

Abstract
Ovarian cancer is the sixth most prevalent cancer in women and the most lethal of the gynecologic malignancies. Treatments have comprised the use of immunotherapeutic agents as well as oncolytic viruses, with varying results for reasons that remain to be clarified. To better understand the mechanisms which may help predict treatment outcome, we have evaluated innate immune signaling in select ovarian cancer cell-lines, governed by the Stimulator of Interferon Genes (STING) which controls self or viral DNA triggered cytokine production. Our results indicate that STING dependent signaling is habitually defective in majority of ovarian cancer cells examined, frequently through the suppression of STING and/or the cyclic dinucleotide (CDNs) enzyme Cyclic GMP-AMP synthase (cGAS) expression, by epigenetic processes. However, STING-independent, dsRNA activated innate immune cytokine production, which require RIG-I/MDA5, were largely unaffected. Such defects enabled ovarian cancer cells to avoid DNA-damaged-mediated cytokine production which would alert the immunosurveillance system. Loss of STING signaling also rendered ovarian cancer cells highly susceptible to viral oncolytic γ34.5 deleted-HSV1 (Herpes simplex virus) infection in vitro and in vivo.
Keywords: STING, cGAS, ovarian cancer, oncoviral therapy, γ34.5 deleted-HSV1, innate immunity
Introduction
Cancer is an important world public health problem and ovarian cancer is the sixth most prevalent cancer in women and the most lethal of the gynecologic malignancies (1-3). Approximately 90% of ovarian tumors are epithelial in origin but most carcinomas follow a pathway of dissemination that involves intravasation into the bloodstream followed by extravasation at distant tissue sites. After spread to lymphatic system, metastasis occurs by dissemination of cancer cells from the primary tumor site into the ascites of the peritoneal space, followed by secondary tumor development in abdominal organs (4,5). Current standard treatment consists of surgery followed by chemotherapy but more than 65% of patients will eventually relapse (6). Thus, there is an urgent need to find more effective therapeutic approaches for the treatment of recurrent or drug-resistant ovarian cancer.
Oncolytic DNA and RNA viruses are under evaluation as a cancer therapy and a number of clinical trials are in progress, with varying degrees of success (7). The mechanisms explaining viral oncolysis remain to be fully evaluated but likely include cellular defects in innate immunity and/or translation control, since these molecular flaws confer advantages to tumor growth and survival (8-10). Recently, Amgen talimogene laherparepvec (T-VEC) a herpes simplex virus type 1 (HSV-1)–based oncolytic viruses engineered to express granulocyte-macrophage colony-stimulating factor (GM-CSF), was approved in USA to treat advanced melanoma (3, 11-14). However, a lack of prognostic insight into whether a particular tumor will respond to select virotherapies is clearly apparent.
Innate immune sensors responsible for detecting microbial infection and generating host defense responses are known to comprise the Toll-like receptor (TLR) and the RIG-I/MDA5 (RLR) pathways (15). The RLR pathway plays a key role in detecting RNA viruses and facilitating adaptive immune responses to eradicate such infectious agents (16). Conversely, the host has devised alternate ways to detect DNA based microbes such as members of the Herpes virus family. For example, the cellular protein referred to as Stimulator of Interferon Genes (STING) is now known to control cytosolic DNA induced innate immune signaling. STING is the sensor for cyclic dinucleotides species [CDNs], including the 2’-3’cGAMP (c[G(2’,5’)pA(3’,5’)p]) produced by a cellular nucleotidyltransferase referred to as cGAS (cyclic GMP-AMP synthase) following its association with cytosolic DNA species (17). CDNs such as cyclic di-AMP (c-di-AMP) directly generated by intracellular bacteria are also potent activators of STING-dependent cytokine production (18,19). Generally, the cytosol is a DNA free zone and so innate immune responses can be triggered in the cytoplasm by the genomes of invading DNA microbes or even self-DNA leaked from the nucleus or possibly mitochondria (17,20). For example, DNA damage is known to cause the leakage of chromatin into the cytosol and even create cytoplasmic micronuclei, which are capable of activating STING signaling. When stimulated with dsDNA, STING rapidly undergoes translocation from the endoplasmic reticulum (ER), along with TBK1, to perinuclear-associated endosomal regions, containing NF-kB and IRF3 that translocate to nucleus and activate type I interferon responses (21,22). STING signaling has been shown to be essential for protecting cells against a variety of pathogens and likely plays a key role in sensing DNA damage and for producing cytokines that alert the immune system to the damaged location (23). STING has further been described as playing an essential role in type I IFN-dependent and activation of cytotoxic T cell anti-tumor responses (24,25).
Our group recently shown that innate immune signaling mediated by STING is impaired in colon cancer cells as well as melanoma (9,10). Possibly, these events may enable tumor cells to escape DNA-damage activated immune responses, as described. In many cases, this inhibition occurred by silencing STING and/or cGAS through processes involving epigenetic hypermethylation. Suppression of STING signaling may be a key requirement for tumor cell escape and for the development of malignant disease. Paradoxically, these same defects in STING signaling may render cancer cells highly susceptible to oncolytic viral infections (9,10). Therefore, the analysis of STING expression in different types of cancers may shed light into the causes of malignant disease as well as facilitate the development of diagnostic and prognostic tools and new therapies that may help cancer treatment.
In this study, we examined the expression and activation of STING pathway in tumor cells established from human ovarian cancers at various stages. Our results indicated that STING and/or cGAS was frequently absent the majority of cell lines examined, with concomitant loss of cytosolic DNA signaling and cytokine production. In contrast, the majority of ovarian cancer lines were readily able to produce cytokines in response to Poly(I:C), indicating that RLR pathway was intact. Defects in STING signaling also facilitated HSV1Δγ34.5 (γ34.5 deleted-HSV1) infection. Collectively, our data indicates that the STING pathway is suppressed in a majority of ovarian cancers and these defects render cancer cells more susceptible to oncolytic virus treatment. Thus, evaluating the expression levels of both STING and cGAS may provide a better prognostic assay that could help predict the outcome of oncoviral therapy in ovarian cancer and others.
Material and Methods
Materials
All reagents were from Invitrogen, Thermo Fisher Scientific or Sigma-Aldrich unless specified.
Cell culture and treatments
Normal human ovarian surface epithelial cells (HOSE) and human ovarian cancer cell lines (ES2, OVCAR3 and SK-OV-3) were purchased from and authenticated by Lonza and ATCC, respectively. Dr. Xiang-Xi Xu kindly provided other ovarian cancer cell lines (A1847, A2780, OVCAR4, OVCAR5, OVCAR8, OVCAR10, PEO1 and UPN251) (Supplementary Table S1) (26). HOSE was cultured in their appropriate growth media from Lonza and ovarian cancer cell lines in RPMI supplemented with 10% fetal bovine serum. hTERT-BJ1 telomerase fibroblasts (hTERT) were originally from Clontech and cultured in 4:1 ratio of DMEM:Medium 199 supplemented with 10% fetal bovine serum (FBS), 4 mM L-glutamine, and 1 mM sodium pyruvate at 37° C in a 5% CO2-humidified atmosphere. dsDNA and polyI:C transfection was done with lipofectamine following manufactures instructions. 5-aza-2’-deoxycytidine (5AZADC) treatment was done at 10 uM for indicate period of time.
Immunoblot analysis
Equal amounts of proteins were resolved on SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). After blocking with 5% blotting-grade blocker (Bio-Rad) in PBS-T buffer (Phosphate-buffered saline [PBS] with 0.1% Tween 20), membranes were incubated with the indicated primary antibodies. Anti-STING rabbit polyclonal antibody were prepared as described previously (21). Other antibodies used in this paper were as follows: β-actin (Sigma, A5441), phospho-IRF3 (Cell Signaling Technology, 4947), IRF3 (Cell Signaling Technology, 4302), phospho-p65 (Cell Signaling Technology, 3031), p65 (Cell Signaling Technology, 3987), phospho-TBK1 (Cell Signaling Technology, 5483), TBK1 (Abcam, ab40676), phospho-STAT1 (Cell Signaling Technology, 9167), STAT1 (Cell Signaling Technology, 14994). The membrane was washed three times using PBS-T buffer and then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit or mouse immunoglobulin G (IgG) (Promega). After three washes, Super Signal West Pico or Femto (Thermo Fisher Scientific) was used to develop the signal, and the membranes were exposed to Premium X-ray film (Phenix).
IFNβ ELISA analysis
IFNβ ELISA was performed using the Human IFNβ ELISA Kit (PBL InterferonSource) following the manufacturer’s protocol.
Quantitative Real-time PCR (qPCR)
Total RNA was reverse-transcribed using QuantiTect Reverse Transcription Kit (Qiagen) following the manufacturer’s instructions. qPCR was performed with the TaqMan gene Expression Assay (Applied Biosystems) to analyze the expression of cGAS, IFNβ, CXCL10, IFIT1 and CCL5 genes using StepOnePlus real-time PCR system (Applied Biosystems). GAPDH was used for normalization.
Immunofluorescence microscopy
Cells were cultured and treated on poly-D-lysine-coated round coverslips (BD Biosciences). Cell were fixed with 4% paraformaldehyde/PBS for 15 minutes at 37° C and permeabilized with 0.2% Triton X-100/PBS for 10 minutes at room temperature (RT). After blocking with 10% BSA/PBS for 30 min, the coverslips were incubated with the indicated primary antibodies in 4% BSA/PBS overnight at 4° C in a wet chamber. Immunostaining was performed with rabbit-anti-STING polyclonal (21), rabbit-anti-IRF3 (Santa Cruz Biotechnology), or rabbit-anti-p65 (Cell Signaling). After washing six times with PBS, the coverslips were incubated with fluorescence conjugated secondary antibody (Alexa Fluor 488-goat anti-rabbit IgG) for 2 h at RT in a wet chamber. After washing six times with PBS, the coverslips were mounted onto glass slides with ProLong Gold antifade reagent (Invitrogen). Images were taken with Leika LSM confocal microscope at the Image Core Facility, University of Miami (Miami, FL).
Immunohistochemistry and Histological Analysis
Tissue microarray was purchased from Pantomics. Immunohistochemistry staining was performed with cGAS or STING antibody following standard protocol. The score for the extent of the IHC-stained area was set as 0 for no IHC signal at all, 1 for <10%, 2 for 10% to 50%, and 3 for >50% of tumor cells stained. The score for IHC intensity was also scaled as 0 for no IHC signal, 1 for weak, 2 for moderate, and 3 for strong. The final score used in the analysis was calculated by multiplying the extent and intensity score, with a maximum score of 9. Staining was considered positive if scored above 3.
Virus amplification, purification, titration, and infection
HSV1Δγ34.5 was kindly provided by Dr. Bernard Roizman (27). Virus was amplified in Vero cells and purified by sucrose gradient ultracentrifugation following standard protocol. Plague assay using serial diluted virus was performed in Vero cells following standard protocol for virus titration. Normal and ovarian cells were infected with HSV1Δγ34.5 at M.O.I (multiplicity of infection) = 1 or 10 for 1 hour, washed, and then incubated with fresh medium (DMEM/10% FBS) for 24h. The infected cells were freeze-thawed three times to release virus from the cells. Vero cells were incubated with the supernatants from those infected cells for 1 h and then cultured in 1% low-melting agarose (Invitrogen)/DMEM/5% FBS to avoid a secondary colony. After 48h incubation we removed the overlaid gel and fixed/stained the plates with 0.1% crystal violet/30% methanol solution to visualize and count plaques. Cell viability was analyzed by trypan blue staining 24 h after infection. Ovarian cells were also infected with HSV1 expressing luciferase (HSV-luc), kindly provided by Dr. David A. Leib, at M.O.I. 1 or 10 for 24 h, and viral propagation was analyzed by luciferase assay (RLU, relative light units).
Mouse treatment
Balb/C nu/nu mice were purchased from Charles River and maintained in the institutional Division of Veterinary Resources. All experiments were performed with Institutional Animal Care and Use Committee (IACUC) approval and in compliance with IACUC guidelines. Tumor cells were introduced in the flanks of Balb/c nude mice by subcutaneous injection of 2E6 of the appropriate tumor cells and tumors allowed to develop to an average diameter of approximately 0.5 cm. HSV1Δγ34.5 was then injected into the tumors every other day for a total of 3 times at the appropriate dosage (i.e., 50 μl at 1E8 PFU). PBS was used as vehicle control. Effects on tumor growth were monitored every other day using a digital caliper. Mice were euthanized when tumor diameter exceeds 10 mm.
Statistical analysis
All statistical analysis was performed by the Student t test unless specified. The data were considered to be significantly different when P < 0.05.
Results
Impairment of STING and cGAS expression and activity in ovarian cancer cells
STING signaling has found to be suppressed in a number of cancer types including colorectal carcinoma and melanoma (9,10,28), suggesting that this pathway may play an important role in helping to prevent cellular transformation. To extend our studies, we examined the expression and activation of the STING pathway in 11 cell lines established from human ovarian cancer at various stages. We first evaluated STING expression by Immunoblot in these cell lines and showed that STING was reduced in 3 of the 11 cell lines examined (A1847, A2780 and ES2) (Fig. 1A). However, robust STING signaling requires 2’3’cGAMP generated by cGAS upon association with cytosolic DNA. Thus, to complement this analysis we also examined cGAS expression in the cells and observed that the expression of this synthase was diminished in 7 of 11 ovarian cancer cell lines (Fig. 1A). Indeed, some cells had greatly decreased cGAS and STING expression (A1847, A2780 and ES2). To complement this approach, we transfected the cells with dsDNA90 to activate STING or dsRNA (polyI:C) to activate RIG-I/MDA5 signaling. Using control hTERT cells and HOSE (human ovarian surface epithelial) cells, we confirmed that dsDNA90 transfection was able to induce IFNβ production (Fig. 1B). However, all 11 ovarian cancers analyzed responded poorly to dsDNA90-mediated cytokine production. In contrast, 9 of the 11 ovarian cancer cell lines produced varying levels of type I IFN in response to polyI:C, indicating that the RLR pathway is preserved in most of the cases examined (Fig. 1B). We noticed that some cells that contained both STING and cGAS were also unresponsive to cytosolic DNA (OVCAR4 and PEO1) suggesting that cytosolic DNA signaling may be impaired at various points of this pathway (Fig. 1B). A similar profile was observed following evaluation of IFNβ and CXCL10 stimulation by qPCR (Fig. 1C and D). Our results suggest that the cytosolic DNA induced STING-dependent signaling pathway is frequently impaired in ovarian cancer cells, but not dsRNA signaling.
Figure 1.
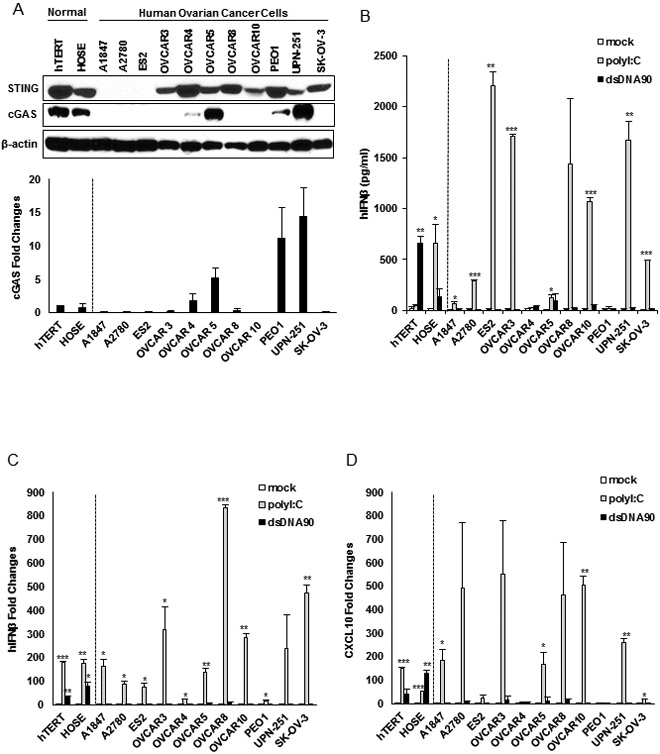
STING-mediated dsDNA-induced innate immune activation is impaired in the majority of human ovarian cancer cell lines. A, Immunoblot of STING and cGAS in hTERT fibroblasts, normal human ovarian surface epithelial (HOSE) and a series of human ovarian cancer cell lines. cGAS expression was also analyzed by qPCR (bottom). B, ELISA analysis of human IFNβ production in the media of cells (same as in A) transfected with 3 μg/ml polyI:C or dsDNA90 or mock transfected for 16 hours. C, qPCR analysis of human IFNβ expression in cells (same as in A) transfected with 3 μg/ml polyI:C or dsDNA90 or mock transfected for 6 hours. D, qPCR analysis of human CXCL10 expression in cells (same as in C). Data are representative of at least two independent experiments. Error bars indicate SD. *p < 0.05, **p < 0.01, and ***p < 0.001; Student’s t test.
Defective STING signaling in ovarian cancer cells
The presence of cytosolic dsDNA rapidly promotes the translocation of STING from the endoplasmic reticulum (ER), along with TBK1, to perinuclear-associated endosomal regions containing NF-kB and IRF3 that translocate to nucleus to activate transcription (21). This process accompanies the phosphorylation and degradation of STING, probably to avoid chronic cytokine production, which could lead to inflammation (21-23). To therefore further evaluate where the observed defects in STING signaling occurred, we examined STING translocation by immunofluorescence. However, only 4 out of 8 ovarian cancer cells exhibited evidence of STING translocation (OVCAR4, OVCAR10, PEO1 and UPN 251) (Fig. 2A). Three cell lines exhibited no STING translocation (SKOV3, OVCAR8, OVCAR3) after DNA stimulation. This may be explained by the fact that cGAS expression was absent in these three cell-lines (Fig. 2D). Thus, STING would not be expected to translocate since there would be a lack of CDN’s required to trigger STING trafficking. Moreover, as a consequence, STING in these cells did not appear to undergo phosphorylation, as determined by immunoblot (Fig. 2D). Previous data has shown that phosphorylation of STING occurs after trafficking, typically to negatively regulate STING activity (21,29).
Figure 2.
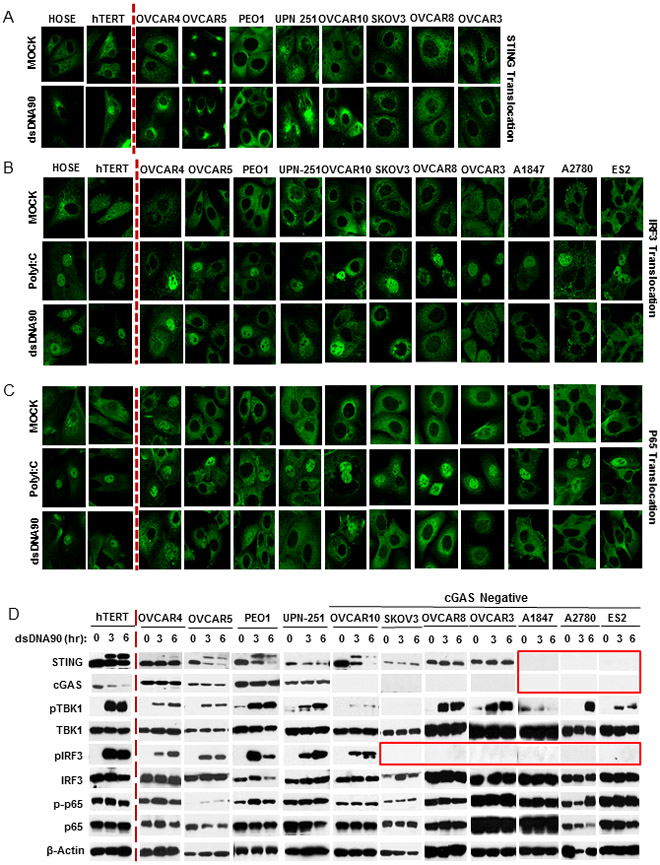
dsDNA-induced STING signaling pathway is defective in human ovarian cancer cell lines. A, Immunofluorescence microscopy analysis of STING translocation in normal and human ovarian cancer cell lines transfected with 3 μg/ml dsDNA90 or polyI:C or mock transfected for 3 hours. B, Immunofluorescence microscopy analysis of IRF3 translocation in normal and human ovarian cancer cell lines. C, Immunofluorescence microscopy analysis of p65 translocation in cells (same as in B). D, Immunoblot analysis of STING signal activation in cells (same as above) transfected with 3 μg/ml dsDNA90 for the indicated time periods.
In control hTERT cells, TBK1 and the transcription factors IRF3 and NF-κB (p65 subunit) translocated in response to cytosolic DNA and were appropriately phosphorylated (Fig. 2 B, C and D). A similar pattern was observed for IRF3 in some of the ovarian cancer cells (OVCAR4, OVCAR5, PEO1 and UPN-251) where cGAS and STING expression were conserved. However, p65 nuclear translocation was not apparent any of the cell lines examined, even in cells where expression of STING and cGAS was evident, for reasons yet to be clarified (Fig. 2C). The transcription of type I IFN requires both IRF3 and NF-κB and reduced function of either of these factors would perhaps help explain the loss of type I IFN production observed in Fig 1. Cytosolic DNA signaling activates STING and invokes the phosphorylation of TBK1, which is required to phosphorylate IRF3. In the presence of cytosolic DNA, we observed the phosphorylation of TBK1 in cells where IRF3 was also concomitantly phosphorylated, as expected (OVCAR4, OVCAR5, PEO1 and UPN-251). However residual phosphorylation of TBK1 appeared evident even in the absence of STING or cGAS (A1847, A2780 and ES2) for reasons that also presently remain to be determined (Fig. 2D). Possibly, cytosolic DNA may associate with other cellular molecules which could influence TBK1 phosphorylation (30,31). However, no phosphorylation or translocation of IRF3 was observed in such cells (Fig. 2B and D). This may be due to the absence of STING, which is required to facilitate TBK1 trafficking to interact with transcription factor targets such as IRF3. Without STING, TBK1 can likely not access IRF3 even when phosphorylated by as yet undefined kinases (22). Although cGAS was not detected in OVCAR10 by either Immunoblot or qPCR (Fig. 1A), dsDNA stimulated STING signaling (including translocation and phosphorylation) was partially preserved, perhaps due to residual cGAS activity, not readily evident by this immunoblot analysis. Collectively, our data indicates that ovarian cancer cells exhibit a variety of defects in the STING signaling pathway, which clearly affects innate immune signaling. This includes loss of STING expression (A1847, A2780 and ES2), cGAS expression (SKOV3, OVCAR8, OVCAR3, A1847, A2780 and ES2) or loss of STING translocation, perhaps explainable by the absence of cGAS and CDN production (SKOV3, OVCAR8, OVCAR3). Loss of cGAS or STING function led to diminished IRF3 phosphorylation and translocation (Fig. 2B and D). In addition, all the ovarian cancer cells examined exhibited a defect in NF-ĸB translocation, unlike control cells (HOSE and hTERT) (Fig 2C). In contrast, IRF3 and NF-ĸB translocation was evident in the majority of ovarian cancer cells examined, in response to poly I:C treatment (Fig. 2B and C). Thus, these transcription factors are functional in response to RIG-I/MDA5 stimulation. The observed defects in IRF3 and NF-ĸB activity therefore appears to be specific for cytosolic DNA-mediated innate immune signaling.
Epigenetic suppression of STING and cGAS.
We have previously shown that loss of STING expression involves epigenetic silencing of transcription via methylation (9,10). Cytidine analogs such as 5-aza-2’-deoxycytidine (5AZADC) (referred to as decitabine) are being evaluated in the clinic as anti-cancer treatments since they inhibit the responsible DNA methyltransferases and frequently rescue the transcription of tumor suppressors (32). To evaluate whether loss of cGAS or STING expression involved hypermethylation of the promoter region, A1847 or ES2 cells, which exhibited loss of both cGAS and STING were treated with 5-azadeoxycytidine (5AZADC). Cells were then treated with dsDNA to see if the STING signaling pathway was re-activated. Immunoblot analysis of treated or untreated cell lysates indicated that cGAS and STING expression were rescued after 5AZADC treatment (Fig. 3A). Moreover, in the presence of dsDNA, STING signaling was rescued, as shown by TBK-1 phosphorylation, IRF3 phosphorylation and translocation (Fig. 3A and B), type I IFN and IFIT1 transcription (Fig. 3C and D). Azacytidine treatment of cells lacking only cGAS (OVCAR3 and SK-OV-3) similarly reversed the expression of this synthase. Consequently, STING signaling was reconstituted in both OVCAR3 and SK-OV-3 (Supplementary figure 1). Thus, loss of cGAS and STING expression can be explained in large part via hypermethylation of the corresponding promoter regions.
Figure 3.
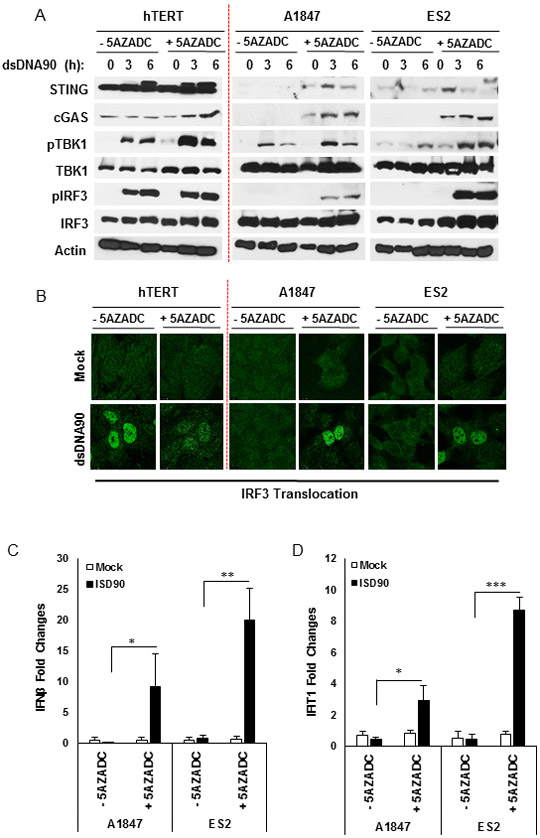
DNA demethylation recapitulated STING and cGAS expression in human ovarian cancer cell lines. A, Immunoblot analysis of STING signal activation in ovarian cancer cells mock or treated with 10 μM 5-aza-2’-deoxycytidine (5AZADC) for 4 days, followed by dsDNA90 transfection at 3 μg/ml for the indicated time periods. B, Immunofluorescence microscopy analysis of IRF3 translocation in ovarian cancer cells treated with 5AZADC (same as above) followed by dsDNA90 transfection at 3 μg/ml for 3 hours. C, Ovarian cancer cells mock treated or treated with 10 μM 5AZADC for 4 days, followed by dsDNA90 transfection at 3 μg/ml for 6 hours. human IFN-β expression was analyzed by qPCR. D, qPCR analysis of human IFIT1 expression in ovarian cancer cells (same as in C). Data are representative of at least two independent experiments. Error bars indicate SD. *p < 0.05, **p < 0.01, and ***p < 0.001; Student’s t test.
Patient-derived ovarian cancer exhibits loss of STING cGAS expression.
To extend our analysis, we next examined, by immunohistochemistry (IHC), STING expression in additional patient-derived ovarian cancer samples (paraffin-embedded tissue microarray; TMA; OVC1021, Pantomics). Principally, we had noted that STING and/or cGAS was observed to be absent in about 54% of the above ovarian cancer cell lines analyzed (Fig. 1A). For this study, we analyzed 4 normal ovarian tissues and 94 ovarian cancer tissues classified in stages I to III according of tumor severity (Fig. 4). We observed that normal ovarian tissue expressed both STING and cGAS, predominantly in ovarian surface epithelial cells. However, in stage I, STING and cGAS was observed in only 44% of ovarian cancer tissues analyzed (cGAS was absent in 28% of ovarian cancer tissues; STING was absent in 17% of tissues; and both STING and cGAS were absent in 11% of tissues). In more advanced stages of ovarian cancers, loss of both STING and cGAS were more pronounced (50% in stage II~III and 51% in stage III) (Fig. 4). Overall, majority of cancer tissues lacked either STING (21%) or cGAS (21%), and 39% of cancer tissues had reduced expression of both STING and cGAS (Fig. 4). Thus, IHC analysis of biopsied tissue for STING/cGAS expression could plausibly provide an assay which may help predict disease outcome in the future, or the response to therapies currently being used to treat ovarian-related malignant disease.
Figure 4.
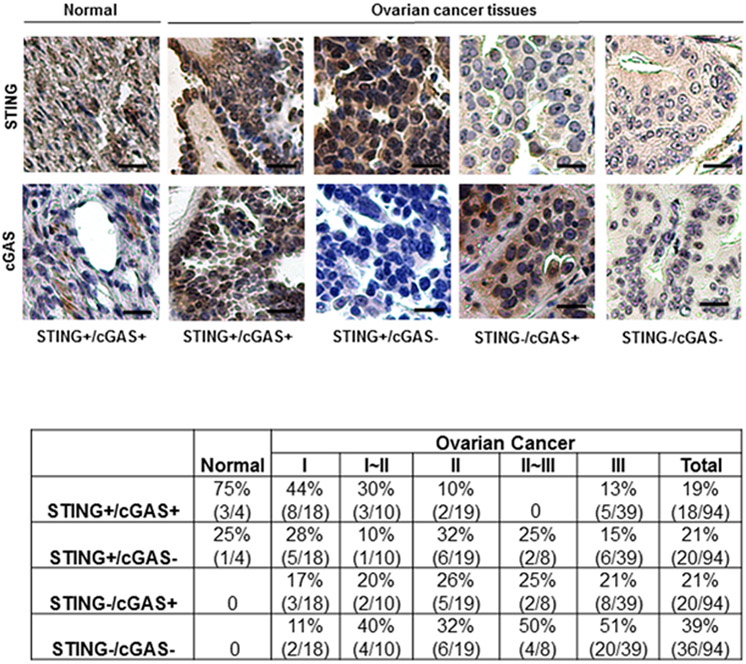
STING and cGAS expression were suppressed in high percentage of human ovarian cancer. IHC analysis of STING and cGAS in 98 human ovarian TMA containing normal/benign and cancer tissues. Representative images of normal human epidermal and human ovarian cancer tissues stained for STING and cGAS. Images are shown at 200x. Bar size, 50 μm. STING and cGAS expression status is summarized and shown in the bottom.
Cancer cells with defective STING-signaling are susceptible to viral oncolysis
Loss of STING signaling has been shown to impede DNA-damage induced cytokine production which may plausibly allow tumor cells to escape the immunosurveillance system (33). In addition, we have previously found evidence that loss of STING signaling may render cells extremely susceptible to infection by DNA microbes, such as Herpes simplex virus (HSV) (21,29). HSV1 is composed of approximately 375 Kb dsDNA90 and has been evaluated in clinical trials as a therapeutic agent for the treatment of cancer (34-36). The oncolytic herpes virus referred to as T-Vec (Imlygic, talimogene laherparepvec) was recently approved in the USA to treat advanced melanoma (13). Oncolytic viruses have also been used to treat ovarian cancer (12). Based on our knowledge that STING is required for effective host defense against HSV1 infection, we previously observed that colorectal carcinoma and melanoma cells with defective STING signaling were sensitive to viral oncolytic therapy (9,10). Given this, we investigated whether ovarian cancer cell lines lacking STING signaling would also respond favorably to viral oncolytic therapy. First, we infected or hTERT control cells or a variety of ovarian cancer cells with HSV1Δγ34.5. Infected cell protein (ICP) λ34.5 inhibits innate immune signaling and cellular translation and HSV1 strains lacking this protein do not replicate efficiently or lyse normal cells due to host defense counter measures being able to impede viral replication (27,37). γ34.5 deleted-HSV1 does not repress innate immune signaling and potently triggers STING-dependent innate immune response, including type I IFN production and other cytokines (38). T-Vec, presently being used in the clinic also lacks γ34.5 and is similar to the HSV1 used in our studies. Tumor cells, in contrast, remain sensitive to HSV lacking γ34.5 (HSV1Δγ34.5), likely due to defects in anti-virus countermeasures. Given our data, one plausible explanation would be that tumor cells with defective STING signaling are sensitive to HSV1Δγ34.5, while those with intact signaling may be more refractory. Thus, an analysis of STING/cGAS expression in patient tumor biopsies may help predict treatment outcome to certain oncolytic viral therapies.
This study indicated that HSV1Δγ34.5 infection activated pIRF3 and pSTAT1, with some cytokine production (IFIT1) only in hTERT control cells and ovarian cancer cells (OVCAR5, OVCAR8 and PEO1) that expressed STING and cGAS (Fig. 1A and Fig. 5A). Our data confirms low level type I IFN production and other select cytokines such as CCL5 in the ovarian cancer lines examined (Fig. 5 B and D), and correlates with a lack of dsDNA-dependent STING activity observed in Figure 1. Nevertheless, in cells where IRF3 was activated, IFIT1 levels were also observed to be high (Fig 5.A and C). However, alternate cancer lines that exhibited some response to dsDNA mediated IRF3 phosphorylation (UPN-251 and OVCAR10; Fig. 2D) failed to robustly exhibit IRF3 phosphorylation following infection with HSV1Δγ34.5 (Fig. 5A). Surprisingly, an analysis of viral replication indicated that HSV1Δγ34.5 failed to replicate efficiently in these cells (UPN-251 and OVCAR10) (Fig. 6A and B; arrows). Thus, it is possible that a failure to efficiently replicate, for reasons that remain unclear, could explain a lack of STING-triggered innate IRF3 activation (Fig. 5A-D). Several other cell-lines exhibited low level HSV1Δγ34.5 replication (OVCAR8, OVCAR10, PEO1, UPN-251, SKOV-3) (Fig. 6A and B). Generally, these cells contained some IRF3 activity but a lack of NF-kB activity (Fig. 2 C and D). Perhaps as a consequence, lack of viral replication was observed to correlate with low levels of cellular lysis following infection (Fig. 6C). Significantly, ovarian cancer cells that lacked both cGAS and STING (A2780, ES2) enabled efficient viral replication, as determined by plaque assay, plausibly since these cells had severely defective innate immune activity (Fig. 6A and B; boxed). Cell viability was also noted to be less than 10% for cells lacking both STING and cGAS (A2780, ES2, A1847) (Fig. 6C; boxed). Although high HSV1Δγ34.5 titer was not detected from infected A1847 cells, defected in STING signaling, this cell-line was highly susceptible to HSV1Δγ34.5 infection and more than 70% of the cells succumbed to HSV1Δγ34.5 infection within 24 hours (Fig. 6A and C). We infected all the ovarian cell lines with HSV1 expressing y34.5 (HSV-luc) and analyzed the infection by luciferase assay (Supplementary figure 2). HSV-luc was able to infect both cells expressing STING/cGAS and deficient cells. UPN-251, that express both STING and cGAS, presents significant higher HSV-luc infection. This data suggests that STING/cGAS pathway cannot efficiently control non-deficient HSV replication. However, ovarian cancer cells exhibiting loss of both cGAS and STING were highly sensitive to HSV1Δγ34.5 replication and viral oncolysis. Thus, assays able to measure cGAS and STING expression in biopsied samples may be of some use in possibly predicting the outcome of select viral oncolytic therapy.
Figure 5.
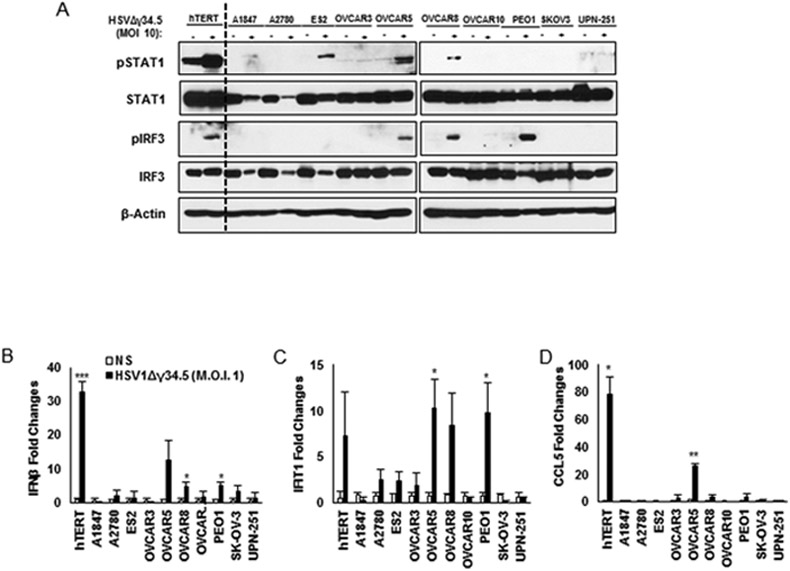
DNA virus infection (HSV1Δγ34.5) does not activate tipe I IFN response in STING/cGAS-deficient cells. A, Normal and human ovarian cancer cell lines were infected with HSV1Δγ34.5 at M.O.I. 10 for 1 hour and analysed 4 hours post infection by immunoblot (pSTAT1, STAT1, pIRF3, IRF3 and β-actin). Same cells as above were infected with HSV1Δγ34.5 at M.O.I. 1 for 1 hour and samples collected 6 hours post infection to check RNA expression by qPCR for IFNβ (B), IFIT1 (C) and CCL5 (D). Data are representative of at least two independent experiments. Error bars indicate SD. *p < 0.05, **p < 0.01, and ***p < 0.001; Student’s t test.
Figure 6.
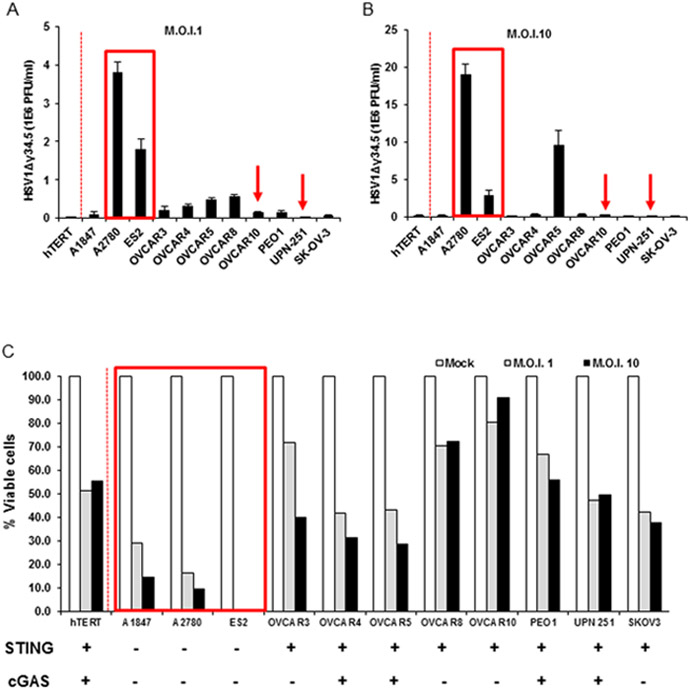
STING signal defect leads ovarian cancer cells more susceptible to HSV1Δγ34.5. Normal human hTERT cells and human ovarian cancer cell lines were infected with HSV1Δγ34.5 at the M.O.I. 1 (A) or M.O.I. 10 (B) for 1 hour and titration of HSV1Δγ34.5 was analyzed by standard plaque assay in Vero cells 24 hours later. (C) Cells (same as in A) were infected with HSV1Δγ34.5 at M.O.I. 1 and 10 for 1 hour, and cell viability was analyzed by trypan blue staining 24 hours later. Data are representative of at least two independent experiments.
To evaluate whether 5AZADC treatment could rescue the activation of STING signaling following HSV1Δγ34.5 infection, we treated three different cell lines (A1847, OVCAR3 and SK-OV-3) for 4 days with 5AZADC. cGAS expression was rescued in all cell-lines treated (Supplementary Fig. 3). We were able to detect pIRF3 phosphorylation predominantly in treated OVCAR3 and SK-OV-3 cells. This was reflected in observing an increase in IFIT1 expression following virus infection. Thus, further evaluation of the consequences of rescuing STING signaling on viral replication and ultimately oncolysis is warranted.
In vivo analysis of cancer cells with defective STING-signaling responses to oncolytic therapy
Our results indicate that ovarian cancer cells with defective STING signaling, especially loss of both cGAS and STING expression, are highly susceptible to HSV1Δγ34.5 infection in vitro. To evaluate whether cells lacking cGAS and STING are also more sensitive to HSV1Δγ34.5 oncolysis in vivo, we subcutaneously inoculated nude mice with ovarian cancer cells with OVCAR5 (cGAS/STING positive, exhibiting some cytokine activity following infection) or ES2 (STING and cGAS negative that were sensitive to HSV1Δγ34.5 replication [Fig. 5]). HSV1Δγ34.5 was then administered intratumorally and tumor growth was monitored (Fig. 7A). Our data indicated that OVCAR5 cells responded poorly to HSV1Δγ34.5 oncolytic treatment (Fig. 7B). This would be consistent with the moderate viral cytopathic effect we observed in vitro on OVCAR5 cells. In contrast, tumors derived from ES2 cells (lacking both STING and cGAS expression) were noted to show significant decrease in tumor size following HSV1Δγ34.5 oncolytic treatment (Fig. 7C). ES2 tumor volumes decreased rapidly following HSV1Δγ34.5 treatment and three out of five tumors completely disappeared 2 weeks after treatment. In contrast, in PBS treated mice, tumors developed aggressively and the animals had to be euthanized within 3 weeks following tumor injection. These data correlate with in vitro findings and indicate that defects in STING pathway may help predict the efficacy of oncolytic DNA based virotherapy for ovarian and other types of cancer.
Figure 7.
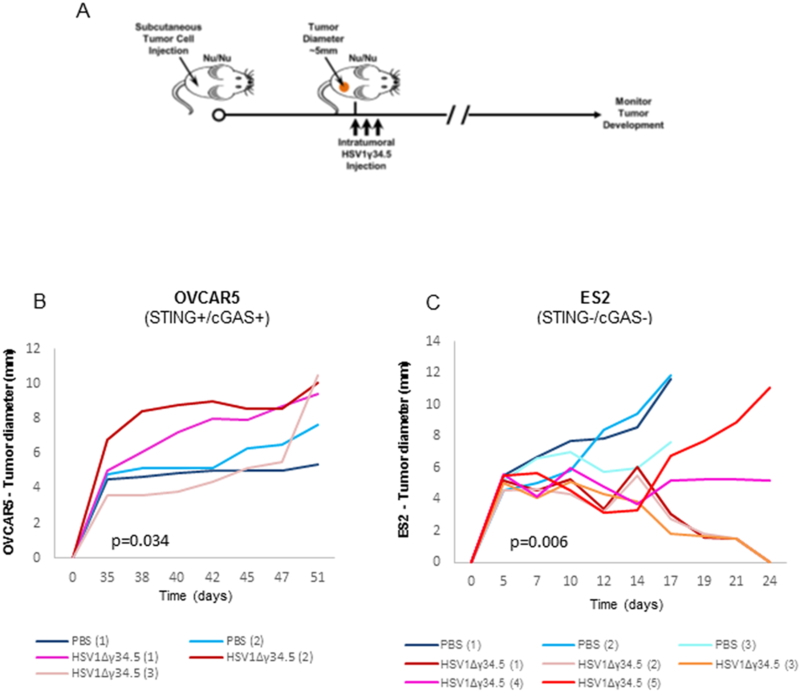
Increased HSV1Δγ34.5 oncolytic effect was observed in ovarian cancer cells with impaired STING signal in vivo. A, Scheme of HSV1Δγ34.5 treatment on a xenograft tumor in nude mice. B-C, The indicated xenograft tumors were generated in the right flank of nude BALB/c mice. When tumors reached 5 mm in diameter, they were injected every other day, for a total of three times, with 1E8 PFU HSV1Δγ34.5 in 50 μl PBS or 50 μl PBS only, and tumor growth was measured every other day. Statistical analysis was carried out comparing the two treatment groups at the last time point using the unpaired Student’s t test. p values are as indicated.
Discussion
Ovarian cancer is amongst the most lethal of the gynecologic malignancies, compounded by patient diagnosis frequently occurring at advanced stages of disease, precipitous metastasis appearing in the peritoneal cavity, and resistance to established chemotherapy and immunotherapy (4,39). To better understand the causes, with a focus on understanding innate immune regulation, we have started to examine the activity of the STING signaling pathway in such cancers (9,10). Compounds that stimulate STING signaling have been shown to exert potent anti-tumor activity since STING activated cytokine production is essential for the generation of robust anti-tumor T cell responses (25,40). Loss of STING signaling has been shown to prevent DNA damage-mediated cytokine production which likely helps tumor cells escape the immunosurveillance system (9,33). Loss of STING signaling may also help to explain resistance to radiation therapy (24,41-43). Our data indicates that the majority of ovarian cancer cells exhibited loss of STING-dependent cytosolic DNA triggered cytokine production. However, the dsRNA-dependent innate immune pathway, predominantly controlled by the RLR pathway was largely intact. cGAS was absent in approximately 64% (7/11) of the cell lines examined, among which 43% (3/7) lacked both STING and cGAS expression.
The suppression of cGAS/STING expression commonly occurred through epigenetic silencing of the promoter regions, as determined through being able to rescue expression using demethylating agents. It is tempting to speculate that the clinical efficacy of epigenetic modifiers presently being used in the clinic may exert their influence, in part, through reconstitution of the STING signaling pathway. The resurrection of STING signaling may generate cytokines that could facilitate anti-tumor immune responses and perhaps improve radiation and chemotherapeutic treatment (24,41-43). A key observation in the ovarian tumor cells was loss of NF-kB activity in response to cytoplasmic dsDNA signaling, even in samples that exhibited cGAS and STING expression. However, NF-kB activity occurred in response to RNA-triggered immune responses. Thus, the defect in NF-kB activity was again STING-dependent, for reasons that presently remain unclear. IRF3 activity was noted to function in some of the cell-lines in a STING dependent manner and was likely responsible for of the gene induction observed in some of the cell-lines. However, IRF3 did not function in the majority of the cell lines, in response to cytosolic DNA treatment (while IRF3 did function in response to dsRNA signaling). Curiously, TBK1 phosphorylation was seen in cells that lacked cGAS and STING, although such cells did not exhibit IRF3 or NF-kB activity. Possibly, other DNA-triggered pathways may influence these events (30,31). Since both NF-kB and IRF3 activity are required for efficient type I IFN production, loss of function of one of these transcription factors may help explain a defect in IFN production. Collectively, our data demonstrates selective defects in cytosolic DNA but not RNA-dependent innate immune signaling in ovarian cancer cells. The mechanisms of suppression involve epigenetic silencing of cGAS and STING expression and loss of NF-kB activity. Our study also included screening a panel of primary patient ovarian cancer tissues classified in stages I to III according of tumor severity. STING or cGAS expression was found to occur in all stages of human ovarian cancer, though was more pronounced in advanced stages. We thus conclude that STING signaling is frequently suppressed in ovarian cancer. This may enable such cells to escape the immunosurveillance system as described. Our findings may also help explain resistance to radiation treatment and plausibly immunotherapy (24,41-43). Thus, the analysis of STING/cGAS expression in clinical tumor samples could help to predict tumor severity and treatment outcome in the future.
STING controls key innate immune responses triggered by dsDNA, comprising the genome of DNA microbes including DNA viruses such as HSV1 or bacteria, as well as self-DNA leaked from the nucleus of DNA damaged cells (18,19,21,29). Thus, we postulated that given our findings, cancer cells with defective STING signaling may be more susceptible to oncolytic DNA viruses, such as HSV1Δγ34.5 (9,10). The first oncolytic herpes virus based therapeutic, T-Vec (IMLYGIC®, talimogene laherparepvec - Amgen) was recently approved in USA to treat advanced melanoma. IMLYGIC has been genetically modified and has lost the γ34.5 gene, though expresses GM-CSF (3,13). Our results indicated that HSV1Δγ34.5 preferentially replicated in ovarian cancer cells that lost STING function. However ovarian cancer cells with loss of both cGAS and STING expression that exhibited no IRF3 or NF-kB activity in response to cytosolic DNA were highly susceptible to viral oncolysis. Therefore, evaluating the expression levels of STING/cGAS in biopsied specimens may help improve response rates to virotherapy.
The importance of both intrinsically (tumor cells) and extrinsically (antigen presenting cells - APC’s) activated STING-dependent cytokine production in controlling anti-tumor immunity is becoming increasingly evident. For example, the therapeutic intratumoral administration of CDNs repress tumor growth, presumably through direct activation of STING in the tumor microenvironment leading to dendritic cell-dependent CTL production, is being evaluated in Phase I trials (25,44,45). STING signaling may also play a role in facilitating the anti-tumor effects of checkpoint inhibitors such as PD-1 (46,47). The immunological effects of chemotherapeutic agents such as cisplatin and etoposide are likely, in part, related to the STING-signaling pathway. Such drugs cause DNA leakage into the cytosol to trigger STING signaling intrinsically (10,48). Recent studies have demonstrated that STING signaling is also essential for efficient radiation therapy, which exerts its immunostimulatory effects predominantly through augmenting STING activity (24,41-43). Thus, loss of this extrinsic STING activity in APC’s may prevent anti-tumor immunotherapeutic efficacy. In this manuscript, we focused on the importance of intrinsic STING activity in tumor response to oncolytic viral therapy. As we show, functional intrinsic STING signaling in a tumor cell impedes the effectiveness of viral replication and oncolytic activity. Conversely, in cells lacking STING signaling, greater oncolytic activity is observed. This may provide a larger amount of tumor cell lysate for engulfment and antigen cross-presentation for T-cell priming, which is dependent on extrinsic STING (49). Possibly, viral oncolytics used in conjunction with checkpoint inhibitors could provide a powerful immuno-therapeutic strategy. In conclusion, a better comprehension of STING signaling in cancer may provide reliable prognosis and improved accuracy in predicting disease outcome as well as responses to cancer treatments.
Supplementary Material
Implications:
STING signaling evaluation in tumors may help predict disease outcome and possibly dictate the efficacy of oncoviral and other types of cancer therapies.
Acknowledges
We thank Dr. Xiang-Xi Xu for providing the ovarian cancer cell lines.
Financial Support: This work was supported by the following founding sources: University of Miami/Sylvester Comprehensive Cancer Center Grant entitled “Analysis of STING and Evaluation of STING Based Therapeutics in Ovarian Cancer” and NIH/NCI (RO1CA194404-01). Queiroz was partially supported by Science without Borders fellowship from the National Council for Scientific and Technological Development (CNPq/Brazil).
Footnotes
Disclosure of Potential Conflict of Interests: No potential conflicts of interest were disclosed.
References
- 1.Cannistra SA. Cancer of the ovary. N Engl J Med 2004;351(24):2519–29 doi 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin 2008;58(2):71–96 doi 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 3.Li S, Tong J, Rahman MM, Shepherd TG, McFadden G. Oncolytic virotherapy for ovarian cancer. Oncolytic Virother 2012;1:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lengyel E Ovarian cancer development and metastasis. Am J Pathol 2010;177(3):1053–64 doi 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naora H, Montell DJ. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nat Rev Cancer 2005;5(5):355–66 doi 10.1038/nrc1611. [DOI] [PubMed] [Google Scholar]
- 6.Fung-Kee-Fung M, Oliver T, Elit L, Oza A, Hirte HW, Bryson P. Optimal chemotherapy treatment for women with recurrent ovarian cancer. Curr Oncol 2007;14(5):195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell SJ, Barber GN. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018;33(4):599–605 doi 10.1016/j.ccell.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol 2012;30(7):658–70 doi 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep 2016;14(2):282–97 doi 10.1016/j.celrep.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia T, Konno H, Barber GN. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res 2016;76(22):6747–59 doi 10.1158/0008-5472.CAN-16-1404. [DOI] [PubMed] [Google Scholar]
- 11.Hartkopf AD, Fehm T, Wallwiener D, Lauer U. Oncolytic virotherapy of gynecologic malignancies. Gynecol Oncol 2011;120(2):302–10 doi 10.1016/j.ygyno.2010.10.031. [DOI] [PubMed] [Google Scholar]
- 12.Hartkopf AD, Fehm T, Wallwiener M, Lauer U. Oncolytic Viruses to Treat Ovarian Cancer Patients - a Review of Results From Clinical Trials. Geburtshilfe Frauenheilkd 2012;72(2):132–6 doi 10.1055/s-0031-1298281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 2015. doi 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 14.Lawler SE, Chiocca EA. Oncolytic Virus-Mediated Immunotherapy: A Combinatorial Approach for Cancer Treatment. J Clin Oncol 2015;33(25):2812–4 doi 10.1200/JCO.2015.62.5244. [DOI] [PubMed] [Google Scholar]
- 15.Kawasaki T, Kawai T, Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev 2011;243(1):61–73 doi 10.1111/j.1600-065X.2011.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. Viral RNA detection by RIG-I-like receptors. Curr Opin Immunol 2015;32:48–53 doi 10.1016/j.coi.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol 2015;15(12):760–70 doi 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013;339(6121):786–91 doi 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011;478(7370):515–8 doi 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hornung V, Hartmann R, Ablasser A, Hopfner KP. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol 2014;14(8):521–8 doi 10.1038/nri3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008;455(7213):674–8 doi 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 2013;155(3):688–98 doi 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahn J, Barber GN. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr Opin Immunol 2014;31:121–6 doi 10.1016/j.coi.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014;41(5):843–52 doi 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014;41(5):830–42 doi 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson SW, Laub PB, Beesley JS, Ozols RF, Hamilton TC. Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Cancer Res 1997;57(5):850–6. [PubMed] [Google Scholar]
- 27.Chou J, Roizman B. Herpes simplex virus 1 gamma(1)34.5 gene function, which blocks the host response to infection, maps in the homologous domain of the genes expressed during growth arrest and DNA damage. Proc Natl Acad Sci U S A 1994;91(12):5247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song S, Peng P, Tang Z, Zhao J, Wu W, Li H, et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci Rep 2017;7:39858 doi 10.1038/srep39858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009;461(7265):788–92 doi 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakad R, Schumacher B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front Genet 2016;7:147 doi 10.3389/fgene.2016.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci U S A 2013;110(8):2969–74 doi 10.1073/pnas.1222694110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seelan RS, Mukhopadhyay P, Pisano MM, Greene RM. Effects of 5-Aza-2’-deoxycytidine (decitabine) on gene expression. Drug Metab Rev 2018;50(2):193–207 doi 10.1080/03602532.2018.1437446. [DOI] [PubMed] [Google Scholar]
- 33.Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun 2014;5:5166 doi 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolodkin-Gal D, Edden Y, Hartshtark Z, Ilan L, Khalaileh A, Pikarsky AJ, et al. Herpes simplex virus delivery to orthotopic rectal carcinoma results in an efficient and selective antitumor effect. Gene Ther 2009;16(7):905–15 doi 10.1038/gt.2009.44. [DOI] [PubMed] [Google Scholar]
- 35.Harrington KJ, Hingorani M, Tanay MA, Hickey J, Bhide SA, Clarke PM, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res 2010;16(15):4005–15 doi 10.1158/1078-0432.CCR-10-0196. [DOI] [PubMed] [Google Scholar]
- 36.Kasuya H, Kodera Y, Nakao A, Yamamura K, Gewen T, Zhiwen W, et al. Phase I Dose-escalation Clinical Trial of HF10 Oncolytic Herpes Virus in 17 Japanese Patients with Advanced Cancer. Hepatogastroenterology 2014;61(131):599–605. [PubMed] [Google Scholar]
- 37.Mulvey M, Camarena V, Mohr I. Full resistance of herpes simplex virus type 1-infected primary human cells to alpha interferon requires both the Us11 and gamma(1)34.5 gene products. J Virol 2004;78(18):10193–6 doi 10.1128/JVI.78.18.10193-10196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abe T, Harashima A, Xia T, Konno H, Konno K, Morales A, et al. STING recognition of cytoplasmic DNA instigates cellular defense. Mol Cell 2013;50(1):5–15 doi 10.1016/j.molcel.2013.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Norouzi-Barough L, Sarookhani MR, Sharifi M, Moghbelinejad S, Jangjoo S, Salehi R. Molecular Mechanisms of Drug Resistance in Ovarian Cancer. J Cell Physiol 2017. doi 10.1002/jcp.26289. [DOI] [PubMed] [Google Scholar]
- 40.Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol 2015;36(4):250–6 doi 10.1016/j.it.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 2017;8:15618 doi 10.1038/ncomms15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017;548(7668):466–70 doi 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017;548(7668):461–5 doi 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 2015;11(7):1018–30 doi 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A 2015;112(50):15408–13 doi 10.1073/pnas.1512832112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med 2015;7(283):283ra52 doi 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foote JB, Kok M, Leatherman JM, Armstrong TD, Marcinkowski BC, Ojalvo LS, et al. A STING Agonist Given with OX40 Receptor and PD-L1 Modulators Primes Immunity and Reduces Tumor Growth in Tolerized Mice. Cancer Immunol Res 2017;5(6):468–79 doi 10.1158/2326-6066.CIR-16-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahn J, Ruiz P, Barber GN. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J Immunol 2014;193(9):4634–42 doi 10.4049/jimmunol.1401337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahn J, Xia T, Rabasa Capote A, Betancourt D, Barber GN. Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer Cell 2018;33(5):862–73 e5 doi 10.1016/j.ccell.2018.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.