

Abstract
The host immune responses that mediate Chlamydia-induced chronic disease sequelae are incompletely understood. The role of TNF-α, TNF receptor 1(TNFR1), and TNF receptor 2 (TNFR2), in Chlamydia pneumoniae (CPN)-induced atherosclerosis was studied using the high-fat diet-fed male C57BL/6J mouse model. Following intranasal CPN infection, TNF-α knockout (KO), TNFR1 KO, TNFR2 KO, and TNFR 1/2 double-knockout, displayed comparable serum anti-chlamydial antibody response, splenic antigen-specific cytokine response, and serum cholesterol profiles compared to wild type (WT) animals. However, atherosclerotic pathology in each CPN-infected KO mouse group was reduced significantly compared to WT mice, suggesting that both TNFR1 and TNFR2 promote CPN-induced atherosclerosis.
Keywords: Chlamydia pneumoniae, atherosclerosis, pathogenesis, tumor necrosis factor, tumor necrosis factor receptor 1, tumor necrosis factor receptor 2
1. Introduction
We have demonstrated that tumor necrosis factor α (TNF-α) production mediates chronic oviduct pathology following Chlamydia muridarum infection in female mice [1]. TNF-α has been shown to signal via two cognate receptors, TNF receptor superfamily member 1a (TNFR1) and TNF receptor superfamily member 1b (TNFR2); we showed that both contribute significantly to the upper genital tract pathology induced by C. muridarum infection [2]. Chlamydia muridarum is a murine pathogen that is closely related to C. trachomatis, a pathogen that induces chronic oviduct pathology following intravaginal (i.vag) infection in humans (reviewed in [3]). However, intravaginal C. trachomatis infection in mice does not induce robust chronic oviduct pathologies [3], thus posing difficulty in validating the findings of C. muridarum pathogenesis using the human pathogenic strain C. trachomatis in the mouse model. In this context, we noted that respiratory infection with the closely related human pathogen Chlamydia pneumoniae (CPN) has been correlated strongly with exacerbation of atherosclerosis, a chronic pathological sequel to the infection in human individuals (reviewed in [4]). Whereas a direct cause-and-effect relationship in humans has yet to be conclusively determined, rabbits or male C57BL/6 mice fed a high-fat (HF) diet and concomitantly infected intravascularly (i.v.) or intranasally (i.n.), respectively, with CPN display exacerbation of atherosclerosis [5, 6]. Using the HF diet-fed male C57BL6 mouse model, a contribution of TNFR1 in CPN-induced exacerbation of atherosclerosis has been demonstrated [7]. Whereas TNFR1 is expressed on most nucleated cells of the body, TNFR2 is more restricted in expression to certain cell types including T-lymphocytes (reviewed in [8]). We have also shown previously that C. pneumoniae-induced enhancement of atherosclerosis in the HF diet-fed C57BL/6 mouse model is mediated by CD8+ T cells [9], thus suggesting that TNFR2 may play a role in CPN-induced exacerbation of atherosclerotic pathology. However, the role of TNFR2, or the contribution of the two TNF receptors in relation to the ligand, TNF-α, in CPN-induced atherosclerosis had not been examined previously.
In this study, we evaluated the effects of i.n. C. pneumoniae infection on atherosclerosis development using mice with a gene deficiency of TNF-α (TNF KO), TNFR1 KO, TNFR2 KO, or TNFR1 & 2 double knockout (TNFR1/2 DKO), in comparison to corresponding wild type C57BL/6J (WT) mice.
2. Materials and methods
2.1. Bacteria.
Chlamydia pneumoniae AR39 (C. pneumoniae or CPN; obtained from Dr. Lee Ann Campbell, University of Washington, Seattle) was grown on confluent HEp-2 cell monolayers as described previously [9]. Cells were lysed using a sonicator and elementary bodies purified on Renograffin (Isovue) gradients. Aliquots of bacteria were stored at −80°C in sucrose-phosphate-glutamine buffer (SPG; Sucrose 218mM, KH2PO4 3.8mM, K2HPO47.2mM, L-Glutamate 4.9mM, pH 7.2 buffer).
2.2. Animals.
TNF KO, TNFR1 KO, TNFR2 KO, and TNFR1/2 DKO mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred in the Midwestern University animal facility. Corresponding wild type male 6–8 week-old C57BL/6J (WT) mice were purchased from the Jackson Laboratory. All experiments described in this manuscript were approved by the Institutional Animal Care and Use Committee of Midwestern University.
2.3. C. pneumoniae infection.
On day 0, six-eight week-old male mice were anesthetized with isofluorane and challenged i.n. (10 µl/nostril) with 1X107 IFU/mouse CPN in buffer (SPG) or mock-infected with SPG alone. Mice were challenged i.n. with the same inoculum again on days 15 and 30. From the day of first challenge, a high-fat diet (HFD; 15% fat, 1.25% cholesterol and 0.5% cholate) [7] was initiated and continued until the end of the experiment on day 105.
2.4. Antigen-specific cytokine response.
Antigen (Ag)-specific cytokine production from splenocytes was measured as described previously [2]. Fourteen days after the initial i.n inoculation with CPN or SPG alone (mock), mice were euthanized. Single cell suspensions of splenocytes (106 cells/well) were stimulated in 96-well culture plates with 106 IFU of UV-inactivated CPN, the unrelated protein bovine serum albumin (BSA), or media alone, and incubated for 72 hr. Supernatants were collected and assayed for IFN-γ and TNF-α production by enzyme-linked immunosorbent assays (ELISA) using eBioscience™ Mouse ELISA Ready-SET-Go™ Kit(Invitrogen, Carlsbad, CA).
2.5. Humoral response.
On day 90 of the experiment, sera from the animals were analyzed by ELISA as described previously [9]. Microtiter plates coated overnight with UV-inactivated CPN (106 IFU/well) were probed with serial dilutions of serum followed by goat anti-mouse total Ab, IgG1, or IgG2c detection antibody followed by substrate per manufacturer’s instructions (5300–05B, Southern Biotechnology Associates, AL). Absorbance at 405 nm was measured using a microplate reader (Biotek Instruments, VT). Reciprocal serum dilutions corresponding to 50% maximal binding were used to obtain titers.
2.6. Serum total cholesterol.
Mice were anesthetized using 3% isoflurane at day 105 following initial infection and blood was collected from the submandibular vein. Total serum cholesterol level was analyzed using a kit (Catalog # ab65390, Abcam, Cambridge, MA).
2.7. Assessment of atherosclerotic lesions in the aorta.
Mice were anesthetized at day 105 following initial infection. After perfusion of the heart with 10ml PBS containing 100 IU heparin followed by 10 ml 2% paraformaldehyde, the aortas were excised from the aortic arch to the iliac bifurcation. Adventitial fat was removed and specimens were fixed in 2% paraformaldehyde. Whole aortas were opened longitudinally and mounted en face, stained for lipids with Oil Red O. Images were acquired using a Bioreader-FZ-beta ELISPOT instrument (Biosys, Germany) with inverted optics. The ratio of the area covered by the atherosclerotic lesions to the total surface area of aortic tissue was quantified using ImageJ software (National Institutes of Health, USA). The percent lesion area was calculated using the formula: (pixels in oil red O-stained region) × 100/ (pixels in the entire surface area of aortic tissue) as previously reported [9].
2.8. Statistical analyse.
Statistical analyses were performed using Analysis of variance (ANOVA; Systat software). P-values ≤ 0.05 were considered significant. All experiments were repeated once to confirm findings.
3. Results
3.1. Cellular cytokine responses following infection.
Fourteen days following primary CPN inoculation, splenic antigen (Ag) specific cytokine production was evaluated. As shown in Figure 1A, in vitro CPN-stimulated splenocytes from CPN-infected WT mice exhibited significantly greater IFN-γ and TNF-α production compared to those from mock (SPG alone)–infected WT or TNF KO animals. CPN-infected TNF KO, TNFR1 KO, or TNFR 1/2 DKO mice displayed significantly enhanced IFN-γ production compared to CPN-infected WT or TNFR2 KO animals. However, CPN-infected TNFR2 KO mice displayed comparable IFN-γ production to infected WT animals. Splenocytes from CPN-infected TNFR1 KO mice also displayed significantly increased IFN-γ production compared to CPN-infected TNF KO animals.
Figure 1. Cellular and humoral immune response to infection.
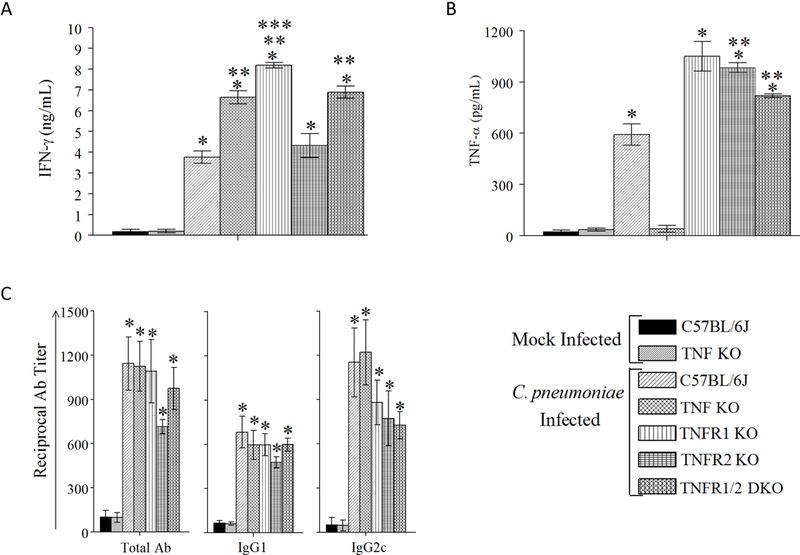
Groups of 6–8-week-old male WT, TNF KO, TNFR1 KO, TNFR2 KO, or TNFR 1/2 DKO were infected on day 0, 15, and 30 with CPN. Separate groups of WT and TNF KO mice were infected with SPG buffer (mock). A & B, Splenic Ag-specific IFN-γ (A) and TNF-α (B) response was measured on day 14 in four mice per group after primary inoculation. Mean ± SEM of cytokine concentrations are shown. * Significant (p≤0.05; ANOVA) between indicated group and mock-infected WT or TNF KO animals. ** Significant (p≤0.05; ANOVA) between indicated group and CPN-infected WT animals. *** Significant (p≤0.05; ANOVA) between indicated group and CPN-infected TNF KO animals. Results are representative of two independent experiments. C, Anti-CPN total Ab, IgG2c, and IgG1 responses analyzed in four to sixteen mice per group on day 90 after primary inoculation. Mean ± SEM of reciprocal 50% binding antibody titers are shown. * Significant (p≤0.05; ANOVA) between indicated group between indicated group and mock-infected WT or TNF KO animals. Results are representative of two independent experiments.
We also measured TNF-α production from CPN-stimulated splenocytes from the different groups of mice (Fig 1B). Splenocyte supernatants from CPN-infected WT, TNFR1 KO, TNFR2 KO, and TNFR 1/2 DKO mice stimulated in vitro with CPN displayed significantly greater TNF-α levels compared to those from mock-infected WT or the expectedly undetectable levels in mock-infected or CPN-infected TNF KO animals. Notably, splenocyte supernatants from CPN-infected TNFR1 KO, TNFR2 KO, and TNF 1/2 DKO mice stimulated in vitro with CPN displayed significantly greater TNF-α levels compared to CPN-infected WT animals. IFN-γ or TNF-α production from CPN-stimulated splenocytes of uninfected animals, or from splenocytes stimulated in vitro with unrelated antigen BSA, or media alone, was minimal. These results demonstrate the induction of a robust Ag-specific Th1 type cellular cytokine response following i.n. CPN infection in all the KO mouse groups at levels comparable or greater than that in the WT mice.
3.2. Humoral response following immunization.
Ninety days following primary CPN inoculation, serum anti-CPN total antibody (Ab), IgG1, and IgG2c response was measured. As shown in Figure 1C, anti-CPN total Ab, IgG1, or IgG2c antibody response in CPN-infected WT, TNF KO, TNFR1 KO, TNFR2 KO, and TNFR 1/2 DKO mice was comparable to each other, and significantly increased compared to mock-infected WT or TNF KO animals. These data indicate that all groups of animals displayed comparable sero-conversion following infection.
3.3. Atherosclerotic lesion development in animals following infection.
On day 105 following primary CPN inoculation, serum cholesterol and aortic atherosclerotic pathology was evaluated. As shown in Figure 2A, all groups displayed comparable serum cholesterol levels on day 105, indicating there were no detected differences in dietary consumption or lipid metabolism. Importantly, as shown in Fig 2B and 2C, CPN-infected WT mice displayed significantly enhanced atherosclerotic pathology compared to mock-infected WT or TNF KO mice, or CPN-infected TNF KO, TNFR1 KO, TNFR2 KO, and TNFR 1/2 DKO mice. The pathology in CPN-infected TNF KO, TNFR1 KO, TNFR2 KO, and TNFR 1/2 DKO mice was comparable to mock-infected WT or TNF KO mice. These results demonstrate that TNFR1 and TNFR2 contribute to atherosclerotic pathology exacerbated by CPN in HF diet-fed mice. Additive effects of TNFR1 and TNFR2 to pathology, if any, were not detectable in the TNFR1/2 DKO mice. This was probably in part because the pathology in CPN-infected TNFR1 or TNFR2 KO animals was comparable to that in CPN-infected mice lacking the ligand TNF-α (TNF KO), or in mock-infected WT or TNF KO animals; as such, this model may not have allowed us to resolve further reductions in pathology.
Figure 2. Atherosclerotic pathology in aorta.
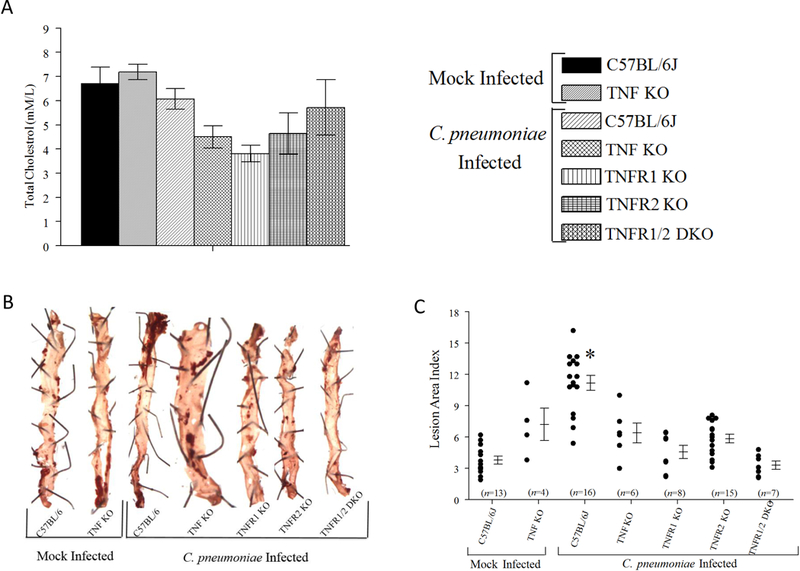
Groups of 6–8-week-old male WT, TNF KO, TNFR1 KO, TNFR2 KO, or TNFR 1/2 DKO were infected on day 0, 15, and 30 with CPN. Separate groups of WT and TNF KO mice were infected with SPG buffer (mock). Mice were bled and euthanized on day 105 after primary inoculation for serum cholesterol assay and pathology estimation. A, Mean ± SEM of serum cholesterol levels in each group is shown. B, Representative images of qualitative pathology estimation using oil red O staining. The pins used to secure tissues to the gel are also seen in the pictures. C, Quantitative estimation of atherosclerotic pathology. Each marker represents an individual aorta/animal. Mean ± SEM for each group are also shown. The number of animals within each group are shown on the X-axis. * Significant (p≤0.05; ANOVA) between CPN-infected WT and all other groups. Results are pooled from two independent experiments.
4. Discussion
In this study, we have demonstrated that deficiency of TNF-α, or either of its cognate receptors, TNFR1 and TNFR2, leads to significant and comparable reduction in CPN-induced exacerbation of atherosclerotic pathology. The serum antibody response and splenic cellular IFN-γ and TNF-α response in each group of CPN-infected gene knock out mice examined was comparable or 10 enhanced to that in CPN-infected WT animals, with the obvious exception of TNF-α production that was absent in TNF KO mice. The pathology in CPN-infected TNF KO, TNFR1 KO, TNFR2 KO, or TNFR 1/2 DKO mice was significantly reduced when compared to CPN-infected WT animals, and to levels found in mock-infected WT or TNF KO animals. Importantly, TNFR1 and TNFR2 appear to act in an inter-dependent fashion based on the observation that deficiency of either TNFR1 or TNFR2 individually in CPN-infected animals reduced pathology to levels found in uninfected animals.
CPN-specific IFN-γ production from splenocytes was significantly enhanced in infected TNF KO, TNFR1 KO, and TNFR1/2 DKO mice when compared to CPN-infected WT animals. IFN-γ is a quintessential cytokine responsible for protective immunity against chlamydial infections (reviewed in [10]); therefore, the compensatory increase of IFN-γ may appear to correlate with the reduced pathology in these animals. However, CPN-infected TNFR2 KO mice displayed significant reduction in pathology with no enhancement of IFN-γ. Furthermore, we demonstrated previously that C. muridarum shedding was not affected in TNFR 1/2 DKO mice which displayed a significant compensatory enhancement of Ag-specific IFN-γ production [2]. Taken together, these findings suggest the likelihood that the reduction in pathology in the KO animals in this study is primarily due to the deficit in TNF-α or the cognate receptors, not to increased Ag-specific IFN-γ production. The levels of TNF-α also were significantly enhanced in the CPN-infected KO mouse groups, with the obvious exception of TNF KO animals, compared to WT animals, suggesting a compensatory enhancement of the ligand due to lack of negative feedback regulation or to reduced re-uptake of cytokine in the absence of the receptor(s). Importantly, this also indicates that the significant reduction of pathology found in the receptor-deficient animals was not due to a deficiency of the cognate ligand TNF-α. Our results also suggest that both TNFR1 and TNFR2 contribute comparably to the CPN-induced atherosclerosis. One caveat of this interpretation is that the moderate, albeit statistically significant, exacerbation of atherosclerosis by C. pneumoniae infection, beyond that induced by the high fat diet, in this wild type C57BL/6 mouse model may have limited the ability to resolve a difference between TNF receptors. In comparison, much greater disease pathology develops in mice with a gene deficiency in Apolipoprotein E or low-density lipoprotein that are fed a Western diet; however, these gene-deficient animal models may not reflect a wild-type metabolic environment. Future experiments in those models or prolonging the observation period in the wild type C57BL/6 mouse model may allow us to observe differential effects of the two TNF receptors towards C. pneumoniae-induced atherosclerosis.
In this study, we confirm our observation regarding the contribution of TNFR1 and TNFR2 to C. muridarum-induced pathology [2] and extend them with the use of a human pathogenic strain, C. pneumoniae AR39, that infects a different mucosal compartment and causes chronic pathology in a different tissue location. On a broader note, the findings of this study add to the limited, but growing body of evidence for a role of TNFR2 in coronary artery disease and atherosclerosis [11, 12, 13, 14]. To this end, due to restricted cellular distribution of TNFR2, the finding that TNFR2 contributes significantly to pathogenesis is promising from a therapeutic intervention standpoint to reduce the clinically relevant pathologies using antagonists [8, 15].
ACKNOWLEDGMENTS
This work was supported by Midwestern University College of Health Sciences Master’s thesis funds to MTZ, Summer Research Fellowship funds to JTC, American Heart Association Midwest Affiliate grant 13SDG17310011, National Institutes of Health grants 1R15AI101920 and 2R15AI101920, and Midwestern University Faculty Startup Grant to AKM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Murthy AK, Li W, Chaganty BK, Kamalakaran S, Guentzel MN, Seshu J, et al. Tumor necrosis factor alpha production from CD8+ T cells mediates oviduct pathological sequelae following primary genital Chlamydia muridarum infection. Infect Immun 2011;79:2928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manam S, Thomas JD, Li W, Maladore A, Schripsema JH, Ramsey KH, et al. Tumor necrosis factor (TNF) receptor superfamily member 1b on CD8+ T Cells and TNF receptor superfamily member 1a on non-CD8+ T Cells contribute significantly to upper genital Tract pathology following chlamydial infection. J Infect Dis 2015;211:2014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murthy AK, Li W, Ramsey KH. Immunopathogenesis of chlamydial infections. Curr Top Microbiol Immunol 2018;412:183–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grayston JT, Belland RJ, Byrne GI, Kuo CC, Schachter J, Stamm WE, et al. Infection with Chlamydia pneumoniae as a cause of coronary heart disease: the hypothesis is still untested. Pathog Dis 2015;73:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muhlestein JB. Chlamydia pneumoniae-induced atherosclerosis in a rabbit model. J Infect Dis 2000;181 Suppl 3:S505–7. [DOI] [PubMed] [Google Scholar]
- 6.Saikku P, Laitinen K, Leinonen M. Animal models for Chlamydia pneumoniae 255 infection. Atherosclerosis 1998;140 Suppl 1:S17–9. [DOI] [PubMed] [Google Scholar]
- 7.Campbell LA, Nosaka T, Rosenfeld ME, Yaraei K, Kuo CC. Tumor necrosis factor alpha plays a role in the acceleration of atherosclerosis by Chlamydia pneumoniae in mice. Infect Immun 2005;73:3164–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov 2010;9:482–93. [DOI] [PubMed] [Google Scholar]
- 9.Zafiratos MT, Manam S, Henderson KK, Ramsey KH, Murthy AK. CD8+ T cells mediate Chlamydia pneumoniae-induced atherosclerosis in mice. Pathog Dis 2015;73:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunham RC, Rey-Ladino J. Immunology of Chlamydia infection: implications for a Chlamydia trachomatis vaccine. Nat Rev Immunol 2005;5:149–61. [DOI] [PubMed] [Google Scholar]
- 11.Carlsson AC, Jansson JH, Soderberg S, Ruge T, Larsson A, Arnlov J. Levels of soluble tumor necrosis factor receptor 1 and 2, gender, and risk of myocardial infarction in Northern Sweden. Atherosclerosis 2018;272:41–6. [DOI] [PubMed] [Google Scholar]
- 12.Carlsson AC, Ruge T, Kjoller E, Hilden J, Kolmos HJ, Sajadieh A, et al. 10-year associations between tumor necrosis factor receptors 1 and 2 and cardiovascular events in patients with stable coronary heart disease: A CLARICOR (effect of clarithromycin 277 on mortality and morbidity in patients with ischemic heart disease) trial substudy. J Am Heart Assoc 2018;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim HL, Lee JP, An JN, Kim JH, Lim WH, Seo JB, et al. Soluble tumor necrosis factor receptors and arterial stiffness in patients with coronary atherosclerosis. Am J Hypertens 2017;30:313–8. [DOI] [PubMed] [Google Scholar]
- 14.Nicolaou A, Zhao Z, Northoff BH, Sass K, Herbst A, Kohlmaier A, et al. Adam17 deficiency promotes atherosclerosis by enhanced TNFR2 signaling in mice. Arterioscler Thromb Vasc Biol 2017;37:247–57. [DOI] [PubMed] [Google Scholar]
- 15.Vanamee ES, Faustman DL. TNFR2: A novel target for cancer immunotherapy. Trends Mol Med 2017;23:1037–46. [DOI] [PubMed] [Google Scholar]