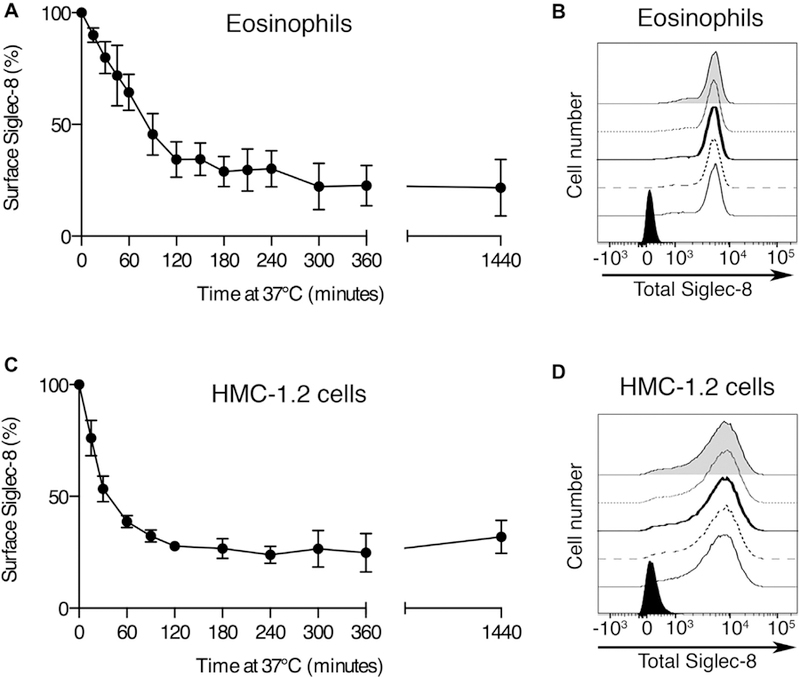
Kinetic analysis of Siglec-8 endocytosis versus shedding. Siglec-8 loss from the cell surface of human primary eosinophils (A) and HMC-1.2 mast cells (C) was measured by the loss of anti–Siglec-8 mAb labeling from the cell surface. Data represent the means ± SDs of 4 independent experiments. Surface Siglec-8 that remained cell-associated (intracellular or remaining on the cell surface) in eosinophils (B) and HMC-1.2 cells (D) was measured using a fluorophore-conjugated anti–Siglec-8 antibody. Remaining fluorescence at times 0 minute (normal line), 30 minutes (dashed line), 60 minutes (bold line), 90 minutes (dotted line), and 120 minutes (gray filled histogram) of incubation at 37°C was detected by flow cytometry. The black histogram represents labeling with the fluorophore-conjugated mouse IgG1 control. Data are representative of results from 3 and 4 independent experiments, respectively.
Mechanisms of Siglec-8 endocytosis
Receptors may be internalized via various pathways, including those mediated by clathrin or lipid rafts/caveolae as well as phagocytosis. The dependence of Siglec-8 internalization on each of these endocytic pathways was investigated on the basis of the sensitivities of each of these pathways to pharmacological or chemical inhibition. Hypertonic sucrose has been shown to impede clathrin-coated pit formation and has been used to disrupt clathrin-mediated endocytosis.14–16 Solutions made hypertonic by the addition of sucrose (at 500 mM, but not at 250 mM or lower concentrations) significantly prevented Siglec-8 endocytosis (Fig 2, A). However, hypertonic sucrose solutions may also disrupt other endocytic pathways.17 Therefore, more selective inhibitors of each of the endocytic pathways were used. Amantadine inhibits the formation of clathrin-coated vesicles and thereby prevents clathrin-mediated endocytosis.18–21 Treatment of eosinophils with 500 to 1000 μM amantadine significantly decreased Siglec-8 endocytosis (Fig 2, B). Dynasore is a noncompetitive inhibitor of dynamin activity, which is required for clathrin-coated pit formation and clathrin-mediated endocytosis.22,23 Treatment with dynasore also inhibited Siglec-8 endocytosis, suggesting a role for dynamin in this process (Fig 2, C). The polyene antibiotic nystatin has been shown to inhibit lipid raft/caveolae-dependent endocytosis by sequestering cholesterol from these membrane domains.24,25 Interestingly, 50 μM nystatin treatment decreased Siglec-8 endocytosis, suggesting that Siglec-8 may be internalized via a hybrid pathway or parallel pathways (Fig 2, D). However, treatment of eosinophils with LY2940002, a phosphatidylinositol 3-kinase inhibitor shown to prevent both pinocytosis and phagocytosis,25,26 exerted no effect on Siglec-8 internalization, indicating that these pathways play no role in the internalization of the receptor (Fig 2, E).
FIG 2.
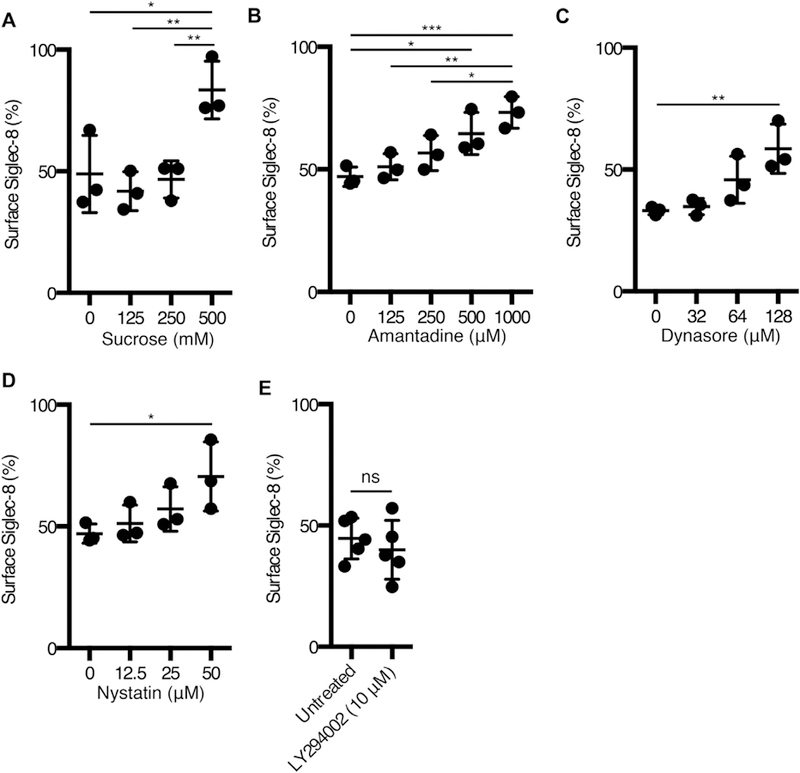
Siglec-8 endocytosis occurs via a clathrin- and dynamin-dependent pathway. Siglec-8 surface expression on eosinophils was measured as in Fig 1 after 120 minutes of incubation at 37°C after preincubation with the indicated concentrations of sucrose (A), amantadine (B), dynasore (C), nystatin (D), or LY294002 (E) and is expressed as a percentage of surface expression at 0 minute. Data represent the means ± SDs of 3 (A, B, C, and D) or 5 (E) independent experiments. *P < .05, **P < .01, and ***P < .001.
The Siglec-8 ITIM is necessary for maximal Siglec-8 endocytosis
It is presumed that Siglec-8 signaling through its intracellular immunoreceptor tyrosine-based inhibitory and switch motifs (ITIM and ITSM, respectively) leads to the internalization of the receptor. However, the motif and signaling molecules that are necessary for this process have not been identified. To examine the contributions of each motif on receptor endocytosis, we introduced Siglec-8 or mutated versions in which the tyrosine residues in the motifs have been replaced by phenylalanine residues into the HEK293T cell line by transduction (Fig 3, A). We observed that the replacement of the tyrosine in the ITIM (Y447F) significantly reduced Siglec-8 endocytosis (Fig 3, B; Y447F vs wild-type, Y447/470F vs Y470F). However, there was no effect on Siglec-8 endocytosis associated with the replacement of the tyrosine in the ITSM (Y470F; Y470F vs wild-type, Y447/470F vs Y447F). Together, these data suggest that the ITIM—and not the ITSM—is responsible for receptor endocytosis upon ligation.
FIG 3.
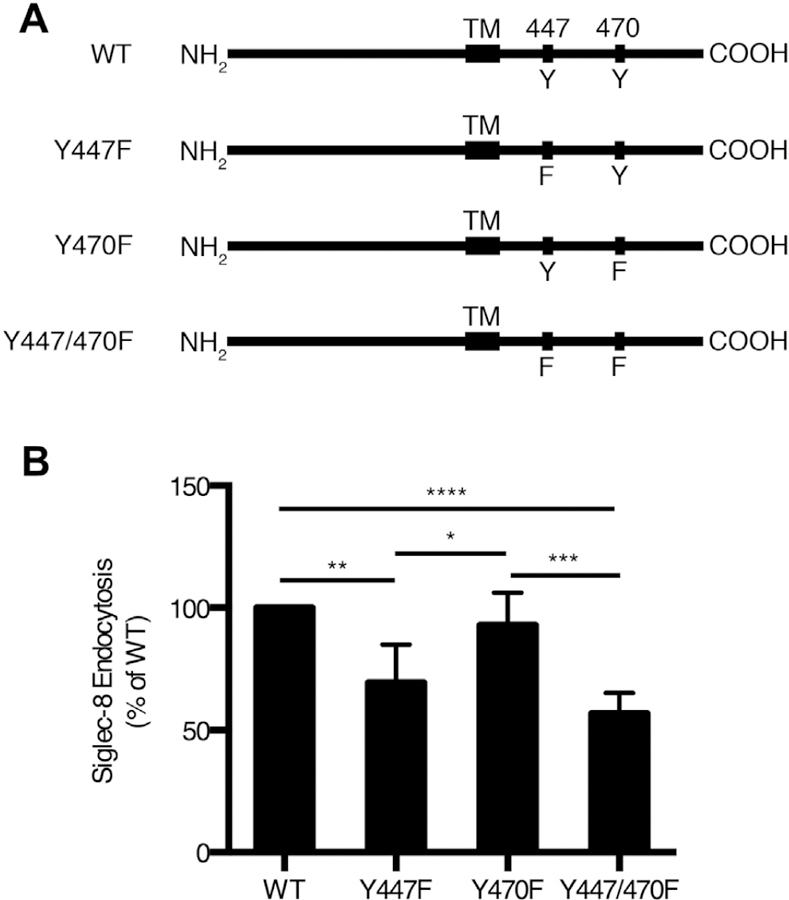
The ITIM but not the ITSM is necessary for maximal Siglec-8 endocytosis. A, Schematic representations of WT and mutant forms of Siglec-8 in which the tyrosine residues in the ITIM (residues 445–450) and/or the ITSM (residues 468–473) have been mutated to phenylalanine residues. B, Siglec-8 endocytosis was examined in HEK293T cells transduced with WT or mutant forms of Siglec-8. Siglec-8 endocytosis is expressed as a percentage of that observed in HEK293T cells transduced with WT Siglec-8. WT, Wild-type. Data represent the means ± SDs of 5 independent experiments. *P < .05, **P < .01, ***P < .001, and ****P < .0001.
Siglec-8 is shuttled to the cell surface via a pathway distinct from that underlying its endocytosis
By using pharmacological inhibitors and disruptors of various signaling molecules and cytoskeletal elements, we investigated the roles of these molecules in Siglec-8 surface expression dynamics. Treatment with the actin monomer–sequestering agent latrunculin B or the actin polymerization–inducing agent jasplakinolide potently inhibited Siglec-8 endocytosis, indicating that Siglec-8 is internalized in a manner dependent on actin rearrangement (Fig 4, A). In contrast, nocodazole, which prevents microtubule polymerization, exerted no inhibitory effect. To identify signaling molecules that may be involved in this endocytic pathway, we pharmacologically inhibited PKC, tyrosine kinases, Src family kinases (SFKs), and protein tyrosine phosphatases (PTPs). Treatment with the PKC inhibitor bisindolylmaleimide I (BIM) also prevented endocytosis of the receptor. In addition, treatment with the tyrosine kinase inhibitor genistein significantly blocked Siglec-8 endocytosis. On the basis of canonical molecular mediators of ITIM signaling,27 we expected to find that SFKs and PTPs were involved in Siglec-8 endocytosis; however, no effect was observed with the SFK inhibitor PP1 or the PTP inhibitor sodium orthovanadate.
FIG 4.

Siglec-8 is internalized and shuttled to the cell surface via independent pathways. Eosinophils were pretreated with a panel of inhibitors at the indicated concentrations. Their effects on Siglec-8 endocytosis (A) or Siglec-8 shuttling to the cell surface (B) were assessed at 120 minutes. Siglec-8 shuttling was measured by detecting surface Siglec-8 that was not bound by the unconjugated primary antibody initially. C, Eosinophils were pretreated with the indicated concentrations of sucrose as in Fig 2. Total surface Siglec-8 was divided into an original surface pool and a shuttled pool at 120 minutes and are represented in a stacked column, normalized to total Siglec-8 surface levels at 0 minute. D, Stacked columns of the original surface pool of Siglec-8 and the shuttled pool at various durations of incubation at 37°C in the presence or absence of LY294002 and BIM (both at 10 μM). Data represent the means ± SDs of 3 or more independent experiments. *P < .05, **P < .01, ***P < .001, and ****P < .0001 relative to untreated eosinophils.
Because this method of tracking Siglec-8 endocytosis only labels the pool of Siglec-8 molecules initially on the cell surface, we used an additional sequential labeling step to detect Siglec-8 newly expressed on the cell surface during the incubation. This technique involved using unlabeled anti–Siglec-8 mAb to ligate the receptor to induce and measure endocytosis and only then using directly labeled antibody of the same clone to measure newly expressed surface Siglec-8. Indeed, this technique indicates the expression of low levels of unbound Siglec-8 at the cell surface after 120 minutes and after as little as 5 minutes of incubation at 37°C (see Fig E3 in this article’s Online Repository at www.jacionline.org and data not shown). As expected, the binding of directly conjugated anti–Siglec-8 to cells that had been preincubated with unconjugated mAb and maintained at 4°C to prevent endocytosis and shuttling did not significantly differ from staining with an isotype control antibody (Fig E3). To explore the mechanisms involved in shuttling Siglec-8 to the cell surface, we again relied on pharmacological inhibition. Shuttling of Siglec-8 to the cell surface occurred independently of cycloheximide treatment, indicating that this was not Siglec-8 synthesized de novo, and was dependent on microtubule assembly, actin rearrangement, dynamin function, and the activities of tyrosine kinases and phosphatidylinositol 3-kinase (Fig 4, B). Although these data do not rule out recycling of Siglec-8 back to the surface following disengagement from the unlabeled antibody in an intracellular compartment, the inhibition of endocytosis and concurrent promotion of shuttling to the cell surface observed with hypertonic sucrose solution (an average increase of 25% in Siglec-8 surface expression at 120 minutes with 500 mM sucrose) or BIM treatment (an average increase of 13%–18% at 30–180 minutes) indicate that the recycling pathway is not likely to be the sole contributor to this pool of Siglec-8 on the cell surface (Fig 4, C and D).
Siglec-8 is internalized to the lysosome within human eosinophils
Following endocytosis, internalized receptors may follow one of several pathways into distinct intracellular compartments or back to the cell surface via a recycling pathway. The compartment to which Siglec-8 localizes was investigated by confocal microscopy. Surface Siglec-8 was first labeled with a fluorophore-conjugated mAb. Lysosomes were labeled with DQ Green BSA, a self-quenched dye conjugate of BSA that fluoresces when the BSA is cleaved by proteases and the quenching is relieved. Siglec-8 was observed at the surface of cells in the absence of endocytosis and, to a small degree, at or near the surface of cells in which endocytosis was allowed to occur (Fig 5, asterisks). Colocalization of the internalized Siglec-8 signal with DQ Green BSAwas observed, indicative of trafficking to the lysosomal compartment (Fig 5, arrows).
FIG 5.
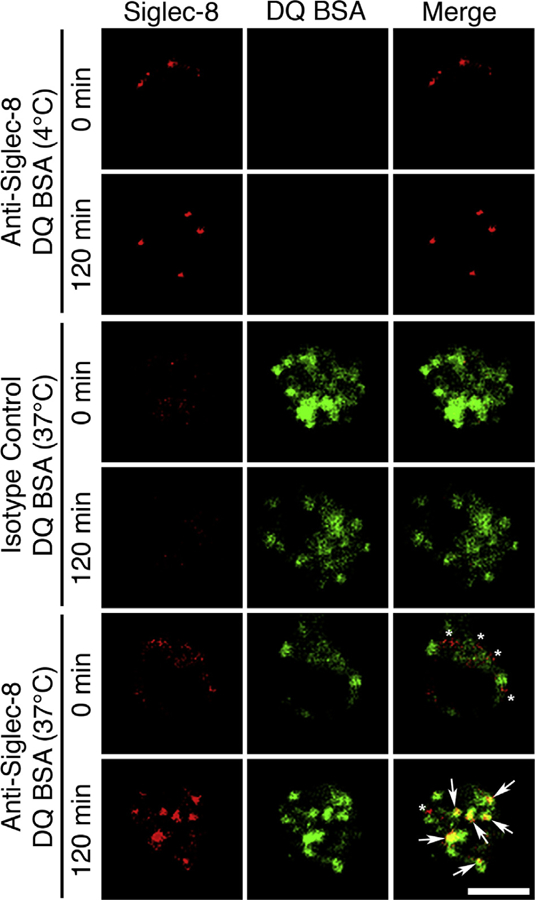
Internalized Siglec-8 localizes to the lysosome. Siglec-8 at the eosinophil cell surface was labeled red with an Alexa Fluor 647–conjugated mAb, and endocytosis of the receptor was permitted at 37°C (120 minutes) or prevented with no incubation at 37°C (0 minute). A matched fluorophore-conjugated isotype-matched mAb was used as a negative staining control in the same manner (middle panel). DQ Green BSA (DQ BSA) was taken up by the cells during a 120-minute incubation to selectively label the proteolytically active lysosomal compartment in green. An incubation with DQ BSA at 4°C only (DQ BSA [0 minute]) was used as a negative staining control (top panel). Colocalization of internalized Siglec-8 and the lysosomal marker (bottom panel) is indicated by yellow fluorescence signal in the merged image (arrows). Siglec-8 at or near the cell surface and distinct from the lysosomal compartment is observed at both 0 and 120 minutes (asterisks). Images are representative of 3 independent experiments. Scale bar, 5 μm.
Sialylated cis ligands on the eosinophil surface partially mask Siglec-8 from interaction with a high-avidity ligand
Many siglecs are known to interact in cis with sialylated glycans at the cell surface that block interactions with lower affinity ligands, thereby setting a threshold for productive siglec signaling.28–32 However, no such interaction had been demonstrated for Siglec-8. To investigate the possible interaction of Siglec-8 with masking cis ligands, a high-avidity synthetic ligand (biotinylated 1-MDa polyacrylamide decorated with 6′-O-sulfo-3′-sialyl-LacNAc) was used to bind Siglec-8 through its ligand-binding domain.33 In addition, the ligand, eosinophils, neither, or both were pretreated with sialidase to remove sialic acid groups necessary for siglec binding. The amount of biotinylated synthetic ligand on the cell surface initially or following incubations of various durations at 37°C was measured by flow cytometry using fluorophore-conjugated streptavidin. Sialidase pretreatment of the ligand decreased surface binding, as expected, whereas sialidase treatment of the eosinophil surface increased synthetic ligand binding by approximately 50%, demonstrating that at least one-third of the surface Siglec-8 pool is prevented from interacting with this high-avidity ligand by sialylated cis ligands (see Fig E4, A, in this article’s Online Repository at www.jacionline.org). In addition, internalization of Siglec-8–bound synthetic ligand is significantly greater in sialidase-treated eosinophils than in untreated eosinophils (Fig E4, B) but is proportional to the initial amount of ligand bound (Fig E4, C).
Targeting of an immunotoxin to Siglec-8 eliminates the need for cytokine priming for induction of death in eosinophils and allows effective killing of malignant mast cells
Signaling through Siglec-8 is well known to induce cell death in eosinophils, especially those that have been primed with IL-5, GM-CSF, or IL-33.6,7 However, in the absence of priming or a secondary cross-linking antibody, anti–Siglec-8 mAb fails to consistently induce substantial eosinophil cell death.6,7 Thus, we sought to develop an agent capable of more consistently causing eosinophil cell death even in the absence of cellular preactivation. To this end, we chose to deliver the ribosome-inactivating protein saporin to eosinophils via Siglec-8 endocytosis. Eosinophils were incubated in the presence or absence of IL-5 and anti–Siglec-8 mAb (2C4) or isotype control with or without saporin conjugation. These treatments were compared with eosinophils that were incubated in the absence of any antibody. After 18 to 24 hours, cell death was assessed using annexin V/DAPI staining. As expected, no statistically significant improvement was achieved in the presence of IL-5 between unconjugated and saporin-conjugated 2C4 due to the consistently high levels of cell death seen with both antibodies (Fig 6, A and B). However, saporin-conjugated 2C4 was significantly and more consistently effective at causing eosinophil cell death in the absence of IL-5. Importantly, this was not due to a nonspecific effect of the saporin because the saporin-conjugated isotype mAb did not induce cell death in the absence of IL-5. In addition, saporin-conjugated anti–Siglec-8 exhibited no off-target cell toxicity when tested separately on donor-matched neutrophils (see Fig E5 in this article’s Online Repository at www.jacionline.org).
FIG 6.

A toxin-conjugated mAb targeted to Siglec-8 causes cell death of eosinophils and a malignant mast cell line in vitro. A and B, Eosinophils were primed with 15 ng/mL IL-5 or left unprimed and concurrently treated with equimolar amounts of anti–Siglec-8 mAb (anti-Sig8), isotype control mAb, saporin (SAP)-conjugated 2C4, or saporin-conjugated isotype control mAb or were left untreated for 18 to 24 hours. C and D, HMC-1.2 cells were preincubated with the indicated mAbs or conjugates, washed extensively, and incubated for 7 to 13 days. Cell death was measured using FITC-labeled annexin V and DAPI staining and flow cytometry. Fig 6, A and C, Representative flow plots for each treatment. Percentages of live cells are shown. Fig 6, B and D, Percentages of viable (annexin V–negative, DAPI-negative) cells for each treatment normalized to the untreated group for each priming condition. Ab, Antibody; FITC, fluorescein isothiocyanate. Error bars represent the means ± SDs (B and D) of 12 and 8 independent experiments, respectively. *P < .05, ***P < .001, and ****P < .0001.
In mast cells, Siglec-8 signaling does not lead to cell death but rather inhibits IgE receptor signaling-dependent mediator release.8 However, the selective targeting of a toxin to malignant mast cells via Siglec-8 may provide a means to eliminate such cells. Thus, we examined whether this immunotoxin could also induce the death of human mast cell leukemia HMC-1.2 cells. Following a brief preincubation with the antibodies with or without saporin conjugation, the cells were cultured and regularly assessed for viability and surface Siglec-8 levels as an indicator of protein synthesis. After 1 day of incubation, saporin-mediated inhibition of protein synthesis in the immunotoxin treatment group was already highly significant (see Fig E6, A, in this article’s Online Repository at www.jacionline.org). Loss of cell viability in this treatment group was slightly delayed and became significant on day 2 (Fig E6, B). To examine whether rare live populations of cells would eventually overcome the treatment effect, we assessed cell viability after at least a week of incubation. However, even at that time point, the vast majority of malignant mast cells treated with the immunotoxin were apoptotic, and there was no evidence of cells overcoming the treatment (Fig 6, C and D).
DISCUSSION
Siglec-8 is considered a potential therapeutic target for allergic disease due to its expression on eosinophils, mast cells, and basophils and its roles in inducing cell death in activated eosinophils and preventing FcεRI signaling-induced mediator release in mast cells.34 However, its potential as a target via which to deliver toxins, chemotherapeutic agents, or drugs had not been explored previously and is especially relevant for mast cells and non–cytokine-primed eosinophils that do not consistently undergo apoptosis with Siglec-8 mAb exposure alone. In this study, it was determined using primary human cells and cell lines that Siglec-8 is internalized and trafficked into the lysosomal compartment via both clathrin-mediated and lipid raft/caveolae-mediated pathways that are dependent on actin cytoskeletal rearrangement and the activities of tyrosine kinases and PKC (Fig 7). Although Siglec-8 endocytosis was found to be relatively slow and endocytosis was partially hindered by the presence of sialylated masking cis ligands on the surface of eosinophils, this endocytic pathway was found to be suitable for targeting an immunotoxin to eosinophils and malignant mast cells. This approach effectively and consistently induced extensive cell death of eosinophils in vitro without prior cellular activation. Notably, for the first time, this approach has been successfully used to target and kill mast cell leukemia cells.
FIG 7.
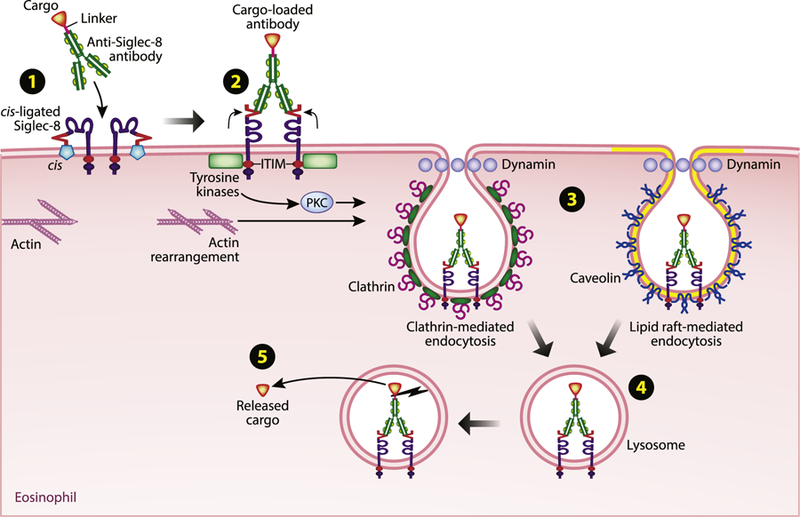
Schematic diagram showing a summary of mechanisms and pathways by which a toxic payload can be delivered into an eosinophil via binding to and inducing the internalization of surface Siglec-8. (1) At baseline, Siglec-8 interacts with sialylated cis ligands on the surface of eosinophils that prevent interaction with trans activating ligands. (2) Antibody ligation of Siglec-8 activates signaling from the cytoplasmic motifs of the receptor. Signaling from the ITIM is involved in the induction of endocytosis, which involves actin rearrangement and the activities of tyrosine kinases and PKC. (3) Siglec-8 endocytosis proceeds via both clathrinmediated and lipid raft/caveolae-mediated pathways. (4) Internalized Siglec-8 localizes to the lysosomal compartment. (5) The linker between the cargo and the anti–Siglec-8 antibody is cleaved, and the cargo is released within the eosinophil to exert its effects. Illustration by Jacqueline Schaffer. PKC, Protein kinase C.
Murine Siglec-F is a functionally similar paralog to Siglec-8 in that it also binds preferentially to the glycan 6′-sulfo-sialyl Lewis X,35 has a similar but distinct pattern of expression (on eosinophils, alveolar macrophages, and intestinal tuft cells, rather than eosinophils, mast cells, and basophils),36,37 and promotes eosinophil cell death.38–40 Because of this, it is often used as a surrogate for Siglec-8 in the mouse. The endocytic pathway used by Siglec-F has been studied,41 and a key similarity between that pathway and the one characterized here for Siglec-8 is that both siglecs are internalized slowly. In fact, relatively slow endocytosis seems to be a feature of many CD33-related siglecs. For example, CD33, Siglec-5, Siglec-F, and Siglec-9 are all internalized slowly when cross-linked with antibody, and, similar to our findings regarding Siglec-8, this endocytosis is dependent on the ITIM in the cytoplasmic domains of CD33, Siglec-F, and Siglec-9.41–44 Although CD33 endocytosis is promoted by treatment with a PTP inhibitor and diminished by treatment with a SFK inhibitor,44 suggesting that tyrosine phosphorylation of the ITIM controls its endocytosis, no significant effect on Siglec-8 endocytosis was seen with similar inhibitor treatments. SFK inhibition also failed to diminish Siglec-F internalization, although tyrosine kinase inhibition prevented its endocytosis,41 as was seen with Siglec-8 as well (Fig 4, A).
These findings highlight yet another key difference between Siglec-8 and Siglec-F that may limit the usefulness of Siglec-F as a surrogate to study Siglec-8 biology. Siglec-F is internalized in a clathrin- and dynamin-independent manner,41 whereas Siglec-8 endocytosis is dependent on both these components but is also internalized in a lipid raft/caveolae-dependent manner. Although there is abundant evidence linking endocytosis to the organization of signaling events,45 it remains to be determined whether the endocytosis of Siglec-8 via each of these pathways leads to distinct signaling pathways or functional consequences. Indeed, it remains unknown whether Siglec-8 endocytosis is necessary to achieve some of its physiologic functions, such as apoptosis induction.
It is important to note that 2 splice variants of Siglec-8 exist and are expressed by human eosinophils, one of which has a truncated cytoplasmic domain that lacks the ITIM and ITSM found in the full-length form of the receptor.5,46 Critically, both the antibody and synthetic ligand used in this study would bind both variants of Siglec-8. Although we found that not all surface Siglec-8 is internalized in response to ligation, it is possible that some or all the remaining surface Siglec-8 is the truncated form of the receptor that lacks the cytoplasmic signaling motifs that may be necessary for endocytosis.
Because Siglec-8 measurement and ligation were inextricably linked by using an antibody or synthetic ligand, it remains possible that Siglec-8 is constitutively internalized with the kinetics and trafficking we observed. Consistent with this possibility, the siglec CD22 undergoes constitutive endocytosis that is not affected by binding with a multivalent glycan ligand.47 However, the fact that total surface expression of Siglec-8 did not remain constant following antibody ligation indicates that endocytosis of Siglec-8 was ligation-dependent rather than constitutive (Fig 4, C and D). The precise signaling mechanism leading to Siglec-8 endocytosis remains incompletely defined. However, genistein and BIM both prevent endocytosis of Siglec-8, implicating tyrosine kinases and PKC in this effect. Actin cytoskeletal rearrangement also played a key role in endocytosis, as demonstrated by treatment with the actin monomer–sequestering and actin filament–stabilizing agents latrunculin B and jasplakinolide. Detecting surface Siglec-8 after various periods of incubation that was not bound by the initial detecting antibody allowed the determination of how much Siglec-8 was shuttled to the surface. Although some components were involved in both endocytosis and shuttling (actin rearrangement and tyrosine kinases), certain components were involved only in shuttling (microtubule polymerization and phosphatidylinositol 3-kinase) or endocytosis (PKC), indicating that these 2 processes are regulated independently. It is important to note that although off-target effects have been noted for several of these inhibitors at higher concentrations, they were used here at concentrations that were appropriate for their intended protein targets. More uncertainty surrounds the specificity of BIM in this process, however. The IC50 of BIM on internalization was approximately 5 μM (data not shown), suggesting that it may be acting on an atypical PKC isoform, although effects on other PKC isoforms, PKG, or myosin light chain kinase cannot be ruled out.
Frequently, other siglecs interact in cis with sialylated ligands that can prevent their interaction with ligands in trans.28–32 These cis interactions may be overcome with a higher-affinity trans ligand or by unmasking the siglec, for example, through cellular activation.48 Data in Fig E4 show for the first time that Siglec-8 is partially masked by such cis ligands. Although unmasking with sialidase is not essential for Siglec-8 to interact with the polymeric synthetic glycan ligand used in this study, determining the nature of this interaction and disrupting it may potentiate the effects of targeting strategies and enable less high-avidity synthetic ligands to be used for this purpose in the future. Given the strong preference of Siglec-8 to recognize specific sulfated α2,3-linked sialylated structures, such cis ligands are likely to contain similar features, although none has yet been detected.49–51
There remains a clinical need for improved, targeted therapies for diseases involving eosinophils and mast cells, such as chronic eosinophilic leukemia, hypereosinophilic syndrome, and systemic mastocytosis. Although some therapeutic mAbs, such as benralizumab, an antibody that targets IL-5Rα, have shown promise in depleting eosinophils and basophils,52 none of these therapies also target mast cells. In addition, there is no curative therapy for systemic mastocytosis.2,53 Therefore, it was of interest to determine whether Siglec-8 endocytosis could be leveraged therapeutically in these cell types. To examine the efficacy of delivering a toxic payload to human eosinophils and mast cell leukemic cells through Siglec-8, a saporin-conjugated anti–Siglec-8 mAb was tested in vitro. Saporin is a type-I ribosome-inactivating protein that lacks a B chain responsible for binding to a target cell surface for internalization that is found in type-II ribosome-inactivating proteins like ricin.54 However, conjugation of saporin to an antibody or ligand for an endocytic receptor allows saporin to be internalized to exert its function. Therefore, a saporin anti–Siglec-8 conjugate seemed to be an ideal candidate to induce the cell death of Siglec-8–expressing cells that do not otherwise undergo Siglec-8– induced apoptosis, such as non–cytokine-primed eosinophils and mast cells.6,8 Indeed, we found that the saporin-antibody conjugate caused consistently high levels of cell death in eosinophils that had not been primed with IL-5, whereas few of these cells would undergo cell death with the anti–Siglec-8 mAb alone (Fig 6, A and B). Furthermore, the saporin-antibody conjugate induced high levels of cell death in the HMC-1.2 human mast cell leukemia cell line (Fig 6, C and D). An additional concern is the type of cell death that these cells undergo. The induction of necrosis or other nonapoptotic cell death of mast cells might cause a dangerous release of preformed mediators in vivo. In contrast, this should not occur during apoptotic cell death. The detection of phosphatidylserine on the outer membrane leaflet by annexin V staining well before loss of membrane integrity by DAPI nuclear staining suggests that HMC-1.2 cells treated with the saporin-antibody conjugate underwent apoptotic cell death. This is consistent with prior publications using other saporin-conjugated antibodies and cell types where apoptotic cell death was observed.55–57 Brentuximab vedotin, a formulation that links the microtubule-disrupting agent monomethyl auristatin E to an mAb specific for the CD30 protein expressed by most neoplastic mast cells, has been investigated in a small number of patients with systemic mastocytosis.58 Encouragingly, there was no evidence of mast cell mediator release upon treatment in this study. This represents the first time that Siglec-8 has been leveraged to selectively induce mast cell death. This demonstrates that Siglec-8 may be a suitable therapeutic target on eosinophils and mast cells in a range of diseases that involve these cell types. Future efforts will include the use of transgenic mice expressing Siglec-8 in appropriate cellular compartments to explore the effectiveness of Siglec-8 targeting in preclinical models of eosinophil- and mast cell–associated diseases.
METHODS
Eosinophil inhibitor treatments
To assess the involvement of various proteins in Siglec-8 endocytosis and shuttling to the cell surface, eosinophils were treated with the following compounds at the indicated concentrations: sucrose, amantadine, nystatin, nocodazole, latrunculin B, genistein, sodium orthovanadate (all from Sigma), dynasore, cycloheximide (both from Santa Cruz Biotechnology, Santa Cruz, Calif), jasplakinolide (Calbiochem, San Diego, Calif), LY294002, PP1, and BIM (all from Selleckchem, Houston, Tex). Cells were pretreated with the agents for 30 minutes at 37°C before Siglec-8 ligation and maintained in the presence of these agents thereafter. Percent inhibition of endocytosis or shuttling was calculated relative to the untreated control sample:
Siglec-8 synthetic glycan ligand binding to eosinophils and sialidase treatments
The 6′-O-sulfo-3′-sialyl-N-acetyl-D-lactosamine (LacNAc)-polyacrylamidebiotin probe was generously provided by Prof. Nicolai Bovin (Russian Academy of Sciences, Moscow, Russia).E1 To demonstrate the necessity of the sialylation of the ligand and as a control for sialidase treatment of cells, the ligand was treated with 50 mU/mL of sialidase from V cholerae generously provided by R. Schnaar of the Johns Hopkins School of Medicine (Baltimore, Md) for 60 minutes at 37°C. Eosinophils were incubated with the sialidase-treated or untreated synthetic ligand at 5 μg/mL at 4°C for 20 minutes to allow binding, then the excess ligand was washed off. After an incubation at 37°C to permit internalization, the remaining synthetic ligand on the cell surface was detected with allophycocyanin-conjugated streptavidin (2 μg/mL; BD Biosciences). To eliminate sialylation on any masking cis ligands, eosinophils were pretreated with 50 mU/mL sialidase for 60 minutes at 37°C and washed before testing synthetic ligand binding.
Extended Data
FIG E1.
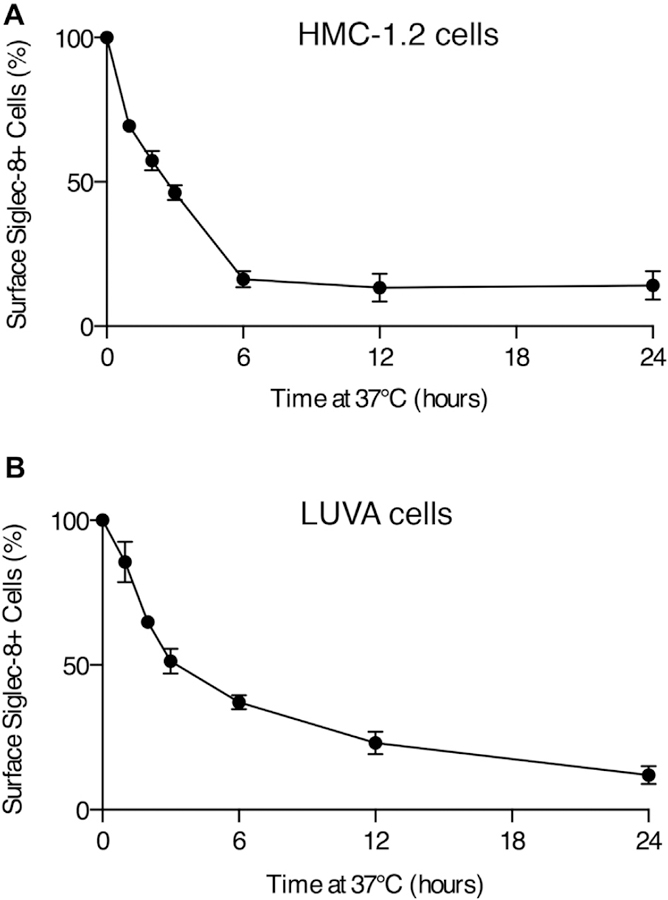
Loss of Siglec-8 from the surface of HMC-1.2 and LUVA mast cells. The percentage of Siglec-8+ HMC-1.2 (A) and LUVA (B) cells was assessed by flow cytometry after antibody ligation of the receptor and incubation at 37°C. The Siglec-8+ gate was positioned on the basis of negative isotype staining. Data represent the means ± SDs of 4 independent experiments.
FIG E2.
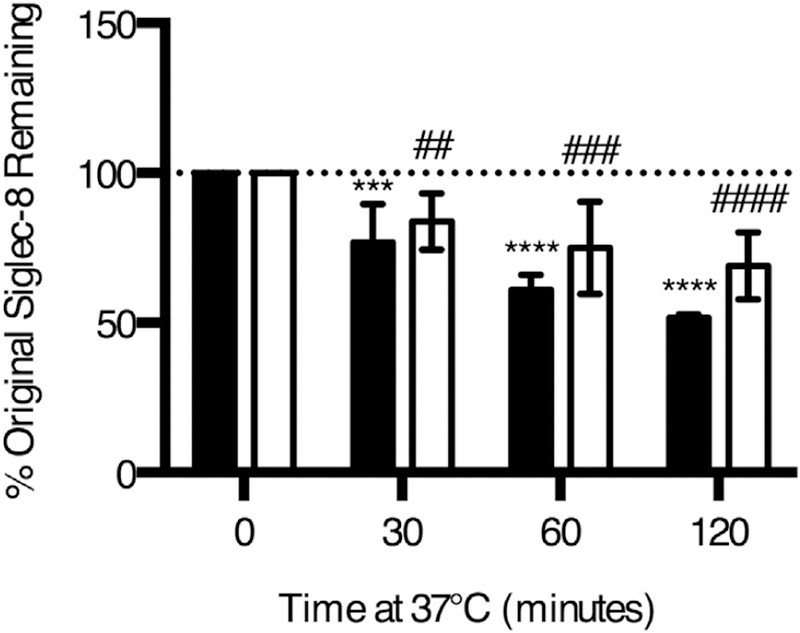
Ligation-induced Siglec-8 endocytosis is not unique to the 2C4 mAb clone. Eosinophils were incubated with anti–Siglec-8 (either clone 2C4 [black columns] or 1H10 [white columns]) for 20 minutes at 4°C. Excess antibody was washed off, and the cells were incubated at 37°C for the indicated durations to permit endocytosis. Antibody remaining at the surface of cell was detected with a secondary antimouse IgG1 antibody. Data represent the means ± SDs of 4 independent experiments. ***P < .001; ****P < .0001 relative to eosinophils incubated with 2C4 mAb without any incubation at 37°C. ##P < .01; ###P < .001; ####P < .0001 relative to eosinophils incubated with 1H10 mAb without any incubation at 37°C.
FIG E3.

Siglec-8 shuttled to the cell surface can be detected using a sequential labeling step. Eosinophils were labeled with unconjugated anti–Siglec-8 mAb (2C4) and incubated at 37°C to permit endocytosis as in Fig 1. Following detection of remaining anti–Siglec-8 at the cell surface with a FITC-conjugated secondary mAb (‘‘Original’’), Siglec-8 newly expressed at the cell surface (‘‘Shuttled’’) was detected using anti–Siglec-8 (2C4) directly conjugated to Alexa Fluor 647. Control antibodies of the same isotype (iso) are included as negative staining controls. Loss of FITC signal and gain of Alexa Fluor 647 signal at 120 minutes when using anti–Siglec-8 mAbs in both channels represent receptor endocytosis and shuttling to the cell surface. Effective blockade of Alexa Fluor 647–conjugated anti–Siglec-8 binding can be seen at 0 minute when anti–Siglec-8 mAbs are used for both channels (no additional Alexa Fluor 647 signal). FITC, Fluorescein isothiocyanate.
FIG E4.

Sialylated cis ligands on eosinophils limits binding and internalization of an artificial Siglec-8 glycan ligand. A biotinylated synthetic Siglec-8 ligand-decorated polyacrylamide polymer was used to label Siglec-8 through its ligand-binding domain. A, Biotinylated 1-MDa polyacrylamide decorated with 6′-O-sulfo-3′-sialyl-LacNAc binding to the cell surface after various durations of incubation at 37°C was detected by flow cytometry using fluorophore-conjugated streptavidin. Sialidase from V cholerae was used to desialylate the ligand (dark gray columns), the eosinophil cell surface (black columns), or both (light gray columns) before binding of Siglec-8 with the artificial ligand. Untreated cells and ligand are represented by the white columns. B, Endocytosis of the Siglec-8 ligand in untreated (unfilled circles) or sialidase-treated (filled squares) eosinophils was calculated as the loss of surface-bound ligand normalized to the amount of ligand bound by untreated eosinophils at time 0. *P < .05; **P < .01; ***P < .001; ****P < .0001 relative to untreated eosinophils incubated for the same duration at 37°C. C, Endocytosis of the Siglec-8 ligand in untreated (unfilled circles) or sialidase-treated (filled squares) eosinophils was calculated as the loss of surface-bound ligand normalized to the amount of ligand bound at time 0 by eosinophils within each treatment group. Data represent the means ± SDs of 8 independent experiments.
FIG E5.
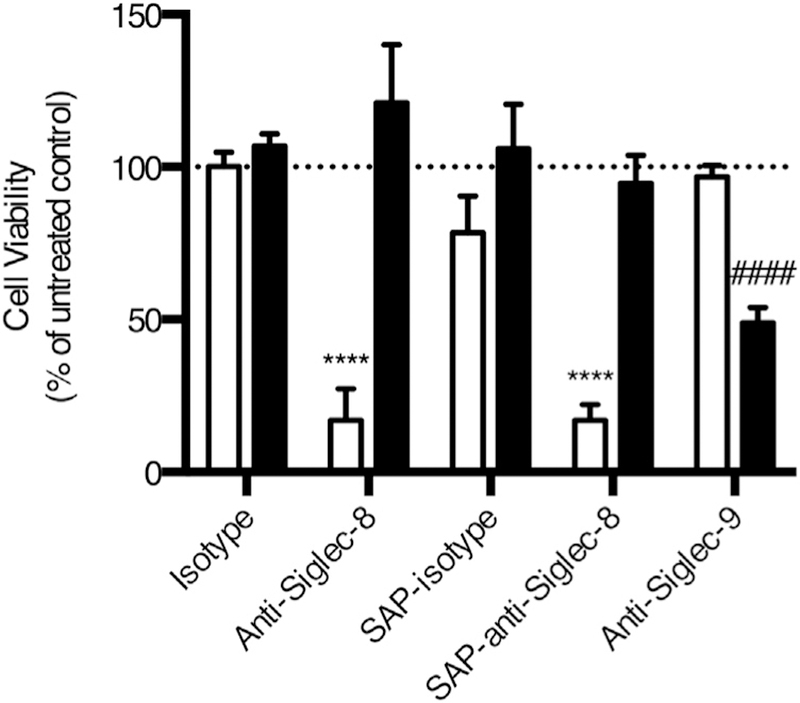
Anti–Siglec-8 and saporin-conjugated anti–Siglec-8 induce cell death in eosinophils but not in neutrophils. Eosinophils and neutrophils were isolated from the same donors. Eosinophils were incubated in 30 ng/mL IL-5. Neutrophils were incubated in 25 ng/mL GM-CSF. Eosinophil (white columns) and neutrophil (black columns) viability was assessed following treatment with anti–Siglec-8 with or without saporin (SAP) conjugation, isotype controls, or anti–Siglec-9 (clone K8, also mouse IgG1) as a positive control for siglec-mediated neutrophil cell death. ****P < .0001 relative to isotype-treated eosinophils. ####P < .0001 relative to isotype-treated neutrophils.
FIG E6.
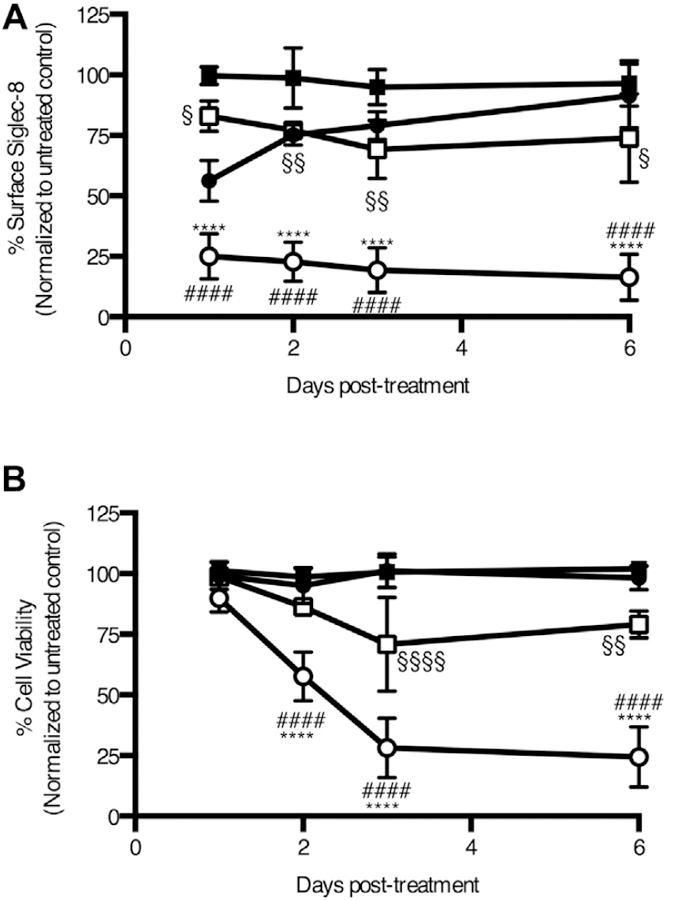
Timeline of the effects of saporin-conjugated anti–Siglec-8 on HMC-1.2 cells. Following treatment with anti–Siglec-8 with (unfilled circles) or without (filled circles) saporin conjugation or isotype control mAb with (unfilled squares) or without (filled squares) saporin conjugation, surface Siglec-8 expression (A) and viability (B) were assessed daily. Cell viability was assessed with annexin V and DAPI staining. ****P < .0001 relative to anti–Siglec-8; ####P < .0001 relative to saporin-conjugated isotype control; §P < .05; §§P < .01; §§§§P < .0001 relative to isotype control. Data represent the means ± SDs of 4 independent experiments.
Key messages.
Siglec-8 is internalized in human eosinophils and mast cells and trafficked to the lysosomal compartment.
The intracellular ITIM, actin rearrangement, tyrosine kinase and PKC activities, clathrin, lipid rafts, and sialylated cis ligands control Siglec-8 endocytosis.
Antibody-toxin targeting via Siglec-8 effectively kills human eosinophils and mast cell leukemia cells in vitro.
Acknowledgments
We thank Drs Peter Valent, John Steinke, Larry Borish, and Nick Lu for providing us with cell lines used in this study, and Dr Nenad Tomasevic for providing 1H10 anti–Siglec-8 antibody. We also thank Dr Ronald Schnaar for providing V cholerae sialidase and the other members of the National Institutes of Health-funded LIDPEG (grant no. P01HL107151) for helpful discussions on the design and analysis of this work. We thank Jacqueline Schaffer for her contribution of Fig 7.
This work was supported by the National Heart, Lung, and Blood Institute (grant no. P01HL107151 to B.S.B.), the National Institute of Allergy and Infectious Diseases (grant no. AI072265 to B.S.B. and grant no. T32AI083216 to J.A.O.), and the National Cancer Institute (Cancer Center Support grant no. NCI CA060553 to the Center for Advanced Microscopy at Northwestern University).
Abbreviations used
- BIM
Bisindolylmaleimide I
- DAPI
4′-6-Diamidino-2-phenylindole
- ITIM
Immunoreceptor tyrosine-based inhibitory motif ITSM: Immunoreceptor tyrosine-based switch motif
- PI3K
Phosphatidylinositol 3-kinase
- PTP
Protein tyrosine phosphatase SFK: Src family kinase
- Siglec
Sialic acid–binding immunoglobulin-type lectin
Footnotes
Disclosure of potential conflict of interest: J. A. O’Sullivan and B. S. Bochner receive grant support from the National Institutes of Health (NIH). B. S. Bochner has current or recent consulting or scientific advisory board arrangements with, or has received honoraria from, Sanofi-Aventis, TEVA, AstraZeneca, and Allakos and owns stock in Allakos and Glycomimetics; receives publication-related royalty payments from Elsevier and UpToDate, and is a coinventor on existing Siglec-8–related patents and thus may be entitled to a share of royalties received by Johns Hopkins University on the potential sales of such products; and is also a cofounder of Allakos, which makes him subject to certain restrictions under university policy. The terms of this arrangement are being managed by the Johns Hopkins University and Northwestern University in accordance with their conflict of interest policies. The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Klion AD. Eosinophilic myeloproliferative disorders. Hematology Am Soc Hematol Educ Program 2011;2011:257–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med 2015;373:163–72. [DOI] [PubMed] [Google Scholar]
- 3.Kikly KK, Bochner BS, Freeman SD, Tan KB, Gallagher KT, D’Alessio KJ, et al. Identification of SAF-2, a novel siglec expressed on eosinophils, mast cells, and basophils. J Allergy Clin Immunol 2000;105:1093–100. [DOI] [PubMed] [Google Scholar]
- 4.Hudson SA, Herrmann H, Du J, Cox P, Haddad el B, Butler B, et al. Developmental, malignancy-related, and cross-species analysis of eosinophil, mast cell, and basophil Siglec-8 expression. J Clin Immunol 2011;31:1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foussias G, Yousef GM, Diamandis EP. Molecular characterization of a Siglec8 variant containing cytoplasmic tyrosine-based motifs, and mapping of the Siglec8 gene. Biochem Biophys Res Commun 2000;278:775–81. [DOI] [PubMed] [Google Scholar]
- 6.Nutku E, Aizawa H, Hudson SA, Bochner BS. Ligation of Siglec-8: a selective mechanism for induction of human eosinophil apoptosis. Blood 2003;101: 5014–20. [DOI] [PubMed] [Google Scholar]
- 7.Na HJ, Hudson SA, Bochner BS. IL-33 enhances Siglec-8 mediated apoptosis of human eosinophils. Cytokine 2012;57:169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yokoi H, Choi OH, Hubbard W, Lee HS, Canning BJ, Lee HH, et al. Inhibition of FcepsilonRI-dependent mediator release and calcium flux from human mast cells by sialic acid-binding immunoglobulin-like lectin 8 engagement. J Allergy Clin Immunol 2008;121:499–505.e1. [DOI] [PubMed] [Google Scholar]
- 9.Yu H, Gonzalez-Gil A, Wei Y, Fernandes SM, Porell RN, Vajn K, et al. Siglec-8 and Siglec-9 binding specificities and endogenous airway ligand distributions and properties. Glycobiology 2017;27:657–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsumoto K, Schleimer RP, Saito H, Iikura Y, Bochner BS. Induction of apoptosis in human eosinophils by anti-Fas antibody treatment in vitro. Blood 1995;86:1437–43. [PubMed] [Google Scholar]
- 11.Butterfield JH, Weiler D, Dewald G, Gleich GJ. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res 1988;12:345–55. [DOI] [PubMed] [Google Scholar]
- 12.Laidlaw TM, Steinke JW, Tinana AM, Feng C, Xing W, Lam BK, et al. Characterization of a novel human mast cell line that responds to stem cell factor and expresses functional FcepsilonRI. J Allergy Clin Immunol 2011;127:815–22, e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Na HJ, Hamilton RG, Klion AD, Bochner BS. Biomarkers of eosinophil involvement in allergic and eosinophilic diseases: review of phenotypic and serum markers including a novel assay to quantify levels of soluble Siglec-8. J Immunol Methods 2012;383:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heuser JE, Anderson RG. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol 1989;108: 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen SH, Sandvig K, van Deurs B. Clathrin and HA2 adaptors: effects of potassium depletion, hypertonic medium, and cytosol acidification. J Cell Biol 1993;121:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inal J, Miot S, Schifferli JA. The complement inhibitor, CRIT, undergoes clathrin-dependent endocytosis. Exp Cell Res 2005;310:54–65. [DOI] [PubMed] [Google Scholar]
- 17.Carpentier JL, Sawano F, Geiger D, Gorden P, Perrelet A, Orci L. Potassium depletion and hypertonic medium reduce ‘‘non-coated’’ and clathrin-coated pit formation, as well as endocytosis through these two gates. J Cell Physiol 1989; 138:519–26. [DOI] [PubMed] [Google Scholar]
- 18.Schlegel R, Dickson RB, Willingham MC, Pastan IH. Amantadine and dansylcadaverine inhibit vesicular stomatitis virus uptake and receptor-mediated endocytosis of alpha 2-macroglobulin. Proc Natl Acad Sci U S A 1982;79:2291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phonphok Y, Rosenthal KS. Stabilization of clathrin coated vesicles by amantadine, tromantadine and other hydrophobic amines. FEBS Lett 1991;281: 188–90. [DOI] [PubMed] [Google Scholar]
- 20.Perry DG, Daugherty GL, Martin WJ II. Clathrin-coated pit-associated proteins are required for alveolar macrophage phagocytosis. J Immunol 1999; 162:380–6. [PubMed] [Google Scholar]
- 21.Eckels PC, Banerjee A, Moore EE, McLaughlin NJ, Gries LM, Kelher MR, et al. Amantadine inhibits platelet-activating factor induced clathrin-mediated endocytosis in human neutrophils. Am J Physiol Cell Physiol 2009;297:C886–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirchhausen T, Macia E, Pelish HE. Use of dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. Methods Enzymol 2008; 438:77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 2006;10:839–50. [DOI] [PubMed] [Google Scholar]
- 24.Ros-Baro A, Lopez-Iglesias C, Peiro S, Bellido D, Palacin M, Zorzano A, et al. Lipid rafts are required for GLUT4 internalization in adipose cells. Proc Natl Acad Sci U S A 2001;98:12050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov AI. Pharmacological inhibition of endocytic pathways: Is it specific enough to be useful? Methods Mol Biol 2008;440:15–33. [DOI] [PubMed] [Google Scholar]
- 26.Araki N, Johnson MT, Swanson JA. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol 1996;135:1249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrow AD, Trowsdale J. You say ITAM and I say ITIM, let’s call the whole thing off: the ambiguity of immunoreceptor signalling. Eur J Immunol 2006;36: 1646–53. [DOI] [PubMed] [Google Scholar]
- 28.Freeman SD, Kelm S, Barber EK, Crocker PR. Characterization of CD33 as a new member of the sialoadhesin family of cellular interaction molecules. Blood 1995;85:2005–12. [PubMed] [Google Scholar]
- 29.Hanasaki K, Varki A, Powell LD. CD22-mediated cell adhesion to cytokine-activated human endothelial cells: positive and negative regulation by alpha 2–6-sialylation of cellular glycoproteins. J Biol Chem 1995;270: 7533–42. [DOI] [PubMed] [Google Scholar]
- 30.Razi N, Varki A. Masking and unmasking of the sialic acid-binding lectin activity of CD22 (Siglec-2) on B lymphocytes. Proc Natl Acad Sci U S A 1998;95: 7469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Razi N, Varki A. Cryptic sialic acid binding lectins on human blood leukocytes can be unmasked by sialidase treatment or cellular activation. Glycobiology 1999;9:1225–34. [DOI] [PubMed] [Google Scholar]
- 32.Collins BE, Blixt O, Han S, Duong B, Li H, Nathan JK, et al. High-affinity ligand probes of CD22 overcome the threshold set by cis ligands to allow for binding, endocytosis, and killing of B cells. J Immunol 2006;177:2994–3003. [DOI] [PubMed] [Google Scholar]
- 33.Hudson SA, Bovin NV, Schnaar RL, Crocker PR, Bochner BS. Eosinophil-selective binding and proapoptotic effect in vitro of a synthetic Siglec-8 ligand, polymeric 6′-sulfated sialyl Lewis x. J Pharmacol Exp Ther 2009; 330:608–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bochner BS. ‘‘Siglec’’ting the allergic response for therapeutic targeting. Glycobiology 2016;26:546–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tateno H, Crocker PR, Paulson JC. Mouse Siglec-F and human Siglec-8 are functionally convergent paralogs that are selectively expressed on eosinophils and recognize 6′-sulfo-sialyl Lewis X as a preferred glycan ligand. Glycobiology 2005;15:1125–35. [DOI] [PubMed] [Google Scholar]
- 36.Zhang JQ, Biedermann B, Nitschke L, Crocker PR. The murine inhibitory receptor mSiglec-E is expressed broadly on cells of the innate immune system whereas mSiglec-F is restricted to eosinophils. Eur J Immunol 2004;34:1175–84. [DOI] [PubMed] [Google Scholar]
- 37.Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016;529:226–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao H, Kano G, Hudson SA, Brummet M, Zimmermann N, Zhu Z, et al. Mechanisms of Siglec-F-induced eosinophil apoptosis: a role for caspases but not for SHP-1, Src kinases, NADPH oxidase or reactive oxygen. PLoS One 2013;8:e68143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang M, Angata T, Cho JY, Miller M, Broide DH, Varki A. Defining the in vivo function of Siglec-F, a CD33-related Siglec expressed on mouse eosinophils. Blood 2007;109:4280–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmermann N, McBride ML, Yamada Y, Hudson SA, Jones C, Cromie KD, et al. Siglec-F antibody administration to mice selectively reduces blood and tissue eosinophils. Allergy 2008;63:1156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tateno H, Li H, Schur MJ, Bovin N, Crocker PR, Wakarchuk WW, et al. Distinct endocytic mechanisms of CD22 (Siglec-2) and Siglec-F reflect roles in cell signaling and innate immunity. Mol Cell Biol 2007;27:5699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biedermann B, Gil D, Bowen DT, Crocker PR. Analysis of the CD33-related siglec family reveals that Siglec-9 is an endocytic receptor expressed on subsets of acute myeloid leukemia cells and absent from normal hematopoietic progenitors. Leuk Res 2007;31:211–20. [DOI] [PubMed] [Google Scholar]
- 43.Walter RB, Raden BW, Kamikura DM, Cooper JA, Bernstein ID. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin-induced cytotoxicity. Blood 2005;105:1295–302. [DOI] [PubMed] [Google Scholar]
- 44.Walter RB, Raden BW, Zeng R, Hausermann P, Bernstein ID, Cooper JA. ITIM-dependent endocytosis of CD33-related Siglecs: role of intracellular domain, tyrosine phosphorylation, and the tyrosine phosphatases, Shp1 and Shp2. J Leukoc Biol 2008;83:200–11. [DOI] [PubMed] [Google Scholar]
- 45.Polo S, Di Fiore PP. Endocytosis conducts the cell signaling orchestra. Cell 2006; 124:897–900. [DOI] [PubMed] [Google Scholar]
- 46.Aizawa H, Plitt J, Bochner BS. Human eosinophils express two Siglec-8 splice variants. J Allergy Clin Immunol 2002;109:176. [DOI] [PubMed] [Google Scholar]
- 47.O’Reilly MK, Tian H, Paulson JC. CD22 is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B cells. J Immunol 2011;186:1554–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol 2007;7:255–66. [DOI] [PubMed] [Google Scholar]
- 49.Kiwamoto T, Katoh T, Evans CM, Janssen WJ, Brummet ME, Hudson SA, et al. Endogenous airway mucins carry glycans that bind Siglec-F and induce eosinophil apoptosis. J Allergy Clin Immunol 2015;135:1329–40, e1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.North SJ, von Gunten S, Antonopoulos A, Trollope A, MacGlashan DW Jr, Jang-Lee J, et al. Glycomic analysis of human mast cells, eosinophils and basophils. Glycobiology 2012;22:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Propster JM, Yang F, Rabbani S, Ernst B, Allain FH, Schubert M. Structural basis for sulfation-dependent self-glycan recognition by the human immune-inhibitory receptor Siglec-8. Proc Natl Acad Sci U S A 2016;113:E4170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Busse WW, Katial R, Gossage D, Sari S, Wang B, Kolbeck R, et al. Safety profile, pharmacokinetics, and biologic activity of MEDI-563, an anti-IL-5 receptor alpha antibody, in a phase I study of subjects with mild asthma. J Allergy Clin Immunol 2010;125:1237–44.e2. [DOI] [PubMed] [Google Scholar]
- 53.Gotlib J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med 2016;374:2530–41. [DOI] [PubMed] [Google Scholar]
- 54.de Virgilio M, Lombardi A, Caliandro R, Fabbrini MS. Ribosome-inactivating proteins: from plant defense to tumor attack. Toxins (Basel) 2010;2:2699–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bergamaschi G, Perfetti V, Tonon L, Novella A, Lucotti C, Danova M, et al. Saporin, a ribosome-inactivating protein used to prepare immunotoxins, induces cell death via apoptosis. Br J Haematol 1996;93:789–94. [DOI] [PubMed] [Google Scholar]
- 56.Bolognesi A, Tazzari PL, Olivieri F, Polito L, Falini B, Stirpe F. Induction of apoptosis by ribosome-inactivating proteins and related immunotoxins. Int J Cancer 1996;68:349–55. [DOI] [PubMed] [Google Scholar]
- 57.Polito L, Bortolotti M, Mercatelli D, Battelli MG, Bolognesi A. Saporin-S6: a useful tool in cancer therapy. Toxins (Basel) 2013;5:1698–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Borate U, Mehta A, Reddy V, Tsai M, Josephson N, Schnadig I. Treatment of CD30-positive systemic mastocytosis with brentuximab vedotin. Leuk Res 2016;44:25–31. [DOI] [PubMed] [Google Scholar]
REFERENCE
- E1.Hudson SA, Bovin NV, Schnaar RL, Crocker PR, Bochner BS. Eosinophil-selective binding and proapoptotic effect in vitro of a synthetic Siglec-8 ligand, polymeric 6′-sulfated sialyl Lewis x. J Pharmacol Exp Ther 2009;330:608–12. [DOI] [PMC free article] [PubMed] [Google Scholar]