

Abstract
Localized expression of effector molecules can initiate anti-tumor responses through engagement of specific receptors on target cells in the tumor microenvironment. These locally induced responses may also have a systemic effect, clearing additional tumors throughout the body. In this study, to evoke systemic anti-tumor responses, we utilized charge-altering releasable transporters (CART) for local intratumoral delivery of mRNA coding for co-stimulatory and immune-modulating factors. Intratumoral injection of the CART-mRNA complexes resulted in mRNA expression at the site of administration, transfecting a substantial proportion of tumor-infiltrating dendritic cells, macrophages, and T cells in addition to the tumor cells, resulting in a local anti-tumor effect. Using a two-tumor model, we further show that mRNA therapy locally administered to one tumor stimulated a systemic anti-tumor response, curing both tumors. The combination of OX40L-, CD80-, and CD86-encoding mRNA resulted in the local upregulation of pro-inflammatory cytokines, robust local T cell activation, and migration of immune cells to local draining lymph node or to an anatomically distant tumor. This approach delayed tumor growth, facilitated tumor regression, and cured tumors in both A20 and CT26 tumor models. These results highlight mRNA-CART therapy as a viable approach to induce systemic anti-tumor immunity from a single localized injection.
Introduction
With recent technological advances in oligonucleotide delivery, mRNA therapy in living animals has become a feasible strategy to treat a variety of pathologies (1). All mRNAs are polyanions that vary only in sequence and length, whereas, the proteins that they encode can elicit countless functions. mRNA therapies offer many of the attractive features of conventional DNA and protein based therapy without some of the associated risks and limitations such as insertional mutagenesis (2–4), cost, in vitro instability, short half-life, and inherent immune-activating (immunogenicity) properties (5). Furthermore, the transient nature of mRNA lends itself to temporary expression of target proteins, which in many therapeutic contexts is advantageous over permanent protein expression (6). Most DNA-based gene therapy and vaccination strategies are based on viral delivery of the DNA payload, however, the inherent immunogenicity (7) of the viral delivery vehicles is often more pronounced leading to an anti-viral response rather than a response to the antigen it delivers. While “naked” mRNA is potentially recognized by intracellular toll-like receptor 7 molecules, and thus can be mildly immunogenic (8), nanoparticle-based delivery vehicles hide the mRNA from systemic recognition. Upon cellular internalization, the vehicle and its cargo must escape the endosome and release the mRNA to the cytoplasm to enable protein expression. In the cytoplasm, the immunogenicity concerns of mRNA are minimized by introducing structural modifications to the mRNA (9). Effective mRNA delivery remains a major technical barrier, only successfully overcome by a few approaches, including lipid nanoparticles (LNPs), charge-altering releasable transporters (CARTs), and a few other systems (10).
Intratumoral delivery of immunostimulatory mRNA ensures transfection of tumor-infiltrating cells which can then initiate a robust, global anti-tumor response by either bolstering the ongoing anti-tumor immune response, or by enhancing local antigen presentation in the tumor and the tumor draining lymph node (11,12). The virtually unlimited number of encodable proteins, ease of synthesis and scalability of mRNA production enable delivery of a multitude of genes which can synergistically overcome the immunosuppressive environment and/or enhance the ongoing anti-tumor immune response. Systemic administration of immune modulators often leads to autoimmune responses (13–16), therefore it is appealing to deliver mRNA “prodrugs” that express non-secreted immunostimulatory proteins. Ideally, mRNA prodrugs and the vectors that deliver them would demonstrate minimal inherent immunogenic properties but convey target-specific immunogenicity by the introduction of tumor-specific antigens or immunostimulatory modulators. However, the most common LNP-based delivery vehicles are inherently immunogenic (17,18), which can trigger undesirable off-target effects such as a vaccination effect against the delivered mRNA encoded immunostimulatory molecules it delivers (19,20). Regardless, nanoparticle-based approaches for transient mRNA expression of immunomodulatory genes within the tumor microenvironment represents a novel, safe, and versatile immunotherapeutic approach.
We recently reported (Haabeth, et al, 2018) an mRNA-based cancer vaccination strategy using CARTs (21–23), where we were able to cure mice with large established tumors. Importantly, this work showed that CARTs themselves have little inherent immunogenicity but allows tailored modulation of immune responses by the incorporation of adjuvants into the mRNA-containing electrostatic complex. Furthermore, unlike LNP formulations, fluorophores are easily covalently attached to CART delivery vehicles (BDK-CART), (McKinlay et al, 2018, Benner et al, 2018) which can be utilized to correlate location of transfected cells (either anatomical, or within specific cell populations) with gene expression, thus providing insight into the mechanistic basis of effective therapeutic outcomes. In this study we utilized the BDK-CART-mRNA delivery platform to induce local expression of genes encoding the well described immunomodulatory molecules CD70 (11), OX40L(24,25), CD80(26,27), CD86, IL-12(28–30), and IFNγ (31), both individually and combinatorial. We used a two-tumor model to demonstrate that local administration of mRNA coding for immunomodulatory molecules can elicit a systemic response that can cure both the treated (local) and untreated (distal) tumors.
Material and Methods
Mice and cells lines
Eight- to twelve-week-old female Balb/c mice were purchased from The Jackson Laboratory and housed in the Laboratory Animal Facility of the Stanford University Medical Center. All experiments were approved by the Stanford Administrative Panel on Laboratory Animal Care and conducted in accordance with Stanford University Animal Facility and NIH guidelines. The A20 and CT26 cell line was obtained from ATCC (ATCC no. TIB-208). A20 is a Balb/c B cell lymphoma line derived from a spontaneous neoplasm found in an old Balb/cAnN mice, expressing MHC class I and class II H-2d molecules.
CT26 is an N-nitroso-N-methylurethane-(NNMU) induced, undifferentiated colon carcinoma cell line. Mycoplasm testing of cell lines was performed with three-month intervals using a MycoAlert Detection Kit (Lonza, Basel, Switzerland). All experiments were performed within five cell passages after thawing. A20 cells (5 × 106) and CT26 (5 × 105) were implanted at two subcutaneous sites on the right and left sides of the abdomen. Treatment began when tumors reached 7 mm to 10 mm in largest diameter. mRNA-CARTs were injected intratumorally into one tumor site at indicated doses every other days for 3 injection (days 7,11 and 15 after tumor inoculation) unless otherwise specified. Tumor size was monitored with a digital caliper (Mitutoyo) every 2 to 3 days and expressed as volume (length × width × height). Mice are sacrificed when tumor size reached 1.5 cm in the largest diameter as per guidelines.
CART
CART D13:A11 was prepared as previously described (22) using benzyl alcohol as an initiator and matched reported characterization. Fluorescent BDK-CART was also prepared as previously described (21,23) using the difluoroboron dibenzoylmethane initiator prepared according to a procedure described by Fraser (32). Endgroup analysis of the protected polymer showed block lengths of 12 dodecyl carbonate monomers and 14 cationic aminoester monomers.
General CART formulation Methods
For all experiments, CARTs were formulated with mRNA at a 10:1 cation:anion ratio assuming full protonation of the CART and full deprotonation of the oligonucleotide. Formulations were carried out in acidic PBS (pH adjusted to 5.5 by addition of 0.1M HCl) before injection (in vivo) or addition to treatment wells (in vitro).
CART in vitro Transfection
For in vitro transfections, cells were seeded at 40,000 cells/well in 24-well plates (for HeLa). Immediately prior to treatment, cells were washed and re-suspended in serum-free media. CART formulations were made using the general method above. In a standard in vitro experiment, 0.42ug of mRNA (2.1uL of a 0.2 mg/mL stock) was added to 5.71uL of PBS (pH 5.5). To this was added 0.59uL of CART (from a 2mM stock), and this was mixed for 20 seconds. 2.5uL of this formulation was added to each of three wells, resulting in a final mRNA dose of 125 ng/well. This was incubated for 8 hours after which gene expression was determined by antibody staining or direct flow cytometric analysis.
Flow Cytometry
The following fluorochrome conjugated rat anti-mouse monoclonal antibodies (mAbs) were used for flow cytometry: CD4-Pe, IFNγ-PE, CD11c-Pe, Perforin-PE, FoxP3-Pe, CD49b-Pe, CD154-Pe, CD137-Pe, CD4-BV-605, B220-FITC, CD69-FITC, Granzyme B-FITC,, CD25-FITC, CD8-Pe-Cy7, F4/80-APC, Ki-67-APC, CD11b-PerCP-Cy5.5, CD3 Alexa Fluor 700,, CD357-BV-711, and rat isotype controls for the listed fluorochromes and antibodies. The following rat anti-mouse unconjugated antibodies were used: anti-CD16/32. Staining using two brilliant violet (BV) fluorochromes was stained in Brilliant stain buffer according to manufacturer’s protocol (BD biosciences). For intracellular staining, cells were treated with golgistop (BD biosciences) for 5hrs prior to staining. Cells were fixed and permeabilized according to manufacturer protocol (BD biosciences and eBiosciences FoxP3 staining kit). Antibodies were purchased from either BD Biosciences, Invitrogen, or eBioscience. All surface marker staining was done for 20 minutes at room temp. Cells were surface stained in wash buffer (phosphate-buffered saline [PBS], 0.5% Bovine Serum Albumin (Sigma), and 0.01% sodium azide), either fixed in 2% paraformaldehyde or runned fresh, and analyzed by flow cytometry on an FACSCalibur or LSR II system (BD Biosciences). Data were analyzed using either Cytobank (Cytobank Inc.) or Flowjo version 10.0 (Flowjo).
In vivo Bioluminescence
For bioluminescence assessment, mice were anesthetized with isoflurane gas (2% isoflurane in oxygen, 1 l/min) during injection and imaging procedures. i.p. injections of d-Luciferin (Biosynth AG) were done at a dose of 150 mg/kg, providing a saturating substrate concentration for Fluc enzyme (luciferin crosses the blood-brain barrier). Mice were imaged in a light-tight chamber using an in vivo optical imaging system (IVIS 100; Xenogen Corp.) equipped with a cooled charge-coupled device camera. During image recording, mice inhaled isofluorane delivered via a nose cone, and their body temperature was maintained at 37°C in the dark box of the camera system. Bioluminescence images were acquired between 10 and 20 minutes after luciferin administration. Mice usually recovered from anesthesia within 2 minutes of imaging.
mRNA
Firefly Fluc (Fluc), and secreted alkaline phosphatase (SEAP) (control mRNA) mRNA were purchased from Trilink BioTechnologies. All purchased mRNAs were capped and polyadenylated and contain 5-methoxyuridine modification. CD70 (NCBI Reference Sequence: NM_011617.2), OX40L (NCBI Ref. Seq. Accession number: NM_011659.2), IFNγ (NCBI Reference Sequence: NM_008337.4), IL-12 single chain construct (monomerized by introduction of a protein linker between the p35 (NCBI Reference Sequence: NM_001159424.2) and p40 (NCBI Reference Sequence: NM_001303244.1) protein chains, CD80 (NCBI Reference Sequence: NM_001359898.1) and CD86 (NCBI Reference Sequence: NM_019388.3) mRNA was produced in house by cloning the genes into T7 promotor containing pcDNA3.1(+) plasmids. Plasmids were linearized using DraIII or SmaI restriction enzyme (New England biolabs) and extracted using phenol:chloroform extraction with subsequent alcohol precipitation. mRNA was then transcribed from the linearized plasmids using INCOGNITO™ T7 ARCA 5mC- & Ψ-RNA Transcription Kit (Cellscript).
Tumor supernatant
Tumors were excised and treated with 1 ml RPMI 1640 medium (Gibco) supplemented with 1 mg ml−1 collagenase type IV from Clostridium histolyticum and 0.3 mg ml−1 DNase I from bovine pancreas (Sigma), at 37 °C for 30 min. Dissolved tumors were passed through a stainless-steel sieve (Sigma) and centrifuged 300 g for 7 min. The cell pellet was analyzed by flow cytometry. Cell-free supernatant was pressed through a 0.45μm syringe filter (PALL Corporation) and kept at −70 °C until analysis by Luminex technology.
Luminex – eBioscience/Affymetrix Magnetic bead Kits:
Cytokine levels in tumor supernatant and serum was assayed on the Luminex Platform. This assay was performed in the Human Immune Monitoring Center at Stanford University. Mouse 38-plex kits were purchased from eBiosciences/Affymetrix and used according to the manufacturer’s recommendations with modifications as described below. Briefly: Beads were added to a 96 well plate and washed in a Biotek ELx405 washer. Samples were added to the plate containing the mixed antibody-linked beads and incubated at room temperature for 1 hour followed by overnight incubation at 4°C with shaking. Cold and Room temperature incubation steps were performed on an orbital shaker at 500–600 rpm. Following the overnight incubation plates were washed in a Biotek ELx405 washer and then biotinylated detection antibody added for 75 minutes at room temperature with shaking. Plate was washed as above and streptavidin-PE was added. After incubation for 30 minutes at room temperature wash was performed as above and reading buffer was added to the wells. Each sample was measured in duplicate. Plates were read using a Luminex 200 instrument with a lower bound of 50 beads per sample per cytokine. Custom assay control beads by Radix Biosolutions were added to all wells.
Statistical analysis
Prism software (GraphPad; La Jolla, CA) was used to analyze tumor growth and to determine statistical significance of differences between groups by applying an non-parametric Mann-Whitney U test. P values <0.05 were considered significant. The Kaplan-Meier method was employed for survival analysis.
Results
CARTs efficiently deliver mRNA to cells in the tumor microenvironment
The tumor microenvironment, like other localized tissue spaces (organs, etc…) is a dynamic entity, with resident cells migrating to other areas in the body. It is therefore important to monitor the trafficking of cells that were treated within the microenvironment as they migrate elsewhere. However, tracking the migration of cells transfected in vivo with reporter genes such as mCherry and eGFP is impractical because expression levels are below the detection limits of flow-cytometric analysis. Therefore, to determine the specific localization, populations, and lineages of cells transfected upon intratumoral injection of CART-mRNAs, we used difluoroboron-beta-diketonate fluorophore functionalized CARTs (BDK-CART)(21,23). This fluorophore has a high fluorescence quantum yield (32) and was readily installed on every CART molecule by using it as an initiator for ring-opening polymerization, resulting in approximately 1000 fluorophores per mRNA molecule in the CART-mRNA NP complexes based on formulation stoichiometry. The BDK fluorophore shares excitation and emission properties with the well-known Pacific Blue fluorochrome and is easily detected by flow cytometry. To validate this approach, we injected an established subcutaneous tumor with BDK-CART/irrelevant mRNA nanoparticles. FACS analysis revealed that 37 percent of the A20 tumor cell population was transfected with BDK-CART NPs (Figure 1A). Moreover, Phenotypic identification revealed that CARTs successfully 10 percent of the infiltrating CD4 T cells, 7 percent of the infiltrating CD8 T cells, 18 percent of the infiltrating DCs, and 28 percent of the infiltrating macrophages (Figure 1A). These data indicate that CARTs effectively transfect multiple immunomodulatory cell subsets within the tumor, and thus provide a means for the in-situ delivery and expression of molecules encoded by the appropriate mRNA. The flexibility and generality of the mRNA delivery strategy enables direct modification the presence of effector molecules within the tumor microenvironment. In a proof of principle study, we generated mRNAs encoding six well described immunomodulators; CD70, OX40L, CD80, CD86, IL-12, and IFNγ. In vitro transfection of HeLa cells with each of the six individual CART-mRNA NPs revealed that all mRNAs were delivered and functionally expressed by the transfected cells (Figure 1B and data not shown). Moreover, in a proof of principle experiment we injected CD80, CD86 and OX40L mRNA-CARTs into tumors and assayed, by flow cytometry, the relative expression of these molecules on tumor infiltrating monocytes, T cells, and dendritic cells, compared to an un-treated contralateral tumor (Supplementary Figure S1). As expected, we only detected upregulated CD80, CD86, and OX40L on the protein level for a small proportion of the parent populations. This observation is reflected by the relative number of cells belonging to each subset we transfect (Figure 1A). However, we were able to detect expression in all of the three tested tumor infiltrating subsets (Supplementary Figure S1). These studies suggest that the levels of immunomodulatory molecules within the tumor microenvironment can be increased, which can in turn lead to a systemic adaptive immune response.
Figure 1. Highly efficient local delivery of immunomodulatory encoded mRNA mediates eradication of the treated tumor.
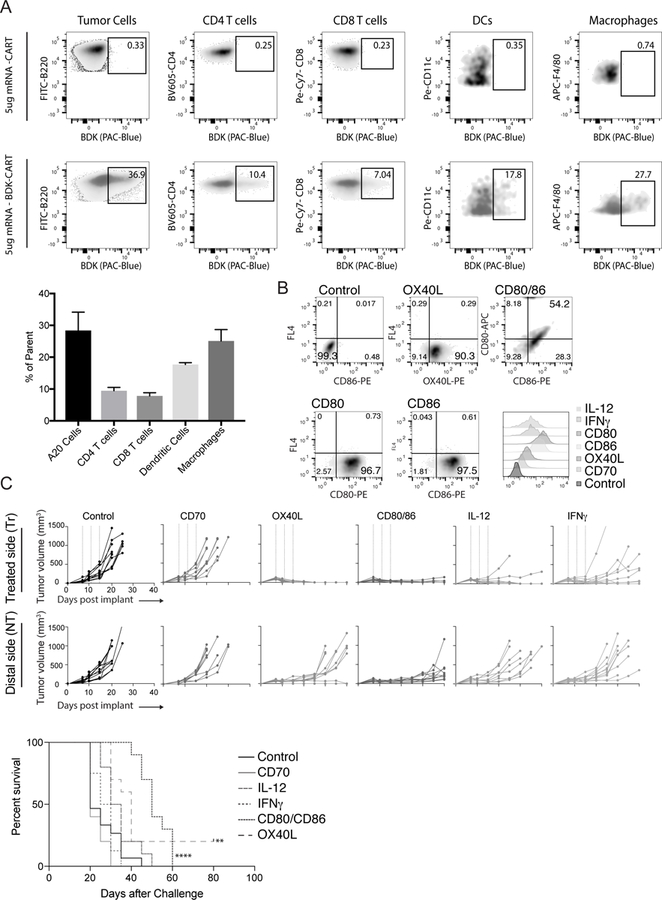
12 established A20 tumors were injected with either fluorophore (BDK) or non-fluorophore mRNA-CART. Phenotypic surface marker expression and BDK was used to identify transfected cells (A) Top: Established A20 tumors were injected with non-fluorophore mRNA-CART. Gates indicate background staining. Bottom: Established A20 tumors were injected with (BDK) mRNA-CART. Gates indicates percentage of parent population transfected with mRNA-CARTs. Bar graph: average percent transfected cells in the injected tumor. (B) Expression of immunomodulatory mRNA in HeLa cells 12 hrs after treatment with mRNA-CARTs. (C) Tumor growth and survival of Ctr., CD70, OX40L, CD80/86, IL-12, and IFNγ mRNA-CART treated animals. Mice with tumors >15mm in the largest diameter were euthanized. Survival of OX40L, and CD80 and CD86 mRNA-CART treated mice were compared to control mRNA-CART treated. Survival significance was calculated using Log Rank (Mantel-Cox) test. Ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001 ****, P < 0.0001. Data representative of 2–4 independent experiments.
These studies suggest that the levels of immunomodulatory molecules within the tumor microenvironment could be increased, which could in turn lead to a systemic adaptive immune response.
In situ mono-therapy with OX40L, CD80/86, or mRNA-CARTs clears treated tumor and slows the growth of distal tumors
Balb/c mice with established, subcutaneous A20 B cell lymphoma tumors were treated three times with local injections of CARTs loaded with 5ug mRNA encoding an irrelevant protein (secreted alkaline phosphatase; SEAP), CD70, OX40L, IL 12, IFNγ or 5ug of both CD80 and CD86. Growth of the treated and the distal tumor was monitored for up to 40 and 80 days after tumor inoculation, respectively. SEAP control mRNA was generated in the same way as all of our functional mRNAs. Thus, the observed difference between control treated and the other treatment groups should not be due to the inherent properties of the mRNAs, but rather the properties of the proteins they encode. Complete clearance of the treated tumor was observed in 100 percent of the OX40L treated mice. Moreover, CD80/86 and IL12 mRNA-CART treatment cured nearly all of the treated tumors, while control mRNA and CD70 mRNA had no effect and IFNγ mRNA-CART only showed a partial response (Figure 1C). While effects of single mRNAs (monotherapies) were most pronounced within the treated tumor, in two mice, OX-40L monotherapy stimulated an effective systemic response that cleared both tumors. Importantly, because the monotherapy triggered rapid involution of many of the treated tumors, repeated intratumoral injections became technically difficult because of the very low residual tumor volume. Regardless, significant growth delay of the distal, non-treated tumor was observed with CD80/86, OX40L, and IL12 mRNA-CART administration (Figure 1C). Monotherapy with CD80/86, OX40L, or IL12 mRNA-CART elicited strong local anti-tumor immunity, with delayed tumor growth at the distal site, however, to achieve more robust systemic immunization, more potent approaches appear to be required.
In situ therapy with a combination of OX40L and CD80/86 or OX40L and IL-12 mRNA-CARTs results in global anti-tumor immunity.
Next, we sought to optimize the induction/stimulation of antitumor immune responses by combining two or more mRNAs in the same CART complexes. Since monotherapy with OX-40L proved to be highly immunostimulatory (Figure 1C), we combined OX40L with CD80 and CD86, OX40L with IL-12, and OX40L with IFNγ. Mice bearing two A20 lymphoma tumors of comparable size were treated with mRNA-CART NPs formulated with 5ug mRNA per component (three treatments in four-day intervals). The combinations of OX40L with CD80 and CD86, and OX40L with IL-12 dramatically increased both survival and delayed the tumor growth in uncured animals. However, while the IFNγ /OX40L combination therapy induced a strong local therapeutic effect, it did not increase the survival, nor did it delay the tumor growth of the non-treated tumor compared to OX40L monotherapy (Figure 1C and Figure 2A). On the other hand, IL-12/OX40L and CD80/CD86/OX40L mRNA therapies induced complete tumor eradication of both tumors in 40 percent of the mice. Although IL-12/OX40L and CD80/CD86/OX40L mRNA therapies achieved similar overall responses, CD80/CD86/OX40L mRNA-CART therapy induced a significant delay in tumor growth. Moreover, CD80/CD86/OX40L mRNA-CART that had completely rejected the tumor were re-challenged with 107 A20 tumor cells and monitored for tumor development and overall survival. The re-challenged mice completely rejected the second challenge, while the previously unchallenged and untreated mice developed tumors indicating the induction of long-lasting tumor-specific immune responses in these mice (Supplemental Figure S2). To determine the general applicability of this approach, treatment was performed on established subcutaneous CT26 colon carcinoma tumors. Again, combining OX40L, CD80, and CD86 mRNA-CART, we were able to completely cure 40 percent of the mice, with a significant delay in tumor outgrowth in the rest of the mice (Figure 2B).
Figure 2. OX40L plus CD80 and CD86 mRNA-CART treatment evokes tumor-specific systemic anti-tumor immunity.
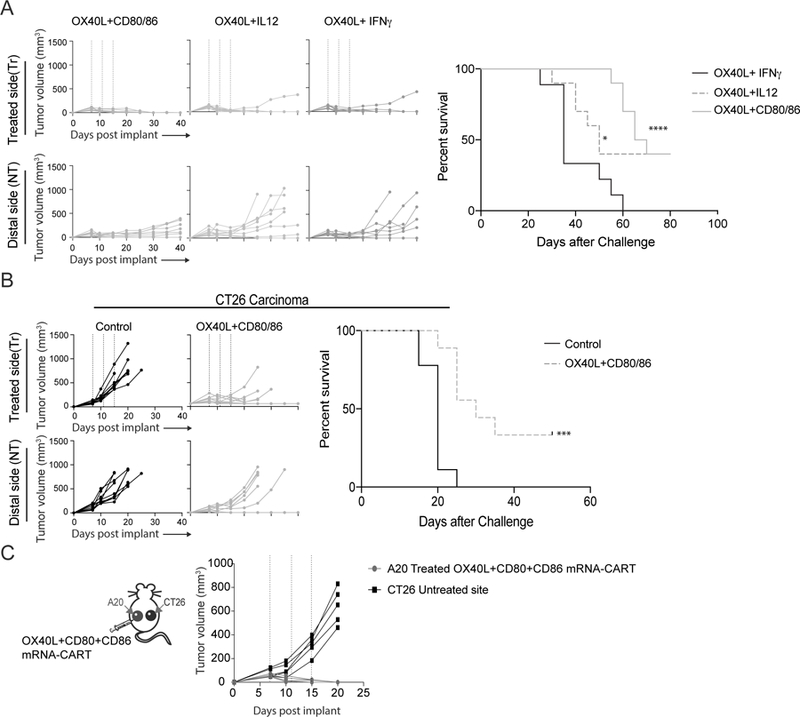
(A) Mice harboring two A20 tumors were treated intratumorally in one of the tumors with either OX40L plus CD80 and CD86 mRNA-CARTs, OX40L plus IL-12 mRNA-CARTs, or OX40L plus IFNγ mRNA-CARTs. Tumor growth of both tumors and overall survival was monitored for 40 and 80 days, respectively. (B) Mice harboring two CT26 colon carcinoma tumors were treated intratumorally in one of the tumors with either OX40L plus CD80 and CD86 mRNA-CARTs, or control mRNA-CARTs. Tumor growth of both tumors and overall survival was monitored for 40 and 50 days, respectively. (C) Mice harboring one A20 tumor and one CT26 tumor were treated intratumorally in the A20 tumor with OX40L plus CD80 and CD86 mRNA-CARTs. Tumor growth was measured over the course of 20 days. (A-C) Mice with tumors >15mm in largest diameter were euthanized. Survival of OX40L plus CD80 and CD86, and OX40L and IL-12 mRNA-CART treated mice were compared to OX40L and IFNγ mRNA-CART treated mice. Survival significance was calculated using Log Rank (Mantel-Cox) test. Ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001 ****, P < 0.0001. Data representative of 2 independent experiments.
In situ therapy with a combination of OX40L and CD80/86 incite tumor-specific responses restricted to related tumors.
It is feasible that some of the therapeutic distal effects observed upon local administration of mRNA-CARTs could arise from circulation/leakage of mRNA-CART complexes to the distal tumor or its draining lymph nodes. Furthermore, locally induced cytokines may enter circulation and non-specifically enhance effector functions at the distal tumor. To investigate the possibility of indirect, distal immunostimulatory effects, we designed a variation for the two-tumor model, in which the Balb/c mice were inoculated with the A20 B cell lymphoma tumor on one flank, and CT26 colon carcinoma-tumor on the contralateral flank. The established A20 tumor was treated intratumorally with the combination therapy using OX40L/CD80/CD86 mRNA-CART (three times, in four-day intervals). Growth of both tumors was monitored for 20 days post tumor inoculation (Figure 2C). In this experiment, we observed complete regression of the treated A20 tumor, with no effect on the growth of the contralateral CT26 tumor. This experiment clearly indicates that the anti-tumor immunity brought about by local treatment is specific to the phenotype of the injected tumor. Thus i.t. immunomodulation strategy does not seem to incite non-specific systemic immunostimulation/inflammation, but triggers a focused, local immune response which lead to systemic tumor-specific immunity.
In situ therapy with a combination of OX40L and CD80/86 promotes local activation of T cells and secretion of pro-inflammatory cytokines that can be traced to the local draining lymph node and the distal non-treated tumor.
To better understand the local and systemic effects of our treatment, we assayed the change in T cell activation, effector function and change in the tumor microenvironment-secreted cytokines. Balb/c mice bearing two A20 tumors on opposite flanks were injected with either 5ug OX40L, CD80 and CD86 mRNA-CART or 5ug control mRNA-CART on one flank. After 36 hours serum and tumor extracellular matrix was collected. In addition, single-cell suspensions from the treated tumor, non-treated tumor, and draining lymph nodes were prepared. Flow cytometry analysis revealed that for the treated groups, compared to non-treated controls, both the local and distal tumors, as well as the draining LNs showed activated CD4 T cells, CD8 T cells, and natural killer cells (NK cells). This included upregulation of activation markers such as CD137, CD69, CD357, and CD25; cytotoxicity markers such as perforin and granzyme B; and the proliferation marker Ki-67. Furthermore, compared to the control group, we observed a significant downregulation of the T regulatory T cell marker FOXP3 and the immunosuppressive marker CTLA-4 (CD152). The enhanced local immune response observed in the treated cohort clearly demonstrates a proinflammatory shift in the tumor microenvironment of both tumors as well as in the secondary lymphatic tissues (Figure 3 A-D). We observed significant, but various degrees of upregulation of activation markers on CD4 and CD8 T cells within the treated tumor (Figure 3A-B), the local draining LN (Figure 3D), and the distal tumor. The relative upregulation of activation markers at the distal site was more pronounced for CD8 T cells as compared to CD4 T cells. In general, both CD4 and CD8 T cells showed a more activated phenotype at the treated tumor compared to the distal tumor (Figure 3A-B). However, the cytotoxicity markers Perforin and Granzyme B were expressed at similar levels on CD4 and CD8 T cells in both tumors. Moreover, compared to control treated animals, CD49b positive NK cells were highly activated as assessed by Granzyme B expression in both the treated and the distal tumor in OX40L, CD80 and CD86 mRNA-CART treated animals. The phenotypical characterization of tumor-infiltrating lymphocytes indicated an enhanced immune response resulting from our treatment strategy. This argument is supported by the observed changes in locally secreted cytokines. Quantitative comparison of secreted cytokines within the tumor microenvironments of the control and treated animals (measured for both tumors and sera) reveals a strong induction of a local proinflammatory milieu in the group of treated mice (Figure 3E). Cytokine quantification from the treated and distal tumors shows an upregulation within both tumors, although at a higher magnitude within the treated tumor. On the other hand, serum levels of tested cytokines were similar between treated and controls. Thus, from a safety perspective, local treatment with OX40L, CD80 and CD86 mRNA- CARTs seems to effectuate a highly localized inflammatory response with no signs of systemic adverse effects. Hence, we propose that the observed systemic tumor-specific effects are mediated by migrating cells and not by bystander activation through elevated systemic cytokine concentrations. However, we did observe decreased systemic levels of the immunosuppressive cytokines TGFb and IL10 which may indicate a therapy induced conversion of the cells that secrete these cytokines. Importantly, when assayed 22 days after the last treatment, we did not observe any difference in serum levels of the canonical pro-inflammatory cytokines IFNα, IL6, TNFα, IFNγ, IL1α, or IL1β, between the treated group and aged match un-treated controls (Supplementary Figure S3). Thus, we do not observe any signs of delayed adverse effects with our treatment.
Figure 3. In situ therapy with a combination of OX40L and CD80/86 induces local activation of T cells and secretion of pro-inflammatory cytokines that can be traced to the local draining lymph node and the distal non-treated tumor.
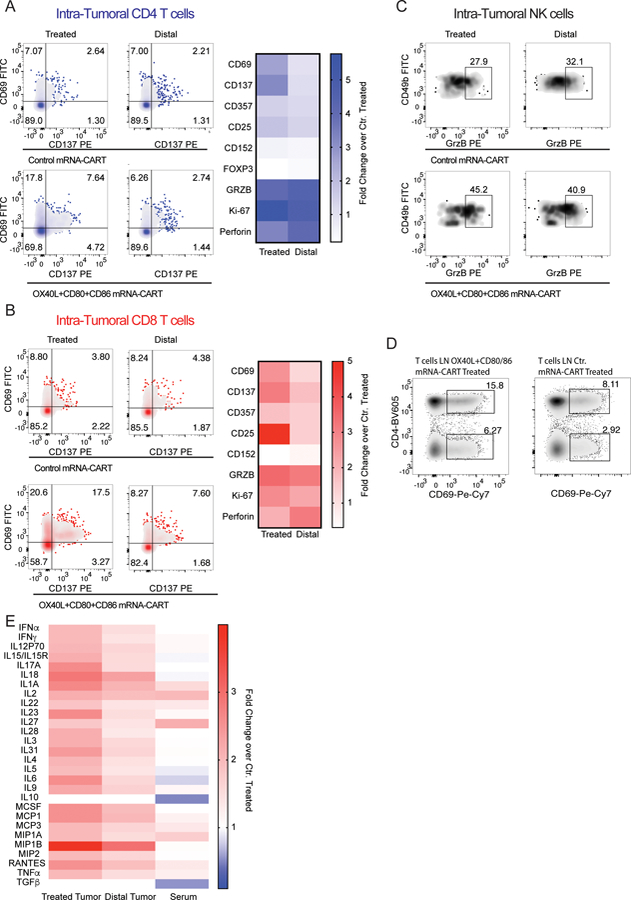
Balb/c mice bearing two A20 tumors on opposite flanks were injected with either 5ug OX40L, CD80 and CD86 mRNA-CART or 5ug control mRNA-CART on one flank. 36 hours later, serum, and tumor extracellular matrix was collected. In addition, single cell suspension of treated, non-treated, and draining lymph nodes were generated (A) Left: Representative plots of activation marker CD137 and CD69 expression by intratumoral CD4 T cells from treated and distal tumors of control mRNA-CART and OX40L, CD80 and CD86 mRNA-CART treated mice. Right: fold change in expression of activation and cytotoxic markers by intratumoral CD4 T cells in both treated and untreated tumors from OX40L, CD80 and CD86 mRNA-CART treated mice compared to tumors from control mRNA-CART treated mice. (B) Left: Representative plots of activation marker CD137 and CD69 expression by intratumoral CD4 T cells from treated and distal tumors of control mRNA-CART and OX40L, CD80 and CD86 mRNA-CART treated mice. Right: fold change in expression of activation and cytotoxic markers by intratumoral CD8 T cells in both treated and untreated tumors from OX40L, CD80 and CD86 mRNA-CART treated mice compared to tumors from control mRNA-CART treated mice. (C) Representative plots of cytotoxicity marker granzyme B on intratumoral CD49b positive Natural Killer cells from treated and distal tumors of control mRNA-CART and OX40L, CD80 and CD86 mRNA-CART treated mice. (D) Representative plots of activation marker CD69 expression by tumor draining lymph node CD4 T cells from treated and distal tumors of control mRNA-CART and OX40L, CD80 and CD86 mRNA-CART treated mice. (E) Fold change in secreted cytokine protein levels in the local tumor microenvironment of both treated and untreated tumors, and serum from OX40L, CD80 and CD86 mRNA-CART treated mice, compared to tumors and serum from control mRNA-CART treated mice. Data representative of 2 independent experiments.
CART delivery is extremely local solely transfecting cells in the injected tumor
To investigate the biodistribution of CARTs upon local IT injection and to determine if expression of immunostimulatory proteins could occur at distal sites, we performed experiments using firefly luciferase (Fluc)- encoding mRNA and fluorophore-labelled CARTs (BDK-CARTs). This was designed to provide direct insight into cellular transfection as opposed to the phenotypic readout from the A20/CT26 two tumor model. In this experiment, mice bearing two A20 tumors were injected with 5ug Fluc mRNA-BDK-CARTs and monitored by bioluminescence imaging for expression of Fluc at 12hrs intervals up to 96 hrs. At all timepoints, no reporter gene expression was detectable outside of the tumor (Figure 4A). At 48hrs, a group of mice were sacrificed, and single-cell suspension of injected tumor, draining LN, spleen, and distal tumor was generated. A group of mice injected with non-fluorophore labelled CARTs were used as a negative control to normalize for the BDK signal. At the 48 hr time-point, cells were assayed by flow cytometry to detect BDK positive cells. Surprisingly, no signal was detected in cells from the distant tumor or any of the other organs.
Figure 4. OX40L and CD80/86 mRNA-CARTs mobilize cells to migrate to the local draining lymph node and distal tumor.
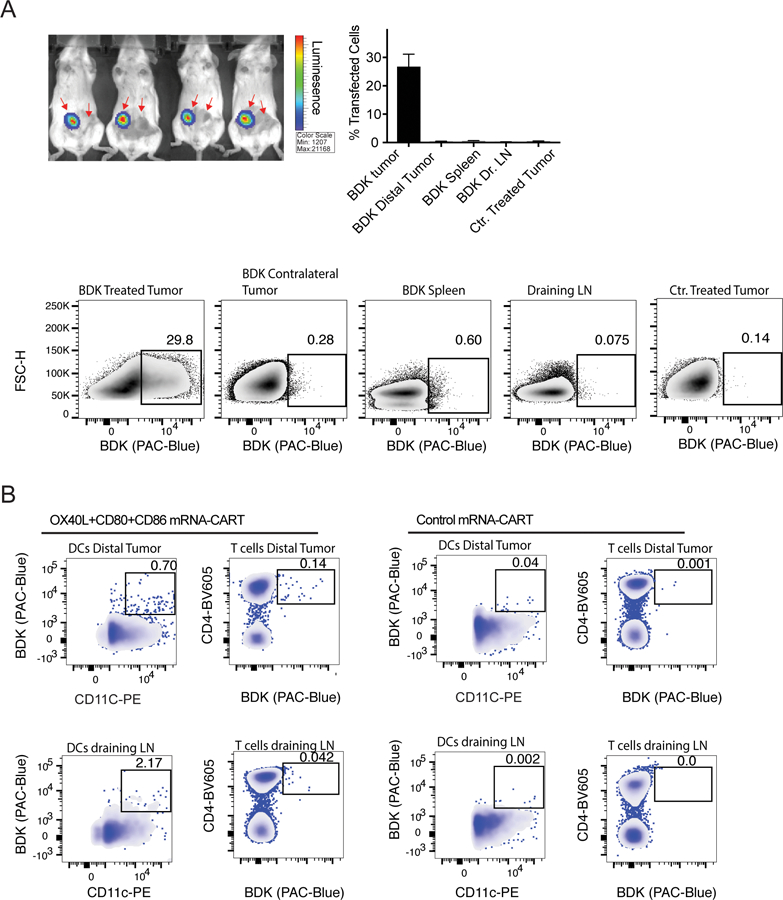
(A) Mice harboring two A20 tumors were injected with firefly luciferase (Fluc) mRNA-BDK-CART into one tumor. Top left: Whole body bioluminescence 24hrs after injection. Top right: frequency of transfected cells in injected tumor, distal tumor, draining LNs, and spleen from Fluc mRNA-BDK-CART injected mice 24hrs after injection. Tumor from Fluc mRNA-CART without the BDK fluorophore was included as a negative ctr. Bottom: representative density plots of data presented in top right histogram. (B) Mice harboring two A20 tumors were injected with OX40L, CD80 and CD86 mRNA-BDK-CART or Control mRNA-BDK-CART into one tumor. Data shows representative density plot of BDK positive Dendritic cells (DCs) and CD4 T cells in the distal tumor and the injected tumor draining lymph node 48hrs after treatment. Gates indicate locally transfected cells that have migrated either to the local draining lymph node or the distal tumor. Mice injected with control mRNA-BDK-CARTs were included as ctr. Data representative of 3 independent experiments.
OX40L and CD80/86 mRNA-CARTs mobilize cells to migrate to the local draining lymph node and distal tumor
To further study the mechanistic basis of the observed systemic anti-tumor immunity after local mRNA-CART treatment, we assayed distribution/localization of locally transfected cells after treatment with 5ug OX40L, CD80 and CD86 mRNA-BDK-CARTs. Interestingly, we were able to detect BDK-positive dendritic cells and T cells in both the local draining lymph nodes and the distal tumors 48hrs after treatment (Figure 4B). This observation indicates that using mRNA delivery to increase expression of co-stimulatory molecules in the local tumor microenvironment may mobilize a small fraction of cells to enter the circulation and exert their effector functions throughout the body. Notably, the observed migration of cells induced by immunostimulatory proteins encoded mRNA-CARTs as opposed to the absence of migration when we use control mRNA or reporter gene (fLuc) encoded mRNA-CARTs, indicate that changing the local tumor microenvironment may enable cells to become activated and migrate.
Discussion
With increasing understanding of immunomodulatory networks operational within tumors, new and improved immunotherapeutic approaches are being implemented in cancer care. With these advances, more precise tools are required to facilitate therapeutic utilization of new discoveries. Thus, there is a high demand for versatile therapeutic platforms that can combine multiple drugs or targeted effects, while minimizing toxicities. Local mRNA delivery using non-toxic delivery vehicles meets these requirements; mRNA can be coded to produce a vast array of proteins, that can be effectively delivered and expressed alone or in a combinatorial fashion using the readily tunable CART delivery platform. Thus, mRNA provides the technological basis to deliver a wide variety of antigens, modulators and cell-signaling factors in a single molecule.
Local injections may overcome some of the potential toxicity associated with systemic delivery, but few strategies ensure a truly local delivery and deposition of the therapeutic substance. Although injected locally, delivery vehicles such as LNPs tend to accumulate in distal organs including the liver (33), which may cause unwanted side effects. Among the most problematic is the potential toxicity of LNP components, including cationic lipids, phospholipids or combinations thereof (20,34). In contrast to the LNP delivery vehicles, CARTs degrade to neutral by-products (22) and are uniquely specific to the injected tissue with no detectable leakage. This work details immunological trafficking pathways initiated by local/in situ treatment, but which remarkably lead to a robust, global, therapeutic response. This is the first report of a nanoparticle mediated mRNA-based immunostimulatory intervention against the tumor microenvironment. Thus, CARTs represent a novel and truly unique means of local gene-therapy. Our data shows that highly efficient intratumoral delivery of immunomodulatory particles encoding mRNA is a fast, safe, versatile, robust, and flexible strategy to induce specific anti-tumor responses against disseminated tumors in a variety of solid cancer types.
Furthermore, the CART platform can be modified with fluorophores that enable monitoring the migration of cells throughout complex immunomodulatory pathways. This study demonstrates the therapeutic potential and potency of in situ gene therapy using mRNA encoding well known immunomodulatory molecules. OX40 is mainly expressed by CD4 T cells, and recent publications have highlighted OX40 ligation as a potent means of inhibiting CD4 Treg function and enhancing effector cell functions of conventional CD4 T cells(24,25). The classical co-stimulatory molecules CD80 and CD86 interact with CD28 molecules on T cells to ensure potent activation (35–37). Several reports have demonstrated the necessity for CD80 and CD86 expression on professional antigen presenting cells (APCs) for T cell activation and propagation (36–40). Interleukin-12 (IL-12) is a heterodimeric pro-inflammatory cytokine that induces the production of interferon-γ (IFN-γ), which induces the differentiation of T helper 1 (Th1) cells, and forms a link between the innate and adaptive immune responses (28–30). Recombinant IL-12 has shown remarkable properties as an anti-tumor agent in preclinical models (41,42). However, in addition to the toxic side effects of high dose IL-12 (43), a caveat to the systemic administration of immunostimulatory mediators such as IL-12 the inability to reach high enough local concentrations within tumors to impart a meaningful effect on resident immune cells. Notwithstanding these complications, the impressive preclinical results with Il-12 gene therapy merits new approaches whereby cytokine production would only occur in close proximity to the tumor, thereby maintaining low serum IL-12 concentrations and reducing systemic toxicity (44). We find that the intratumoral induction/production of OX40L with either IL-12 or CD80 and CD86 is sufficient to overcome the local immunosuppressive environment of a tumor and surpass the threshold for effective systemic anti-tumor immunity.
Importantly, the immunomodulatory molecules chosen in this study represents only a small fraction of the molecules available. Thus, combining our mRNA delivery platform and the array of immunomodulatory molecules available, including the potential for multiplexed gene delivery, represents a robust strategy to therapeutically reverse intratumoral immunosuppression and elicit curative anti-tumor immune responses. We believe that this strategy is a general approach for personalized therapy against a wide variety of tumors either as a stand-alone therapy or in combination with existing treatments.
Supplementary Material
Significance Statement.
The mRNA-CART system is a highly effective delivery platform for delivering immunostimulatory genes into the tumor microenvironment for potential therapeutic development.
Acknowledgment
We are grateful for the technical help, guidance, and flow cytometry assistance provided by Debra Czerwinski, and the synthetic work by Kayvon Pedram.
Funding
This work was supported by: NIH R35CA197353 (to R.L.) National Science Foundation Grant NSF CHE-1607092 (to R.M.W.), NIH Grants NSF CHE-848280 and NIH-CA031845 (to P.A.W.) and the Child Health Research Institute at Stanford University and the SPARK Translational Research Program in the Stanford University School of Medicine (R.M.W, P.A.W, R.L). Support through the Norwegian Cancer Society (NCS) and the Norwegian Health Authorities (HSØ) is further acknowledged (O.A.W.H. and A.T.). Support through the Stanford Cancer Translational Nanotechnology Training T32 Training Grant T32 CA196585 funded by the National Cancer Institute (T.R.B) and the Stanford Center for Molecular Analysis and Design (CMAD) is further acknowledged (C.J.M.). Flow cytometry data was collected on an instrument in the Stanford Shared FACS Facility obtained using NIH S10 Shared Instrument Grant S10RR027431–01. Luminex cytokine data was collected by the Stanford Human Immune Monitoring Center.
Footnotes
Conflict of interest: The authors declare that there is no conflict of interest regarding the publication of this article.
References
- 1.Stanton MG. Current Status of Messenger RNA Delivery Systems. Nucleic acid therapeutics 2018;28:158–65 [DOI] [PubMed] [Google Scholar]
- 2.Guan S, Rosenecker J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene therapy 2017;24:133–43 [DOI] [PubMed] [Google Scholar]
- 3.Sahin U, Kariko K, Tureci O. mRNA-based therapeutics--developing a new class of drugs. Nat Rev Drug Discov 2014;13:759–80 [DOI] [PubMed] [Google Scholar]
- 4.Hackett PB, Largaespada DA, Switzer KC, Cooper LJ. Evaluating risks of insertional mutagenesis by DNA transposons in gene therapy. Translational research : the journal of laboratory and clinical medicine 2013;161:265–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pisal DS, Kosloski MP, Balu-Iyer SV. Delivery of therapeutic proteins. Journal of pharmaceutical sciences 2010;99:2557–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramaswamy S, Tonnu N, Tachikawa K, Limphong P, Vega JB, Karmali PP, et al. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc Natl Acad Sci U S A 2017;114:E1941–E50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramamoorth M, Narvekar A. Non viral vectors in gene therapy- an overview. J Clin Diagn Res 2015;9:GE01–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kariko K, Muramatsu H, Ludwig J, Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic acids research 2011;39:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. Journal of virology 2012;86:2900–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li B, Zhang X, Dong Y. Nanoscale platforms for messenger RNA delivery. Wiley interdisciplinary reviews Nanomedicine and nanobiotechnology 2018:e1530. [DOI] [PMC free article] [PubMed]
- 11.Van Lint S, Renmans D, Broos K, Goethals L, Maenhout S, Benteyn D, et al. Intratumoral Delivery of TriMix mRNA Results in T-cell Activation by Cross-Presenting Dendritic Cells. Cancer immunology research 2016;4:146–56 [DOI] [PubMed] [Google Scholar]
- 12.Aznar MA, Tinari N, Rullan AJ, Sanchez-Paulete AR, Rodriguez-Ruiz ME, Melero I. Intratumoral Delivery of Immunotherapy-Act Locally, Think Globally. Journal of immunology 2017;198:31–9 [DOI] [PubMed] [Google Scholar]
- 13.McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DC, et al. Survival, Durable Response, and Long-Term Safety in Patients With Previously Treated Advanced Renal Cell Carcinoma Receiving Nivolumab. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33:2013–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33:2004–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez-Rodriguez E, Rodriguez-Abreu D, Spanish Group for Cancer I-B. Immune Checkpoint Inhibitors: Review and Management of Endocrine Adverse Events. The oncologist 2016;21:804–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sgambato A, Casaluce F, Sacco PC, Palazzolo G, Maione P, Rossi A, et al. Anti PD-1 and PDL-1 Immunotherapy in the Treatment of Advanced Non- Small Cell Lung Cancer (NSCLC): A Review on Toxicity Profile and its Management. Current drug safety 2016;11:62–8 [DOI] [PubMed] [Google Scholar]
- 17.Bahl K, Senn JJ, Yuzhakov O, Bulychev A, Brito LA, Hassett KJ, et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol Ther 2017;25:1316–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carstens MG, Camps MG, Henriksen-Lacey M, Franken K, Ottenhoff TH, Perrie Y, et al. Effect of vesicle size on tissue localization and immunogenicity of liposomal DNA vaccines. Vaccine 2011;29:4761–70 [DOI] [PubMed] [Google Scholar]
- 19.Judge A, McClintock K, Phelps JR, Maclachlan I. Hypersensitivity and loss of disease site targeting caused by antibody responses to PEGylated liposomes. Mol Ther 2006;13:328–37 [DOI] [PubMed] [Google Scholar]
- 20.Reichmuth AM, Oberli MA, Jaklenec A, Langer R, Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Therapeutic delivery 2016;7:319–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKinlay CJ, Benner NL, Haabeth OA, Waymouth RM, Wender PA. Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc Natl Acad Sci U S A 2018 [DOI] [PMC free article] [PubMed]
- 22.McKinlay CJ, Vargas JR, Blake TR, Hardy JW, Kanada M, Contag CH, et al. Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc Natl Acad Sci U S A 2017;114:E448–E56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benner NL, Near KE, Bachmann MH, Contag CH, Waymouth RM, Wender PA. Functional DNA Delivery Enabled by Lipid-Modified Charge-Altering Releasable Transporters (CARTs). Biomacromolecules 2018 [DOI] [PMC free article] [PubMed]
- 24.Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, et al. Eradication of spontaneous malignancy by local immunotherapy. Science translational medicine 2018;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. The Journal of clinical investigation 2013;123:2447–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smythe JA, Fink PD, Logan GJ, Lees J, Rowe PB, Alexander IE. Human fibroblasts transduced with CD80 or CD86 efficiently trans-costimulate CD4+ and CD8+ T lymphocytes in HLA-restricted reactions: implications for immune augmentation cancer therapy and autoimmunity. Journal of immunology 1999;163:3239–49 [PubMed] [Google Scholar]
- 27.Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science 1993;259:368–70 [DOI] [PubMed] [Google Scholar]
- 28.Burkart C, Mukhopadhyay A, Shirley SA, Connolly RJ, Wright JH, Bahrami A, et al. Improving therapeutic efficacy of IL-12 intratumoral gene electrotransfer through novel plasmid design and modified parameters. Gene therapy 2018;25:93–103 [DOI] [PubMed] [Google Scholar]
- 29.Trinchieri G, Rengaraju M, D’Andrea A, Valiante NM, Kubin M, Aste M, et al. Producer cells of interleukin-12. Immunology today 1993;14:237–8 [DOI] [PubMed] [Google Scholar]
- 30.Medzhitov R Toll-like receptors and innate immunity. Nature reviews Immunology 2001;1:135–45 [DOI] [PubMed] [Google Scholar]
- 31.Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 2016;16:131–44 [DOI] [PubMed] [Google Scholar]
- 32.Zhang G, Chen J, Payne SJ, Kooi SE, Demas JN, Fraser CL. Multi-emissive difluoroboron dibenzoylmethane polylactide exhibiting intense fluorescence and oxygen-sensitive room-temperature phosphorescence. J Am Chem Soc 2007;129:8942–3 [DOI] [PubMed] [Google Scholar]
- 33.Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release 2015;217:345–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solmesky LJ, Shuman M, Goldsmith M, Weil M, Peer D. Assessing cellular toxicities in fibroblasts upon exposure to lipid-based nanoparticles: a high content analysis approach. Nanotechnology 2011;22:494016. [DOI] [PubMed] [Google Scholar]
- 35.Freeman GJ, Freedman AS, Segil JM, Lee G, Whitman JF, Nadler LM. B7, a new member of the Ig superfamily with unique expression on activated and neoplastic B cells. Journal of immunology 1989;143:2714–22 [PubMed] [Google Scholar]
- 36.Freeman GJ, Gray GS, Gimmi CD, Lombard DB, Zhou LJ, White M, et al. Structure, expression, and T cell costimulatory activity of the murine homologue of the human B lymphocyte activation antigen B7. The Journal of experimental medicine 1991;174:625–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gimmi CD, Freeman GJ, Gribben JG, Sugita K, Freedman AS, Morimoto C, et al. B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc Natl Acad Sci U S A 1991;88:6575–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hathcock KS, Laszlo G, Dickler HB, Bradshaw J, Linsley P, Hodes RJ. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science 1993;262:905–7 [DOI] [PubMed] [Google Scholar]
- 39.Freeman GJ, Borriello F, Hodes RJ, Reiser H, Gribben JG, Ng JW, et al. Murine B7–2, an alternative CTLA4 counter-receptor that costimulates T cell proliferation and interleukin 2 production. The Journal of experimental medicine 1993;178:2185–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VA Jr., Lombard LA, et al. Cloning of B7–2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science 1993;262:909–11 [DOI] [PubMed] [Google Scholar]
- 41.Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, et al. New insights into IL-12-mediated tumor suppression. Cell death and differentiation 2015;22:237–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiss JM, Subleski JJ, Wigginton JM, Wiltrout RH. Immunotherapy of cancer by IL-12-based cytokine combinations. Expert opinion on biological therapy 2007;7:1705–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarmiento UM, Riley JH, Knaack PA, Lipman JM, Becker JM, Gately MK, et al. Biologic effects of recombinant human interleukin-12 in squirrel monkeys (Sciureus saimiri). Laboratory investigation; a journal of technical methods and pathology 1994;71:862–73 [PubMed] [Google Scholar]
- 44.Mendiratta SK, Quezada A, Matar M, Wang J, Hebel HL, Long S, et al. Intratumoral delivery of IL-12 gene by polyvinyl polymeric vector system to murine renal and colon carcinoma results in potent antitumor immunity. Gene therapy 1999;6:833–9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.