

Abstract
Lung adenocarcinomas are characterized by mutations in the receptor tyrosine kinase (RTK)/Ras/Raf pathway, with up to 75% of cases containing mutations in known driver genes. However, the driver alterations in the remaining cases are yet to be determined. Recent exome sequencing analysis has identified SOS1, encoding a guanine nucleotide exchange factor, as significantly mutated in lung adenocarcinomas lacking canonical oncogenic RTK/Ras/Raf pathway mutations. Here, we demonstrate that ectopic expression of lung adenocarcinoma-derived mutants of SOS1 induces anchorage-independent cell growth in vitro and tumor formation in vivo. Biochemical experiments suggest that these mutations lead to over-activation of the Ras pathway, which can be suppressed by mutations that disrupt either the Ras-GEF or putative Rac-GEF activity of SOS1. Transcriptional profiling reveals that the expression of mutant SOS1 leads to the upregulation of MYC target genes and genes associated with Ras transformation. Furthermore, we demonstrate that an AML cancer cell line harboring a lung adenocarcinoma-associated mutant SOS1 is dependent on SOS1 for survival and is also sensitive to MEK inhibition. Our work provides experimental evidence for the role of SOS1 as an oncogene and suggests a possible therapeutic strategy to target SOS1-mutated cancers.
Keywords: SOS1, oncogene, lung adenocarcinoma, RTK/Ras/Raf
Introduction
Lung cancers, the second most common form of cancer and the leading cause of cancer death in men and women, are marked by mutations along the receptor tyrosine kinase (RTK)/Ras/Raf pathway. In lung adenocarcinoma, which comprises approximately 40% of lung cancer, mutations and amplifications in the EGFR and KRAS genes alone are found in ~50% of cases and are generally mutually exclusive (1–3). Other genetic aberrations in the RTK/Ras/Raf pathway include lower-frequency NRAS, HRAS, BRAF, and MAP2K1 mutations (1, 3–5), MET amplifications and mutations (6, 7), as well as EML4-ALK (8), ROS1 (9–11), RET (11–13), and NTRK fusions (14).
In the most comprehensive study to date, whole exome sequencing of 660 lung adenocarcinoma/normal paired DNA samples revealed 242 cases without a canonical oncogenic driver (15). Analysis of these oncogene-negative cases for significantly mutated genes revealed recurrent mutations in additional genes with a clear linkage to the RTK/Ras/Raf pathway, such as ARHGAP35, RASA1, SOS1, and VAV1 (15). The discovery of recurrent SOS1 mutations was a testament to the statistical discovery power of a larger sample set, as a previous sequencing study of lung adenocarcinomas had failed to reveal mutations in this candidate oncogene (16). Furthermore, a recent TCGA paper also reports the discovery of SOS1 mutations in 1% of lung adenocarcinoma samples, 1% of uterine carcinomas, and <1% in other cancers, highlighting the potential to discover rarer oncogenic events through additional sequencing of samples (17).
In this study, we analyzed the functional impact of mutations in Son of Sevenless 1 (SOS1) identified in oncogene-negative lung adenocarcinoma on oncogenic growth and signaling. SOS1 is a guanine nucleotide exchange factor (GEF) for Ras proteins and catalyzes the exchange of GDP for GTP, activating Ras (18, 19). The SOS1 protein is composed of several domains: a Dbl homology (DH) domain with putative Rac-GEF activity (20, 21); a pleckstrin homology (PH) domain, which forms an autoinhibitory domain together with DH; a Ras exchange motif (REM), which acts as an allosteric activator; and a CDC25-homologous catalytic domain (Figure 1A) (22). SOS1 mutations found in oncogene-negative lung adenocarcinomas span all domains of the gene, with a hotspot mutation at the asparagine 233 residue (Figure 1A). Germline SOS1 mutations are also found in RASopathies, including Noonan syndrome (Figure 1A) (23, 24) as well as hereditary gingival fibromatosis type I (25). In both of these inherited diseases, mutations in SOS1 are believed to be gain-of-function and lead to greater activation of the Ras pathway.
Figure 1: SOS1 mutations drive anchorage-independent growth in vitro and in vivo.
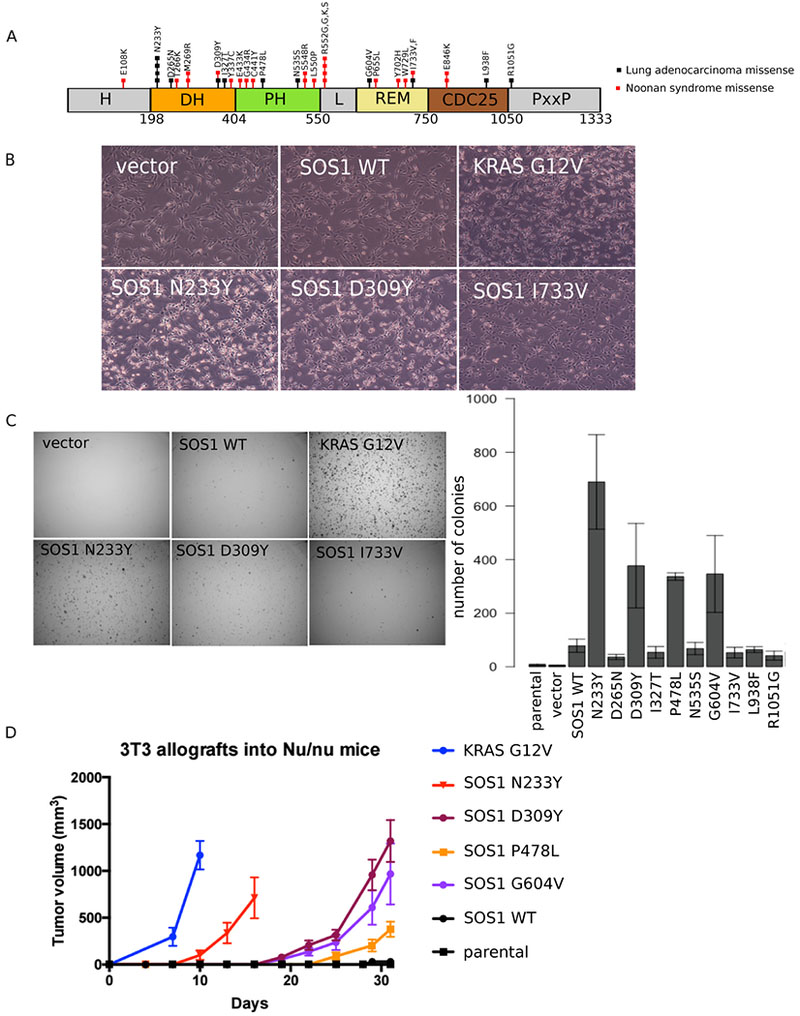
A: SOS1 mutations found in oncogene-negative lung adenocarcinomas and Noonan Syndrome.
B: NIH-3T3 cells expressing different SOS1 mutants were imaged at 40×.
C: NIH-3T3 cells expressing SOS1 mutants were grown in soft agar and imaged after 3 weeks at 6.3×. Colonies were quantified using Cell Profiler. Images are representative of at least three biological replicates, with three technical replicates each. Error bars show SEM from at least three biological replicates. Western blots of SOS1 expression levels across all cells are shown in Figure 2B.
D: NIH-3T3 cells expressing different SOS1 mutants were injected into 4-week old Nu/J female mice. Tumor size was measured every three days using calipers. Tumor volume was calculated as 0.5 × (longer diameter) × (shorter diameter)2. KRAS G12V mutant cells were included as a positive control.
Although SOS1 somatic mutations are statistically significant in lung adenocarcinoma, it has remained to be established experimentally whether these mutations are driver or merely passenger events. In addition, the mechanism of action of any potentially dominantly acting SOS1 mutants has remained unclear. Here, we use a combination of in vitro and in vivo models to study the phenotypic effects of SOS1 mutations, undertake biochemical and transcriptomic studies to better understand the mechanism by which mutant SOS1 acts, and test an inhibitor against cells expressing mutant SOS1 to examine potential therapeutic strategies. Altogether, our study implicates SOS1 as an oncogenic driver in human cancer, suggests a targeted therapeutic opportunity for patients with SOS1 mutations, and increases our understanding of how tumors may develop from normal cells.
Materials and Methods:
Cell culture:
NIH-3T3 cells were purchased from the American Type Culture Collection, were grown in DMEM supplemented with 10% bovine calf serum (BCS) and Antibiotic-Antimycotic from Gibco, and were not used past the 10th passage. OCI-AML3, OCI-AML5, and PC-9 cells were obtained from the Cancer Cell Line Encyclopedia (26). OCI-AML3 and OCI-AML5 were grown in MEM-α supplemented with 20% FBS and Antibiotic-Antimycotic. OCI-AML5 were additionally supplemented with 10 ng/mL of GM-CSF as suggested by the Leibniz Institute DSMZ—German Collection of Microorganisms and Cell Cultures. PC-9 cells were grown in RPMI 1640 supplemented with 10% FBS and Antibiotic-Antimycotic. AALE cells were grown in Small Airway Epithelial Growth Media (SAGM) from Lonza.
Antibodies:
Antibodies against SOS1 (12409), vinculin (4650), MEK 1/2 (4694), p-MEK 1/2 (9121), ERK 1/2 (9107), p-ERK (4370), AKT1 (2967), and p-AKT (4058) were purchased from Cell Signaling Technology. The β-actin (sc47778) antibody was purchased from Santa Cruz Biotechnology.
Drugs:
Erlotinib (S1023) and trametinib (S2673) were purchased from Selleck Chemicals and reconstituted in DMSO.
Vectors and Mutagenesis:
SOS1 cDNA was originally obtained from the Harvard PlasmID Database in a pENTR223.1 vector. The full-length cDNA was confirmed by sequencing. Gateway cloning was performed to insert SOS1 into the pLEX_307 expression vector (Addgene 41392), where it is expressed from the EF1α promoter.
Mutagenesis primers were designed using the Agilent QuickChange Primer Design tool. The sequences can be found in Table S1. Site-directed mutagenesis using the QuickChange II XL or QuickChange Lightning from Agilent was performed on SOS1 in pENTR223.1 which was then transferred into the pLEX_307 expression vector by Gateway cloning.
Cellular transfection and infection:
Plasmids were co-transfected with psPAX2 and pMD2.G vectors into HEK-293T cells using the CalPhos Mammalian Transfection Kit (Takara) to make lentivirus. Cells were infected with the virus and selected with 2 μg/mL puromycin beginning two days after infection.
SOS1 plasmid transfection into NIH-3T3 cells was performed using either the CalPhos Mammalian Transfection Kit (Takara) or the Lipofectamine LTX Reagent with PLUS Reagent (ThermoFisher Scientific 15338100).
Ras pull-down assays:
Plasmids were transfected into NIH-3T3 cells using either the CalPhos Mammalian Transfection Kit (Takara) or the Lipofectamine LTX with Plus Reagent (Thermo Fisher Scientific 15338100). Cells were allowed to recover ~24 hours after transfection and serum-starved for ~24 hours, after which total lysates were collected and processed for Ras-GTP pull-down following the Ras Pull-down Activation Assay Biochem Kit from Cytoskeleton (BK008).
Anchorage-independent growth assay:
For NIH-3T3 cells, 2 mL of 0.75% select agar in DMEM, 10% BCS, Antibiotic-Antimycotic were used to fill the base of 6-well plates and allowed to solidify. Approximately 25,000 cells were resuspended in 1 mL of 0.33% select agar in DMEM, 10% BCS, Antibiotic-Antimycotic, which was then added on top of the base layer. For AALE cells, 2 mL of 0.75% select agar in SAGM minus EGF were used to fill the base of 6-well plates and allowed to solidify. Approximately 40,000 cells were resuspended in 1mL of 0.33% select agar in SAGM minus EGF, which was then added on top of the base layer. Three weeks after seeding cells in soft agar, wells were imaged at 6.3× and the number of colonies was quantified using CellProfiler.
Allograft experiments:
All animal experiments were compliant with institutional requirements. Cohorts of three 4-week old Nu/J female mice were injected subcutaneously into both flanks with 200,000 NIH-3T3 parental cells or cells expressing SOS1 WT, SOS1 N233Y, SOS1 D309Y, SOS1 P478L, or SOS1 G604V. Tumors were measured every three days using calipers and tumor volume was calculated as 0.5 × (longer diameter) × (shorter diameter)2.
Western blots:
Lysates were collected in 1% NP-40, 150 mM NaCl, 50 mM Tris-HCl pH 8.0, 10% glycerol, 0.1% SDS, and 0.5% Na-deoxycholate supplemented with the protease and phosphatase inhibitor HALT (Thermo Fisher Scientific). Pre-aliquoted SDS and DTT from New England Biolabs (B7703) were added to the lysate, and proteins were denatured at 100˚C for five minutes. Proteins were then separated by SDS-PAGE and transferred to a nitrocellulose membrane via either the iBlot dry transfer system (Invitrogen) or overnight at 30 V in transfer buffer containing 10% MeOH and 1× Tris-Glycine. Antibody binding was detected using the LI-COR Odyssey IR imaging system (LI-COR Biosciences).
RNA sequencing and analysis:
RNA was extracted from stably-infected cell lines using the Qiagen RNeasy kit with on-column DNase I treatment. One microgram of total RNA was used for poly-A selection with the NEBNext PolyA mRNA Magnetic Isolation Module (E7490) followed by library construction using the NEBNext Ultra Directional RNA Library Prep kit (E7420).
Paired-end sequencing was performed on an Illumina MiSeq using a 150 cycle V3 kit (MS-102-3001). Alignment against the mouse genome (GRCm38) was performed using the STAR aligner (27), and reads were quantified using HTSeq (28). Each gene was then fit with a generalized linear model (GLM) using DESeq2 (29). Gene set enrichment analysis was performed with normalized gene expression values using GenePattern (30).
SOS1 knockdown:
RNA hairpins (shRNAs) were designed based on SOS1 target sequences from the Broad Institute’s Genetics Perturbations Platform. The sequences can be found in Table S2. The shRNAs were cloned into the pLKO.1 expression vector and were transfected with psPAX2 and pMD2.G into HEK-293T cells to make lentivirus. Cells were infected with the virus and were selected with puromycin beginning two days post-infection. Cell viability was assessed eight days post infection.
Drug treatment of cells:
Cells were seeded in 96-well Corning Costar assay white clear-bottom microplates overnight in 50 μL media. The following day, 50 μL of drug (erlotinib or trametinib) at varying concentrations were added to the cells. Cell viability was assessed three days after drug treatment.
Cell viability measurements:
ATP concentration measured using the CellTiter-Glo (CTG) luminescence assay from Promega (PAG7572) was used as a proxy for cell viability. Briefly, cells were seeded in 96-well Corning Costar assay white clear-bottom microplates in 100 μL per well. CTG solution was diluted at 4x in PBS and 100 μL of the diluted solution was added to each well. The plate was then left in the dark for 10 minutes, after which luminescence was assessed using the Infinite 200 Pro plate reader from Tecan Life Sciences. Integration time was one second.
Data availability:
RNA sequencing data are deposited at NCBI Gene Expression Omnibus (accession number GSE122132).
Results
Lung cancer-derived SOS1 mutants induce transformation in vitro and in vivo
To better understand the effects of SOS1 mutations identified in oncogene-negative lung adenocarcinomas (Figure 1A), we generated NIH-3T3 cell lines expressing lentivirally transduced wild-type or mutant SOS1. We noticed a change in the morphology of cells expressing some of the SOS1 mutants; for example, NIH-3T3 cells harboring the recurrent N233Y mutation closely resembled cells expressing KRAS G12V with more refractive cell bodies, suggesting the cells are rounder, and with thinner projections compared to control cells expressing vector or wildtype SOS1 (Figure 1B). Among the other SOS1 mutants, cells expressing the D309Y mutant were also morphologically similar to KRAS G12V cells, whereas cells expressing the I733V mutant did not undergo a morphological change and resembled control cells (Figure 1B).
In order to determine whether NIH-3T3 cells expressing SOS1 mutants exhibit an oncogenic phenotype, we tested their ability to undergo anchorage-independent colony formation in soft agar. We observed that cells expressing several of the mutants, including the recurrent N233Y mutation as well as the D309Y, P478L, and G604V mutations, formed significantly more colonies compared to wildtype cells in soft agar, ranging from a 48- to 100-fold increase over cells expressing a vector control (Figure 1C; see Figure 2B for expression levels). Overexpression of wildtype SOS1 in NIH-3T3 cells resulted in a smaller 10-fold increase in soft agar colony formation relative to vector control, with similar results for the other SOS1 mutants tested: D265N, I327T, N535S, I733V, L938F, and R1051G (Figures 1C and 2B).
Figure 2: Ras and MAPK pathway are more highly active in SOS1 mutant cells.
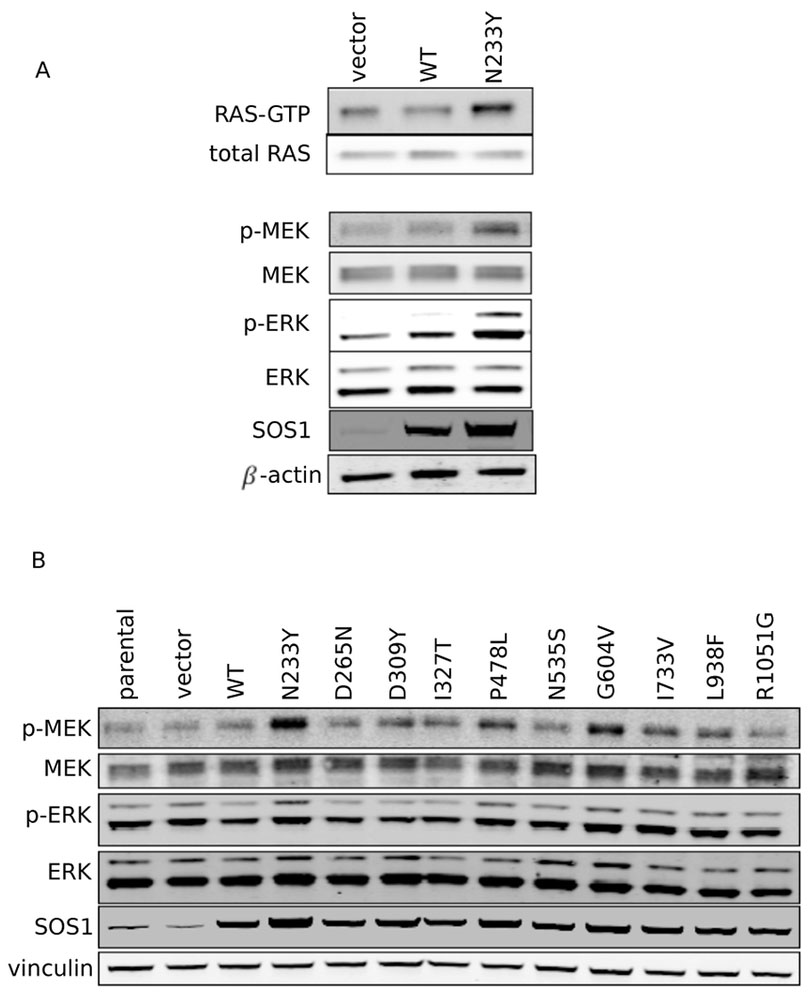
A: Western blot showing RAS-GTP pulldown in serum-starved SOS1 wildtype and N233Y mutant cells, together with total RAS control. Levels of MEK, p-MEK, ERK, and p-ERK are also shown, together with SOS1 and beta-actin controls.
B: Western blot showing levels of MEK, p-MEK, ERK and p-ERK across SOS1-mutant NIH-3T3 cells, together with SOS1 and vinculin controls.
To test the ability of mutant SOS1 to transform human lung epithelial cells, we also generated immortalized tracheobronchial epithelial cells (AALE cells) expressing lentivirally transduced wild-type or mutant SOS1 and tested their ability to undergo anchorage-independent growth in soft agar. We observed a similar trend in soft agar colony formation in AALE cells as in NIH-3T3 cells, with SOS1 N233Y cells forming the most number of colonies (Figure S1A). These results suggest that SOS1 has properties of a proto-oncogene and that the N233Y, D309Y, P478L, and G604V mutations exhibit phenotypic characteristics of oncogenic mutants.
Next, we tested the ability of cells expressing SOS1 mutants to form tumors in vivo. We injected nude mice subcutaneously with NIH-3T3 cells expressing the SOS1 mutants that induced colony formation in soft agar and measured the tumor volume every 3-4 days. Cells expressing the SOS1 N233Y mutant showed the fastest growth, with palpable tumors appearing ten days after injection. The other SOS1 mutants tested in vivo also formed visible tumors, though at a later 19 to 23 days after injection (Figure 1D). In contrast, the parental cells never formed tumors within the 76 days that the mice were monitored, while cells expressing wild-type SOS1 started to form tumors only after 41 days. From these results, we conclude that SOS1 is a potent oncogene and that several mutations, including the recurrent N233Y mutation, can drive tumor formation in vivo.
Analysis of effector pathways associated with oncogenic SOS1 mutants
We next sought to better understand the signaling pathways that may promote tumorigenicity in SOS1 mutant cells. Since SOS1 is a guanine nucleotide exchange factor for RAS, we assessed for RAS activity by performing a pulldown of RAS-GTP with the Ras-binding domain of RAF1. Under serum-starved conditions, we observed that SOS1 N233Y cells have higher levels of RAS-GTP compared to both the parental and SOS1 WT cells (Figure 2A). We then analyzed the activity of kinases downstream of the Ras pathway and found that MEK and ERK were more highly phosphorylated in SOS1 N233Y cells compared to the parental and SOS1 WT cells.
We next assessed the activity of kinases downstream of the Ras pathway across all NIH-3T3 cell lines under normal serum conditions (10%) and observed that SOS1 mutants that were able to promote anchorage-independent growth also induced higher levels of MEK phosphorylation (Figure 2B). We also observed increased MEK phosphorylation from AALE cells grown in SAGM media (Figure S1B). Altogether, our pulldown of RAS-GTP in control and SOS1 N233Y cells, as well as our subsequent analysis of phosphorylation of kinases downstream of RAS in all SOS1 mutant NIH-3T3 and AALE cells, are consistent with the possibility that the tumorigenic phenotype of specific SOS1 mutants is due to an increased activation of RAS by SOS1.
We then assessed the transcriptional consequences of ectopic expression of activated SOS1. We performed RNA-sequencing of NIH-3T3 cells transduced with vector control, SOS1 wildtype (WT), SOS1 N233Y, and KRAS G12V, with three biological replicates for each condition. Clustering the samples based on gene expression, we observed that cells expressing SOS1 N233Y have a transcriptional profile that resembles cells expressing KRAS G12V more than cells transduced with SOS1 WT or vector control (Figure 3A). To assess pathway differences between cells expressing SOS1 N233Y versus wild-type SOS1, we performed gene set enrichment analysis of the RNA-sequencing data. We observed that in SOS1 N233Y cells, there is an enrichment for gene signatures associated with Kras transformation and MYC targets (Figure 3B). Other gene sets enriched in the SOS1 N233Y cells include the KEGG Ribosome and Reactome peptide chain elongation sets (Table S3). This differential expression can be confirmed on the level of specific genes, including Hmga2, Tgfb1, and Gdnf, which are upregulated in NIH-3T3 cells transformed with active Kras, and Eif4e, Eif2s1, and Uba2, which are targets of MYC (Figure 3C). Taken together, our RNA sequencing results indicate the SOS1 N233Y mutation has a transcriptional effect on cells that is similar to that of the KRAS G12V mutation, and SOS1 N233Y cells likely drive tumorigenesis by upregulating similar pathways and genes as RAS-mutant cells.
Figure 3: The expression profile of SOS1 N233Y NIH-3T3 cells resembles KRAS G12V NIH-3T3 cells.
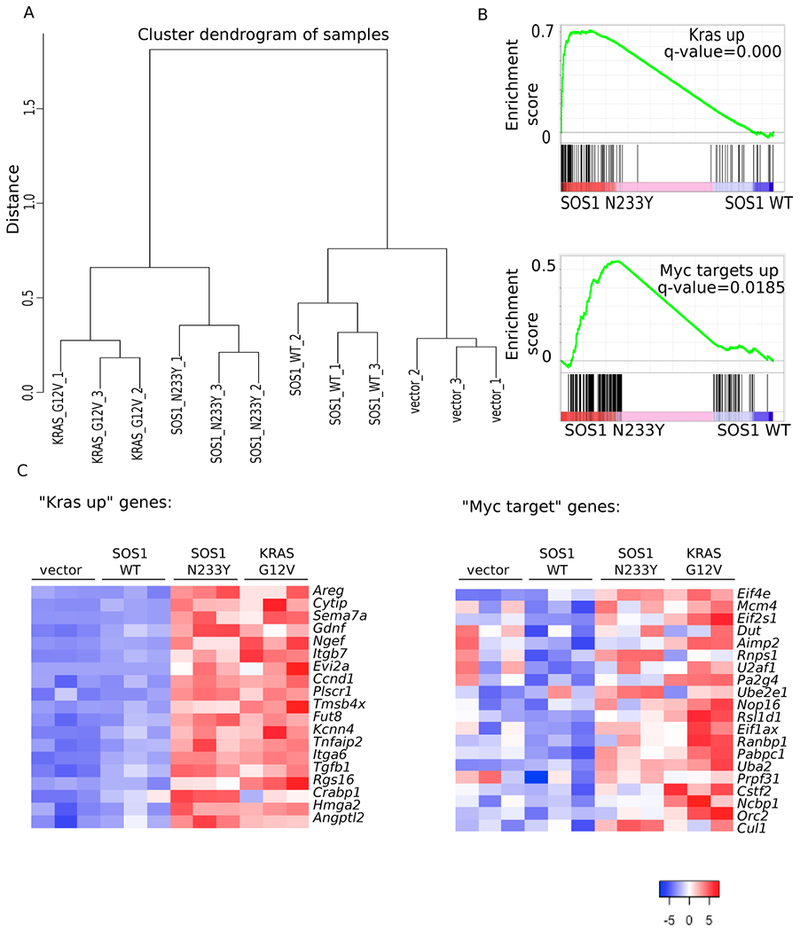
A: RNA sequencing was performed on three biological replicates of vector-control, SOS1 wildtype, SOS1 N233Y, and KRAS G12V NIH-3T3 cells. Normalized read counts were used to compute the distance between the samples based on Pearson correlation coefficient, and complete linkage was used to create clusters for the dendrogram.
B: Gene set enrichment analysis was performed using normalized read counts. “Kras up” is the dataset from (63). “Myc targets up” is the dataset from the Molecular Signatures Database Hallmark MYC V1 collection (64).
C: Heat map showing standardized t-statistics (Tij) of normalized expression values for each sample per gene. , where xij=normalized expression value for sample i and gene j, =sample mean for normalized expression of gene j, sj=sample standard deviation for normalized expression of gene j, n=number of samples. “Kras up” refers to the set of genes shown that are also in the “Kras up” set from Figure 3B and (63). “Myc target” refers to the set of genes shown that are also in the “Myc targets up set” from Figure 3B and (64).
The RAS-GEF and RAC-GEF activity of SOS1 is required for oncogenic transformation
To better understand the mechanism by which SOS1 mutations drive an oncogenic phenotype, we decided to test the domains required for transformation by the SOS1 N233Y mutant. We introduced second-site mutations in three locations: F929A, which disrupts the catalytic CDC25 domain (31); W729E, which disrupts the allosteric REM domain (22); and 351-LHYFELL-357→351-IIIRDII-357, which disrupts the putative Rac-GEF region in SOS1 (32) (Figure 4A). Our results indicate that second-site mutations at all three sites abolish the ability of the SOS1 N233Y mutant to induce anchorage-independent growth of NIH-3T3 in soft agar (Figure 4B). Due to the known and putative functions of the secondary sites, we were interested in whether there were alterations in the activity of kinases downstream of Ras in the cells. We observed a decrease in the level of p-MEK and p-ERK in the double mutant cells compared to cells with the N233Y mutation alone (Figure 4C). For control, we also performed second-site mutagenesis at H349Q, a site in the DH domain that is conserved across other species (Figure S2A). We observed similar numbers of colonies formed for SOS1 N233Y H349Q compared to SOS1 N233Y alone (Figure S2B). Furthermore, we observed similar levels of MEK phosphorylation in SOS1 N233Y H349Q cells as in SOS1 N233Y cells (Figure S2C). Altogether, our experiments disrupting functional sites in SOS1 as well as our control experiment suggest the ability of SOS1 to act as both a RAS-GEF and putative RAC-GEF is potentially critical to its oncogenic potential and further support our observations that the oncogenic phenotype we observe is a result of increased RAS activity.
Figure 4: Ras-GEF and putative Rac-GEF function are required for SOS1 N233Y to transform NIH-3T3 cells.
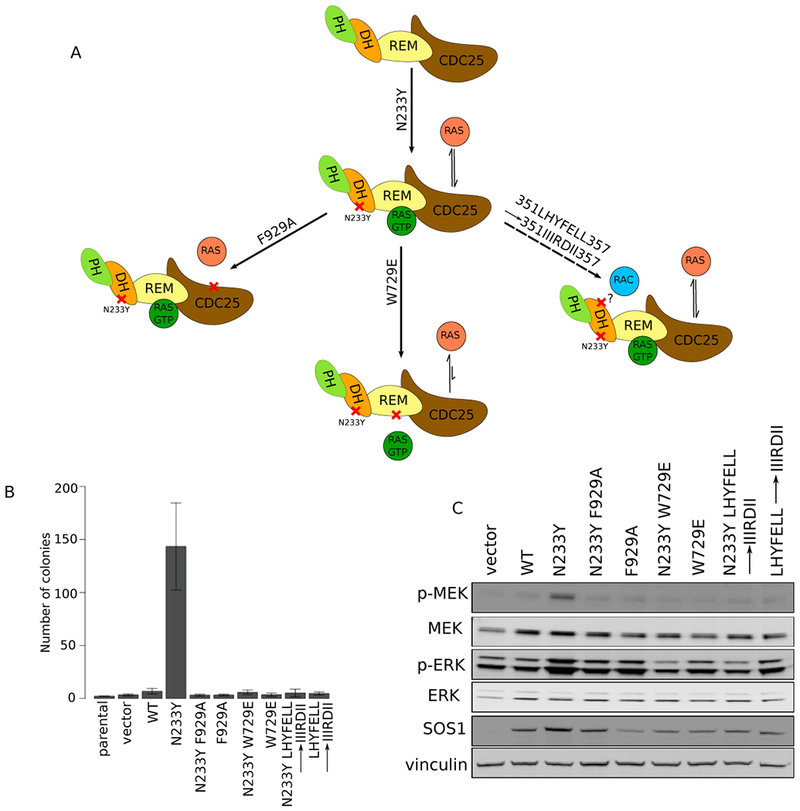
A: Model of SOS1 domain organization and interactions with partner signaling molecules. Second-site mutations were generated at the catalytic CDC25 (F929A), allosteric activating REM (W729E), and putative RacGEF (351LHYFELL357→351IIIRDII357) domains. Color scheme matches Figure 1A. Diagrams based on (22).
B: NIH-3T3 cells expressing SOS1 single and double mutants were grown in soft agar and imaged after 3 weeks at 6.3×. Colonies were quantified using Cell Profiler. Images are representative of at least three biological replicates, with three technical replicates each. Error bars show SEM from at least three biological replicates.
C: Western blot of MAP kinase signaling in NIH-3T3 cells expressing single and double mutants of SOS1.
SOS1 mutant cells depend on SOS1 for growth and are sensitive to MEK inhibition
Given the oncogenic impact of SOS1 mutations, we sought to determine whether patient-derived human cancer cells harboring SOS1 mutations are dependent on SOS1 for growth. To address this question, we first analyzed SOS1 dependency across cell lines using recently published data from a large-scale effort to identify cancer vulnerabilities using genome-wide short-hairpin RNA (shRNA) screens in 501 cell lines (33). We identified an acute myeloid leukemia (AML) cell line, OCI-AML5, with a SOS1 N233Y mutation, as having a significant dependency on SOS1 (Figure 5A; only AML cell lines are shown). We proceeded to validate this result using OCI-AML3, an AML line that is wildtype for SOS1 but harbors an NRAS Q61L mutation, as a control. We observed that SOS1 mutant OCI-AML5 cells are significantly more sensitive to shRNA knockdown of SOS1 compared to the SOS1 wildtype OCI-AML3 cells (Figure 5B) and that there is a slight decrease in the levels of p-MEK in OCI-AML5 cells upon SOS1 knockdown (Figure 5C). These results indicate that cancer cells with SOS1 N233Y mutation may be dependent on SOS1 for growth and that this dependency may be due to a decrease in MAPK signaling.
Figure 5: SOS1 N233Y mutant OCIAML5 cells are dependent on SOS1 for growth, and are sensitive to the MEK inhibitor trametinib.

A: SOS1 dependency scores from shRNA knockdown in AML cell lines were ranked (data from (33)). Cell lines with scores <=−2 are considered dependent on SOS1. OCI-AML5 has a SOS1 N233Y mutation and is labeled in red. OCI-AML3 has an NRAS Q61L mutation and is third from top.
B: SOS1 knockdown using shRNAs. Three different shRNAs targeting SOS1 were used, with at least three biological replicates performed for each shRNA. Cell viability is normalized to shGFP control.
C: Western blot of SOS1, p-MEK, and MEK, with vinculin control, in parental and SOS1-knockdown OCI-AML3 (NRAS mutant) and OCI-AML5 (SOS1 mutant) cells.
D: OCI-AML3, OCI-AML5, and PC-9 (EGFR mutant) cells were tested for their sensitivity to erlotinib (10−4 to 10 μM) and trametinib (10−6 to 10 μM). Cell viability is normalized to DMSO-treated cells. Error bars represent SEM from at least three biological replicates, with six technical replicates each.
E: NIH-3T3 cells expressing the SOS1 N233Y mutant were grown in soft agar under different concentrations of trametinib and imaged after 3 weeks at 6.3×, with cells expressing KRAS G12V as a control. Colonies were quantified using Cell Profiler and normalized to DMSO-treated colonies. Error bars represent SEM.
Next, we hypothesized that SOS1 mutant OCI-AML5 cells may also be dependent on kinases downstream of SOS1 and RAS. To test this hypothesis, we treated cells with the MEK inhibitor trametinib or, as a negative control, with the EGFR inhibitor erlotinib. OCI-AML3 and PC-9 cells, a lung adenocarcinoma cell line with an activating EGFR ∆E746-A750 mutation, were also treated as controls. We observed that only PC-9 cells were sensitive to erlotinib while all three cell lines were sensitive to trametinib (Figure 5D). Our findings indicate that SOS1 mutant cancer cells can be dependent on the activity of kinases downstream of RAS for growth and suggest the possibility of using trametinib to reduce the growth of such cells.
While OCI-AML5 cells are sensitive to trametinib, we wondered whether the sensitivity was specific to the SOS1 N233Y mutation. To learn more, we decided to test the anchorage-independent growth ability of SOS1 N233Y or control KRAS G12V NIH-3T3 cells under different concentrations of trametinib. We observed that while trametinib can abolish anchorage-independent growth in both cell types, SOS1 N233Y cells appeared to be more trametinib-sensitive compared to KRAS G12V cells (Figure 5E). In contrast, both cell lines continued to form colonies in soft agar in the presence of erlotinib (Figure S3). From these studies, we conclude that cells whose transformation is dependent on the SOS1 N233Y mutation are sensitive to the MEK inhibitor trametinib.
Discussion:
In this study, we demonstrate that SOS1 mutations identified in oncogene-negative lung adenocarcinoma patients promote a tumorigenic phenotype, activate the Ras pathway, and upregulate MYC target genes. We further demonstrate that SOS1 mutant cells are dependent on SOS1 for growth and that they are sensitive to the MEK inhibitor trametinib. Previous studies of SOS1 mutations have primarily focused on germline mutations found in Noonan syndrome (23, 24) and hereditary gingival fibromatosis type I (25). The connection between activating SOS1 mutations and Noonan syndrome previously led our laboratory to perform a study where targeted sequencing of SOS1 was performed on 810 primary cancers of various histologies; however, SOS1 was not significantly mutated in this cohort (16). We find here that SOS1, which is significantly mutated in lung adenocarcinoma (15), is an oncogene and that mutations in SOS1 are capable of driving tumor formation.
SOS1 is a highly conserved guanine nucleotide exchange factor with multiple domains, including the DH, PH, REM, and CDC25 domains. The H, DH, and PH domains form an autoinhibitory module that blocks the allosteric REM domain, suppressing activation of the catalytic CDC25 domain and thereby inhibiting SOS1 function (22). The SOS1 N233Y mutation in the DH domain could activate the SOS1 protein by relieving this autoinhibition and allowing effector RAS-GTP molecules to bind more efficiently to the REM domain, activating the CDC25 domain to exchange GDP for GTP on Ras. Activation of SOS1 then leads to the oncogenic phenotype we observe, including greater activation of RAS (Figure 2A) and greater phosphorylation of MEK downstream of RAS (Figures 2B and S1B). Our experiments demonstrating loss of anchorage-independent growth due to second-site mutations at F929A, which eliminates the catalytic activity in the CDC25 domain (31), and W729E, which prevents REM from allosterically binding RAS-GTP (22), further support the notion that functional Ras-GEF activity is necessary for mutant SOS1 oncogenicity (Figures 4B and 4C).
Alternatively, previous studies suggest another mechanism for the oncogenic phenotype: One mechanism reported to be responsible for SOS1 recruitment to the membrane is PIP2 tethering by the PH domain of SOS1 (34, 35). Membrane localization aids in the release of the autoinhibitory domain of SOS1 and leads to downstream signaling. This leads to the possibility that SOS1 N233Y mutant protein may localize to the membrane with greater affinity compared to the SOS1 wildtype protein, thereby enabling greater activation of RAS and downstream pathways, leading to the observed phenotypes. Nonetheless, regardless of the precise mechanism by which SOS1 N233Y leads to an oncogenic phenotype, our experiments indicate that it does so by activating the Ras pathway.
In addition to the established functions of the DH, PH, REM, and CDC25 domains, SOS1 also contains putative Rac guanine nucleotide exchange factor (Rac-GEF) activity within the DH domain (20, 21). Though direct contact between SOS1 and Rac has not been established, previous studies have revealed that a complex consisting of SOS1, E3B1 and EPS8 has Rac-GEF activity in vitro (36). Furthermore, it has been hypothesized that mutating amino acids 351-357 (LHYFELL) in the SOS1 DH domain would disrupt Rac-GEF activity (32), as it does in corresponding sites of the Dbl proto-oncogene (37) as well as in the Rac-GEF Vav (38). Our second-site mutagenesis results (LHYFELL→IIIRDII) suggest SOS1 Rac-GEF function is necessary for the oncogenic properties of the SOS1 N233Y mutation (Figure 4B and 4C) and that Ras-GEF activity alone may not be sufficient for tumorigenesis. This result is in agreement with previous studies showing the Rac-GEF region to be necessary for the transforming abilities of myristoylated SOS1 (21), the necessity for Rac1 in Kras-driven lung cancer models (39), and the necessity for the Rac-GEF Tiam1 in Ras-driven skin cancer (40). Previous studies have also demonstrated that Rac signals downstream to the MAPK pathway through PAK (41–43) and that this signal is required for Ras-driven skin tumors (44). Therefore, the increased p-MEK levels we observe in the SOS1 mutant NIH-3T3 cells compared to control cells may be a result of both Ras and Rac signaling. The results from our second-site mutagenesis experiments of SOS1 N233Y support this hypothesis as disruptions to the RAS-GEF catalytic site (F929A), allosteric site (W729E), or putative RAC-GEF site (LHYFELL→IIIRDII) are associated with a decreased level of p-MEK in the SOS1 N233Y mutant NIH-3T3 cells (Figure 4C).
A potential confounder for the results observed in our second-site mutation experiments in the Rac-GEF domain may be that the SOS1 LHYFELL mutant protein has an abnormal conformation, especially since the mutation in this domain stretches across several (seven) amino acids. No structural studies have yet been performed to determine the conformation of this mutant protein, so it is also possible that the phenotype we observe from the double mutant is a result of protein misfolding rather than an indication of SOS1 RAC-GEF activity being required for the oncogenic phenotype observed.
While our study did not focus on mutations found in Noonan Syndrome (NS), two of the mutations we studied, D309Y and I733V, are shared between cancer and NS (with I733V being I733F in NS). The D309Y mutation is oncogenic in our assays whereas the I733V mutation is not (Figures 1C, 1D, 2B, S1A, and S1B), suggesting that a subset of SOS1 mutations found in Noonan Syndrome are capable of driving tumor formation. This result is consistent with the increased incidence of cancer in patients with Noonan Syndrome (45–47).
The upregulation of KRAS and MYC target genes that we observe in the transcriptional profile of SOS1 N233Y cells is consistent with the function of SOS1 as a regulator of RAS and the ability of RAS signaling to enhance MYC protein stability (48, 49). The genes upregulated in SOS1 N233Y cells include Eif4e and Eif2s1, which encode translation initiation factors that are regulated by MYC. It is particularly interesting that Eif4e is normally the least abundant translation initiation factor in most cell types (50–52) but has been shown to be overexpressed in many cancers, including breast (53–55), head and neck (56, 57), and colorectal cancers (58, 59). It is proposed that Eif4e overexpression causes enhanced translation of mRNAs with a high degree of secondary structure (52, 60). Hence, further studies using ribosomal profiling or mass spectrometry could determine the differences in translated protein levels between SOS1-mutant and wildtype cells and could further help us understand the mechanisms by which SOS1-mutant cells are tumorigenic.
Our results showing the dependency of SOS1-mutant OCI-AML5 cells on SOS1 and their sensitivity to the MEK inhibitor trametinib opens up the possibility of treatment options for SOS1-mutant cancers in the future (Figures 5B, 5C, and 5D). This is further supported by the ability of trametinib to suppress SOS1-mutant induced transformation in NIH-3T3 cells (Figure 5E). Interestingly, we observed that RAS-mutant OCI-AML3 cells are more sensitive to trametinib than SOS1-mutant OCI-AML5 cells, which, in turn, are more sensitive than EGFR-mutant PC-9 cells. This is in agreement with previous studies demonstrating the sensitivity of OCI-AML3 cells to MEK inhibitors (61, 62). Additionally, this pattern may be explained by the fact that, relative to MEK, both SOS1 and EGFR are further upstream in the MAPK signaling pathway.
Though the RTK/Ras/Raf pathway is frequently mutated across cancers, the genetic and molecular underpinnings of a large percentage of cancers have yet to be uncovered. Our study allows us to better understand the biology and impact of SOS1 mutations in cancer, which could enable more targeted therapies for individuals harboring these mutations, including the possibility of treatment with MEK inhibitors.
Supplementary Material
Implications:
This study demonstrates that SOS1 mutations found in lung adenocarcinoma are oncogenic and that MEK inhibition may be a therapeutic avenue for the treatment of SOS1-mutant cancers.
Acknowledgements:
The authors thank Rameen Beroukhim, David Pellman, Alex Toker, and Kwok Wong for discussions and advice on this work. We would also like to thank many members of the Meyerson lab, especially Josh Campbell, Hugh Gannon, Jon Goldstein, Heidi Greulich, Tikvah Hayes, and Xiaoyang Zhang, for reagents, advice, and help. This work was supported by the NSF Graduate Research Fellowship (D.C.); the NIH Pathway to Independence Award K99 CA208028 (P.S.C.); and the National Cancer Institute grant 1R35CA197568, the American Cancer Society Research Professorship, and the Norman R. Seaman Endowment Fund for Lung Cancer (M.M).
Financial support: We acknowledge support from National Cancer Institute (1R35CA197568), the American Cancer Society Research Professorship, and the Norman R. Seaman Endowment Fund for Lung Cancer to M. Meyerson; the NSF Graduate Research Fellowship to D. Cai; and, the NIH Pathway to Independence Award K99 (CA208028) to P.S. Choi.
Footnotes
Conflict of interest: Matthew Meyerson receives funding from Bayer Pharmaceuticals.
References:
- 1.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dogan S, Shen R, Ang DC, Johnson ML, D’Angelo SP, Paik PK, et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(22):6169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arcila ME, Drilon A, Sylvester BE, Lovly CM, Borsu L, Reva B, et al. MAP2K1 (MEK1) Mutations Define a Distinct Subset of Lung Adenocarcinoma Associated with Smoking. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(8):1935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawrence RE, Salgia R. MET molecular mechanisms and therapies in lung cancer. Cell Adh Migr. 2010;4(1):146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65(4):1479–88. [DOI] [PubMed] [Google Scholar]
- 8.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6. [DOI] [PubMed] [Google Scholar]
- 9.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(8):863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rimkunas VM, Crosby KE, Li D, Hu Y, Kelly ME, Gu TL, et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(16):4449–57. [DOI] [PubMed] [Google Scholar]
- 11.Suehara Y, Arcila M, Wang L, Hasanovic A, Ang D, Ito T, et al. Identification of KIF5B-RET and GOPC-ROS1 fusions in lung adenocarcinomas through a comprehensive mRNA-based screen for tyrosine kinase fusions. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(24):6599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome research. 2012;22(3):436–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nature medicine. 2012;18(3):375–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doebele RC, Davis LE, Vaishnavi A, Le AT, Estrada-Bernal A, Keysar S, et al. An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101. Cancer Discov. 2015;5(10):1049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nature genetics. 2016;48(6):607–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swanson KD, Winter JM, Reis M, Bentires-Alj M, Greulich H, Grewal R, et al. SOS1 mutations are rare in human malignancies: implications for Noonan Syndrome patients. Genes, chromosomes & cancer. 2008;47(3):253–9. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173(2):321–37 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogge RD, Karlovich CA, Banerjee U. Genetic dissection of a neurodevelopmental pathway: Son of sevenless functions downstream of the sevenless and EGF receptor tyrosine kinases. Cell. 1991;64(1):39–48. [DOI] [PubMed] [Google Scholar]
- 19.Simon MA, Bowtell DD, Dodson GS, Laverty TR, Rubin GM. Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell. 1991;67(4):701–16. [DOI] [PubMed] [Google Scholar]
- 20.Chen PC, Wakimoto H, Conner D, Araki T, Yuan T, Roberts A, et al. Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J Clin Invest. 2010;120(12):4353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nimnual AS, Yatsula BA, Bar-Sagi D. Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science. 1998;279(5350):560–3. [DOI] [PubMed] [Google Scholar]
- 22.Sondermann H, Soisson SM, Boykevisch S, Yang SS, Bar-Sagi D, Kuriyan J. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell. 2004;119(3):393–405. [DOI] [PubMed] [Google Scholar]
- 23.Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nature genetics. 2007;39(1):70–4. [DOI] [PubMed] [Google Scholar]
- 24.Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nature genetics. 2007;39(1):75–9. [DOI] [PubMed] [Google Scholar]
- 25.Hart TC, Zhang Y, Gorry MC, Hart PS, Cooper M, Marazita ML, et al. A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. American journal of human genetics. 2002;70(4):943–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nature genetics. 2006;38(5):500–1. [DOI] [PubMed] [Google Scholar]
- 31.Hall BE, Yang SS, Boriack-Sjodin PA, Kuriyan J, Bar-Sagi D. Structure-based mutagenesis reveals distinct functions for Ras switch 1 and switch 2 in Sos-catalyzed guanine nucleotide exchange. J Biol Chem. 2001;276(29):27629–37. [DOI] [PubMed] [Google Scholar]
- 32.Qian X, Vass WC, Papageorge AG, Anborgh PH, Lowy DR. N terminus of Sos1 Ras exchange factor: critical roles for the Dbl and pleckstrin homology domains. Mol Cell Biol. 1998;18(2):771–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell. 2017;170(3):564–76 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gureasko J, Galush WJ, Boykevisch S, Sondermann H, Bar-Sagi D, Groves JT, et al. Membrane-dependent signal integration by the Ras activator Son of sevenless. Nat Struct Mol Biol. 2008;15(5):452–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat Cell Biol. 2007;9(6):706–12. [DOI] [PubMed] [Google Scholar]
- 36.Innocenti M, Tenca P, Frittoli E, Faretta M, Tocchetti A, Di Fiore PP, et al. Mechanisms through which Sos-1 coordinates the activation of Ras and Rac. J Cell Biol. 2002;156(1):125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hart MJ, Eva A, Zangrilli D, Aaronson SA, Evans T, Cerione RA, et al. Cellular transformation and guanine nucleotide exchange activity are catalyzed by a common domain on the dbl oncogene product. J Biol Chem. 1994;269(1):62–5. [PubMed] [Google Scholar]
- 38.Kuhne MR, Ku G, Weiss A. A guanine nucleotide exchange factor-independent function of Vav1 in transcriptional activation. J Biol Chem. 2000;275(3):2185–90. [DOI] [PubMed] [Google Scholar]
- 39.Kissil JL, Walmsley MJ, Hanlon L, Haigis KM, Bender Kim CF, Sweet-Cordero A, et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007;67(17):8089–94. [DOI] [PubMed] [Google Scholar]
- 40.Malliri A, van der Kammen RA, Clark K, van der Valk M, Michiels F, Collard JG. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature. 2002;417(6891):867–71. [DOI] [PubMed] [Google Scholar]
- 41.Coles LC, Shaw PE. PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene. 2002;21(14):2236–44. [DOI] [PubMed] [Google Scholar]
- 42.Frost JA, Steen H, Shapiro P, Lewis T, Ahn N, Shaw PE, et al. Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 1997;16(21):6426–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slack-Davis JK, Eblen ST, Zecevic M, Boerner SA, Tarcsafalvi A, Diaz HB, et al. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J Cell Biol. 2003;162(2):281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, Pedersen E, Basse A, Lefever T, Peyrollier K, Kapoor S, et al. Rac1 is crucial for Ras-dependent skin tumor formation by controlling Pak1-Mek-Erk hyperactivation and hyperproliferation in vivo. Oncogene. 2010;29(23):3362–73. [DOI] [PubMed] [Google Scholar]
- 45.Jongmans MC, van der Burgt I, Hoogerbrugge PM, Noordam K, Yntema HG, Nillesen WM, et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur J Hum Genet. 2011;19(8):870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kratz CP, Franke L, Peters H, Kohlschmidt N, Kazmierczak B, Finckh U, et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer. 2015;112(8):1392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strullu M, Caye A, Lachenaud J, Cassinat B, Gazal S, Fenneteau O, et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet. 2014;51(10):689–97. [DOI] [PubMed] [Google Scholar]
- 48.Marampon F, Ciccarelli C, Zani BM. Down-regulation of c-Myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol Cancer. 2006;5:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999;3(2):169–79. [DOI] [PubMed] [Google Scholar]
- 50.Duncan R, Milburn SC, Hershey JW. Regulated phosphorylation and low abundance of HeLa cell initiation factor eIF-4F suggest a role in translational control. Heat shock effects on eIF-4F. J Biol Chem. 1987;262(1):380–8. [PubMed] [Google Scholar]
- 51.Hiremath LS, Webb NR, Rhoads RE. Immunological detection of the messenger RNA cap-binding protein. J Biol Chem. 1985;260(13):7843–9. [PubMed] [Google Scholar]
- 52.Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N. eIF4E--from translation to transformation. Oncogene. 2004;23(18):3172–9. [DOI] [PubMed] [Google Scholar]
- 53.Kerekatte V, Smiley K, Hu B, Smith A, Gelder F, De Benedetti A. The proto-oncogene/translation factor eIF4E: a survey of its expression in breast carcinomas. Int J Cancer. 1995;64(1):27–31. [DOI] [PubMed] [Google Scholar]
- 54.Li BD, Liu L, Dawson M, De Benedetti A. Overexpression of eukaryotic initiation factor 4E (eIF4E) in breast carcinoma. Cancer. 1997;79(12):2385–90. [PubMed] [Google Scholar]
- 55.Nathan CO, Carter P, Liu L, Li BD, Abreo F, Tudor A, et al. Elevated expression of eIF4E and FGF-2 isoforms during vascularization of breast carcinomas. Oncogene. 1997;15(9):1087–94. [DOI] [PubMed] [Google Scholar]
- 56.Nathan CO, Franklin S, Abreo FW, Nassar R, De Benedetti A, Glass J. Analysis of surgical margins with the molecular marker eIF4E: a prognostic factor in patients with head and neck cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1999;17(9):2909–14. [DOI] [PubMed] [Google Scholar]
- 57.Nathan CO, Franklin S, Abreo FW, Nassar R, de Benedetti A, Williams J, et al. Expression of eIF4E during head and neck tumorigenesis: possible role in angiogenesis. Laryngoscope. 1999;109(8):1253–8. [DOI] [PubMed] [Google Scholar]
- 58.Rosenwald IB, Chen JJ, Wang S, Savas L, London IM, Pullman J. Upregulation of protein synthesis initiation factor eIF-4E is an early event during colon carcinogenesis. Oncogene. 1999;18(15):2507–17. [DOI] [PubMed] [Google Scholar]
- 59.Xu T, Zong Y, Peng L, Kong S, Zhou M, Zou J, et al. Overexpression of eIF4E in colorectal cancer patients is associated with liver metastasis. Onco Targets Ther. 2016;9:815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koromilas AE, Lazaris-Karatzas A, Sonenberg N. mRNAs containing extensive secondary structure in their 5’ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J. 1992;11(11):4153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milella M, Kornblau SM, Estrov Z, Carter BZ, Lapillonne H, Harris D, et al. Therapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemia. J Clin Invest. 2001;108(6):851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang W, Konopleva M, Burks JK, Dywer KC, Schober WD, Yang JY, et al. Blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase and murine double minute synergistically induces Apoptosis in acute myeloid leukemia via BH3-only proteins Puma and Bim. Cancer Res. 2010;70(6):2424–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chiaradonna F, Gaglio D, Vanoni M, Alberghina L. Expression of transforming K-Ras oncogene affects mitochondrial function and morphology in mouse fibroblasts. Biochim Biophys Acta. 2006;1757(9–10):1338–56. [DOI] [PubMed] [Google Scholar]
- 64.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data are deposited at NCBI Gene Expression Omnibus (accession number GSE122132).