

Abstract
Diphenyl phosphate (DPHP) is an aryl phosphate ester (APE) used as an industrial catalyst and chemical additive and is the primary metabolite of flame retardant APEs, including triphenyl phosphate (TPHP). Minimal DPHP-specific toxicity studies have been published despite ubiquitous exposure within human populations following metabolism of TPHP and other APEs. Therefore, the objective of this study was to determine the potential for DPHP-induced toxicity during embryonic development. Using zebrafish as a model, we found that DPHP significantly increased the distance between the sinus venosus and bulbus arteriosis (SV-BA) at 72 h postfertilization (hpf) following initiation of exposure before and after cardiac looping. Interestingly, pretreatment with D-mannitol mitigated DPHP-induced effects on SV-BA length despite the absence of DPHP effects on pericardial area, suggesting that DPHP-induced cardiac defects are independent of pericardial edema formation. Using mRNA-sequencing, we found that DPHP disrupts pathways related to mitochondrial function and heme biosynthesis; indeed, DPHP significantly decreased hemoglobin levels in situ at 72 hpf following exposure from 24 to 72 hpf. Overall, our findings suggest that, similar to TPHP, DPHP impacts cardiac development, albeit the potency of DPHP is significantly less than TPHP within developing zebrafish.
Graphical Abstract
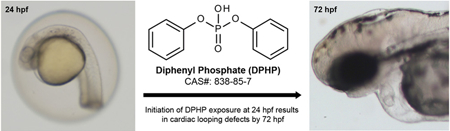
INTRODUCTION
Diphenyl phosphate (DPHP) is an aryl phosphate ester (APE) used as an industrial catalyst and chemical additive and is the primary metabolite of flame retardant APEs, including triphenyl phosphate (TPHP).1 TPHP is primarily metabolized to DPHP via cleavage of an ester bond between the phosphate group and one of three benzene rings.2 Metabolism of TPHP to DPHP is mediated by cytochrome P450 enzymes within the liver and serum.3 DPHP can also be generated from metabolism of ethylhexyl diphenyl phosphate and resorcinol bis(diphenylphosphate), both of which are used as APE flame retardants and plasticizers.4,5
Following the phase-out of certain polybrominated diphenyl ethers (PBDEs), the use of APE-based flame retardants (FRs) within the United States has increased in order to maintain compliance with California-specific flammability standards in a wide range of consumer products. As a result, TPHP and DPHP have been found within indoor dust, as both TPHP and DPHP are additive FRs that, similar to PBDEs, migrate out of end-use products into surrounding environmental media.6 TPHP and DPHP have been detected within human samples,7,8 including urine of pregnant women9 and infants,10–12 underscoring the need for generation of developmental toxicity data. Interestingly, higher concentrations of DPHP have been detected in urine samples collected from women, a finding that is strongly associated with altered total thyroxine levels13 and TPHP-induced disruption of thyroid function within animal- and cell-based systems.14–16
Although a growing body of scientific literature has focused on the potential toxicity of TPHP, minimal DPHP-specific toxicity studies have been published despite ubiquitous exposure within human populations following metabolism of TPHP and other APE-based FRs. One study found that subcutaneous exposure of mice from postnatal days 1−10 to lower doses of DPHP (2 μg/day) resulted in adverse effects on lipid metabolism, whereas exposure to higher doses of DPHP (200 μg/day) resulted in a decrease in metabolites related to pyruvate metabolism and the tricarboxylic acid cycle.17 Interestingly, this study also found that 600 mg/kg DPHP was more acutely toxic to mice and rats compared to the same dose of TPHP. However, within chicken embryonic hepatocyte cultures, TPHP was cytotoxic at nominal concentrations <10 μM whereas DPHP was not cytotoxic at all concentrations tested (up to 1000 μM).18 While multiple nuclear receptors (e.g., estrogen receptor α, peroxisome proliferator-activated receptor γ, and pregnane X receptor) may be potential TPHP targets, DPHP exhibits limited to no affinity to these same receptors.19 Within humans, developing embryos will most likely be exposed to both TPHP and DPHP, as adult pregnant women will metabolize TPHP and other APEs to DPHP. While uptake of these two compounds into cells and tissues most likely vary based on physicochemical properties, the potential for human exposure is high since TPHP is ubiquitous within indoor and outdoor environments such as ambient air. Moreover, TPHP and DPHP likely have different half-lives and persistence in humans since TPHP is significantly more lipophilic (log Kow = 4.59)20 than DPHP (log Kow = 1.4),21 suggesting that the potential for TPHP and/or DPHP-induced effects during prenatal development is driven by chemical-specific distribution and partitioning following ingestion and/or inhalation.
As it is currently unknown whether DPHP affects developing embryos, the objective of this study was to determine the potential for DPHP-induced toxicity during embryonic development. Using zebrafish as a model, we relied on phenotyping, mRNA-sequencing, and hemoglobin quantification in situ to determine the potential impacts of DPHP on cardiac morphogenesis, the embryonic transcriptome, and red blood cell formation, respectively, following embryonic exposure from 24 to 72 h postfertilization (hpf). Finally, we quantified embryonic doses of DPHP and TPHP at 30, 48, and 72 hpf following initiation of DPHP exposure at 24 hpf.
METHODS
Animals.
Adult wildtype (5D) zebrafish were maintained and bred on a recirculating system using previously described procedures.22 Adult breeders were handled and treated in accordance with Institutional Animal Care and Use Committee-approved animal use protocols (#20150035 and #20180063) at the University of California, Riverside.
Chemicals.
TPHP (99.5% purity) was purchased from ChemService, Inc. (West Chester, PA U.S.A.); DPHP (>99% purity), butafenacil (99.3% purity), and D-mannitol (>98% purity) were purchased from Millipore Sigma (St. Louis, MO U.S.A.); fenretinide (>99.3% purity) and HX 531 (>99.8% purity) were purchased from Bio-Techne Corp. (Minneapolis, MN U.S.A.). TPHP, butafenacil, D-mannitol, fenretinide, and HX 531 stock solutions were prepared by dissolving chemicals in high performance liquid chromatography-grade dimethyl sulfoxide (DMSO) and stored at room temperature within 2 ml amber glass vials with polytetrafluoroethylene-lined caps. Working solutions were freshly prepared by spiking stock solutions into particulate-free water from our recirculating system (pH and conductivity of ∼7.2 and ∼950 μS, respectively), resulting in 0.1% DMSO within all vehicle control and treatment groups. Working solutions of DPHP were freshly prepared by adding neat standard directly to particulate-free water from our recirculating system.
Diphenyl Phosphate Exposures for Phenotyping.
Newly fertilized eggs were collected immediately after spawning and incubated in groups of approximately 100 per 100 × 15 mm polystyrene Petri dish within a light- and temperature-controlled incubator until exposure initiation. A working solution of 1000 μM DPHP was freshly prepared as described above and then serially diluted by 2-fold to 500, 250, and 125 μM DPHP. Viable embryos were transferred to 35 × 10 mm polystyrene Petri dishes containing 1.5 mL of control or treatment solution, resulting in 30 embryos per dish (one dish per treatment). To identify potential sensitive windows of DPHP-induced toxicity during pharyngula (24−48 hpf), embryos were exposed to system water alone or DPHP from 24 to 72 hpf, 30 to 72 hpf, or 48 to 72 hpf. All dishes were covered with a plastic lid and incubated under static conditions at 28 °C under a 14 h/10 h light−dark cycle until exposure termination. At 72 hpf, embryos were assessed for survival and hatch rate, and these were thhen fixed in 4% paraformaldehyde in 1× phosphate buffered saline (PBS) overnight at 4 °C, transferred to 1× PBS, and stored at 4 °C for no longer than one month until whole-mount immunohistochemistry.
Whole-Mount Immunohistochemistry.
Fixed embryos were labeled with a 1:20 dilution of zn-8 IgG1 (Developmental Studies Hybridoma Bank, University of Iowa) and 1:500 dilution of Alexa Fluor 488-conjugated goat antimouse IgG1 (Invitrogen) following previously described protocols23 to visualize both chambers of the embryonic heart. zn-8-labeled embryos were oriented in right lateral recumbency and imaged at 4× or 8× magnification using a Leica MZ10 F stereomicroscope equipped with a GFP filter and DMC2900 camera. Body length, pericardial area, and yolk sac area were quantified within ImageJ (https://imagej.nih.gov/ij/) using images acquired under transmitted light. The length between where blood enters the atrium (the sinus venosus [SV]) and the point where blood leaves the ventricle (the bulbus arteriosis [BA]) was quantified within ImageJ using images derived from zn-8-labeled embryos. A line was measured from the SV to the BA, as SV-BA length provides a quantitative indicator of heart tube looping.28
Quantification of Embryonic Doses of DPHP and TPHP.
Embryonic doses of DPHP and TPHP were quantified following exposure to DPHP from 24 to 30 hpf, 24 to 48 hpf, and 24 to 72 hpf. Briefly, 24-hpf embryos were transferred to 35 × 10 mm polystyrene Petri dishes (30 embryos per dish; four dishes per treatment), containing either system water or 1000 μM DPHP, and then incubated until 30, 48, or 72 hpf as described above. For each replicate pool (4 pools per treatment), 30 embryos were placed into a 2 ml cryovial, immediately snap-frozen in liquid nitrogen, and stored at −80 °C until analysis. Immediately prior to extraction, samples were spiked with deuterated TPHP (d15-TPHP) and deuterated DPHP (d10-DPHP). Analytes were extracted and quantified similar to previously published methods.22 Method detection limits (MDLs) were defined as three times the standard deviation of lab blanks (if present) or three times the noise. The MDLs for DPHP and TPHP were 2.75 ng and 2.21 ng, respectively.
Fenretinide, HX 531, and Mannitol Pretreatments.
Since TPHP may interact with the retinoic acid receptor/retinoid X receptor (RAR/RXR) signaling pathway22,24 in embryonic zebrafish, we tested whether, similar to TPHP, pretreatment with fenretinide (a pan-RAR agonist) or HX 531 (a pan-RXR antagonist) mitigates and enhances DPHP-induced cardiotoxicity in zebrafish, respectively. Embryos (30 embryos per treatment; 1.5 mL of treatment solution per 35 × 10 mm polystyrene Petri dish; one dish per treatment) were first exposed to vehicle (0.1% DMSO), 2 μM fenretinide, or 5 μM HX 531 from 24 to 30 hpf as described above. At 30 hpf, embryos were transferred to clean Petri dishes, rinsed twice with molecular-grade water, and then incubated in 1.5 mL of either vehicle (0.1% DMSO), 20 μM TPHP, or 1000 μM DPHP until 72 hpf; for DPHP treatments, 100% DMSO was spiked into working solutions to yield a final concentration of 0.1% DMSO. At 72 hpf, survival and hatch rate were recorded, and embryos were fixed and stained as described above.
D-Mannitol has been previously used to raise the osmolarity of water surrounding developing zebrafish, leading to a decrease in chemically induced pericardial edema.25 Therefore, embryos were pretreated with system water or 175 mM D-mannitol from 24 to 30 hpf (30 embryos per treatment; 1.5 mL of treatment solution per 35 × 10 mm polystyrene Petri dish; one dish per treatment). At 30 hpf, embryos were transferred to clean Petri dishes, washed twice with moleculargrade water, and treated with vehicle (0.1% DMSO), 20 μM TPHP, or 1000 μM DPHP in the presence or absence of 175 mM D-Mannitol until 72 hpf, resulting in a total of six treatment groups. At 72 hpf, survival and hatch rate were recorded, and embryos were fixed and stained as described above.
Hemoglobin Staining.
Zebrafish embryos were exposed to vehicle (0.1% DMSO), DPHP (250, 500, or 1000 μM), or 0.09 μM butafenacil (a positive control and potent inducer of anemia in embryonic zebrafish)26 from 24 to 72, 30 to 72, or 48 to 72 hpf as described above. To quantify hemoglobin levels within developing embryos, 72-hpf embryos were stained with o-dianisidine using previously described protocols.26 All embryos were oriented in dorsal recumbency and imaged against a black background under transmitted light at 3.2× magnification using a Leica MZ10 F stereomicroscope equipped with a DMC2900 camera. The intensity of o-dianisidine within the pericardial region was quantified using ImageJ.
mRNA-Sequencing.
To assess potential effects of DPHP on the embryonic transcriptome, embryos were exposed to system water or 1000 μM DPHP (30 embryos per Petri dish; six Petri dishes per treatment and time-point) from 24 to 30 hpf or 24 to 48 hpf as described above. At 30 and 48 hpf, two Petri dishes containing 25 embryos each were pooled into one cryovial and snap-frozen in liquid nitrogen, resulting in 50 embryos per vial and three replicate cryovials per treatment per time-point. All samples (12 total) were stored at −80 °C until total RNA extraction. Embryos were homogenized in 2 ml cryovials using a PowerGen Homogenizer (ThermoFisher Scientific), and following homogenization, an SV Total RNA Isolation System (Promega) was used to extract total RNA from each replicate sample following the manufacturer’s instructions. RNA quantity and quality were confirmed using a Qubit 4.0 Fluorometer (ThermoFisher Scientific) and 2100 Bioanalyzer system (Agilent), respectively. On the basis of sample-specific Bioanalyzer traces, the RNA Integrity Number (RIN) was >8 for all RNA samples used for library preparations. Libraries were prepared using a Quantseq 3′ mRNA-Seq Library Prep Kit FWD (Lexogen), and mRNA-sequencing was performed as previously described.22
Raw Illumina (fastq.gz) sequencing files (12 files) are available via NCBI’s BioProject database under BioProject ID PRJNA511154, and a summary of sequencing run metrics are provided in Table S1 (>90% of reads were ≥ Q30 across all runs). All 12 raw and indexed Illumina (fastq.gz) sequencing files were downloaded from Illumina’s BaseSpace and uploaded to Bluebee’s genomics analysis platform (www.bluebee.com) to align reads against zebrafish genome assembly GRCz10. After combining treatment replicate files, a DESeq2 application within Bluebee (Lexogen Quantseq DE 1.2) was used to identify significant treatment-related effects on transcript abundance (relative to controls) based on a false discovery rate (FDR) p-adjusted value ≤0.1. Using DESeq2-identified transcripts with a FDR p-adjusted value ≤0.1, downstream analyses were performed within Qiagen’s Ingenuity Pathway Analysis (IPA) as previously described.22 In addition, significantly affected transcripts were imported into the Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.8 for Gene Ontology (GO) enrichment analysis.
Statistical Analyses.
For phenotypic measurements (pericardial area, body length, yolk sac area, and SV-BA length), analytical chemistry, and hemoglobin levels, a general linear model (GLM) analysis of variance (ANOVA) (α = 0.05) was performed using SPSS Statistics 24, as these data did not meet the equal variance assumption for non-GLM ANOVAs. Treatments groups were compared with vehicle controls using pairwise Tukey-based multiple comparisons of least-squares means to identify significant differences in effects.
RESULTS
Effect of DPHP on SV-BA Length.
Up to the highest concentration tested (1000 μM), DPHP exposure from 24 to 72 hpf, 30 to 72 hpf, or 48 to 72 hpf did not significantly affect hatch rate or survival relative to controls (Figure S1). However, exposure to 1000 μM DPHP from 24 to 72 hpf, 30 to 72 hpf, or 48 to 72 hpf resulted in a significant increase in SV-BA length (Figure 1) in the absence of significant, concentration-dependent effects on pericardial area, body length, and yolk sac area (Figure S2). As adverse DPHP-induced effects on the developing heart could not be measured nor quantified until 72 hpf (posthatch), we were unable to quantify SV-BA length and pericardial area at 30 and 48 hpf. However, exposure to 1000 μM DPHP from 24 to 30 hpf or 24 to 48 hpf did not affect body length or yolk sac area relative to controls (Figure S3), suggesting that DPHP exhibited minimal to no toxicity during pharyngula (24−48 hpf). On the basis of these exposures, a nominal concentration of 1000 μM DPHP was selected for all remaining experiments since this concentration reliably increased SV-BA length relative to controls.
Figure 1.
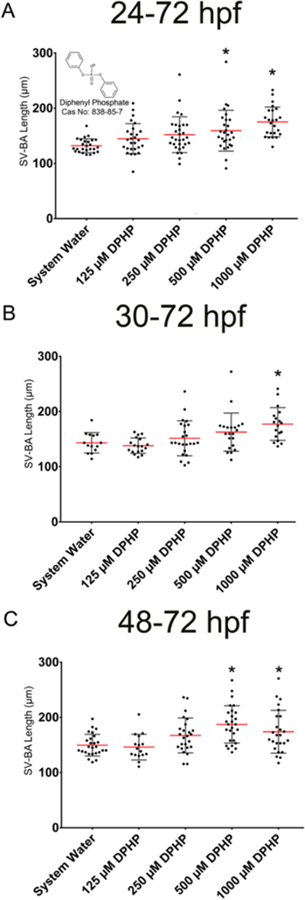
Effect of DPHP on SV-BA length. Mean SV-BA length (±standard deviation) for 72-hpf embryos following exposure from (A) 24 to 72 hpf, (B) 30 to 72 hpf, and (C) 48 to 72 hpf to DPHP (125−1000 μM). Asterisk (*) denotes significant treatment (p < 0.05) relative to controls.
Embryonic Doses of DPHP and TPHP.
Mean embryonic doses of DPHP following a static 24−30 hpf, 24−48 hpf, and 24−72 hpf exposure to 1000 μM DPHP were 23139, 57292, and 56553 ng per 30 embryos, respectively, whereas mean embryonic doses of TPHP within these same treatment groups were below background levels (Figure 2). Within controls, embryonic doses of DPHP and TPHP were either at or below background levels (Figure 2).
Figure 2.
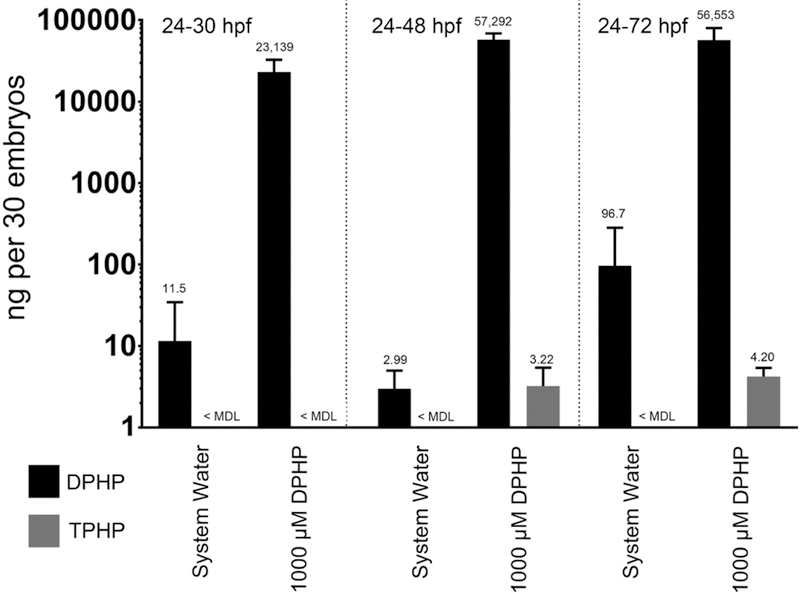
Embryonic doses of DPHP. Mean (±standard deviation) dose (ng) of DPHP detected in homogenates of 30 whole embryos exposed to system water or 1000 μM DPHP from 24 to 30 hpf, 24 to 48 hpf, or 24 to 72 hpf. TPHP doses were at or below background levels across all treatment groups.
Effect of Mannitol, Fenretinide, and HX 531 on DPHP-Induced Cardiotoxicity.
Body length and yolk sac area data for all treatment groups are provided within Figure S4, whereas pericardial area and SV-BA length data for all treatment groups are provided within Figure 3. Pretreatment with 175 mM D-mannitol from 24 to 30 hpf followed by coexposure to 1000 μM DPHP and 175 mM D-mannitol from 30 to 72 hpf resulted in a significant decrease in SV-BA length relative to embryos pretreated with system water followed by exposure to 1000 μM DPHP. Pretreatment with 175 mM D-mannitol from 24 to 30 hpf followed by coexposure to 20 μM TPHP and 175 mM D-mannitol from 30 to 72 hpf significantly decreased pericardial area and SV-BA length relative to TPHP-alone exposures, whereas pretreatment with 2 μM fenretinide from 24 to 30 hpf followed by exposure to 20 μM TPHP from 30 to 72 hpf significantly decreased pericardial area but not SV-BA length relative to TPHP-alone exposures. Pretreatment with 2 μM fenretinide or 5 μM HX 531 did not significantly affect DPHP-induced effects on SV-BA length, and pretreatment with 5 μM HX 531 did not significantly affect TPHP-induced effects on SV-BA length.
Figure 3.
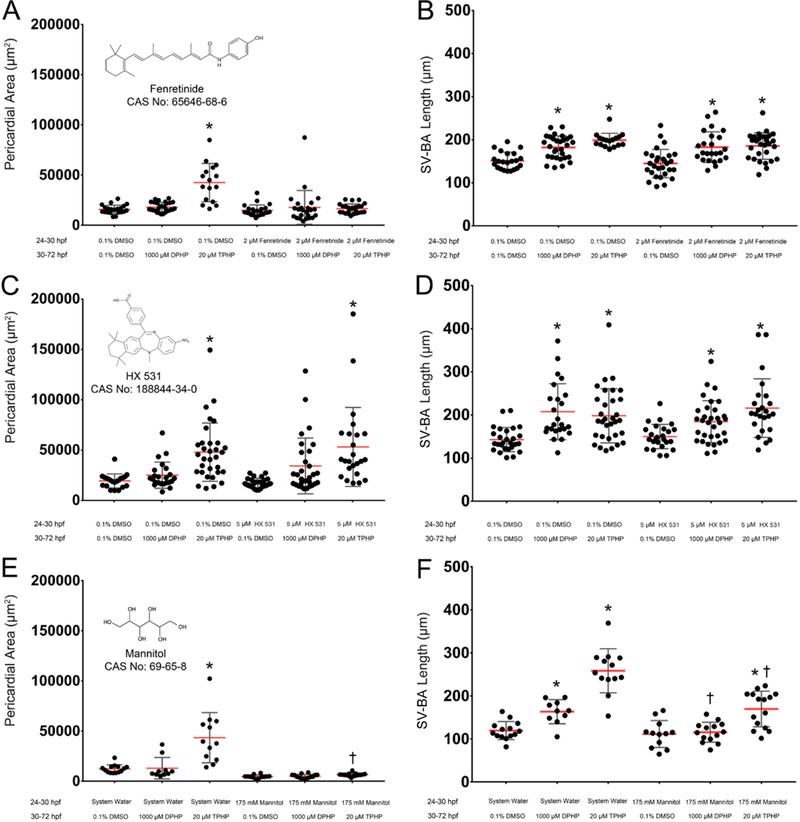
Effect of mannitol, fenretinide, and HX 531 on DPHP-induced cardiotoxicity. Mean pericardial area (±standard deviation) (A, C, E) and mean SV-BA length (±standard deviation) (B, D, F) of 72-hpf embryos pretreated with vehicle (0.1% DMSO), 2 μM fenretinide, 5 μM HX 531, or 175 mM D-mannitol from 24 to 30 hpf and then treated with vehicle (0.1% DMSO), 1000 μM DPHP, or 20 μM TPHP from 30 to 72 hpf. Asterisk (*) denotes significant treatment effect (p < 0.05) relative to vehicle controls (0.1% DMSO), whereas single cross (†) denotes significant coexposure effect (p < 0.05) relative to embryos exposed to TPHP or DPHP alone.
Transcriptional Effects of DPHP at 30 and 48 hpf Following Initiation of Exposure at 24 hpf.
DESeq2 output for 24−30 hpf or 24−48 hpf exposures to 1000 μM DPHP are provided within Tables S2 and S3, respectively. On the basis of these data, initiation of DPHP exposure at 24 hpf resulted in an exposure duration-dependent increase in the total number, magnitude, and statistical significance of affected transcripts relative to controls (Figure 4). On the basis of the lowest p-adjusted value, the most significantly affected transcripts following exposure to 1000 μM DPHP from 24 to 30 and 30−48 hpf were an unannotated mitochondrial gene and apolipoprotein A-Ia, respectively (Tables S2 and S3).
Figure 4.
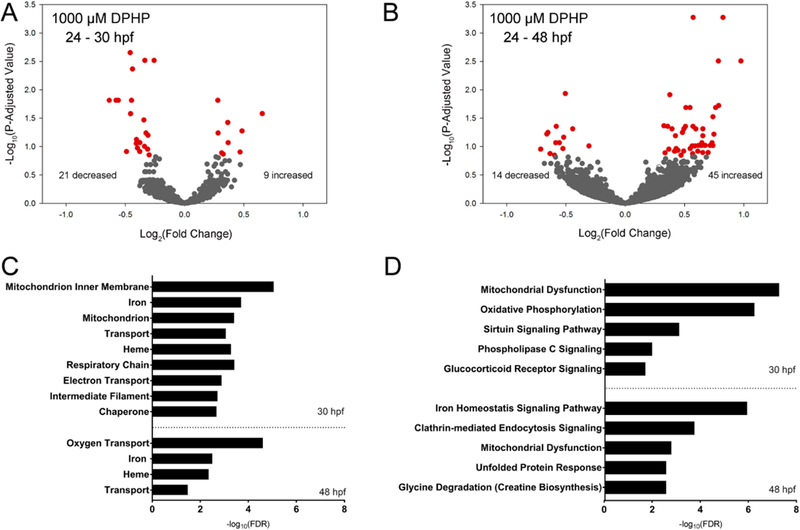
Transcriptional effects of DPHP at 30 and 48 hpf. Volcano plots showing the number of significantly different transcripts (red circles) following exposure to 1000 μM DPHP from 24 to 30 hpf (A) or 24−48 hpf (B); log2-transformed fold change is plotted on the x-axis and log10-transformed p-adjusted value is plotted on the y-axis. DAVID-based (UP_Keyword) processes (C) and IPA-based (canonical) pathways (D) affected by DPHP were identified using a Fisher’s exact p-value ≤ 0.05.
Following automated identification of human, rat, or mouse homologues within IPA, 50% (15 out of 30) and 53% (31 out of 59) of statistically significant transcripts within embryos exposed to 1000 μM DPHP for 30 and 48 hpf, respectively, were included in IPA’s Tox Analysis; the remaining statistically significant transcripts were excluded by IPA’s Tox Analysis due to the absence of human, rat, and/or mouse orthologs within NCBI’s HomoloGene database. A complete list of all significantly affected canonical pathways identified by IPA’s Tox Analysis for 1000 μM DPHP for 30 and 48 hpf are provided within Tables S4 and S5, respectively. We also used DAVID v6.8 to compare against IPA-predicted pathways impacted by DPHP (Tables S6 and S7). Unlike IPA, DAVID relies on zebrafish-specific annotation and, as such, identifies overrepresented GO terms without discarding statistically significant, DESeq2-identified transcripts. On the basis of UniprotKB Keywords27 (UP_Keywords), nine and four processes were significantly altered at 30 and 48 hpf, respectively, based on a Fisher’s Exact Test p-value threshold of 0.05 (Figure 4).
For transcripts significantly impacted by DPHP, curated data available within the Zebrafish Information Network (https://zfin.org/) were queried to identify transcript-specific chromosome locations (including nuclear or mitochondrial) and wildtype embryonic tissue localization. Out of 30 transcripts that were significantly altered at 30 hpf, five transcripts were derived from mitochondrial genes. Indeed, mitochondrial dysfunction and oxidative phosphorylation pathways (based on IPA) and mitochondrial-specific structures and processes (mitochondrion inner membrane, mitochondrion, respiratory chain, and electron transport) (based on DAVID) were significantly altered at 30 hpf. At 48 hpf, mitochondria dysfunction was an altered pathway based on IPA, although no transcripts were derived from mitochondrial genes. At 30 and 48 hpf, red blood cell-related terms (iron, heme, and oxygen transport based on DAVID) and pathways (iron homeostasis signaling pathway based on IPA) were significantly altered.
Effect of DPHP on Hemoglobin Levels.
As expected, butafenacil induced anemia following exposure from 24 to 72 hpf or 30 to 72 hpf, whereas exposures initiated at 48 hpf did not result in adverse effects on hemoglobin levels at 72 hpf (Figure 5). Exposure to 1000 μM DPHP from 24 to 72 hpf resulted in significantly decreased hemoglobin levels compared to negative controls, whereas initiation of exposure to 1000 μM DPHP at 30 and 48 hpf, as well as exposure to 250 or 500 μM DPHP from 24 to 30 hpf, 24 to 48 hpf, or 24 to 72 hpf, did not significantly affect hemoglobin levels (Figure 5).
Figure 5.
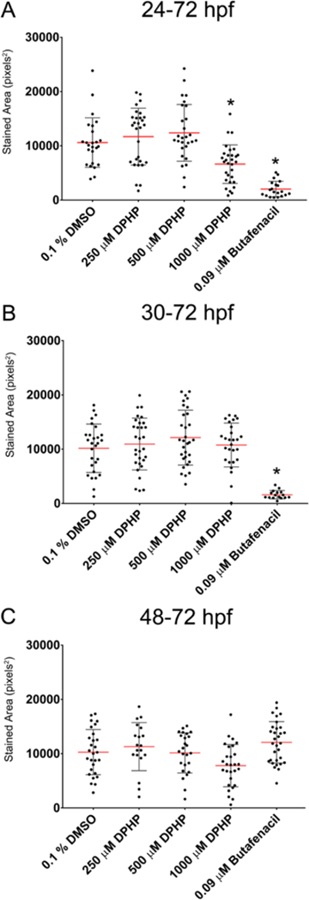
Effect of DPHP on hemoglobin levels. Mean stained area of hemoglobin (±standard deviation) within 72-hpf embryos exposed to vehicle (0.1% DMSO), DPHP (250, 500, or 1000 μM), or 0.09 μM butafenacil (positive control) from 24 to 72 hpf (A), 30 to 72 hpf (B), or 48 to 72 hpf (C). Asterisk (*) denotes significant treatment effect (p < 0.05) relative to vehicle controls (0.1% DMSO).
DISCUSSION
Within this study, we found that exposure to high concentrations of DPHP (1000 μM) blocked cardiac looping and increased SV-BA length in the absence of effects on survival, hatch rate, and gross morphology (body length, pericardial area, and yolk sac area). There is a possibility that, at concentrations higher than 1000 μM, DPHP may adversely affect survival, hatch rate, and/or growth. However, the potency and toxicity of DPHP within developing zebrafish embryos was minimal even at the highest nominal concentration tested (1000 μM). In addition to TPHP,22,24,32 compounds such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD),28 triadimefon,29 and propranolol30 also block cardiac looping and increase SV-BA length within developing zebrafish embryos. TCDD induces developmental toxicity via the aryl hydrocarbon receptor pathway,31 whereas triadimefon alters Ca2+ signaling,29 and propranolol is a β-adrenergic receptor antagonist.30 Therefore, structurally diverse compounds with varying mechanisms of action have the potential to block cardiac looping in developing zebrafish embryos, suggesting that there may be common pathways and/or processes that underlie chemically induced cardiotoxicity during development.
The embryonic dose of DPHP following exposure from 24 to 72 hpf was 56553 ng per 30 embryos. As there was 375000 ng of DPHP within 1.5 mL of 1000 μM DPHP solution, approximately 15% percent of available DPHP was detected within whole embryo homogenates even though DPHP has a log Kow= 1.4. In contrast, based on data available within Mitchell et al. (2018),22 approximately 6% percent of available TPHP (log Kow = 4.59) was detected within whole embryo homogenates following exposure to 20 μM TPHP from 24 to 72 hpf. Therefore, while the embryonic dose of DPHP within this study was significantly higher than the embryonic dose of TPHP in our prior study, the toxicity of DPHP was significantly lower than TPHP despite minor differences in chemical structure (e.g., two vs three benzene rings within DPHP vs TPHP, respectively).
Our lab and others have shown that exposure of developing zebrafish to TPHP prior to cardiac looping results in heart defects and pericardial edema.22,24,32,33 TPHP-induced edema may be due to decreased water export, and other organs may be affected if TPHP disrupts proper kidney development or impairs glomerular function.28 Therefore, TPHP might affect electrolyte/solute reabsorption and/or disturbing osmotic balance both of which may lead to pericardial edema. Interestingly, D-mannitol decreased TPHP-induced pericardial edema and SV-BA length relative to TPHP alone, suggesting that TPHP-induced pericardial edema may be leading to cardiotoxicity. However, D-mannitol also mitigated DPHP-induced effects on SV-BA length (relative to controls) despite the absence of DPHP effects on pericardial area. While there is a possibility that D-mannitol may have decreased DPHP or TPHP uptake into the embryo, our prior studies have demonstrated that coexposure to TPHP in the presence of BMS493, fenretinide, or ciglitazone does not affect TPHP uptake within developing zebrafish embryos.22,24 Moreover, mannitol has been previously used to mitigate TCDD-induced pericardial edema within developing zebrafish embryos25,28 and, to our knowledge, there is no evidence in the published literature to suggest that mannitol interacts with other organic chemicals within solution. Alternatively, our data suggest that DPHP-induced cardiac defects may be independent of edema formation and, rather, a result of toxicity to other tissues and/ or organs within the developing embryo. For example, other studies in zebrafish have suggested that adverse effects of chemical exposure on the developing heart may be mediated by direct injury to the liver, as was previously observed with naproxen.34
The pericardial area has been widely used as a biomarker of cardiotoxicity with embryonic zebrafish.22,24,32 We previously reported that fenretinide (a pan-RAR agonist) blocked TPHP-induced pericardial edema.22 However, despite blocking edema formation, within this study, we found that fenretinide was unable to mitigate DPHP-and TPHP-induced effects on SV-BA length. Similarly, exposure of zebrafish embryos to ethanol results in pericardial edema a phenotype that was mitigated by coexposing with all-trans retinoic acid.35,36 Therefore, RAR/RXR signaling may play a role in water export within the pericardial region, resulting in decreased TPHP-induced pericardial edema with sustained cardiac looping defects. Contrary to TPHP, pretreatment with fenretinide or HX 531 (a pan-RXR antagonist) did not mitigate or enhance DPHP-induced toxicity, suggesting that DPHP-induced cardiotoxicity is not mediated via RAR/RXR signaling.
On the basis of a targeted gene expression panel using chicken embryonic hepatocyte cultures, DPHP exposure significantly altered transcripts related to Phase I or II metabolism, glucose/fatty acid metabolism, lipid/cholesterol metabolism, thyroid hormone pathway, and farnesoid X receptor/liver X receptor signaling.18 However, to our knowledge, no previous studies have evaluated the potential effect of DPHP on the transcriptome within any model system. On the basis of mRNA-sequencing, the most affected processes and pathways within embryonic zebrafish were specific to red blood cells and mitochondria. Indeed, 1000 μM DPHP significantly decreased hemoglobin levels following exposure from 24 to 72 hpf (but not 30−72 hpf or 48−72 hpf or after circulation commences), suggesting that transcriptional profiling at 30 and 48 hpf predicted downstream impacts on red blood cell abundance at 72 hpf. Since we did not quantify the potential for DPHP to affect mitochondrial structure and/or function within this study, follow up studies are needed to determine if DPHP targets mitochondria during embryonic development.
Our data are consistent with other studies demonstrating that DPHP is less cytotoxic than TPHP in vitro. For example, the U.S. Environmental Protection Agency’s ToxCast data (based on cell-free and cell-based systems) show that out of 882 assays, 113 were identified as active hits (12.8%) for TPHP whereas out of 316 assays, only three were identified as active hits (0.95%) for DPHP.37 However, even though TPHP is more potent than DPHP, in utero exposure to TPHP within the general human population is likely minimal since TPHP is rapidly metabolized to DPHP within mammals,2 DPHP is hydrophilic, and DPHP will likely be rapidly excreted via urine within pregnant women.
To our knowledge, this is one of the first studies to investigate the potential of DPHP-induced effects on early embryonic development. Collectively, our data suggest that high concentrations of DPHP (1000 μM) induce cardiotoxicity and hemotoxicity during embryonic development within zebrafish, possibly via mitochondria dysfunction, hepatoxicity, and/or renal toxicity. Interestingly, adverse effects of DPHP on the embryonic transcriptome at 30 and 48 hpf preceded detectable, downstream effects on cardiac development and red blood cell formation phenotypes that may have been driven by mitochondrial dysfunction earlier in development. If persistent, these cardiac and circulatory defects may inhibit proper larval or adult growth, survival, and reproduction. Finally, our data suggest that the potential risk of TPHP in utero may be minimal, as TPHP is rapidly metabolized to DPHP in mammals and DPHP is significantly less potent than TPHP in zebrafish as well as cell-free and cell-based systems. However, our findings suggest that future studies are needed to better understand the potential of DPHP to impair mitochondrial function as well as induce renal toxicity, hepatoxicity, and hemotoxicity within model systems that are more relevant to humans.
Supplementary Material
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.8b07238.
Sequencing run metrics, DESeq2 output, and significantly altered IPA-based pathways and DAVID-based processes (XLSX)
Effect of DPHP on survival rate or hatch rate at 72 hpf, effect of DPHP at 30 and 48 hpf following initiation of exposure at 24 hpf, effect of DPHP on morphological endpoints at 72 hpf, effect of mannitol, fenretinide, and HX 531 on DPHP-induced effects on body length and yolk sac area (PDF)
ACKNOWLEDGMENTS
Fellowship support was provided by UCR’s Graduate Division to A.R. and NRSA T32 Training Program [T32ES018827] to C.A.M. Research support was provided by a National Institutes of Health grant [R01ES027576] and the USDA National Institute of Food and Agriculture Hatch Project [1009609] to D.C.V.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).EPA Chemicals under the Toxic Substance Control Act (TSCA) https://www.epa.gov/chemicals-under-tsca.
- (2).Sasaki K; Suzuki T; Takeda M; Uchiyama M Metabolism of phosphoric acid triesters by rat liver homogenate. Bull. Environ. Contam. Toxicol 1984, 33, 281–288. [DOI] [PubMed] [Google Scholar]
- (3).Van den Eede N; Ballesteros-Gomez A; Neels H; Covaci A Does biotransformation of aryl phosphate flame retardants in blood cast a new perspective on their debated biomarkers? Environ. Sci. Technol 2016, 50 (22), 12439–12445. [DOI] [PubMed] [Google Scholar]
- (4).Dodson RE; Van den Eede N; Covaci A; Perovich LJ; Brody JG; Rudel RA Urinary biomonitoring of phosphate flame retardants: levels in California adults and recommendations for future studies. Environ. Sci. Technol 2014, 48, 13625–13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ballesteros-Gomez A; Van den Eede N; Covaci A In vitro human metabolism of the flame retardant resorcinol bis-(diphenylphosphate) (RDP). Environ. Sci. Technol 2015, 49 (6), 3897–3904. [DOI] [PubMed] [Google Scholar]
- (6).van der Veen I; de Boer J Phosphorus flame retardants: Properties, production, environmental occurrence, toxicity and analysis. Chemosphere 2012, 88 (10), 1119–1153. [DOI] [PubMed] [Google Scholar]
- (7).Hoffman K; Garantziotis S; Birnbaum LS; Stapleton HM Monitoring indoor exposure to organophosphate flame retardants: hand wipes and house dust. Environ. Health Perspect 2015, 123, 160–>165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhao F; Wan Y; Zhao H; Hu W; Mu D; Webster TF; Hu J Levels of blood organophosphorus flame retardants and association with changes in human sphingolipid homeostasis. Environ. Sci. Technol 2016, 50, 8896–8903. [DOI] [PubMed] [Google Scholar]
- (9).Bjornsdotter MK; Romera-Garcia E; Borrull J; de Boer J; Rubio S; Ballesteros-Gomez A Presence of diphenyl phosphate and aryl-phosphate flame retardants in indoor dust from different microenvironments in Spain and the Netherlands and estimation of human exposure. Environ. Int 2018, 112, 59–67. [DOI] [PubMed] [Google Scholar]
- (10).Hoffman K; Butt CM; Chen A; Limkakeng AT; Stapleton HM High exposure to organophosphate flame retardants in infants: associations with baby products. Environ. Sci. Technol 2015, 49, 14554–14559. [DOI] [PubMed] [Google Scholar]
- (11).He C; English K; Baduel C; Thai P; Jagals P; Ware RS; Li Y; Wang X; Sly PD; Mueller JF Concentrations of organophosphate flame retardants and plasticizers in urine from young children in QueenslFand, Australia and associations with environmental and behavioural factors. Environ. Res 2018, 164, 262–>270. [DOI] [PubMed] [Google Scholar]
- (12).Zhang B; Lu S; Huang M; Zhou M; Zhou Z; Zheng H; Jiang Y; Bai X; Zhang T Urinary metabolites of organophosphate flame retardants in 0−5-year-old children: Potential exposure risk for inpatients and home-stay infants. Environ. Pollut 2018, 243, 318–325. [DOI] [PubMed] [Google Scholar]
- (13).Preston EV; McClean MD; Claus Henn B.; Stapleton HM; Braverman LE; Pearce EN; Makey CM; Webster TF. Associations between urinary diphenyl phosphate and thyroid function. Environ. Int 2017, 101, 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kim S; Jung J; Lee I; Jung D; Youn H; Choi K Thyroid disruption by triphenyl phosphate, an organophosphate flame retardant, in zebrafish (Danio rerio) embryos/larvae, and in GH3 and FRTL-5 cell lines. Aquat. Toxicol 2015, 160, 188–196. [DOI] [PubMed] [Google Scholar]
- (15).Kojima H; Takeuchi S; Itoh T; Iida M; Kobayashi S; Yoshida T In vitro endocrine disruption potential of organophosphate flame retardants via human nuclear receptors. Toxicology 2013, 314 (1), 76–83. [DOI] [PubMed] [Google Scholar]
- (16).Liu X; Jung D; Jo A; Ji K; Moon HB; Choi K Longterm exposure to triphenyl phosphate alters hormone balance and HPG, HPI, and HPT gene expression in zebrafish (Danio rerio). Environ. Toxicol. Chem 2016, 35 (9), 2288–2296. [DOI] [PubMed] [Google Scholar]
- (17).Wang D; Zhu W; Chen L; Yan J; Teng M; Zhou Z Neonatal triphenyl phosphate and its metabolite diphenyl phosphate exposure induce sex-and dose-dependent metabolic disruptions in adult mice. Environ. Pollut 2018, 237, 10–17. [DOI] [PubMed] [Google Scholar]
- (18).Su G; Crump D; Letcher RJ; Kennedy SW Rapid in vitro metabolism of the flame retardant triphenyl phosphate and effects on cytotoxicity and mRNA expression in chicken embryonic hepatocytes. Environ. Sci. Technol 2014, 48 (22), 13511–13519. [DOI] [PubMed] [Google Scholar]
- (19).Kojima H; Takeuchi S; Van den Eede N; Covaci A Effects of primary metabolites of organophosphate flame retardants on transcriptional activity via human nuclear receptors. Toxicol. Lett 2016, 245, 31–39. [DOI] [PubMed] [Google Scholar]
- (20).Triphenyl Phosphate pubchem.ncbi.nlm.nih.gov/compound/triphenyl_phosphate (accessed Dec 21st, 2018).
- (21).Diphenyl Phosphate pubchem.ncbi.nlm.nih.gov/compound/13282 (accessed Dec 21st, 2018).
- (22).Mitchell CA; Dasgupta S; Zhang S; Stapleton HM; Volz DC Disruption of nuclear receptor signaling alters triphenyl phosphate-induced cardiotoxicity in zebrafish embryos. Toxicol. Sci 2018, 163 (1), 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yozzo KL; McGee SP; Volz DC Adverse outcome pathways during zebrafish embryogenesis: A case study with paraoxon. Aquat. Toxicol 2013, 126, 346–354. [DOI] [PubMed] [Google Scholar]
- (24).Isales GM; Hipszer RA; Raftery TD; Chen A; Stapleton HM; Volz DC Triphenyl phosphate-induced developmental toxicity in zebrafish: Potential role of the retinoic acid receptor. Aquat. Toxicol 2015, 161, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hill AJ; Bello SM; Prasch AL; Peterson RE; Heideman W Water permeability and TCDD-induced edema in zebrafish early-life stages. Toxicol. Sci 2004, 78 (1), 78–87. [DOI] [PubMed] [Google Scholar]
- (26).Leet JK; Lindberg CD; Bassett LA; Isales GM; Yozzo KL; Raftery TD; Volz DC High-content screening in zebrafish embryos identifies butafenacil as a potent inducer of anemia. PLoS One 2014, 9 (8), No. e104190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Keywords. www.uniprot.org/help/keywords (accessed Dec 21st, 2018).
- (28).Antkiewicz DS; Burns CG; Carney SA; Peterson RE; Heideman W Heart malformation is an early response to TCDD in embryonic zebrafish. Toxicol. Sci 2005, 84 (2), 368–377. [DOI] [PubMed] [Google Scholar]
- (29).Liu HC; Chu TY; Chen LL; Gui WJ; Zhu GN The cardiovascular toxicity of triadimefon in early life stage of zebrafish and potential implications to human health. Environ. Pollut 2017, 231, 1093–1103. [DOI] [PubMed] [Google Scholar]
- (30).Finn J; Hui M; Li V; Lorenzi V; de la Paz N; Cheng SH; Lai-Chen L; Schlenk D Effects of propranolol on heart rate and development in Japanese medaka (Oryzias latipes) and zebrafish (Danio rerio). Aquat. Toxicol 2012, 122–123, 214. [DOI] [PubMed] [Google Scholar]
- (31).Thackaberry EA; Gabaldon DM; Walker MK; Smith SM Aryl hydrocarbon receptor null mice develop cardiac hypertrophy and increased hypoxia-inducible factor-1alpha in the absence of cardiac hypoxia. Cardiovasc. Toxicol 2002, 2, 263–274. [DOI] [PubMed] [Google Scholar]
- (32).McGee SP; Konstantinov A; Stapleton HM; Volz DC Aryl phosphate esters within a major pentaBDE replacement product induce cardiotoxicity in developing zebrafish embryos: potential role of the aryl hydrocarbon receptor. Toxicol. Sci 2013, 133 (1), 144–156. [DOI] [PubMed] [Google Scholar]
- (33).Du Z; Guowei W; Gao S; Wang Z Aryl organophosphate flame retardants induced cardiotoxicity during zebrafish embryogenesis: by disturbing expression of the transcriptional regulators. Aquat. Toxicol 2015, 161 (2015), 25–32. [DOI] [PubMed] [Google Scholar]
- (34).Li Q; Wang P; Chen L; Gao H; Wu L Acute toxicity and histopathological effects of naproxen in zebrafish (Danio rerio) early life stages. Environ. Sci. Pollut. Res 2016, 23 (18), 18832–18841. [DOI] [PubMed] [Google Scholar]
- (35).Ferdous J; Mukherjee R; Ahmed KT; Ali DW Retinoic acid prevents synaptic deficiencies induced by alcohol exposure during gastrulation in zebrafish embryos. NeuroToxicology 2017, 62, 100–110. [DOI] [PubMed] [Google Scholar]
- (36).Marrs JA; Clendenon SG; Ratcliffe DR; Fielding SM; Liu Q; Bosron WF Zebrafish fetal alcohol syndrome model: effects of ethanol are rescued by retinoic acid supplement. Alcohol 2010, 44 (7−8), 707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).ToxCast Dashboard https://www.epa.gov/chemical-research/toxcast-dashboard (accessed Dec 21st, 2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.8b07238.
Sequencing run metrics, DESeq2 output, and significantly altered IPA-based pathways and DAVID-based processes (XLSX)
Effect of DPHP on survival rate or hatch rate at 72 hpf, effect of DPHP at 30 and 48 hpf following initiation of exposure at 24 hpf, effect of DPHP on morphological endpoints at 72 hpf, effect of mannitol, fenretinide, and HX 531 on DPHP-induced effects on body length and yolk sac area (PDF)