

Abstract
The PI3K pathway is mutated and aberrantly activated in many cancers and plays a central role in tumor cell proliferation and survival, making it a rational therapeutic target. To date, however, results from clinical trials with PI3K inhibitors in solid tumors have been largely disappointing. Here, we describe several factors that have limited the success of these agents, including the weak driver oncogenic activity of mutant PI3K, sub-optimal patient selection in trials, drug-related toxicities, feedback upregulation of compensatory mechanisms when PI3K is blocked, increased insulin production upon PI3Kα inhibition, lack of mutant-specific inhibitors, and a relative scarcity of studies using combinations with PI3K antagonists. We also suggest strategies to improve the impact of these agents in solid tumors. Despite these challenges, we are optimistic that isoform-specific PI3K inhibitors, particularly in combination with other agents, may be valuable in treating appropriately selected patients with PI3K-dependent tumors.
Keywords: PI3K, alpelisib, taselisib, targeted therapy, biomarkers
Introduction
The PI3K/AKT/TOR signaling network is commonly altered in several human cancers. Gain-of-function mutations in PIK3CA, the gene encoding the p110α catalytic subunit of PI3K, are among the most common somatic alterations in solid tumors. Other alterations in the pathway include mutations in PIK3R1, encoding the PI3K regulatory subunit p85α, the PI3K effectors AKT1/2/3, and loss of the lipid phosphatases PTEN and INPP4B [reviewed in (1)]. Further, PI3K is aberrantly activated by activated oncogenes and/or amplified/mutated tyrosine kinases such as mutant RAS, ERBB2 (HER2), MET, BCR-ABL, and KIT, among others. The association of these alterations with a transformed phenotype both in vitro and in vivo has led to the development of a plethora of PI3K antagonists. These include pan-PI3K inhibitors, inhibitors of all PI3Ks and mTOR, and other ATP mimetics with variable selectivity to the p110α (PI3Kα) isozyme [reviewed in (2)]. Despite the initial enthusiasm for and significant investment in the development of PI3K inhibitors for solid tumors, they have not yielded the outstanding clinical activity observed with other approved targeted therapies.
In this mini-review, we will present a critical analysis for this modest outcome and speculate on possible directions to improve the therapeutic targeting of this oncogenic pathway, with a focus on PI3Kα (Table 1). Of note, the PI3Kδ inhibitors idelalisib and copanlisib are effective and currently approved for the treatment of non-Hodgkin’s lymphoma and will not be the focus of this review. For a comprehensive review of recent progress in targeting all PI3K isoforms, AKT, and mTOR, we refer the reader Janku et al. (3). We also focus this review on inhibition of PI3K within tumor cells. However, we note that there is increasing evidence that interfering with stromal PI3K activity may contribute to the anti-tumor effects of PI3K inhibitors, particularly through inhibition of angiogenesis (PI3Kα inhibitors) and through modulating the immune system (PI3Kγ/δ inhibitors) [reviewed in (4)].
Table 1.
Clinical obstacles to PI3K inhibitor efficacy and proposed solutions
| Clinical obstacles | Proposed solutions |
|---|---|
| Sub-optimal patient selection | • Selection of patients with tumors harboring activating PIK3CA mutations • Identification of PI3K-dependent cancers (i.e., endocrine-resistant ER+ breast cancers with PIK3CA mutations) • Exclusion of tumors harboring biomarkers of resistance in PIK3CA-mutant tumors (i.e., KRAS, TP53, or FGFR1) • Identification of other genotypes that may benefit from PI3K inhibitors (i.e., PIK3R1 mutations or PIK3CA amplification) |
| Drug-related toxicity limits target inhibition | • Focus on isoform-specific inhibitors • Development of PIK3CA mutant-selective inhibitors |
| Feedback upregulation of compensatory mechanisms | • Use of combinations that limit adaptive response (i.e., with antiestrogens, RTK and PI3Kβ inhibitors, CDK4/6 inhibitors) • Optimizing dosing schedules of combinations to ameliorate toxicities |
| Increase in insulin production upon systemic inhibition of PI3Kα | • Combinations with SGLT2 inhibitors or ketogenic diet • Development of PIK3CA mutant-selective inhibitors |
PIK3CA mutations are weak oncogenes
Genetically engineered mouse models (GEMMs) are a powerful approach used to ‘graduate’ a dominant oncogene as an inducer or driver of a cancer. Indeed, several studies using GEMMs have demonstrated a causal role of mutant PIK3CA in tumor initiation, progression and maintenance in vivo. However, many of these studies have relied on tissue-specific overexpression of mutant p110α (5,6), which in human tumors is not found to be amplified and/or overexpressed. Thus, these models may not represent an otherwise low signaling output and transforming potential of these oncogenes. Indeed, knock-in of Pik3caH1047R into the endogenous Pik3ca locus of mouse ovary cells results in ovarian epithelial hyperplasia, but no invasive cancers. However, concomitant deletion of Pten in mouse ovary leads to the development of serous adenocarcinomas (7). This oncogenic cooperativity is reminiscent of frequent coexistence of PIK3CA mutations with KRAS and/or PTEN alterations in human ovarian (8) and endometrial cancers (9), suggesting that mutations in either PIK3CA of PTEN alone are not sufficient to hyperactivate PIP3-induced signaling. Mutations in Tp53 have also been shown to be required to cooperate with mutant Pik3ca to induce ovarian cancer and breast cancer in GEMMs (10,11). Knockin of Pik3caH1047R in the mouse mammary gland results in hyperplasia and eventual mammary tumorigenesis, but with very long latencies (>12 months) and incomplete penetrance in some models (12,13), suggesting that time is needed for additional mutational events to trigger tumorigenesis. Cancer development is accelerated by estrogen supplementation, leading to estrogen receptor-positive (ER+) mammary tumors, whereas tumors in Pik3caH1047R mice that did not receive exogenous estrogen were predominantly ER-negative (14). Translationally, these data suggest mutant PIK3CA may not be able to induce invasive cancer progression on its own. Thus, although PIK3CA mutations may play a partial role in the progression of carcinomas, its pharmacological inhibition should be coupled with other therapies in order to exert an important antitumor effect.
PIK3CA mutations that occur early in embryonic development lead to tissue overgrowth in a mosaic-like pattern or PROS (for PIK3CA-related overgrowth spectrum), venous malformations, epidermal nevi, and brain malformations associated with epilepsy (15). One of these syndromes is CLOVES, a complex disorder characterized by tissue overgrowth and malformations affecting the epidermis, skeleton, internal organs and central nervous system. PROS disorders do not appear linked to an increased risk of cancer and are not associated with progression to invasive tumors. Further, treatment with PI3Kα inhibitors, at doses much lower than those used in adult cancers, results in marked objective and functional benefits in patients with multiple affected organs (16).
PI3K consists of a catalytic subunit, p110α, and one of several regulatory subunits, a major one being p85α. In the basal state, p85 stabilizes p110α and inhibits its enzymatic activity. Upon stimulation by growth factors, the SH2 domains of p85 bind phosphotyrosine residues on receptor tyrosine kinases (RTKs) or signaling adaptors, such as IRS1, HER3, etc., thus relieving p110α from inhibitory contacts and facilitating its lipid kinase activity at the plasma membrane, where it can access its substrate and receive other inputs from RAS. Oncogenic mutations in PIK3CA have been shown to enhance the natural activation of p110α (17). For example, the helical domain mutation E545K can associate with IRS1 independent of p85 thus increasing response to insulin and IGFs (18,19). Less common deletions in the C2 domain also relieve inhibitory contacts with p85 and enhance p110α activity (20). The most common PIK3CA mutation, H1047R in the kinase domain, has higher affinity for cellular membranes, thus bypassing the requirement for association with RAS and resulting in greater access to the PI3K substrate PIP2 (21).
These data suggest that in cancers with PIK3CA mutations, these alterations are permissive for growth factor signaling but not potent signaling units or driver oncogenes per se. Further circumstantial evidence in support of this notion is the subclonal nature of mutations in the PI3K pathway in metastatic vs. primary lesions from the same patient (22,23). A large study of the evolution of cancer heterogeneity showed that subclonal mutations in the PI3K/AKT pathway were more frequent and less ubiquitously expressed across tumor subclones than subclonal mutations in the more potent RAS/RAF/MEK/ERK pathway (24). This could have a negative impact in patient selection for a clinical trial targeting PIK3CA mutations as these can be missed if a ‘PI3K normal’ metastasis is profiled. On the other hand, the discordant ‘dependence’ on PI3K signaling of these lesions as a result of this heterogeneity may result in muted clinical responses to a PI3K inhibitor. A possible exception to this generalization is breast cancer, where recent genomic analyses suggest that PIK3CA mutations are primarily clonal (24,25).
Sub-optimal patient selection in clinical trials
Trials with PI3K inhibitors have suggested preferential clinical activity in patients with PIK3CA mutant cancers. The phase I study of alpelisib included 134 patients with all cancer types; 64/76 patients in this trial whose tumors were tested contained hotspot PIK3CA mutations in their cancers. The clinical benefit rate was 44% in tumors with PIK3CA mutations vs. 20% among those patients with PIK3CA wild type (WT) cancers (26). In the phase I trial of taselisib, the overall response rate was 36% among patients with PIK3CA-mutant tumors, all with the H1047R variant, vs. 0% in the group with PIK3CA WT tumors (27). BELLE-2 was the first phase III randomized clinical trial comparing fulvestrant and placebo vs. fulvestrant and the pan-PI3K inhibitor buparlisib in patients with ER+ metastatic breast cancer who had progressed on an aromatase inhibitor (28). In the overall group, treatment with buparlisib and fulvestrant resulted in a modest prolongation of PFS by 1.9 months compared to placebo and fulvestrant (6.9 vs. 5.0 months; HR 0.79, 95% CI 0.67–0.89; p<0.01). In the PI3K-activated group (defined as any mutation detected by Sanger sequencing in PIK3CA exons 1, 7, 9 or 20, or PTEN expression by immunohistochemistry in <10% of cells) the PFS in the investigational arm was 6.8 vs 4.0 months in the control arm (HR 0.76, 95% CI 0.60–0.97; p=0.014). In this trial, 446 patients had paired PIK3CA mutation status in tumor and plasma circulating tumor DNA (ctDNA) with 77% concordance between both. Notably, among 307 patients with PIK3CA WT tumor tissue, 64 (21%) had PIK3CA mutations detected in ctDNA, suggesting that the cancer evolved between the original diagnosis and the development of metastatic disease and treatment in this trial. In an exploratory analysis, a significant difference in PFS in the buparlisib vs. the placebo arm was observed in patients with ctDNA PIK3CA mutations (7.0 vs. 3.2 months; HR 0.56, 95% CI 0.39–0.80; p=0.0005) but not those with WT ctDNA.
The recent phase III SOLAR-1 trial of alpelisib + fulvestrant vs placebo + fulvestrant in patients with metastatic ER+ breast cancer showed that PIK3CA-mutant cancers were more likely to respond to alpelisib. In the PIK3CA-mutant cohort (n=341), the median PFS was 11.0 months in the alpelisib arm vs. 5.7 months in the fulvestrant arm (HR 0.65, 95% CI 0.50–0.85), p=0.00065). In contrast, in the PIK3CA-WT cohort (n=231), alpelisib only modestly extended PFS (7.4 months vs 5.6 months; HR 0.85, 95% CI 0.58–1.25) (29). A statistically significant benefit in PFS was also observed for alpelisib in patients with PIK3CA-mutant ctDNA (10.9 months vs 3.6 months; HR 0.55, 95% CI 0.39–0.79; p=0.0005) (30). These data strongly suggest that the development of PI3Kα inhibitors should be focused on PIK3CA-mutant tumors.
However, not all patients with PIK3CA mutations have similar benefit from PI3K inhibitors. In a phase Ib trial by Mayer et al, patients with PI3KCA mutations and concurrent alterations in KRAS, TP53 or FGFR1 did not benefit from alpelisib (31). Larger studies are needed to confirm these associations and to identify other alterations that promote intrinsic resistance. Further, the most frequent mutation in PIK3CA, H1047R, appeared to be associated with higher clinical benefit from alpelisib compared with mutations in the helical domain (31). However, this association was not confirmed in the larger SOLAR-1 trial (29). In most studies, there is a small fraction of patients without detectable PIK3CA hotspot mutations that respond clinically to PI3K inhibitors. The molecular basis for a potential dependence on PI3K signaling by these tumors has not always been investigated. For example, PIK3CA C-terminal truncations and deletion mutants that disrupt the coupling to the p85 regulatory subunit result in hyperactivation of p110α and transformation and have been associated with an excellent clinical response to alpelisib (20). Interestingly, mutations in the corresponding domains of p85 also activate p110α and are oncogenic (32). PIK3CA C2 domain mutations and others outside the ‘hotspots’ as well as PIK3R1 (p85) mutations are generally not included in the DNA sequencing panels used for patient stratification in trials with PI3K inhibitors. All this would suggest that PIK3CA hotspot mutations do not necessarily capture all PI3K-dependent tumor genotypes that can potentially respond to PI3K inhibitors.
Therefore, we propose that only patients with cancers with activating PIK3CA mutations and other lesions conferring PI3K pathway dependence [potentially PIK3R1 mutations (32) or PIK3CA amplifications (33)] should be included in trials with PI3Kα inhibitors. Further selection could be refined by analyzing PIK3CA mutation clonality, determining the full repertoire of activating PIK3CA mutations (both canonical ‘hotspot’ and less frequent recurrent mutations), and identifying biomarkers of intrinsic resistance to PI3Kα inhibitors (Table 1).
Drug-related toxicity limits sustained target inhibition
A major hurdle for the development of PI3K pathway inhibitors has been the inability to achieve optimal drug target blockade in tumors while avoiding undue toxicities in patients. Pan-PI3K inhibitors share common, dose-dependent toxicities such as rash, fatigue, hyperglycemia and diarrhea. In general, toxicity from small molecule PI3K inhibitors depends on their PI3K isozyme specificity. For example, PI3Kα inhibitors are associated with hyperglycemia and rash whereas PI3Kδ inhibitors are associated with gastrointestinal side effects, myelosuppression and transaminitis. This toxicity profile is even broader with pan-PI3K/mTOR inhibitors. At this time, the development of these non-specific drugs, which will not be discussed herein, appears to be stalled.
In BELLE-2, the combination of the pan-PI3K inhibitor buparlisib with fulvestrant was found to be superior to fulvestrant and placebo in patients with PIK3CA-mutated tumors, even though the median time on buparlisib was <2 months (28). Grade 3–4 toxicities were significant and included hyperglycemia (15%), increased ALT (26%), rash (8%), depression (5%) and anxiety (5%), leading to discontinuation of further drug development. Pictilisib, another pan-PI3K inhibitor, was evaluated in the randomized phase II FERGIE trial that compared pictilisib and fulvestrant vs. placebo and fulvestrant. In this study, the investigational arm did not significantly improve progression free survival, possibly due to its suboptimal dosing limited by toxicity. Toxic side effects included included grade 3–4 fatigue (8%) and diarrhea (8%) but rare hyperglycemia and rash (34), further suggesting limited drug-target engagement. Copanlisib, a small molecule with predominant activity against PI3Kα and PI3Kδ, was associated with grade 3–4 hyperglycemia (41%), hypertension (24%), lung infection (16%, including one death on study) and diarrhea (5%) (35). Alpelisib, a PI3Kα selective inhibitor, at a dose of 300 mg daily, had a better safety profile with grade 3–4 diarrhea and hyperglycemia seen in 10% of patients and grade 3–4 elevation of transaminases seen in 5% pf patients (26). The rate of grade 3–4 hyperglycemia was even higher in combination with fulvestrant in the SOLAR-1 trial (36.7%) (29). Taselisib, a PI3Kβ-sparing small molecule, was associated with grade 3–4 hyperglycemia (14.7%), rash (11.8%), diarrhea (5.9%), fatigue (5.9%) and pruritus (5.9%) (27). We speculate that the differences in safety between alpelisib and taselisib are due to inhibition of PI3Kγ/δ by taselisib. Finally idelalisib, a PI3K-δ selective inhibitor, was associated with grade 3–4 diarrhea (13%), increased ALT (13%) and pneumonia (7%) (36). The increased success of p110δ inhibitors in hematological malignancies relative to PI3K inhibitors in solid tumors may be due, in part, to the different toxicity profiles.
Due to the safety profile associated with continuous dosing of PI3Kα inhibitors, preclinical studies have experimented with intermittent dosing schedules. Intermittent high-doses of the PI3Kα/β inhibitor AZD8835 effectively blocked P-AKT, induced apoptosis, and induced regressions in breast cancer xenografts, and facilitated combinations with fulvestrant and palbociclib in vivo (37). Likewise, treatment of tumor-bearing mice with high doses of pictilisb every 3 days enabled it to be combined with a MEK inhibitor and blocked growth of xenografts with KRAS or BRAF and PTEN or PIK3CA alterations (38). Further exploration of intermittent dosing of PI3K inhibitors in the clinic is warranted.The toxicities from PI3K inhibitors have limited the ability of clinical trials with these small molecules to adequately assess the dose required to optimally inhibit PI3K in cancers in situ. On-treatment glucose uptake in primary tumors, as measured by [18F]-FDG-PET, has been used as a surrogate of therapeutic inhibition of PI3K in several early phase studies (26,27,39). Of note, however, the best timing for this pharmacodynamic assessment is not clear and, overall, the observed drug-induced inhibition of FDG uptake has only been partial. Thus, on-target toxicities from PI3K inhibitors and sub-optimal doses and dosing schedules have limited complete and sustained PI3K inhibition and may explain the discrepancies between the results of preclinical studies and those in clinical trials. In addition, the toxicity profile of PI3K inhibitors makes combinations with some other small molecules quite challenging (see below).
Feedback upregulation of compensatory mechanisms
Pharmacological inhibition of the PI3K pathway in cancer cells is followed within hours to days by non-genetic mechanisms of adaptation eventually leading to drug resistance. This adaptation can be explained in good part by RTK-induced activation of PI3K/AKT/TOR, resulting in AKT-mediated phosphorylation of FOXO proteins (Figure 1). In turn, FOXO proteins transcriptionally repress RTKs and/or adaptors that activate PI3K, such as HER3, EGFR, IGF-IR, insulin receptors (InsR), and FGFRs (40,41). Further, AKT activates TORC1 and S6K, which repress IRS1 expression in order to regulate pathway signaling output (42). In addition, activated TORC1, downstream AKT, phosphorylates and activates GRB10, which binds and downregulates InsR (43). Hence, inhibition of PI3K/AKT blocks FOXO phosphorylation and transcriptional repression of RTKs, and leads to derepression of S6K and GRB10, resulting in activation of multiple RTKs and partial maintenance of PIP3 formation. In some luminal breast cancer cells with PIK3CA mutations or with HER2 gene amplification – where PI3K is hyperactivated as a result of signaling by HER2-HER3 dimers – there is re-accumulation of PIP3 mediated by p110β (44). Using ovarian cancer spheroids, Muranen et al. elegantly showed that inhibition of PI3K/mTOR results in death of inner matrix-deprived cells, but cells attached to the matrix survived. This matrix-associated resistance occurs as a result of FOXO-mediated transcription and cap-independent translation of survival factors such as ERα, BCL2 and IGF-IR (45). Another FOXO-mediated adaptive response to PI3K inhibition is upregulation of Rictor, resulting in increased AKT phosphorylation in renal cancer cells (46). Finally, inhibition of PI3K/mTOR increases IRS1-dependent activation of JAK2/STAT5 and secretion of IL-8 in triple negative breast cancer cells and primary tumors, with co-treatment with a JAK inhibitor abrogating this feedback loop (47).
Figure 1. Adaptive upregulation of compensatory pathways limits the efficacy of PI3K inhibitors.
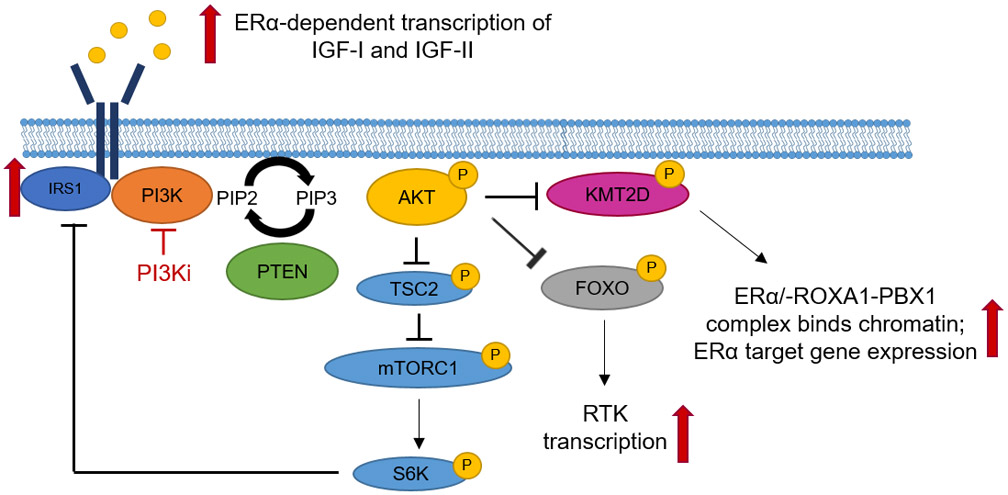
Several adaptive feedback mechanisms that limit complete suppression of PIP3 and the cellular response to PI3K inhibitors have been described. These include FOXO-mediated de-repression of RTKs and de-repression of IRS1, leading to partial maintenance of PIP3. In ER+ breast cancer cells, treatment with PI3K inhibitors induces ERα transcriptional activity via the histone methyltransferase KMT2D.
These compensatory mechanisms following inhibition of PI3K have been extensively investigated in ER+ human breast cancer cells and primary tumors. Treatment with the AKT inhibitor AZD5363 upregulates several RTKs as well ESR1 mRNA and ERα-dependent transcription of IGF-I and IGF-II ligands (48). Bosch et al. reported increased ESR1 mRNA and ER-dependent gene expression signature in tumors from patients treated with the PI3Kα inhibitor alpelisib. These drug-induced transcriptional changes were abrogated by the anti-ER drugs tamoxifen and fulvestrant (49). Finally, Toska et al. elegantly showed that treatment with alpelisib of ER+ breast cancer cells and primary tumors in patients triggers activation of the lysine methyltransferase KMT2D which, in turn, activates ERα transcription by facilitating assembly of an ERα-FOXA1-PBX1 complex (50). Taken together, these data suggest that in patients with ER+ breast cancer, PI3K inhibitors should be developed in combination with endocrine therapy, thus leading to the current registration trials SANDPIPER and SOLAR-1 discussed below.
The adaptive responses to the inhibition of PI3K in cancer cells have suggested other logical combinations with small molecules or antibodies against RTK signaling pathways, including inhibitors of IGF-IR or HER3 (48,51–53). However, because of lack of selectivity of the partner drugs and/or mainly toxicity in patients, these combinations have been challenging. For example, a phase Ib trial of the PI3K inhibitor alpelisib with the the IGF-IR MAb ganitumab (AMG 479; clinicaltrials.gov NCT 01708161) resulted in excessive rash and hyperglycemia and no evidence of clinical activity (https://clinicaltrials.gov/ct2/show/NCT01708161?term=NCT01708161&rank=1). This can probably be explained by elevation of growth hormone (GH) levels upon IGF-IR MAb-mediated blockade of IGF-I, a ligand necessary for negative regulation of GH in the brain. Elevated GH levels result in insulin resistance partially from increase fatty acid efflux from the liver leading to enhanced insulin production (54) . These elevated insulin and IGF-I levels, in turn, may stimulate tumor growth via activation of uninhibited insulin receptors in cancer cells (55), thus limiting any potential clinical effect of the combination.
Significant gastrointestinal and metabolic toxicities were also observed with the combination of alpelisib, trastuzumab, and the HER3 monoclonal antibody LJM716. These toxicities severely limited drug delivery and dose escalation, thus allowing 72% and 83% of the planned doses of alpelisib and LJM716, respectively (56). As mentioned above, intermittent dosing schedules may optimize the therapeutic index of combination therapies, enabling combinations that are unachievable with continuous dosing.
Increase in insulin production upon inhibition of PI3K
The p110α isozyme and AKT2 mediate insulin-driven glucose uptake in muscle, liver and fat cells, mainly attributable to translocation of glucose transporters (GLUT) to the plasma membrane (57). As a result, therapeutic inhibition of PI3K/AKT blocks insulin action, thus preventing glucose uptake in adipose tissue and skeletal muscle, and promoting glycogen breakdown in the liver (Figure 2). This generates hyperglycemia which, in turn, leads to insulin release from the pancreas with potential normalization of glucose levels [reviewed in (58)]. Therefore, a dose dependent increase in the plasma levels of fasting C-peptide and insulin, most of the times associated with hyperglycemia, is an obligatory on-target pharmacodynamic surrogate of PI3K inhibition in trials with PI3K inhibitors (26,59). This obligatory surge in insulin secretion may activate InsR and PI3K, particularly in tumors rich in InsR, and limit the clinical activity of PI3K antagonists. This is supported by the correlation between glucose uptake in primary tumors as measured by [18F]-FDG-PET following treatment with PI3K inhibitors. In the phase Ib trial of letrozole and the pan-PI3K small molecule buparlisib, 50% of patients exhibiting a reduction in FDG tumor uptake derived clinical benefit whereas increased FDG uptake preceded rapid tumor progression (39). These data suggest, first, that an increase in tumor FDG uptake shortly after treatment initiation, potentially explained by an insulin surge following PI3K inhibition in an InsR rich tumor, can be used as a signal for early treatment discontinuation. Second, that the level of on-treatment FDG uptake would reflect the net effect of PI3K inhibition in the cancer counteracted by the insulin-mediated activation of tumor InsR. Thus, the magnitude of inhibition of FDG uptake in tumors on therapy can be used as a metric to score interventions aimed at abrogating the insulin rebound in patients on PI3K inhibitors.
Figure 2. Insulin feedback limits the anti-tumor effects of PI3K inhibitors.
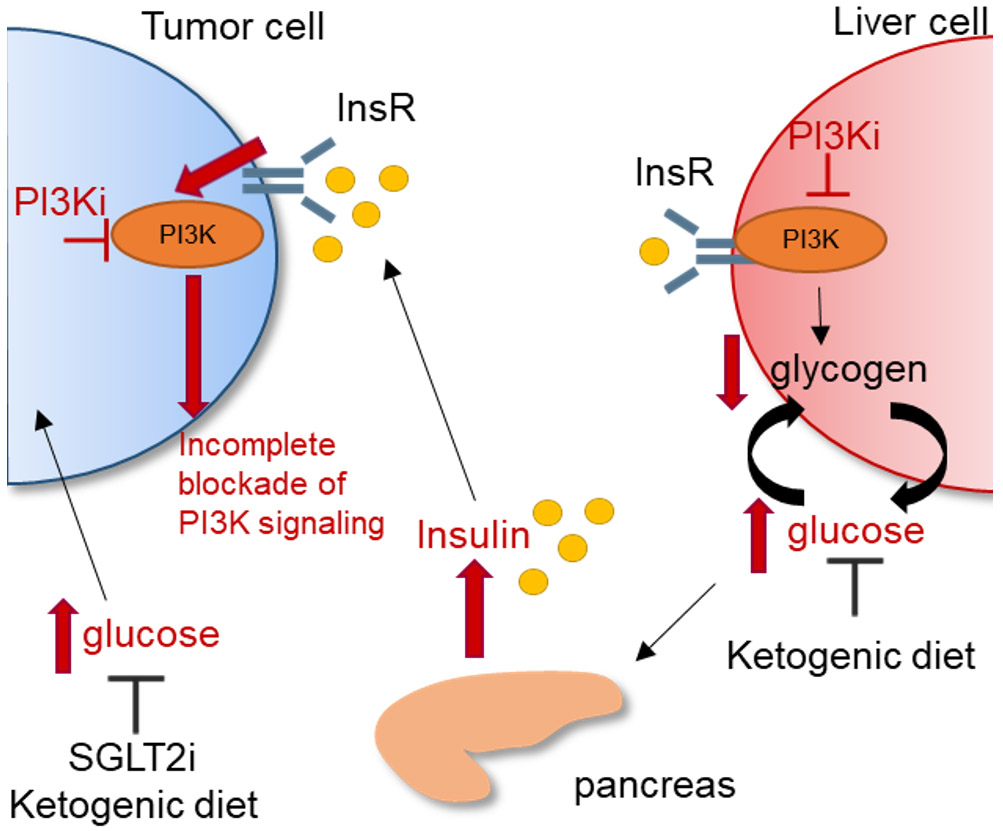
PI3Kα mediates insulin-driven glucose uptake in muscle, liver, and fat cells. Upon inhibition of PI3Kα, glucose uptake in muscle and fat cells is blocked, and breakdown of liver glycogen is stimulated, thus resulting in hyperglycemia. Elevated glucose levels, in turn, drive insulin release from the pancreas. Elevated insulin levels activate the insulin receptor (InsR) in tumor cells, leading to enhanced stimulation of PI3K, and limiting the effects of PI3K inhibitors. Tumors with PIK3CA mutations would be particularly sensitive to insulin. In preclinical models, SGLT2 inhibitors or a ketogenic diet restore homeostasis and synergize with PI3K inhibitors to reduce tumor growth. Red arrows indicate response to PI3K inhibitors.
In a recent paper using pre-clinical animal models, Hopkins et al. reported dietary and pharmacological strategies aimed at preventing insulin feedback that, in turn, enhance the efficacy and reduce the toxicity of PI3K inhibitors. These included the anti-diabetic drug metformin, which increases insulin sensitivity and reduces insulin levels; sodium glucose co-transporter 2 (SGLT2) inhibitors, which reduce glucose reabsorption in kidney tubules; and a ketogenic diet, which depletes glycogen stores and thus limits the acute efflux of glucose from the liver upon inhibition of PI3K (60). SGLT2 inhibitors and a ketogenic diet prevented insulin feedback and enhanced the antitumor effect of PI3K pathway inhibitors in both PIK3CA-mutant and PIK3CA-WT tumors. These combinations remain to be tested in the clinic and are of particular importance considering that, pending their approval, long term use of PI3K inhibitors would be expected to induce insulin resistance and potentially type II diabetes, unless insulin feedback is controlled.
Lack of mutant PIK3CA-specific inhibitors
An outstanding demonstration of the driver oncogenic role of PIK3CA mutations was provided by Juric et al. Upon development of acquired clinical resistance to the PI3Kα inhibitor alpelisib in a patient with PIK3CA mutant breast cancer, these authors identified six distinct subclonal mutations in PTEN in multiple metastatic lesions, all resulting in loss of PTEN function and on a common background of a clonal mono-allelic deletion of PTEN (61). In absence of PTEN, cells become dependent on p110β to maintain PI3K pathway activity when p110α is blocked. In this report, a xenograft derived from a PTEN-null lung metastasis from the patient progressing on alpelisib was sensitive to the combination of alpelisib with the p110β inhibitor AZD6482 (61). This result is remarkable considering the convergent evolution of drug-resistant mutations occurred after treatment with a drug that may not have blocked mutant PIK3CA completely and/or in sustained fashion. This report also suggests that for tumors highly dependent of mutant PIK3CA and PI3K signaling, mutant specific inhibitors should be able to exert an even stronger selective pressure. Theoretically, drugs that specifically target mutant p110α (H1047R, E542K, etc.) should spare endogenous p110α and downstream effectors that maintain normal homeostasis, thus limiting toxicities and permitting higher doses and more complete inhibition of the drug target.
Taselisib is a small molecule inhibitor of p110α that induces ubiquitin-mediated, proteasome-dependent degradation of PIK3CAH1047R in cancer cells in culture and patient-derived xenografts without significant change in WT p110α (Freeman et al. SABCS 2016, S6–04). It spares p110β but also inhibits p110γ and p110δ. This relative selectivity for mutant PIK3CA was recently tested in the SANDPIPER randomized trial in patients with ER+/PIK3CA mutant breast cancer (62). Patients treated with the ER antagonist fulvestrant plus taselisib exhibited a modestly improved progression free survival compared to those treated with fulvestrant plus placebo. Main toxicities included diarrhea, hyperglycemia, rash, stomatitis and colitis, thus limiting the median time on treatment to under 5 months. We speculate the prominent gastrointestinal side effects could have been secondary to inhibition of p110δ as it has been seen in trials with the p110δ inhibitors idelalisib and copanlisib (35,36) and may have compromised selective inhibition of mutant PIK3CA in primary tumors in vivo. Another clinical candidate is the ATP mimetic GDC-0077, with over 300-fold selectivity against p110α (IC50 0.038 nM) over the β, γ and δ class I PI3K isoforms (63). GDC-0077, which also selectively degrades mutant PI3K, has shown remarkable preclinical activity against PIK3CA-mutant breast cancer cells and PDXs (63). GDC-0077 is now in early clinical development as a single agent and in combination with endocrine and other targeted therapies in patients with advanced breast cancer that harbor PIK3CA mutations.
Other mechanisms of resistance
In addition to those described above, other mechanisms of compensation and/or resistance to PI3K inhibitors have been reported, primarily derived from laboratory studies and/or clinical correlations. These include CDK4/6 (64), MYC amplification (65), KRAS mutations (5), PIK3CB (p110β) mutations (66), FGFR1 amplification (31), and overexpression and/or aberrant activation of PIM1 (67), AXL (68), PDK1-SGK1 (69), and SGK3 (70), among others. In this review, however, we have focused on those aspects intrinsic to therapeutic targets in the PI3K pathway that are unique to it and that make the development of current PI3K inhibitors different and perhaps more challenging than that of other molecularly targeted therapies. Therefore, we will not cover these mechanisms of resistance in any detail herein.
Recent advances in targeting the PI3K/AKT pathway in solid tumors
The SOLAR-1 phase III trial was the first to demonstrate a clinically significant effect of PI3Kα inhibition in tumors with PIK3CA mutations (29). We speculate that the apparent success of this trial is likely due to following aspects: 1) use of a potent, isoform-specific PI3K inhibitor; 2) inclusion of PIK3CA-mutant cancers; 3) endocrine-resistant ER+ breast cancers tend to have clonal PIK3CA mutations; and 4) the PI3K inhibitor was given in combination with an antiestrogen, likely dampening feedback compensation. The increased success of alpelsib vs taselisib in a similar setting in the SANDPIPER trial [PFS prolongation of 5.3 months vs 2 months, respectively (62)] may be due to more potent inhibition of PI3Kα by alpelisib, as evidenced by the higher rates of hyperglycemia seen in the SOLAR-1 trial compared to the SANDPIPER trial. The high rates of dose reductions and discontinuations in the PI3K inhibitor arms reported in both trials underscore the remaining challenges associated with long-term systemic inhibition of PI3Kα.
Many of the principles outlined above can also be applied to the development of AKT inhibitors. The AKT inhibitor capivasertib (AZD5363) exhibited significant clinical activity in patients with AKT1-mutant tumors; the majority of the responses were seen in ER+ breast cancers (71). Likewise, the AKT inhibitor ipatasertib (GDC-0068), in combination with the antiandrogen abiraterone, significantly prolonged PFS in prostate cancers with loss of PTEN (72), and also prolonged PFS in combination with paclitaxel in triple-negative breast cancers with alterations in PIK3CA, AKT, or PTEN (73). As with PI3Kα inhibitors, the most common adverse events with AKT-selective inhibitors were hyperglycemia, diarrhea, and rash, pointing to the shared roles of PI3Kα and AKT in physiology.
Conclusions
Several factors have limited the development of PI3K inhibitors, as well as the enthusiasm of the cancer community for this class of drugs. These include 1) adaptive molecular mechanisms upon therapeutic inhibition of PI3K, 2) our inability to specifically inhibit signaling by PIK3CA mutations while sparing endogenous p110α, 3) the limited use of these therapies in rational combinations, several of them informed by a strong mechanistic background, and 4) dose-limiting toxicities that prevent sustained PI3K pathway suppression. Despite these limitations, PI3K inhibitors have already shown clinical activity that is superior to that of single agent trastuzumab (https://clinicaltrials.gov/ct2/show/NCT00842998), a HER2-targeted monoclonal antibody that in combination with chemotherapy has significantly improved the survival of patients with HER2-overexpressing breast cancer (74,75). We posit that moving forward, combination approaches with PI3Kα inhibitors that can be prioritized are those 1) with CDK4/6 inhibitors (64,76,77), 2) with drugs that limit insulin feedback, such as SGLT2 inhibitors, or a ketogenic diet (60), and 3) combinations of p110α and p110β inhibitors (44,78). For now, we believe that trials of PI3Kα specific inhibitors in combination with antiestrogens in patients with PIK3CA-mutant ER+ breast cancer are the best available test of the hypothesis that PIK3CA mutations are a pathogenic driver in cancer. The clinical activity of the PI3Kα inhibitor alpelisib in combination with fulvestrant in patients with advanced ER+ breast cancer who have progressed after primary antiestrogen therapy, reported in the SOLAR-1 trial, provides important evidence that the PI3K pathway is an important therapeutic target in tumors with PI3K pathway dependence.
Statement of Significance.
Despite the modest clinical activity of PI3K inhibitors in solid tumors, there is an increasing understanding of the factors that may have limited their success. Strategies to ameliorate drug-related toxicities, use of rational combinations with PI3K antagonists, development of mutant-selective PI3K inhibitors, and better patient selection should improve the success of these targeted agents against solid tumors.
Footnotes
Conflict of interest disclosure statement: A.B. Hanker receives grant support from Takeda. V. Kaklamani receives grant support from Eisai; she is a speaker for Novartis, Eisai, Genentech, Pfizer, and Genomic Health; she is a consultant for Novartis, Athenex, Eisai, and Amgen. C.L. Arteaga receives grant support from Pfizer, Eli Lilly, Radius, and Takeda; he has served or serves in advisory roles to Symphogen, Daiichi Sankyo, TAIHO Oncology, Novartis, Merck, PUMA Biotechnology, Eli Lilly, Radius, Sanofi, OrigiMed, ABBVIE, and H3Biomedicine; he holds stock options in Provista and Y-Trap. He serves in the Scientific Advisory Board of the Komen Foundation.
REFERENCES
- 1.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2015;15(1):7–24 doi 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayer IA, Arteaga CL. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu Rev Med 2016;67:11–28 doi 10.1146/annurev-med-062913-051343. [DOI] [PubMed] [Google Scholar]
- 3.Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nature reviews Clinical oncology 2018;15(5):273–91 doi 10.1038/nrclinonc.2018.28. [DOI] [PubMed] [Google Scholar]
- 4.Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer discovery 2016;6(10):1090–105 doi 10.1158/2159-8290.Cd-16-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008;14(12):1351–6 doi nm.1890 [pii] 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu P, Cheng H, Santiago S, Raeder M, Zhang F, Isabella A, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat Med 2011;17(9):1116–20 doi 10.1038/nm.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kinross KM, Montgomery KG, Kleinschmidt M, Waring P, Ivetac I, Tikoo A, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest 2012;122(2):553–7 doi 10.1172/JCI59309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer cell 2007;11(4):321–33 doi 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497(7447):67–73 doi 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu R, Baker SJ, Hu TC, Norman KM, Fearon ER, Cho KR. Type I to type II ovarian carcinoma progression: mutant Trp53 or Pik3ca confers a more aggressive tumor phenotype in a mouse model of ovarian cancer. The American journal of pathology 2013;182(4):1391–9 doi 10.1016/j.ajpath.2012.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams JR, Xu K, Liu JC, Agamez NM, Loch AJ, Wong RG, et al. Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer research 2011;71(7):2706–17 doi 10.1158/0008-5472.CAN-10-0738. [DOI] [PubMed] [Google Scholar]
- 12.Tikoo A, Roh V, Montgomery KG, Ivetac I, Waring P, Pelzer R, et al. Physiological levels of Pik3ca(H1047R) mutation in the mouse mammary gland results in ductal hyperplasia and formation of ERalpha-positive tumors. PloS one 2012;7(5):e36924 doi 10.1371/journal.pone.0036924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan W, Stawiski E, Janakiraman V, Chan E, Durinck S, Edgar KA, et al. Conditional activation of Pik3ca(H1047R) in a knock-in mouse model promotes mammary tumorigenesis and emergence of mutations. Oncogene 2013;32(3):318–26 doi 10.1038/onc.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stratikopoulos EE, Kiess N, Szabolcs M, Pegno S, Kakit C, Wu X, et al. Mouse ER+/PIK3CA(H1047R) breast cancers caused by exogenous estrogen are heterogeneously dependent on estrogen and undergo BIM-dependent apoptosis with BH3 and PI3K agents. Oncogene 2019;38(1):47–59 doi 10.1038/s41388-018-0436-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet 2012;90(6):1108–15 doi 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong JP, et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 2018;558(7711):540–6 doi 10.1038/s41586-018-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA). Proceedings of the National Academy of Sciences of the United States of America 2012;109(38):15259–64 doi 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao Y, Wang C, Cao B, Hirsch BM, Song J, Markowitz SD, et al. Gain of interaction with IRS1 by p110alpha-helical domain mutants is crucial for their oncogenic functions. Cancer cell 2013;23(5):583–93 doi 10.1016/j.ccr.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 2007;317(5835):239–42 doi 317/5835/239 [pii] 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- 20.Croessmann S, Sheehan JH, Lee KM, Sliwoski G, He J, Nagy R, et al. PIK3CA C2 Domain Deletions Hyperactivate Phosphoinositide 3-kinase (PI3K), Generate Oncogene Dependence, and Are Exquisitely Sensitive to PI3Kalpha Inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research 2018;24(6):1426–35 doi 10.1158/1078-0432.CCR-17-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci 2015;40(2):88–100 doi 10.1016/j.tibs.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med 2015;21(7):751–9 doi 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer discovery 2015;5(11):1164–77 doi 10.1158/2159-8290.Cd-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 2015;7(283):283ra54 doi 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. New England Journal of Medicine 2017;376(22):2109–21 doi 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 26.Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, et al. Phosphatidylinositol 3-Kinase alpha-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018;36(13):1291–9 doi 10.1200/JCO.2017.72.7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juric D, Krop I, Ramanathan RK, Wilson TR, Ware JA, Sanabria Bohorquez SM, et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer discovery 2017;7(7):704–15 doi 10.1158/2159-8290.CD-16-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baselga J, Im SA, Iwata H, Cortes J, De Laurentiis M, Jiang Z, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology 2017;18(7):904–16 doi 10.1016/S1470-2045(17)30376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.André F, Ciruelos EM, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. LBA3_PRAlpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): Results of the phase III SOLAR-1 trial. Annals of Oncology 2018;29(suppl_8):mdy424.010–mdy424.010 doi 10.1093/annonc/mdy424.010. [DOI] [Google Scholar]
- 30.Juric D, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib + fulvestrant for advanced breast cancer: Subgroup analyses from the phase III SOLAR-1 trial [abstract]. In: Proceedings of the 2018 San Antonio Breast Cancer Symposium; 2018. Dec 5–8; San Antonio, TX. Abstract GS3–08. [Google Scholar]
- 31.Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kalpha-Specific Inhibitor, with Letrozole in ER+/HER2- Metastatic Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(1):26–34 doi 10.1158/1078-0432.CCR-16-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun M, Hillmann P, Hofmann BT, Hart JR, Vogt PK. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proceedings of the National Academy of Sciences of the United States of America 2010;107(35):15547–52 doi 10.1073/pnas.1009652107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Molecular cancer therapeutics 2014;13(5):1117–29 doi 10.1158/1535-7163.Mct-13-0865. [DOI] [PubMed] [Google Scholar]
- 34.Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. The Lancet Oncology 2016;17(6):811–21 doi 10.1016/S1470-2045(16)00106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dreyling M, Santoro A, Mollica L, Leppa S, Follows GA, Lenz G, et al. Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2017;35(35):3898–905 doi 10.1200/JCO.2017.75.4648. [DOI] [PubMed] [Google Scholar]
- 36.Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med 2014;370(11):1008–18 doi 10.1056/NEJMoa1314583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hudson K, Hancox UJ, Trigwell C, McEwen R, Polanska UM, Nikolaou M, et al. Intermittent High-Dose Scheduling of AZD8835, a Novel Selective Inhibitor of PI3Kalpha and PI3Kdelta, Demonstrates Treatment Strategies for PIK3CA-Dependent Breast Cancers. Molecular cancer therapeutics 2016;15(5):877–89 doi 10.1158/1535-7163.Mct-15-0687. [DOI] [PubMed] [Google Scholar]
- 38.Hoeflich KP, Merchant M, Orr C, Chan J, Den Otter D, Berry L, et al. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer research 2012;72(1):210–9 doi 10.1158/0008-5472.Can-11-1515. [DOI] [PubMed] [Google Scholar]
- 39.Mayer IA, Abramson VG, Isakoff SJ, Forero A, Balko JM, Kuba MG, et al. Stand up to cancer phase Ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2014;32(12):1202–9 doi 10.1200/JCO.2013.54.0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer cell 2011;19(1):58–71 doi S1535–6108(10)00433–2 [pii] 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chakrabarty A, Sanchez V, Kuba MG, Rinehart C, Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proceedings of the National Academy of Sciences of the United States of America 2012;109(8):2718–23 doi 1018001108 [pii] 10.1073/pnas.1018001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer research 2006;66(3):1500–8 doi 66/3/1500 [pii] 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011;332(6035):1322–6 doi 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costa C, Ebi H, Martini M, Beausoleil SA, Faber AC, Jakubik CT, et al. Measurement of PIP3 levels reveals an unexpected role for p110beta in early adaptive responses to p110alpha-specific inhibitors in luminal breast cancer. Cancer cell 2015;27(1):97–108 doi 10.1016/j.ccell.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer cell 2012;21(2):227–39 doi 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin A, Piao HL, Zhuang L, Sarbassov dos D, Ma L, Gan B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K-AKT pathway. Cancer research 2014;74(6):1682–93 doi 10.1158/0008-5472.CAN-13-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Muller U, et al. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer cell 2012;22(6):796–811 doi 10.1016/j.ccr.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 48.Fox EM, Kuba MG, Miller TW, Davies BR, Arteaga CL. Autocrine IGF-I/Insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with acquired resistance to estrogen deprivation. Breast cancer research : BCR 2013;15(4):R55 doi 10.1186/bcr3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med 2015;7(283):283ra51 doi 10.1126/scitranslmed.aaa4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017;355(6331):1324–30 doi 10.1126/science.aah6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garrett JT, Sutton CR, Kurupi R, Bialucha CU, Ettenberg SA, Collins SD, et al. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110alpha inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer research 2013;73(19):6013–23 doi 10.1158/0008-5472.CAN-13-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, et al. mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med 2013;5(196):196ra99 doi 10.1126/scitranslmed.3005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Garcia C, Ibrahim YH, Serra V, Calvo MT, Guzman M, Grueso J, et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2012;18(9):2603–12 doi 10.1158/1078-0432.CCR-11-2750. [DOI] [PubMed] [Google Scholar]
- 54.Vijayakumar A, Novosyadlyy R, Wu Y, Yakar S, LeRoith D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm IGF Res 2010;20(1):1–7 doi 10.1016/j.ghir.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H, Pelzer AM, Kiang DT, Yee D. Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer research 2007;67(1):391–7 doi 10.1158/0008-5472.CAN-06-1712. [DOI] [PubMed] [Google Scholar]
- 56.Shah PD, Chandarlapaty S, Dickler MN, Ulaner G, Zamora SJ, Sterlin V, et al. Phase I study of LJM716, BYL719, and trastuzumab in patients (pts) with HER2-amplified (HER2+) metastatic breast cancer (MBC). Journal of Clinical Oncology 2015;33(15_suppl):590 doi 10.1200/jco.2015.33.15_suppl.590. [DOI] [Google Scholar]
- 57.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab 2007;5(4):237–52 doi 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 58.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell 2017;170(4):605–35 doi 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30(3):282–90 doi JCO.2011.36.1360 [pii] 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 60.Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018;560(7719):499–503 doi 10.1038/s41586-018-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature 2015;518(7538):240–4 doi 10.1038/nature13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baselga J, Dent SF, Cortés J, Im Y-H, Diéras V, Harbeck N, et al. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): Primary analysis from SANDPIPER. Journal of Clinical Oncology 2018;36(18_suppl):LBA1006-LBA doi 10.1200/JCO.2018.36.18_suppl.LBA1006. [DOI] [Google Scholar]
- 63.Hong R, Edgar K, Song K, Steven S, Young A, Hamilton P, et al. Abstract PD4–14: GDC-0077 is a selective PI3Kalpha inhibitor that demonstrates robust efficacy in <em>PIK3CA</em> mutant breast cancer models as a single agent and in combination with standard of care therapies. Cancer research 2018;78(4 Supplement):PD4–14 doi 10.1158/1538-7445.SABCS17-PD4-14. [DOI] [Google Scholar]
- 64.Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer cell 2014;26(1):136–49 doi 10.1016/j.ccr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, et al. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer cell 2015;27(6):837–51 doi 10.1016/j.ccell.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakanishi Y, Walter K, Spoerke JM, O’Brien C, Huw LY, Hampton GM, et al. Activating Mutations in PIK3CB Confer Resistance to PI3K Inhibition and Define a Novel Oncogenic Role for p110beta. Cancer research 2016;76(5):1193–203 doi 10.1158/0008-5472.CAN-15-2201. [DOI] [PubMed] [Google Scholar]
- 67.Le X, Antony R, Razavi P, Treacy DJ, Luo F, Ghandi M, et al. Systematic Functional Characterization of Resistance to PI3K Inhibition in Breast Cancer. Cancer discovery 2016;6(10):1134–47 doi 10.1158/2159-8290.CD-16-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elkabets M, Pazarentzos E, Juric D, Sheng Q, Pelossof RA, Brook S, et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer cell 2015;27(4):533–46 doi 10.1016/j.ccell.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kalpha Inhibition. Cancer cell 2016;30(2):229–42 doi 10.1016/j.ccell.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R, Toker A. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Molecular cell 2014;56(4):595–607 doi 10.1016/j.molcel.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hyman DM, Smyth LM, Donoghue MTA, Westin SN, Bedard PL, Dean EJ, et al. AKT Inhibition in Solid Tumors With AKT1 Mutations. Journal of Clinical Oncology 2017;35(20):2251–9 doi 10.1200/jco.2017.73.0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Bono JS, De Giorgi U, Rodrigues DN, Massard C, Bracarda S, Font A, et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clinical cancer research : an official journal of the American Association for Cancer Research 2018. (in press) doi 10.1158/1078-0432.CCR-18-0981. [DOI] [PubMed] [Google Scholar]
- 73.Kim SB, Dent R, Im SA, Espie M, Blau S, Tan AR, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. The Lancet Oncology 2017;18(10):1360–72 doi 10.1016/s1470-2045(17)30450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344(11):783–92. [DOI] [PubMed] [Google Scholar]
- 75.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr., Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005;353(16):1673–84. [DOI] [PubMed] [Google Scholar]
- 76.Jansen VM, Bhola NE, Bauer JA, Formisano L, Lee KM, Hutchinson KE, et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer research 2017;77(9):2488–99 doi 10.1158/0008-5472.CAN-16-2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer research 2016;76(8):2301–13 doi 10.1158/0008-5472.CAN-15-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, et al. Feedback suppression of PI3Kalpha signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer cell 2015;27(1):109–22 doi 10.1016/j.ccell.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]