

Abstract
Purpose:
Myelofibrosis (MF) is a hematopoietic stem cell neoplasm characterized by bone marrow reticulin fibrosis, extramedullary hematopoiesis, and frequent transformation to acute myeloid leukemia. Constitutive activation of JAK/STAT signaling through mutations in JAK2, CALR, or MPL is central to MF pathogenesis. JAK inhibitors such as ruxolitinib reduce symptoms and improve quality of life, but are not curative and do not prevent leukemic transformation, defining a need to identify better therapeutic targets in MF.
Experimental Design:
An shRNA library screen was performed on JAK2V617F-mutant HEL cells. Nuclear-cytoplasmic transport (NCT) genes including RAN and RANBP2 were among top candidates. JAK2V617F-mutant cell lines, human primary MF CD34+ cells, and a retroviral JAK2V617F - driven MPN mouse model were used to determine the effects of inhibiting NCT with selective inhibitors of nuclear export (SINE) compounds KPT-330 (selinexor) or KPT-8602 (eltanexor).
Results:
JAK2V617F-mutant HEL, SET-2, and HEL cells resistant to JAK inhibition are exquisitely sensitive to RAN knockdown or pharmacologic inhibition by KPT-330 or KPT-8602. Inhibition of NCT selectively decreased viable cells and colony formation by MF compared to cord blood CD34+ cells and enhanced ruxolitinib-mediated growth inhibition and apoptosis, both in newly diagnosed and ruxolitinib exposed MF cells. Inhibition of NCT in MF CD34+ cells led to nuclear accumulation of p53. KPT-330 in combination with ruxolitinib normalized white blood cells, hematocrit, spleen size and architecture, and selectively reduced JAK2V617F mutant cells in vivo.
Conclusions:
Our data implicate NCT as a potential therapeutic target in MF and provide a rationale for clinical evaluation in ruxolitinib exposed MF patients.
Keywords: myeloproliferative neoplasms, nuclear export, SINE, RAN, CRM1, XPO1
INTRODUCTION
Myelofibrosis (MF) has the most profound impact on quality of life and survival among the classic BCR-ABL negative myeloproliferative neoplasms (MPNs) (1). MF can present de novo (primary MF) or as secondary arising from polycythemia vera (post-PV MF) or essential thrombocythemia (post-ET MF). Cytopenias, thromboembolic complications, and transformation to acute myeloid leukemia (AML) cause excess mortality compared to age-matched controls, as well as patients with PV or ET (2,3). Morbidity is profound due to debilitating constitutional symptoms such as fatigue, anorexia, night sweats and weight loss (4). Constitutive activation of JAK/STAT signaling through mutations in JAK2 (JAK2V617F, 50–60%), calreticulin (CALR, 20–30%) or MPL (MPLW515L and others, 5–7%) is characteristic of MF (5–10). Most patients have additional mutations, commonly involving genes associated with epigenetic regulation, such as ASXL1, EZH2, TET2, IDH1/2, and DNMT3A (11–15). ASXL1 mutations are associated with inferior overall survival, while patients with CALR mutations exhibit a more indolent clinical course (16).
For many years, MF treatment was limited to cytotoxic chemotherapy to control myeloproliferation, and supportive care, e.g. cytokines, to improve cytopenias. Immunomodulatory drugs such as thalidomide in combination with prednisone were used with modest success (17). The discovery of JAK2V617F in MF led to the clinical development of the JAK1/2 inhibitor ruxolitinib. In two phase 3 studies in intermediate-2 and high risk MF patients, ruxolitinib was superior to placebo or best available therapy, providing the basis for regulatory approval in 2012 (18,19). Moreover, recent updates reported a trend towards improved overall survival, although the studies’ crossover design precludes a definitive conclusion (20,21). While ruxolitinib represents a major advance in MF management, treatment failure is common and progression to AML still occurs (20,21). Additional patients are ineligible for ruxolitinib due to thrombocytopenia, or require dose reductions due to myelosuppression that compromise efficacy. Except in rare cases, ruxolitinib does not significantly reduce the JAK2V617F mutant allele burden, suggesting limited disease modifying potential (22). None of the molecular abnormalities identified in addition to JAK/STAT activating mutations have led to significant therapeutic advances, reflecting the genetic complexity of MF and the fact that many MF mutations are loss-of-function. Allogeneic stem cell transplant remains the only potentially curative therapy, but transplant-related morbidity and mortality are considerable and many patients are ineligible due to age or co-morbidities (23).
To identify new targets in MF, irrespective of somatic mutation status, we performed a short hairpin RNA (shRNA) library screen on the JAK2V617F-mutant HEL human leukemia cell line as a model of JAK/STAT-driven myeloid neoplasia (24). The results of the screen and validation experiments using cell lines, primary patient samples and a mouse model implicate nuclear-cytoplasmic transport (NCT) as a major vulnerability in MF cells that can be targeted with selective inhibitors of nuclear export (SINE) compounds, suggesting a new therapeutic approach for MF, both for newly-diagnosed and ruxolitinib exposed MF patients.
METHODS
Cell lines.
We used human leukemia cell lines HEL (homozygous for JAK2V617F), SET-2 (heterozygous for JAK2V617F), TF1, UT7, and HEL-R (HEL resistant to JAK inhibition, described below). HEL, SET-2, TF1, and UT7 cells were obtained from DSMZ (Braunschweig, Germany), cultured in RPMI medium supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO), 2 mM L-glutamine, 100 units/mL penicillin, and 100 µg/mL of streptomycin. TF1 and UT7 cells were cultured in 10 ng/mL GM-CSF. Cell lines were authenticated (GenePrint 24 kit, Promega, Madison, WI), and compared to the DSMZ Online STR Analysis database. Cells were screened for mycoplasma with the MycoAlert detection kit (Basel, Switzerland) and determined to be negative. HEL cells resistant to JAK inhibition (HEL-R) were generated by long-term culture in increasing concentrations of momelotinib (CYT387), starting at 0.05 µM, with increases at 2-week intervals up to 7 µM (>4-fold above the reported IC50) (25). For the present study, HEL-R cells were maintained in 5 µM ruxolitinib. See Supplemental Methods.
Primary samples.
Blood or bone marrow was subjected to Ficoll separation followed by red blood cell lysis. An AutoMACS Pro Separator (Miltenyi Biotech, Bergisch Gladbach, Germany) was employed to purify CD34+ cells from MF patient samples and cord blood (CB) (St. Louis Cord Blood Bank, SSM Health Cardinal Glennon Children’s Hospital). Primary cells were cultured in RPMI medium supplemented with 10% FBS and cytokines (CC100: SCF, FLT3L, IL-3, IL-6; StemCell Technologies, Vancouver, Canada). We typically culture cells for 24 hours prior to experiments to minimize the influence of drugs present in the plasma. “Ruxolitinib exposed” refers to the status of the patient from which the sample was collected, and is defined by the patient’s physician, with adherence to published guidelines for diagnosis and disease monitoring (26–29). Except where noted as “responsive”, this term includes primary and secondary occurrences of relapse, refractory, and resistant disease, collectively known as failure. Written informed consent was obtained from all donors (The University of Utah IRB #45880). Patient information is summarized in Supplemental Table 1.
shRNA library screen.
Cellecta, Inc. (Mountain View, CA) provided Human Module 1 (HM1) lentiviral shRNA library, containing 27,239 non-control shRNAs targeting 5,034 genes involved in cell signaling, with 5–6 shRNAs per gene. Each shRNA is identifiable by a unique 18-base pair barcode. HEL cells were subjected to a library screen in three independent experiments using previously described methods (24). Briefly, HEL cells were infected with packaged library to yield a transduction rate of ~30% (monitored by flow cytometry for red fluorescent protein 72 hours post infection), equivalent to multiplicity of infection (MOI) of ≤1. Transduced cells were then selected with puromycin (2 µg/ml) for 9 days, as per Cellecta’s protocol. Medium changes and volume expansions were done as necessary to maintain exponential growth. After selection, DNA was extracted, and individual shRNA barcodes were amplified as recommended by Cellecta (Pooled Barcoded Lentiviral shRNA library v5, http://www.cellecta.com/resources/protocols) with ≤14 cycles for each step to minimize biased barcode amplification. Amplicon sizes were confirmed on a 3% agarose gel. PCR products were purified (PCR Clean-Up Kit, Qiagen, Valencia, CA), and subjected to next generation sequencing (NGS) (Illumina HiSeq 2500, rapid mode).
Bioinformatic analysis of the shRNA library.
We previously described the algorithm used for bioinformatics analysis (24). Fastq files were de-convoluted by Cellecta, providing the frequency that reads matched a barcode from the HM1 library for each sample, as well as gene mapping. A median depth of 237–495 reads across the samples was obtained for identified barcodes. Barcode frequencies for each sample were normalized to a total of 20 million reads. The fold-change for each shRNA was calculated as the ratio of observed reads to the Cellecta-provided HiSeq2000 plasmid counts after adjusting numerator and denominator by the addition of 10 to reduce potentially inflated estimates for low counts. Zero-read shRNAs and shRNAs present in only 1/3 replicates were excluded, and the median fold-depletion of those remaining was analyzed. Candidate genes were selected based on a 10-fold depletion in ≥3 shRNAs (Supplemental Table 2). All shRNA plasmids were made by Cellecta (pRSIT12-U6Tet-(sh)-CMV-TetRep-2A-TagRFP-2A-Puro).
Inhibitors.
Karyopharm Therapeutics (Newton, MA) provided KPT-330 (selinexor) and KPT-8602 (eltanexor), CRM1 inhibitors known as selective inhibitors of nuclear export (SINE) compounds (30,31). Ruxolitinib was purchased from Chemietek (Indianapolis, IN). For in vitro analysis, inhibitors were dissolved in DMSO at 10 mM and stored at −20 ºC. For in vivo use, see Supplemental Methods.
Viable cell assay (MTS).
Cells were seeded in triplicate in 96-well plates (cell lines, 5×103 cells/well; primary CD34+ cells, 1–2×104 cells/well) without or with 100 ng/mL Doxycycline (Dox) or indicated inhibitors. Viable cells was measured using CellTiter 96 AQueous One Solution MTS Reagent (Promega) on an Epoch microplate spectrophotometer (BioTek Instruments, Winooski, VT).
Apoptosis assay.
Apoptosis was assayed with allophycocyanin-conjugated annexin V in combination with 7-aminoactinomycin D (BD Biosciences, San Jose, CA) or DAPI (Sigma-Aldrich, St. Louis, MO) on a Guava HT6 (Millipore, Billerica, MA) or a BD FACSCanto flow cytometer.
PCR and shRNA protocols and sequences.
See Supplemental Methods, Supplemental Tables 3-4.
Nucleocytoplasmic fractionation and immunoblot analysis.
Nucleocytoplasmic fractionation was performed as described (24). For antibody information, see Supplemental Table 5.
Colony formation assay.
HEL or SET-2 shRAN cells (200 cells/dish) were plated in duplicate in MethoCult H4230 (StemCell Technologies) without or with 100 ng/mL Dox. Colonies were scored 7 days after plating. Primary MF or CB CD34+ cells (1×103 cells/dish) were seeded in duplicate in MethoCult H4230 with CC100 and indicated inhibitors. Colonies were counted after 10–15 days (Supplemental Table 6).
Bone marrow transduction/transplant and drug delivery.
MF-like disease was induced in Balb/c mice as described (25,32,33). Briefly, Balb/CJ mice were purchased from Jackson Laboratory (Bar Harbor, ME). Donors (6–8 weeks old) were primed with 5-fluorouracil (100 mg/kg, Fresenius Kabi, Warrendale, PA) by IV injection. Recipients were 7–9 weeks old at the time of irradiation and transplant. Complete blood counts were acquired on a Heska HemaTrue instrument (Loveland, CO) and GFP was analyzed on a Guava HT6 flow cytometer. All drugs and vehicles were administered by oral gavage. Details of the dosing schedule are shown in Supplemental Tables 7-8. Bone marrow fibrosis was scored as described (33,34). Bone marrow cellularity was estimated by a hematopathologist on an H&E-stained femur marrow cavity. Cellularity was variable within specimens, so an overall average was provided based multiple medium-power fields encompassing the whole specimen. The myeloid:erythroid (M:E) ratio was estimated on Periodic acid–Schiff-stained femur marrow cavity. A minimum of five high-power fields were evaluated within the more cellular areas to produce an overall average estimate rounded to the nearest integer.
Mouse procedures were performed according to guidelines approved by the Institutional Animal Care and Use Committee at The University of Utah.
Statistics and IC50 calculations.
Results are provided as mean ± SEM. Data were analyzed by one-way ANOVA or a two-tailed Student’s t test using Prism 7.02 (GraphPad, La Jolla, CA). To compare single agent to combination treatment, we performed one way ANOVA (uncorrected Dunn’s test, Kruskal-Wallis test). P < 0.05 was considered to be statistically significant. For HEL and SET-2 cells, a 4-parameter variable-slope regression analysis was used to calculate 50% inhibition concentration (IC50) values; for HEL-R cells, where 50% inhibition was not reached, IC50 values were determined by variable-slope regression analysis.
Synergy analysis used the “response surface” approach and formula [5] of Greco, Bravo and Parsons (35) for two-drug interaction. We set parameters m2 = m1 = m in this equation and solved for E to obtain the following equation:
D1 and D2 are concentrations of Drugs 1 and 2 (Ruxolitinib and KPT-330, or Ruxolitinib and KPT-8602).
E = effect; Ec = Effect of control
The parameters to be estimated are:
IC50,1 and IC50,2, defined as concentrations of Drugs 1 and 2 resulting in 50% inhibition.
m, a shape parameter. (Note again that there could be separate shape parameters for each drug. We have set them equal.)
α, a measure of interaction or synergy, with α > 0 representing positive interaction and α< 0 representing negative interaction.
Due to the normalization setting control equal to 100, for this data Ec= 100.
Synergy analysis was performed using the non-linear mixed effects modeling package “nlme” in “R” statistical software language. The models had random patient effects for IC50,1 and IC50,2 corresponding to differential effectiveness for each patient. All models were fit by maximum likelihood. There was difficulty in fitting the models, most likely due to the small number of drug combinations (due to the limited number of cells available from any given MF patient sample). For this reason, we first fit a model with α = 0, fixed m, and then fit the model to estimate α. We used a likelihood ratio test to evaluate α.
RESULTS
A functional genetic screen identifies NCT as essential for survival of JAK2V617F –mutant HEL cells.
Aberrant JAK/STAT pathway activation is central to the pathogenesis of MF. However, JAK inhibitors, such as ruxolitinib, only reduce symptoms, but are not curative. To identify novel vulnerabilities in MF, irrespective of somatic mutation status, an shRNA library screen was performed on JAK2V617F-mutant HEL cells as a model of JAK/STAT-driven myeloid neoplasia. The shRNA library screen protocol is outlined in Supplemental Figure 1. Barcode abundance in the final population and the original library were compared by next generation sequencing. Significantly depleted shRNAs are those targeting genes with a potentially critical role for HEL cell survival. Candidate genes were prioritized based on two criteria: (i) reduction of barcode abundance ≥10-fold, and (ii) reduction in ≥3 shRNAs targeting the same gene. This customized algorithm identified 72 genes putatively critical to HEL cell survival and/or proliferation. Nuclear-cytoplasmic transport (NCT)-related genes RAN and RANBP2 were among the top 20 candidates (Table 1, Supplemental Table 2), suggesting that HEL cells are dependent on NCT for survival and/or proliferation.
Table 1.
The top 20 hits from the shRNA library screen.
| Rank | Gene Symbol |
Median Fold Depletion |
Gene Name | Group |
|---|---|---|---|---|
| 1 | PSMB2 | 59 | proteasome subunit beta 2 | P |
| 2 | RAN | 50 | RAN family GTPase, Ras superfamily GTPase | N |
| 3 | SHFM1 | 45 | 26S proteasome complex subunit | P |
| 4 | IL28B | 43 | interferon lambda 3 | O |
| 5 | PSMD13 | 41 | proteasome 26S subunit, non-ATPase 13 | P |
| 6 | POLR2F | 40 | RNA polymerase II subunit F | T |
| 7 | RPL11 | 40 | ribosomal protein L11 | T |
| 8 | PSMD2 | 38 | proteasome 26S subunit, non-ATPase 2 | P |
| 9 | SSRP1 | 36 | structure specific recognition protein 1 | T |
| 10 | RPL12 | 36 | ribosomal protein L12 | T |
| 11 | HMGCR | 35 | 3-hydroxy-3-methylglutaryl-CoA reductase | M |
| 12 | RPL6 | 34 | ribosomal protein L6 | T |
| 13 | HNRNPC | 34 | heterogeneous nuclear ribonucleoprotein C (C1/C2) | T |
| 14 | ITK | 33 | IL2 inducible T-cell kinase | O |
| 15 | SIN3A | 33 | SIN3 transcription regulator family member A | T |
| 16 | RANBP2 | 33 | RAN binding protein 2 | N |
| 17 | PSMA3 | 33 | proteasome subunit alpha 3 | P |
| 18 | PSMB7 | 33 | proteasome subunit beta 7 | P |
| 19 | LOC402057 | 32 | unknown gene, NM_001080499.1 | O |
| 20 | PSMB3 | 32 | proteasome subunit beta 3 | P |
| Transcription, Translation (7) | T | |||
| Proteasome (7) | P | |||
| Other (3) | O | |||
| Nuclear-cytoplasmic transport (2) | N | |||
| Metabolism (1) | M |
Cell lines expressing JAK2V617F are dependent on NCT.
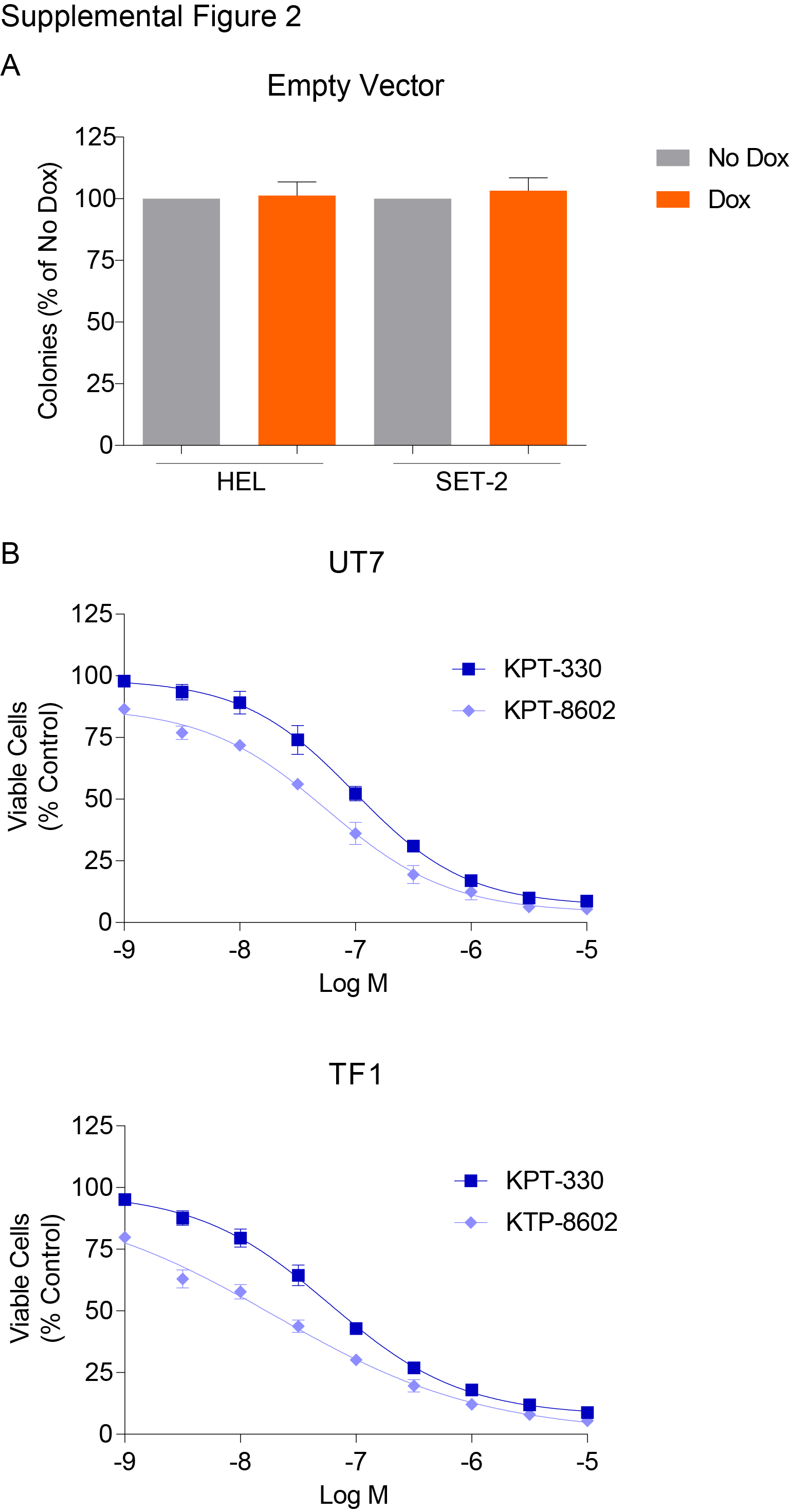
To validate our screen results, we transduced HEL and another JAK2V617F-mutant, SET-2 cells with four different (3 from library, 1 new) Dox-inducible shRNAs targeting RAN and confirmed knockdown (KD) at the mRNA and protein level (Figure 1A,B). RAN KD was associated with a time-dependent reduction of viable cells (Figure 1C,D), and nearly abolished colony formation (Figure 1E,G). RAN KD also strongly increased apoptosis at 72 hours after Dox addition (Figure 1F,H,I,J). We also evaluated two shRNAs targeting RANBP2 (Figure 2; 1 from library, 1 new) and observed reductions in viable cells with both, albeit along a slower timeline than with RAN KD. HEL and SET-2 cells transduced with the empty vector (pRSIT12) did not respond to Dox (colony assay, vs. no Dox: HEL 103.3%, p=0.56, SET-2 101.3%, p=0.8245, Supplemental Figure 2A). These data demonstrate that MPN cells expressing JAK2V617F are dependent on NCT, confirming the results of the shRNA library screen.
Figure 1. Knockdown of RAN inhibits growth and survival of cells expressing JAK2V617F.

HEL cells (left panels) and SET-2 cells (right panels) were engineered to express doxycycline (Dox)-inducible shRNAs specifically targeting RAN. RAN expression was assessed by qRT-PCR and immunoblot (A, B) at 72 hours after adding Dox. Effects of RAN knockdown were measured by MTS assay (C, D), colony formation assay (E, G), and Annexin V flow cytometry (F, H, I, J). Data are from three independent experiments.
*p<0.05, *p<0.01, *p<0.001
Figure 2. Knockdown of RANBP2 inhibits growth and survival of cells expressing JAK2V617F.

HEL cells (A, B) and SET-2 cells (C, D) were engineered to express doxycycline (Dox)-inducible shRNAs specifically targeting RANBP2. RANBP2 expression was assessed by immunoblot (A, C) at 72 hours after adding Dox. Effects of RANBP2 knockdown were measured by viable cell counting (B, D). Data are from two to three independent experiments.
*p<0.01, *p<0.001
To further validate dependence of JAK2V617F-mutant HEL and SET-2 cells on NCT, we used the selective inhibitors of nuclear export (SINE) compounds KPT-330 or KPT-8602 to inhibit NCT. KPT-330 covalently binds cysteine-528 of the chromosome region maintenance 1 protein (CRM1), a key component of NCT, thereby preventing cargo binding and consequently NCT. KPT-8602 is a second-generation inhibitor of CRM1, with similar pharmacokinetics to KPT-330. Unlike KPT-330, KPT-8602 has substantially lower brain penetration, resulting in improved tolerability (31). Both inhibitors are being evaluated in clinical trials for various malignancies. KPT-330 or KPT-8602 treatment (72 hours) reduced viability of HEL and SET-2 cells in a dose-dependent fashion, with IC50 values ~100 nM. (Figure 3A,B). Similarly, TF1 and UT7 cells, JAK2 WT myeloid leukemia lines, were also sensitive to these SINE compounds, with IC50 values between 30–98 nM (Supplemental Figure 2B), similar to other malignant, non-leukemic cell lines (36–38), demonstrating that these inhibitors function through a JAK2-independent mechanism.
Figure 3. Inhibition of NCT by KPT-330 or KPT-8602 reduces survival of cells expressing JAK2V617F.

IC50 of ruxolitinib, KPT-330, and KPT-8602 in HEL (A) and SET-2 (B) cells following 72 hour treatment. C. IC50 of ruxolitinib, KPT-330, and KPT-8602 in HEL-R cells following 72 hour treatment. IC50 values were measured by MTS assay in triplicate 72 hours after addition of inhibitors and data are from two-three independent experiments.
JAK2V617F expressing cells acquire resistance to ruxolitinib and other JAK1,2 inhibitors through reactivation of JAK/STAT signaling, and the same mechanism is operational in vivo (39). We generated HEL cells resistant to JAK1,2 inhibitors (HEL-R) that maintain JAK2, STAT5, and STAT3 phosphorylation despite JAK1,2 inhibition (Supplemental Figure 3A). To determine whether NCT remains a critical target in JAK inhibitor-resistant JAK2V617F expressing cells, we first assessed the effects of RAN KD on HEL-R cells and found a significant increase in apoptosis (Supplemental Figure 3B). We next added SINE compounds to HEL-R cells growing in ruxolitinib. While the IC50 of ruxolitinib in HEL-R exceeded 20 µM, 45-fold higher than that in the parental cells, the IC50 for KPT-330 or KPT-8602 in HEL-R was comparable (<100 nM) to the parental cells, indicating HEL-R cells remain sensitive to NCT inhibition (Figure 3C). Taken together, these data suggest that JAK2V617F expressing MPN cells are highly dependent on NCT and that this dependence extends to JAK1,2 inhibitor-resistant cells.
Inhibition of NCT selectively suppresses primary MF over normal stem/progenitor cells.
We next investigated the NCT dependence of primary MF and normal hematopoietic stem/progenitor cells. CD34+ cells were isolated from MF patients (for clinical details, see Supplemental Table 1) or from cord blood (CB), and treated with vehicle (DMSO), KPT-330, or KPT-8602 in medium containing cytokines. KPT-330 at 100 nM and KPT-8620 at 50 and 100 nM induced significant reductions in viable cells in MF compared to CB, and this was associated with much stronger induction of apoptosis in MF than in CB CD34+ cells (Figure 4A-C). Inhibition of NCT, especially by KPT-8602, also inhibited colony formation by MF cells, with minimal reductions in CB cells, demonstrating selectivity toward MF cells (Figure 4D).
Figure 4. Inhibition of NCT by KPT-330 or KPT-8602 selectively inhibits growth and survival of primary MF compared to cord blood (CB) CD34+ cells.

A. KPT-330 (left panel, n=3 primary MF samples) or KPT-8602 (right panel, n=3 primary MF samples) treatment significantly reduced viable cells of primary MF compared to CB CD34+ cells (n=3 samples) at indicated concentrations as measured in triplicate by MTS assays after 72 h. B,C. KPT-330 or KPT-8602 treatment significantly induces apoptosis in primary MF over CB CD34+ cells (n=3 each, 72 hours). Representative flow plots are shown (B), and bar graphs show the data from three independent experiments (C). D. KPT-330 (n=7) or KPT-8602 (n=3) at indicated concentrations inhibited colony formation of primary MF cells, with minimal effects on CB CD34+ cells (n=3). Statistical comparisons were made between MF vs. CB.
*p<0.05; **p<0.01, ***p<0.001
Inhibition of NCT enhances the effect of ruxolitinib on primary MF cells.
We next performed a series of experiments to investigate the effects of combining ruxolitinib with KPT-330 or KPT-8602 in MF patient samples with different genotypes, including patients who failed ruxolitinib (Supplemental Table 1). We initially incubated primary MF CD34+ cells with graded concentrations of either ruxolitinib alone or in combination with KPT-330 (Figure 5A) or KPT-8602 (Figure 5B). In all six samples tested, inhibition of NCT enhanced the effect of ruxolitinib. We tested for synergy for each KPT inhibitor in combination with ruxolitinib with the response surface approach and formula (5) of Greco, Bravo and Parsons (35) for two-drug interaction. The value of alpha for KPT-330 was 1.265 and the value for KPT-8602 was 8.003. This analysis revealed alpha values greater than 0 for both inhibitors, indicated that KPT-330 and KPT-8602 are synergistic with ruxolitinib against MF cells. Apoptosis induced by 100 nM ruxolitinib was increased by adding 50 nM KPT-330 or KPT-8620, resulting in a significant difference compared to CB cells (Figure 5C,D). Similarly, 1.5 μM ruxolitinib alone reduced MF colony formation by ~70%, compared to ~40% for CB. Single agent KPT-330 and KPT-8602 reduced MF colony formation by ~50%, and ~85%, respectively, compared to ~25% and ~10% for CB (Figure 5E,F). KPT-330 combined with ruxolitinib reduced MF colony formation by >80%, compared to 60% for CB. Profound differential effects were observed for the KPT-8620/ruxolitinib combination, which reduced CB colonies by ~70%, but abolished MF colony formation (Figure 5E,F, Supplemental Table 6). In each annexin V or colony assay (Figure 5 C-F), the combination treatment was more effective against the MF cells than was the ruxolitinib treatment alone (p<0.002 in all tests). Altogether, these data show that KPT-330 or KPT-8602 enhance the effect of ruxolitinib on MF CD34+ cells, with relative preservation of normal CD34+ cells.
Figure 5. Inhibition of NCT by KPT-330 or KPT-8602 enhances effect of ruxolitinib on primary MF CD34+ cells.

Primary MF CD34+ cells were treated with either ruxolitinib alone or in combination with a fixed concentration of KPT-330 at 50 or 100 nM (A) or KPT-8602 at 50 or 100 nM (B) for 72 h. Viable cells were quantified by MTS assay. Each plot represents one sample from one patient. When known, genotypes and treatment status are shown. Inhibition of NCT by KPT-330 at 50 nM (C) or KPT-8602 at 50 nM (D) increased ruxolitinib (100 nM)-induced apoptosis (n=3 each, 72 h). Primary MF or CB CD34+ cells were treated with indicated concentrations of either ruxolitinib alone or in combination with KPT-330 or KPT-8602 for 72 h, and apoptosis was measured with Annexin V. KPT-330 at 100 nM (E) or KPT-8602 at 50 nM (F) enhanced ruxolitinib (1.5 µM)-induced inhibition in colony formation of primary MF vs. CB CD34+ cells (n=3–7). Statistical significance was compared between MF vs. CB (*p<0.05; **p<0.01). Purple asterisks represent combination groups that are statistically distinct (p<0.002 for each) from the ruxolitinib group.
p53 is retained in the nucleus following inhibition of NCT.
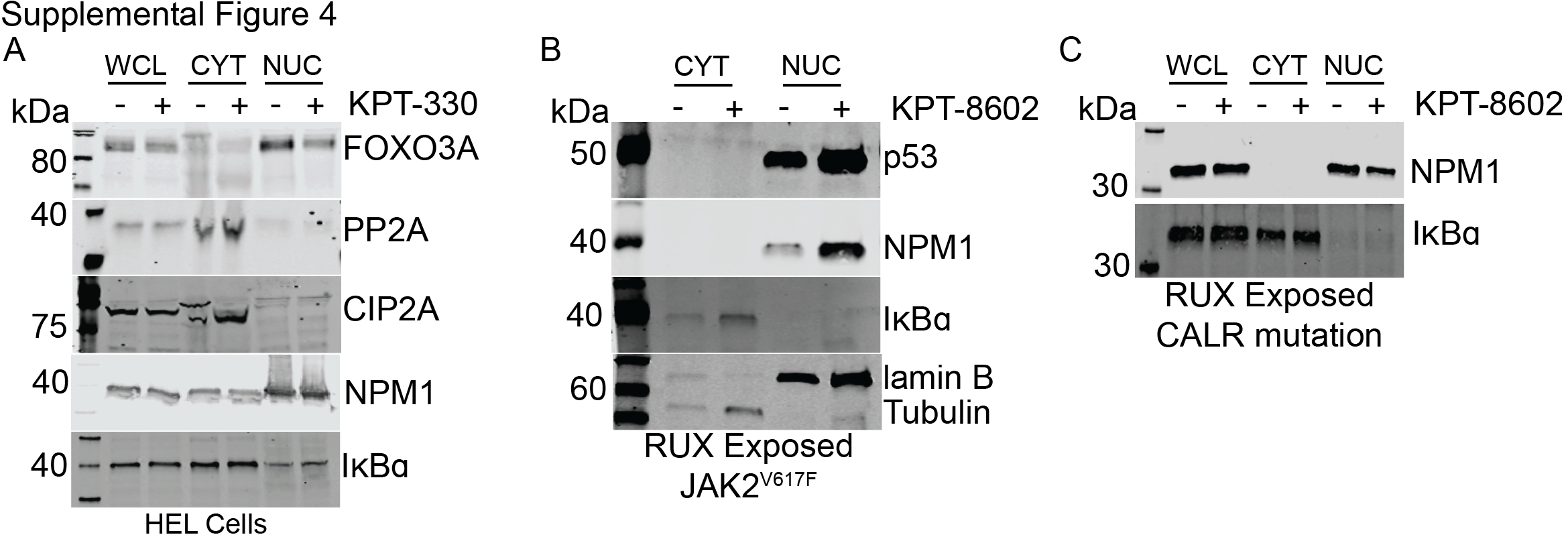
Several studies have analyzed the effects of SINE compounds on the nuclear-cytoplasmic distribution of tumor suppressor proteins and associated their nuclear retention with effects on cell growth and viability (40–44). To determine the effect of KPT-330 and KPT-8620 on major NCT cargo proteins in JAK2V617F expressing cells, we treated HEL cells with either vehicle, KPT-330, or KPT-8602 followed by nucleocytoplasmic fractionation and immunoblot analysis for tumor suppressor proteins previously identified as NCT substrates (Figure 6A,B; Supplemental Figure 4A) (45). Proper fractionation was confirmed by immunoblot for lamin B (nuclear protein marker) and tubulin (cytoplasmic protein marker). NPM1, p53, and IκBα were also analyzed in MF CD34+ cells treated with KPT-8620 (Figure 6C; Supplemental Figure 4B,C). Nuclear localization of p53 was consistently increased, while results for NPM1 varied by sample. IκBα was not detected in the nucleus of any of the patient samples analyzed. These data suggest that KPT-330 and KPT-8602 promote nuclear retention of p53 and certain other tumor suppressor proteins in MPN, consistent with other malignancies, with considerable variations according to sample types and inhibitors.
Figure 6. Inhibition of NCT increases the nuclear localization of tumor suppressor p53.

Whole cell (WCL), cytoplasmic (CYT), and nuclear (NUC) lysates of HEL cells treated with either DMSO vehicle (-) or (A) KPT-330 (100 nM) or (B) KPT-8602 (100 nM) for 24 hours were analyzed for subcellular localization of p53. Primary MF CD34+ cells (C) were similarly treated and analyzed. Patient treatment status and genotype is shown below panel C.
Inhibition of NCT induces hematologic responses in an MPN mouse model.
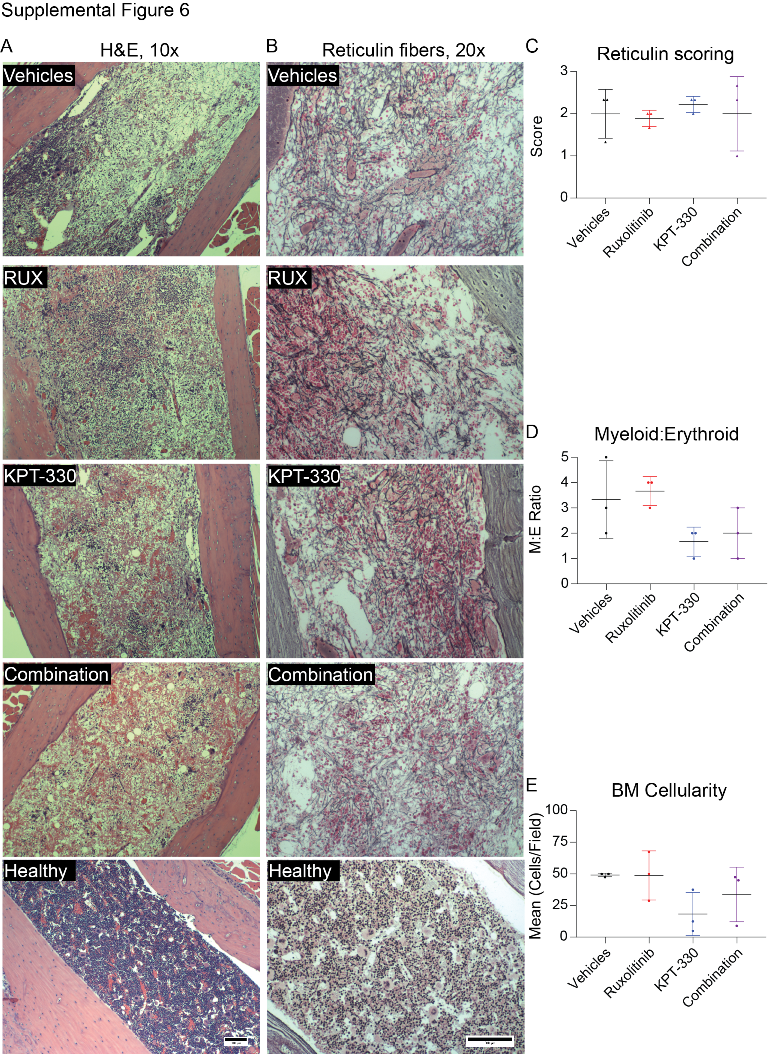
To evaluate the role of NCT in vivo, we tested KPT-330 in a mouse model of JAK2V617F-driven MPN that resembles PV progressing to MF (32). MPN was induced in Balb/c mice by transplanting donor BM infected with JAK2V617F–GFP retrovirus. On day 21 post-transplant, mice were randomized to vehicles, KPT-330 (initial dose: 20 mg/kg, 3x weekly, orally), ruxolitinib (initial dose: 50 mg/kg, twice daily, orally), or ruxolitinib plus KPT-330 using 13–14 mice/group. Blood GFP+ cells ranged from 7–23%, with a median of 14–16% in each of the treatment groups at this point (Supplemental Figure 5). Over the course of the experiment, mice in all groups experienced weight loss and lethality, including the vehicle controls, implicating the vehicles as the cause of toxicity. As a result, KPT-330 was reduced to 10 mg/kg twice weekly, followed by a reduction of ruxolitinib to 50 mg/kg daily (Supplemental Table 7, 8). On treatment day 15, KPT-330 significantly reduced white blood cells and granulocytes (both p<0.05), with a trend toward significant reduction in blood GFP+ cells (p=0.12), while hematocrit was unchanged (Figure 7A-D). The KPT-330/ruxolitinib combination significantly reduced white blood cells, hematocrit, granulocytes, and blood GFP+ cells (all p<0.05). On treatment day 28, the KPT-330 single agent group showed significantly reduced spleen GFP+ cells compared to controls (p<0.05), while ruxolitinib-treated mice developed resistance, as shown by increasing WBC counts and GFP+ cells in peripheral blood, consistent with a previous study (46). In contrast, combination treatment significantly reduced white blood cells, granulocytes, spleen GFP+ cells and spleen weight (p<0.05) (Figure 7A-G). Histopathology revealed that combination treatment also partially restored splenic architecture (Figure 7H), while bone marrow fibrosis persisted in all treatment groups (Supplemental Figure 6A-C). The myeloid:erythroid ratio and overall cellularity were reduced in the KPT-330 and combination groups (Supplemental Figure 6 D,E). Taken together, these data suggest that NCT inhibition, alone and in combination with JAK inhibitors, can reduce disease burden, attenuate MF features, and suppress/delay resistance to JAK inhibitors in vivo.
Figure 7. Inhibition of NCT attenuates MPN and reduces mutant allele burden in a JAK2V617F-driven MPN mouse model.

Mice with established MPN were treated with vehicles, KPT-330, ruxolitinib (RUX), and combination KPT-330 and ruxolitinib (Combination). In panels A-D, for each of the treatments, the left data set represents the results after 14 days, and the right data set after 28 days of treatment. Gray horizontal bars represent the range from two healthy mice for each parameter. (A) White blood cells (WBC), (B) hematocrit (HCT), and (C) granulocytes were measured with a Heska HemaTrue. (D) Green fluorescent protein (GFP) positive cells were measured in the peripheral blood. At 4 weeks (end of treatment) GFP+ cells were measured in (E) spleen (SP) and (F) bone marrow (BM). (G) Spleen weights were determined at the end of treatment. (H) H&E stains of representative spleen sections are shown. The bar represents 1 millimeter. Data were analyzed with one-way ANOVA (Kruskal-Wallis and uncorrected Dunn’s test, with nonparametric analysis) in GraphPad Prism 7.02. *p<0.05 when compared to Vehicles.
DISCUSSION
Ruxolitinib, the first approved JAK kinase inhibitor, is the drug therapy standard for intermediate-2 and high-risk MF. Ruxolitinib ameliorates splenomegaly, improves quality of life, and may prolong survival, but its utility is frequently limited by myelosuppression that necessitates dose reduction or discontinuation (20,21). Moreover, ruxolitinib does not typically reduce mutant allele burden and clonal hematopoiesis persists despite clinical responses (22). Alternative JAK1/2 inhibitors in clinical development may overcome some of ruxolitinib’s shortcomings. Pacritinib, a selective JAK2/FLT3 inhibitor with less myelosuppressive effects than ruxolitinib, was found to be superior to best available therapy in two phase 3 randomized studies that included patients with thrombocytopenia (47,48). Deaths related to bleeding and cardiovascular events lead to temporary full clinical hold by the FDA; additional studies are underway (49). Another JAK1/2 inhibitor, momelotinib, is no longer being developed due to lack of superiority over best available therapy in phase 3 studies, adding to a considerable list of JAK kinase inhibitors that failed in clinical trials (33,50–52). In contrast to ruxolitinib and other clinical candidates, CHZ868 is a type II inhibitor that binds an inactive JAK2 conformation, and a reduction of mutant allele burden was demonstrated in mouse models, but clinical development of this molecule is uncertain (53). The limited disease-modifying ability of ruxolitinib monotherapy led to numerous trials of ruxolitinib-based combinations, some of which have shown activity, for example, the combination of ruxolitinib and 5-azacitidine (54,55). Thus far however, JAK1/2 inhibitors have not solved the clinical challenge of progressive MF.
To identify potential therapeutic targets in MF other than JAK/STAT pathway, we performed a lentiviral shRNA library screen on HEL cells, a myeloid cell line homozygous for JAK2V617F. Although cell lines do not recapitulate many features of primary cells, their rapid proliferation in vitro allows large fold changes to occur over a limited period of time, enabling the identification of vulnerabilities (24). To minimize false positive results, we developed a set of stringent criteria to prioritize candidates and identified genes related to proteasome, transcription, and translation as well as NCT as potentially critical for the survival and/or proliferation of HEL cells. Despite the strong representation of proteasome components amongst the top hits, this lead was not followed, since proteasome inhibitors such as bortezomib were ineffective or even associated with increased disease activity in MF clinical trials (56,57). Given the strong and consistent reduction of shRNAs targeting NCT-related mRNAs, we hypothesized that NCT may represent a hitherto unrecognized vulnerability in MF amenable to targeting with pharmacological inhibitors (SINE compounds).
In initial experiments using shRAN, we demonstrated that HEL and SET-2 cells are highly dependent on RAN and that MF CD34+ cells are much more sensitive to NCT inhibitors than CB CD34+ cells. This was consistent with our previous observations that CB CD34+ cells are less dependent on RAN than chronic myeloid leukemia (CML) CD34+ cells and suggested a therapeutic window for targeting NCT in MF (24). KPT-330 was equally potent against MF cells from newly diagnosed and ruxolitinib exposed patients, including different genotypes, suggesting that targeting NCT in MF would have broad applicability. The differential between CB and MF seemed to be even greater for the next generation compound KPT-8602, which may be clinically relevant, given the reduced central nervous system penetration and reduced side effects of KPT-8602 (31). Although more toxicity towards normal cells was observed with combinations of KPT-330 or KPT-8620 with ruxolitinib, selectivity toward MF vs. CB was consistently maintained. For clinical use, it will be important to optimize dosing and carefully monitor hematologic toxicity. The efficacy of NCT inhibition was confirmed in a mouse model of JAK2V617F -induced MPN. Compared to single agent KPT-330 or ruxolitinib, KPT-330 combined with ruxolitinib appeared to have the most profound effects, including a reduction of JAK2V617F expressing cells in the blood (day 15) and spleen (day 28), and delayed/suppressed emergence of resistance. A limitation of these experiments is the attrition of the cohorts due to toxicity, which was identical across treatment groups, implicating the vehicles in toxicity. The loss of animals and statistical power likely accounts for the relative inconsistency over time. Future optimization in vehicles, dosing, and treatment periods will be required to conclusively assess the efficacy of KPT-330 in this MPN model. Altogether, these data show that the selective ex vivo activity of SINE compounds against MF over normal progenitors is reproducible in vivo. Given the efficacy of NCT inhibition against JAK inhibitor-exposed cells in primary MF samples and in vivo mouse model, evaluation of SINE compounds in relapse and ruxolitinib exposed MF patients is warranted.
Export of proteins from the nucleus to the cytoplasm is an energy-dependent process that involves the formation of a multimeric complex including RAN, RANBP2 and CRM1 (58). A nuclear export signal in cargo proteins promotes formation of a trimeric complex with CRM1 and the GTP-bound form of the small GTPase RAN that shuttles nuclear cargos through the nuclear pore complex into the cytoplasm. The RAN-GTP/RAN-GDP gradient provides the energy for directional cargo transport (59). Overexpression of CRM1 correlates with poor prognosis and reduced survival in many solid and hematologic malignancies, implicating NCT as a potential cancer therapy target (60,61). CRM1 cargos include a number of tumor suppressor proteins, including p53, NPM1, p21, p27, forkhead box O (FOXO) transcription factors, inhibitor of κB (IκB), PP2A and survivin (reviewed by Tan et al (45)). As these tumor suppressor proteins require nuclear localization to exert their functions, nuclear export leads to their functional inactivation and/or proteasome-dependent cytoplasmic degradation. We observed that p53 is consistently retained in the nucleus following treatment with SINE compounds, with variable results for other tumor suppressors such as NPM1, potentially implicating the NCT of these tumor suppressors as a SINE target in MF, similar to other malignancies (45). Differences in the cargos may be related to the genetic heterogeneity of MF.
In summary, our results are the first to demonstrate selective activity of SINE compounds against MF progenitors, including cells from patients who failed ruxolitinib, over normal controls. This provides a strong rationale for testing this therapeutic concept in a clinical trial.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TRANSLATIONAL RELEVANCE:
Myelofibrosis (MF) is a fatal hematopoietic stem cell neoplasm characterized by constitutive activation of JAK/STAT signaling. JAK kinase inhibitors such as ruxolitinib reduce MF symptoms, but like all other drugs used in MF are not curative, with persistence of mutant cells and prompt symptom rebound upon discontinuation. Therefore, more effective drug therapies are needed to improve survival in MF. In this study, using a lentiviral shRNA screen, we have discovered that HEL and SET-2 cell lines and primary MF cells are exquisitely dependent on nuclear-cytoplasmic transport (NCT). Pharmacologic inhibition of NCT by KPT-330 or KPT-8602 selectively suppressed colony formation by MF compared to normal CD34+ cells and enhanced ruxolitinib-mediated growth inhibition and apoptosis. In an MPN mouse model addition of KPT-330 to ruxolitinib improved responses compared to monotherapy, including reductions of mutant cells and restoration of splenic architecture. Our results warrant clinical trials of KPT-330 and/or KPT-8602 in ruxolitinib exposed MF patients.
Acknowledgements
This work was supported by a SWOG HOPE Foundation Award to MWD and R01CA178397 from the National Institutes of Health National Cancer Institute to MWD and TO. The University of Utah Flow Cytometry Facility is supported by the National Cancer Institute through award 5P30CA042014–24 and the National Center for Research Resources of the National Institutes of Health under award 1S10RR026802–01. JK was and DY is supported by the Special Fellow Award from the Leukemia & Lymphoma Society. ST was and ABP is supported by the Research Training Award for Fellows from the American Society of Hematology. AME was supported by the American Society of Hematology Scholar Award.
Disclosure: This work was supported by The SWOG/Hope Foundation, American Society of Hematology, Leukemia & Lymphoma Society, and National Institutes of Health National Cancer Institute.
Abbreviation list:
- MF
Myelofibrosis
- NCT
nuclear-cytoplasmic transport
- SINE
selective inhibitors of nuclear export
- ET
essential thrombocythemia
- shRNA
short hairpin RNA
- MOI
multiplicity of infection
- HM1
Human Module 1
- M:E
myeloid:erythroid
- dox
doxycycline
- CB
cord blood
Footnotes
Conflict of interest statement: MWD’s laboratory is funded by Novartis and Pfizer and DY, ST, ADP, AS, JA, QW, HT, ABP, WH, AE, PC, KG, HR, and JK currently or did previously work in his laboratory. MWD is a consultant/advisory board member of Novartis, Pfizer, Galena Biopharma, ARIAD Pharmaceuticals, Blueprint Medicines, and Incyte. EB and SS are employees of Karyopharm Therapeutics.
REFERENCES
- 1.Tefferi A. Pathogenesis of myelofibrosis with myeloid metaplasia. J Clin Oncol 2005;23(33):8520–30 doi 10.1200/JCO.2004.00.9316. [DOI] [PubMed] [Google Scholar]
- 2.Mesa R, Miller CB, Thyne M, Mangan J, Goldberger S, Fazal S, et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer 2016;16:167 doi 10.1186/s12885-016-2208-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price GL, Davis KL, Karve S, Pohl G, Walgren RA. Survival patterns in United States (US) medicare enrollees with non-CML myeloproliferative neoplasms (MPN). PLoS One 2014;9(3):e90299 doi 10.1371/journal.pone.0090299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel AB, Vellore NA, Deininger MW. New Strategies in Myeloproliferative Neoplasms: The Evolving Genetic and Therapeutic Landscape. Clin Cancer Res 2016;22(5):1037–47 doi 10.1158/1078-0432.CCR-15-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365(9464):1054–61 doi 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 6.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7(4):387–97 doi 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 7.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005;352(17):1779–90 doi 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 8.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3(7):e270 doi 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369(25):2391–405 doi 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369(25):2379–90 doi 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 11.Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 2010;42(8):722–6 doi 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 12.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009;360(22):2289–301 doi 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 13.Tefferi A, Lasho TL, Abdel-Wahab O, Guglielmelli P, Patel J, Caramazza D, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010;24(7):1302–9 doi 10.1038/leu.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adelaide J, Rey J, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009;23(11):2183–6 doi 10.1038/leu.2009.141. [DOI] [PubMed] [Google Scholar]
- 15.Abdel-Wahab O, Pardanani A, Rampal R, Lasho TL, Levine RL, Tefferi A. DNMT3A mutational analysis in primary myelofibrosis, chronic myelomonocytic leukemia and advanced phases of myeloproliferative neoplasms. Leukemia 2011;25(7):1219–20 doi 10.1038/leu.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tefferi A, Lasho TL, Finke CM, Knudson RA, Ketterling R, Hanson CH, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia 2014;28(7):1472–7 doi 10.1038/leu.2014.3. [DOI] [PubMed] [Google Scholar]
- 17.Jabbour E, Thomas D, Kantarjian H, Zhou L, Pierce S, Cortes J, et al. Comparison of thalidomide and lenalidomide as therapy for myelofibrosis. Blood 2011;118(4):899–902 doi 10.1182/blood-2010-12-325589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 2012;366(9):787–98 doi 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 19.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366(9):799–807 doi 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verstovsek S, Mesa RA, Gotlib J, Gupta V, DiPersio JF, Catalano JV, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol 2017;10(1):55 doi 10.1186/s13045-017-0417-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrison CN, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Gisslinger H, Knoops L, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016;30(8):1701–7 doi 10.1038/leu.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deininger M, Radich J, Burn TC, Huber R, Paranagama D, Verstovsek S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015;126(13):1551–4 doi 10.1182/blood-2015-03-635235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rondelli D, Goldberg JD, Isola L, Price LS, Shore TB, Boyer M, et al. MPD-RC 101 prospective study of reduced-intensity allogeneic hematopoietic stem cell transplantation in patients with myelofibrosis. Blood 2014;124(7):1183–91 doi 10.1182/blood-2014-04-572545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khorashad JS, Eiring AM, Mason CC, Gantz KC, Bowler AD, Redwine HM, et al. shRNA library screening identifies nucleocytoplasmic transport as a mediator of BCR-ABL1 kinase-independent resistance. Blood 2015;125(11):1772–81 doi 10.1182/blood-2014-08-588855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood 2010;115(25):5232–40 doi 10.1182/blood-2009-05-223727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pardanani A, Tefferi A. Definition and management of ruxolitinib treatment failure in myelofibrosis. Blood Cancer J 2014;4:e268 doi 10.1038/bcj.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tefferi A, Cervantes F, Mesa R, Passamonti F, Verstovsek S, Vannucchi AM, et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood 2013;122(8):1395–8 doi 10.1182/blood-2013-03-488098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barosi G, Tefferi A, Besses C, Birgegard G, Cervantes F, Finazzi G, et al. Clinical end points for drug treatment trials in BCR-ABL1-negative classic myeloproliferative neoplasms: consensus statements from European LeukemiaNET (ELN) and Internation Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT). Leukemia 2015;29(1):20–6 doi 10.1038/leu.2014.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barbui T, Thiele J, Gisslinger H, Kvasnicka HM, Vannucchi AM, Guglielmelli P, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J 2018;8(2):15 doi 10.1038/s41408-018-0054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azmi AS, Aboukameel A, Bao B, Sarkar FH, Philip PA, Kauffman M, et al. Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology 2013;144(2):447–56 doi 10.1053/j.gastro.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Etchin J, Berezovskaya A, Conway AS, Galinsky IA, Stone RM, Baloglu E, et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 2017;31(1):143–50 doi 10.1038/leu.2016.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bumm TG, Elsea C, Corbin AS, Loriaux M, Sherbenou D, Wood L, et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res 2006;66(23):11156–65 doi 10.1158/0008-5472.CAN-06-2210. [DOI] [PubMed] [Google Scholar]
- 33.Pomicter AD, Eiring AM, Senina AV, Zabriskie MS, Marvin JE, Prchal JT, et al. Limited efficacy of BMS-911543 in a murine model of Janus kinase 2 V617F myeloproliferative neoplasm. Exp Hematol 2015;43(7):537–45 e1-11 doi 10.1016/j.exphem.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005;90(8):1128–32. [PubMed] [Google Scholar]
- 35.Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev 1995;47(2):331–85. [PubMed] [Google Scholar]
- 36.Zheng Y, Gery S, Sun H, Shacham S, Kauffman M, Koeffler HP. KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother Pharmacol 2014;74(3):487–95 doi 10.1007/s00280-014-2495-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendonca J, Sharma A, Kim HS, Hammers H, Meeker A, De Marzo A, et al. Selective inhibitors of nuclear export (SINE) as novel therapeutics for prostate cancer. Oncotarget 2014;5(15):6102–12 doi 10.18632/oncotarget.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Etchin J, Sanda T, Mansour MR, Kentsis A, Montero J, Le BT, et al. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br J Haematol 2013;161(1):117–27 doi 10.1111/bjh.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koppikar P, Bhagwat N, Kilpivaara O, Manshouri T, Adli M, Hricik T, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012;489(7414):155–9 doi 10.1038/nature11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garg M, Kanojia D, Mayakonda A, Said JW, Doan NB, Chien W, et al. Molecular mechanism and therapeutic implications of selinexor (KPT-330) in liposarcoma. Oncotarget 2017;8(5):7521–32 doi 10.18632/oncotarget.13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hing ZA, Fung HY, Ranganathan P, Mitchell S, El-Gamal D, Woyach JA, et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia 2016;30(12):2364–72 doi 10.1038/leu.2016.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferreiro-Neira I, Torres NE, Liesenfeld LF, Chan CH, Penson T, Landesman Y, et al. XPO1 Inhibition Enhances Radiation Response in Preclinical Models of Rectal Cancer. Clin Cancer Res 2016;22(7):1663–73 doi 10.1158/1078-0432.CCR-15-0978. [DOI] [PubMed] [Google Scholar]
- 43.Marcus JM, Burke RT, DeSisto JA, Landesman Y, Orth JD. Longitudinal tracking of single live cancer cells to understand cell cycle effects of the nuclear export inhibitor, selinexor. Sci Rep 2015;5:14391 doi 10.1038/srep14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun H, Hattori N, Chien W, Sun Q, Sudo M, GL EL, et al. KPT-330 has antitumour activity against non-small cell lung cancer. Br J Cancer 2014;111(2):281–91 doi 10.1038/bjc.2014.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan DS, Bedard PL, Kuruvilla J, Siu LL, Razak AR. Promising SINEs for embargoing nuclear-cytoplasmic export as an anticancer strategy. Cancer Discov 2014;4(5):527–37 doi 10.1158/2159-8290.CD-13-1005. [DOI] [PubMed] [Google Scholar]
- 46.Kleppe M, Koche R, Zou L, van Galen P, Hill CE, Dong L, et al. Dual Targeting of Oncogenic Activation and Inflammatory Signaling Increases Therapeutic Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2018;33(1):29–43 e7 doi 10.1016/j.ccell.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mesa RA, Vannucchi AM, Mead A, Egyed M, Szoke A, Suvorov A, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol 2017;4(5):e225–e36 doi 10.1016/S2352-3026(17)30027-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mascarenhas J HR, Talpaz M, Gerds AT, Stein B, Gupta V et al. Results of the Persist-2 phase 3 study of pacritinib (PAC) versus best available therapy (BAT), including ruxolitinib (RUX), in patients (pts) with myelofibrosis (MF) and platelet counts <100,000/ml. Blood 2016;128(22) Abstract LBA-5 [Google Scholar]
- 49.CTI BioPharma Announces Removal Of Full Clinical Hold On Pacritinib. https://wwwprnewswirecom/news-releases/cti-biopharma-announces-removal-of-full-clinical-hold-on-pacritinib-300386115html 2017. [Google Scholar]
- 50.Harrison CN VA, Platzbecker U, et al. Phase 3 randomized trial of momelotinib (MMB) versus best available therapy (BAT) in patients with myelofibrosis (MF) previously treated with ruxolitinib (RUX). J Clin Oncol 2017;35 Abstract 7001. [Google Scholar]
- 51.Pardanani A, Harrison C, Cortes JE, Cervantes F, Mesa RA, Milligan D, et al. Safety and Efficacy of Fedratinib in Patients With Primary or Secondary Myelofibrosis: A Randomized Clinical Trial. JAMA Oncology 2015;1(5):643–51 doi 10.1001/jamaoncol.2015.1590. [DOI] [PubMed] [Google Scholar]
- 52.Mesa RA, Kiladjian JJ, Catalano JV, Devos T, Egyed M, Hellmann A, et al. SIMPLIFY-1: A Phase III Randomized Trial of Momelotinib Versus Ruxolitinib in Janus Kinase Inhibitor-Naive Patients With Myelofibrosis. J Clin Oncol 2017;35(34):3844–50 doi 10.1200/JCO.2017.73.4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyer SC, Keller MD, Chiu S, Koppikar P, Guryanova OA, Rapaport F, et al. CHZ868, a Type II JAK2 Inhibitor, Reverses Type I JAK Inhibitor Persistence and Demonstrates Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2015;28(1):15–28 doi 10.1016/j.ccell.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bose P, Verstovsek S. JAK2 inhibitors for myeloproliferative neoplasms: what is next? Blood 2017;130(2):115–25 doi 10.1182/blood-2017-04-742288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daver N CJ, Pemmaraju N, Jabbour EJ, Bose P, Zhour L et al. Ruxolitinib (RUX) in Combination with 5-Azacytidine (AZA) As Therapy for Patients (pts) with Myelofibrosis (MF). Blood 2016;128:1127. [Google Scholar]
- 56.Mesa RA, Verstovsek S, Rivera C, Pardanani A, Hussein K, Lasho T, et al. Bortezomib therapy in myelofibrosis: a phase II clinical trial. Leukemia 2008;22(8):1636–8 doi 10.1038/leu.2008.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barosi G, Gattoni E, Guglielmelli P, Campanelli R, Facchetti F, Fisogni S, et al. Phase I/II study of single-agent bortezomib for the treatment of patients with myelofibrosis. Clinical and biological effects of proteasome inhibition. American Journal of Hematology 2010;85(8):616–9 doi 10.1002/ajh.21754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, et al. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature 2009;458(7242):1136–41 doi 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dong X, Biswas A, Chook YM. Structural basis for assembly and disassembly of the CRM1 nuclear export complex. Nat Struct Mol Biol 2009;16(5):558–60 doi 10.1038/nsmb.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu C, Figueroa JA, Liu Z, Konala V, Aulakh A, Verma R, et al. Nuclear Export as a Novel Therapeutic Target: The CRM1 Connection. Current Cancer Drug Targets 2015;15(7):575–92. [DOI] [PubMed] [Google Scholar]
- 61.Das A, Wei G, Parikh K, Liu D. Selective inhibitors of nuclear export (SINE) in hematological malignancies. Experimental Hematology & Oncology 2015;4:7 doi 10.1186/s40164-015-0002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.