

Abstract
Purpose:
Endoglin (ENG; CD105) is a co-receptor of the transforming growth factor-β (TGF-β) family that is highly expressed in proliferating endothelial cells. Often coopted by cancer cells, ENG can lead to neo-angiogenesis and vasculogenic mimicry in aggressive malignancies. It exists both as a transmembrane cell surface protein, where it primarily interacts with TGF-β, and as a soluble matricellular protein (sENG) when cleaved by matrix metalloproteinase 14 (MMP14). High ENG expression has been associated with poor prognosis in Ewing sarcoma (ES), an aggressive bone cancer that primarily occurs in adolescents and young adults. However, the therapeutic value of ENG-targeting has not been fully explored in this disease.
Experimental Design:
We characterized the expression pattern of transmembrane ENG, sENG and MMP14 in preclinical and clinical samples. Subsequently, the antineoplastic potential of two novel ENG-targeting monoclonal antibody-drug conjugates (ADCs), OMTX503 and OMTX703, which differed only by their drug payload (nigrin-b A chain and cytolysin, respectively), was assessed in cell lines and preclinical animal models of ES.
Results:
Both ADCs suppressed cell proliferation in proportion to the endogenous levels of ENG observed in vitro. Moreover, the ADCs significantly delayed tumor growth in ES cell line-derived xenografts and patient-derived xenografts (PDXs) in a dose-dependent manner.
Conclusions:
Taken together, these studies demonstrate potent preclinical activity of first-in-class anti-ENG ADCs as a nascent strategy to eradicate ES.
Keywords: Ewing Sarcoma, Endoglin, Antibody-drug-conjugate, PDX, precision medicine
Introduction
Ewing sarcoma (ES) is the second most common primary bone and soft tissue sarcoma of children, adolescents, and young adults. It results from pathognomonic balanced t(11;22)(q24;q12) chromosomal translocations between EWSR1 and one of several genes in the ETS family of transcription factors. Micrometastatic spread is nearly universal at the time of diagnosis, and about one-quarter of ES patients present with radiologically-evident metastatic disease at that time.
Though the long-term survival rate has improved over the last few decades for those who present with localized disease, mainly due to the introduction of multimodal therapeutic strategies, the 5-year survival rate in patients with metastatic disease remains below 20–30% (1). Poor clinical outcomes can, in part, be attributed to tumor cell heterogeneity and varied expression of the fusion-protein expression (2), which influences the epigenomic (3,4), transcriptomic, and proteomic profiles (5) of the cells. Ultimately, those changes regulate cell phenotype and drug sensitivity (3,6).
This implies a critical need to shift from an outdated ‘one fits all’ therapeutic approach to a more customized treatment strategy directed towards the unique molecular traits that exist within each tumor. Toward that end, ADCs have been used with increasing frequency in the last few decades as a precision-guided treatment to combat cancer and are now an essential weapon in the oncologists’ arsenal of antineoplastic therapies (7). ADCs consist of a monoclonal antibody (MAb) joined to a cytotoxic agent by a chemical linker. Among a wide range of cytotoxic molecules, we evaluated the type-2 ribosome-inactivating protein nigrin-b A chain and the microtubule-disrupting synthetic small molecule cytolysin (8,9). Irrespective of the type of cytotoxic payload fused to the MAb, the targeting antibody recognizes tumor tissues by specifically binding to the cognate cell surface protein. Once the ADC is internalized via receptor-mediated endocytosis, the drug is released into the late endosome compartment and becomes active (7,10). The mechanism of action of ADCs can be incredibly specific, and off-target effects can often be limited to tissues that selectively express the target of interest (10).
ENG is a homodimeric glycoprotein that acts as a co-receptor of TGF-β family of proteins. It occurs in two isoforms and exists at the tumor cell surface and tumor microenvironment within the extracellular matrix (11). ENG is also expressed in endothelial cells (ECs), activated macrophages, normal marrow cells, some leukemic cells, and other types of tumor cells. Transmembrane ENG usually exists as part of a multi-protein complex that can include TGF-β receptor 1 (ALK1, ALK5), TGF-β receptor 2, and BMP receptors (12,13). Soluble ENG (sENG), on the other hand, is predominantly distributed in the extracellular matrix (ECM) following cleavage by MMP14 (14). Its expression plays a key role during embryogenesis, angiogenesis, vascular development and homeostasis (15,16). Of particular relevance to our work, ENG is highly expressed by a range of human sarcomas and by human mesenchymal stem cells (MSCs), the putative ES cell of origin (17). Notably, its expression has been linked to cell plasticity, an especially-important finding given that ES is a poorly differentiated neoplasm with no normal tissue counterpart. Further, the sentinel trait of ES plasticity and epigenetic dysregulation appears to facilitate vasculogenic mimicry and promote tumor cell invasiveness (18–20).
Herein, we sought to better understand how ENG contributes to ES biology while teasing apart the direct and indirect contribution of ENG-targeting. Critically, with an eye towards a future clinical development, our research leverages the strength of two independent laboratories to validate the preclinical results. Independently, we establish that human-specific anti-ENG ADCs are superior to unconjugated ENG-targeting therapies in their capacity to suppress cell proliferation and hinder tumor growth. Furthermore, we demonstrate a number of heretofore unreported tumor-centric effects that have a major impact on critical cell signaling pathways. Though naked anti-ENG MAbs (e.g. TRC105) are already being tested across a diverse window of tumor types, our data suggests that ADCs could significantly improve the likelihood of clinical benefit.
Material and Methods
Cell lines and cell cultures
A4573, CADO, ES8, RDES, RM82, TC32, VH64 and TC71 ES cell lines were cultured in complete RPMI medium. Other ES cell lines, A673 and SKNMC were cultured in DMEM 4.5 g/L glucose and EMEM medium, respectively. All the cell culture media were supplemented with 1% penicillin/streptomycin, 1% glutamine and 10% FBS (all from Gibco, Thermo Fisher). All parental cell lines were tested twice per year for mycoplasma contamination using the MycoAlert Detection Kit (Lonza Group Ltd.) according to the manufacturer’s protocol and authenticated using either Short Tandem Repeats (STR) fingerprinting with an AmpFLSTR Identifier kit as previously described or STR analysis (CLS, Cell Lines Service GmbH). Cell line individual characteristics and origin are depicted in Table 1S. All commercially available cell lines were purchased in ATCC, and each were used for no longer than 3 months.
Patient studies
Plasmas from ES patients (n = 27) were kindly provided by Dr. Arjan Lankester from the Leiden University Medical Center. Patient samples (plasma, tumor) were obtained under patient consent and in accordance to Declaration of Helsinki. Plasmas from healthy donors (n = 7) were obtained under patient consent by the Centro de Investigación del Cáncer in Salamanca, Spain. A cohort of 22 ES samples embedded in Tissue-Tek® O.C.T. compound (SAKURA FINETEK USA INC) and an additional cohort of 43 FFPE samples (used to generate TMA) were obtained from the Hospital Universitario Virgen del Rocío-Instituto de Biomedicina de Sevilla Biobank (Andalusian Public Health System Biobank and ISCIII-Red de Biobancos PT13/0010/0056). All identified patients and collected data were in accordance with guidelines of MDACC and IBIS’s institutional review board. Authors followed the recommendations suggested by REMARK guidelines.
Animal Experiments
All experiments were conducted in accordance with protocols and conditions approved by the European guidelines (EU Directive 2010/63/EU) and by the University of Texas MDACC (Houston, Texas) Institutional Animal Care and Committee (eACUF Protocol #00000928-RN01). In vivo binding assay, dose-ranging–study and RM82 xenograft models were approved by the local institution and the Dirección General de la producción Agrícola y ganadera de la Junta de Andalucía in Spain. PDX studies were approved by the local animal care and use committee (Comité Ético de Experimentación Animal at Universidad de Barcelona, protocol 419/15). ES8 xenograft model was approved by the University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee (ACUF Protocol#000928-RN01). Additional material and methods are provided in the supplementary information section.
Results
ENG is heterogeneously expressed in ES cell lines and PDX models
ENG expression was evaluated in a set of ES cell lines, using MSCs and an endothelial cell line (HUVEC) as positive controls. The ES panel comprises cell lines bearing various EWS-ETS fusion variants (Table 1S). ENG mRNA and protein levels varied considerably across the ES cell lines (Figures 1A, 1B). The CADO cell line lacked expression of ENG, both at the mRNA (Figure 1A), and at the protein levels (Figure 1B). This heterogeneous expression of ENG prompted us to analyze the methylation status of the CpG islands present in ENG promoter in ES cell lines. In fact, the ENG promoter was only hypermethylated in the CADO cell line, suggesting that ENG expression is epigenetically regulated in ES (Figure 1C, S1A). In addition, compared to CADO cell line, the ENG was expressed in RM82 (high level) and TC71 (intermediate-low level) cell lines, as confirmed by immunofluorescence (Figure S1B) and FACS analyses (Figure S1C). When cleaved by MMP14, ENG is shed into the extracellular matrix in a soluble form (sENG), which can also be detected in the supernatants of ES cell lines (n = 7) by ELISA (Figure 1D). Here, sENG concentration positively correlated with the mRNA levels of ENG, (Pearson’s correlation: r = 0.7747, p = 0.0408; Figure S1D). sENG was also evaluated in patient-derived plasma from healthy donors and ES patients, however no significant differences were observed between healthy donors/ES-patient, localized/metastatic disease, or low/high tumor volume groups (Figure S2A–C). Finally, no link between sENG and clinical parameters was observed (Figure S2F). One possible explanation for this finding could be that the constitutive concentration of sENG in the organism may be overlapping the differential tumor-derived sENG concentration, as ENG is expressed in physiological conditions.
Figure 1. Heterogeneous expression of ENG/MMP14 in ES cell lines and xenografts.

(A-B) The expression of ENG was heterogeneous amongst ES cell lines at both mRNA (A) and protein (B) levels measured, respectively by qRT-PCR and western blotting (n = 10). (B) Similarly, heterogeneous expression of MMP14 was observed at the protein level as determined by western blot (n = 10). (C) The methylation status of ENG/MMP14 genes in the CADO cell line suggests that these genes are regulated at the epigenetic level (GSE#118872). (D) The soluble form of ENG was detected at the extracellular compartment by ELISA in a set of ES cell lines (n = 7) that express ENG. MCF7 cells are used as a negative control. (E) Cell surface and intracellular expression of endoglin as assessed by flow cytometry against a panel of human ES cells. The OMTX003 vehicle antibody exhibits linear dose-dependent binding and effectively discriminates between medium-low-expressing (TC71 and A673) and high-expressing (ES8, TC32 and A4573) cell lines. (F) Maintained expression of ENG was confirmed in RM82, TC71 and CADO xenograft tumors of ES (n = 3), respectively with high, intermediate-low and negative expression. (G) Heterogeneous expression of ENG was investigated in 9 PDX models of ES, which are screened based on the intensity of stain and % of stained cells. The frequency of ENG expression intensity (high-intermediate-low) was presented on the right table.
In parallel, ENG expression was characterized by flow cytometry analysis in a panel of five ES cell lines (ES8, TC32, TC71, A4573 and A673) using the OMTX003 vehicle antibody at their cellular membrane and intracellular levels. OMTX003 exhibited linear dose-dependent binding and effectively discriminated between low-expressing (TC71 and A673) and high-expressing (ES8, TC32 and A4573) cell lines (Figure 1E). ENG expression was also evaluated by IHC analysis in a set of ES xenograft (n = 3, Figure 1F) and PDX models (n = 9, Figure 1G), altogether confirmed heterogeneous expression of ENG. In the group of PDX tumors, this heterogeneous ENG expression was also confirmed ranging from 11.1% low, 22.2% intermediate to 66.7% high levels (Figure 1G).
MMP14 expression positively correlates with ENG expression in ES cell lines and patients
We investigated the expression of MMP14 within a panel of ES cell lines (Figure 1B and Figure 2A–B) and determined whether this could be useful as a prognostic factor. The CADO cell line, which exhibited no ENG expression (Figure 1B and Figure 2B), showed a lack of MMP14 expression and hypermethylation of its promoter (Figure 1C and S1A). To clarify if MMP14 is the metalloprotease implicated in the extracellular domain cleavage of ENG in ES, we could not find any correlation between MMP14 and ENG at their mRNA expression levels (r = 0.6053; p = 0.1498 by Pearson’s-Figure S2D). However, we observed a positive correlation between the mRNA levels of MMP14 and the concentration of sENG in ES cell line supernatants (n = 5; r = 0.9531; p = 0.0121 by Pearson’s-Figure S2E). Furthermore, frozen tissue samples from one cohort of ES patients (n = 22) were used to evaluate the mRNA expression of ENG and MMP14 (Figure 2C) and FFPE tissues from another cohort (n = 43) of patients were used to evaluate protein expression of ENG and MMP14 (Figure 2D and Table S3) by IHC. High ENG expression was found in 51.16% of the analyzed samples (Figure 2F). In both cohorts, we found a significant correlation between ENG and MMP14 at their mRNA and protein levels (r = 0.8331, p < 0.0001-Figure 2E; and r = 0.4614, p = 0.0021 - Figure 2D, respectively). These results were in agreement with the in vitro data in ES cell lines and PDX models, demonstrating heterogeneous expression of ENG and MMP14 in ES samples. The pattern of ENG and MMP14 protein expression did not correlate with the clinical parameters either in the presence of primary tumor, metastasis or relapse.
Figure 2. MMP14 expression correlates with ENG levels in ES cell lines and patients.

(A-B) MMP14 presents a heterogeneous expression amongst ES cell lines, as determined by qRT-PCR (A). Relative protein expression of both ENG/MMP14 after normalization against GAPDH is depicted in (B). (C) Heterogeneous expression of ENG and MMP14 at the mRNA levels measured by qRT-PCR in a cohort of 22 frozen ES tumor samples. (D) Evaluation of ENG and MMP14 expression by IHC in 43 FFPE ES tumor samples (E). Significant correlation between ENG and MMP14 at the mRNA levels (r = 0.8331, p = 0.0001). (F) Representative images of different intensity scores (0, 2, 3) of ENG/MMP14 expression in ES patients.
ADCs impair in vitro ES cellular proliferation in an ENG expression-dependent manner
Having shown that ENG is highly expressed not only in the endothelium, but also variably-expressed in tumor cells, we sought to isolate the relative contribution of targeting ENG within the vascular stroma vs. tumor. As a non-tumoral in vitro cell-based assay for OMTX003 drug efficacy, HUVEC cells were exposed to this antibody. It was shown previously that OMTX003 binds to human ENG extracellular domain (amino acids 26–584) in a concentration-dependent manner, with high specificity and affinity (Figure S3C). Incubation of OMTX003 with human rhENG or human HT1080 cells showed OD50 at 0.3 and MFI50 at 0.4 nM with ELISA and flow cytometry analyses, respectively (Figure S3C and D).
Incubation of OMTX003 (100 μg/mL, 699.3 nM) with HUVEC cells for 72 hours was able to inhibit tube formation (Figure S4B). ES cell lines presenting the ability to form vasculogenic mimicry (Figure S4A) were also exposed to OMTX003 within the same conditions (Figure S4C), however no effects were observed. Since ENG has been reported to enhance ES cell proliferation, we assumed that the maximal anti-proliferative effect of OMTX003 would occur in ES cells that abundantly express ENG. To test that hypothesis, three ES cell lines with varied ENG expression were chosen for experimental validation: RM82 (high expression), TC71 (medium-low expression) and CADO (no expression) cell lines. Surprisingly, we observed little to no effect of OMTX003 in these cell lines (Figure 3A). Though OMTX003 was able to achieve more than 50% suppression in the ENG-high expressing RM82 cells, the requirement for μmolar range drug concentrations suggested that ENG targeting using naked MAbs such as OMTX003 may not adequately impair ES cell proliferation.
Figure 3. ES cells are affected in vitro by ENG-ADCs treatment within an ENG expression-dependent manner.

(A) OMTX003 was tested on three ES cell lines: RM82, TC71 and CADO, which show differential expression of ENG, in order to evaluate in vitro the effect of this ADC on the proliferation rate of these cell lines. (B) Chemical structure of the OMTX-ADC, ENG MAb candidate lead linked to a cytotoxic agent, nigrin-b A chain (OMTX503) or cytolysin (OMTX703). (C) OMTX503 activity was tested on these cell lines, which resulted in an inhibition of their proliferation rate in an ENG expression-dependent manner. (D) There is a significant correlation between the IC50 of OMTX503 over the ES cell lines RM82, TC71 and CADO and the ENG protein expression of these cell lines (r = −0.9999, p = 0.0082). (E) OMTX003 and OMTX703 were tested during 72 h in WST1 proliferation cell-based assay using ES8 cell line. Only OMTX703 showed a significant inhibition of ES8 cell proliferation in a dose-dependent manner (IC50 = 260 nM).
Considering the promise of ADC technology, we assessed whether drug conjugation to OMTX003 could enhance its anti-proliferative effect in ES cells. Two ADCs were generated by cystein-based conjugation of OMTX003 to either the ribosome-inactivating protein nigrin-b A chain (OMTX503), using the 4-succynimidyloxycarbonyl-α-methyl-α-(2-pyridyl-dithio)toluene (SMPT) chemical linker, or the microtubule disrupting cytotoxic drug cytolysin (OMTX703), using the protease-cleavable vcPABA optimized linker, (Figure 3B). Similarly to OMTX003, both ADCs were highly stable and able to bind with high specificity and affinity to human ENG on the surface of ENG-expressing cells and got internalized efficiently to allow specific intracellular release and activation of their payload moieties (data not shown). As anticipated, OMTX503 demonstrated a compelling anti-proliferative effect in the assessed ES cells: RM82 (high ENG expression; IC50 = 1.18·10−10 M), TC71 (medium-low ENG expression; IC50 = 9.155·10−9 M) and CADO cell lines (no ENG expression; IC50 ≈ 1.738·10−8 M-Figure 3C). As shown in Figure 3D, the sensitivity of ES cell lines to this agent positively correlated with ENG expression (r = −0.9999; p = 0.0082). A parallel evaluation was performed to assess the in vitro efficacy of OMTX703 by choosing ES8 cell line, which presents the highest expression of ENG among other ES cell lines to validate the assay (Figure 1E). This high expression level of ENG in ES8 correlated with efficient internalization (data not shown), efficacy, and cytotoxic effects in vitro through cell proliferation assays (Figure 3E, IC50 = 260.6 nM). Collectively, these data indicate that ADCs impair in vitro ES cell proliferation in an ENG expression-dependent manner.
ENG-targeting ADCs exhibit on-target effects in ES animal models
To initiate animal studies, we determined first whether high ENG expression by ES xenografts (RM82 and CADO) is required to achieve an in vivo effect, as our in vitro data suggested would be the case. Importantly, as assessed by IHC, the expression of ENG in the xenograft tumors was maintained at levels comparable to the in vitro parental cell lines (Figure 1F). As a second pivotal control, the in vivo binding assay confirmed that OMTX003 binds specifically to human ENG, which was only present in tumor cells, but not in the murine-derived microvessels (Figure 4A). A murine OMTX003 MAb version (OMTX003-mu) targeting ENG was also used to detect its binding to murine-derived vessels expressing ENG, as it was observed in the lower panel of Figure 4A. To evaluate the adequate therapeutic window of the drugs, a dose-ranging study (DRS) was performed. OMTX003 (unconjugated MAb) and OMTX503 (nigrin-b A chain-ADC) were tested in vivo in dose-escalated manner and the animal weight loss (defined as a loss >20% of the initial animal weight) was assessed as an indirect metric of general drug toxicity (Figure 4B). The vehicle OMTX003 MAb elicited no toxicity when tested at 10 mg/kg in both RM82, CADO (Figure 4B) and ES8 (Figure 5A–5B) xenograft models. OMTX503 dosing at 0.5 mg/kg also appeared safe. Following euthanasia, careful histopathological analyses of the lung, liver and kidney were performed, showing no significant deviation from normality.
Figure 4. OMTX503 impairs in vivo ES tumor growth at a safe concentration.

(A) In vivo binding assay: OMTX003 specifically binds to RM82 cell membranes, as indicated with a white arrow in the middle image, but not in murine-derived microvessels within the xenograft tumor, (as indicated with a white arrow in the far right image of the panel). In contrast, the murine targeting MAb (OMTX003-mu) binds specifically to the microvessels and is undetected in the membrane of the tumor cells as indicated with a black arrow in the lower image of the panel. (B) Comparison of normalized body weight during drug treatment in RM82 xenografts (n = 3 animals per group). (C-D) Therapeutic effect of OMTX503 in RM82 ES xenografts. Tumor volumes were reported after treatment with OMTX003, OMTX503, PBS as negative control, and irinotecan as positive control (n = 9 animals per group). (C) Curves showing mean tumor volumes for RM82 xenograft tumors during the drug treatments. * represents significance between OMTX503 0.5 mg/kg and the control group: at day 5 p = 0.0008; at day 7 p = 0.0118; at day 10 p= 0.0064 and at day 12 p = 0.0218. (D) Tumor growth Inhibition at day 15 in RM82 xenografts treated with OMTX003, OMTX503 and irinotecan (n = 5, n = 7 and n = 9 animals per group, respectively). (E) Cell viability percentage using H&E stain in different treated RM82 tumors at day 15 with OMTX003 (10 mg/kg, p = 0.010) and OMTX503 (0.5 mg/kg, p = 0.002). * and ** represent the significance of difference between two groups of treated mice, respectively p < 0.05 and p < 0.005 according to two tailed, paired Student’s T-test.
Figure 5. OMTX703 strongly reduces tumor growth in ES8 xenograft models.
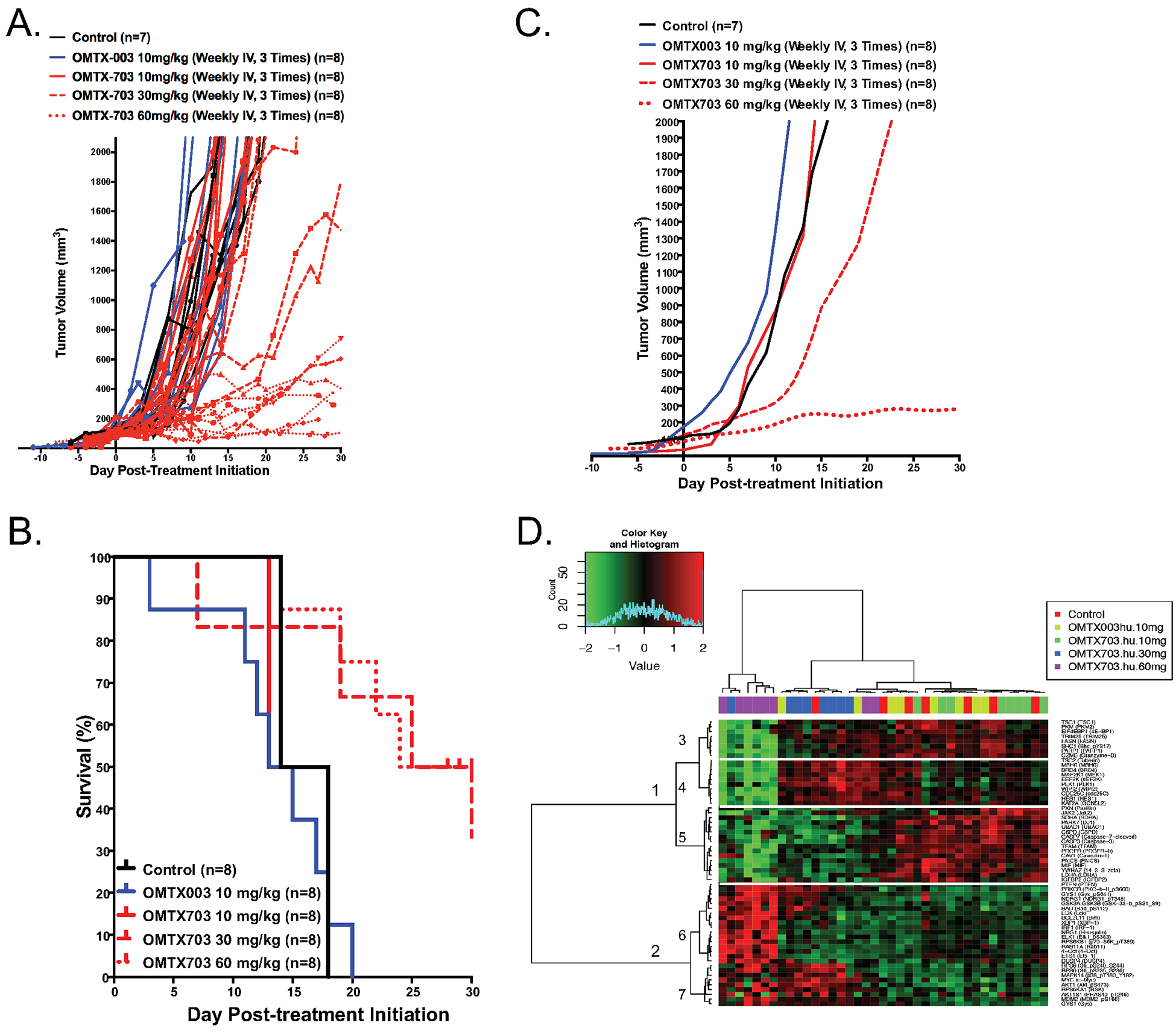
(A) Therapeutic effect of OMTX003 and OMTX703 in ES8 Ewing sarcoma xenografts. Immunocompromised NOD-SCID-IL-2Rgnull/null mice (NSG) were injected with 1·106 ES8 cells at their flank. Treatment with OMTX003 (n = 8), OMTX703 (n = 8 for each dosing group) or control vehicle (n = 7) was started when tumor volumes reached of about 100 mm3. The curves show individual tumor volumes for a period of 30-days drug exposure. (B) The panel shows Kaplan-Meier curves. The p < 0.019 value for differences between the treated OMTX703 (60 mg/kg) and control mice were performed with the log-rank (Mantel-Cox) test. (C) The curves show the smoothed grouped median relative tumor volumes. The OMTX703 (60 mg/kg) treatment show a significant blockade of ES8 tumor growth as compared to control group of mice using unpaired t-test and two-tailed p-value (p < 0.0001). However, no significant effect of low doses OMTX (10 or 30 mg/kg) on this tumor growth. (D) RPPA profiling (GSE#114866) of control (n=7), OMTX003 (10 mg/kg; n = 8), OMTX703 (10 mg/kg; n = 8), OMTX703 (30 mg/kg; n = 8) and OMTX703 (60 mg/kg; n = 8)-treated ES8 xenografts following a 2-week exposure identifies statistically significant 60 proteins at a false discovery rate (FDR) of 0.01.
ENG-targeting ADCs impair ES xenografts tumor growth
The efficacy of OMTX003 and OMT503 was evaluated in RM82 xenografts in parallel with irinotecan (Table 4S), which was used as a positive control of drug efficacy (Figure S5A). The evolution of tumor growth in each individual group was assessed throughout the 21-day treatment period. The irinotecan-treated group was maintained in a long-term survival study until day 41, when all animals relapsed and were sacrificed (Figure S5A). Animal weight was also evaluated during this time as a measure of treatment-induced toxicity (Figure S5B). Notably, significant delays in tumor growth were observed in OMTX503-treated mice by day 13 (p = 0.0218-Figure 4C), and tumor growth inhibition (TGI) was higher than 50% after 15 days post-treatment (Figure 4D and Table 5S). ENG and MMP14 expressions (Figure S5C, S5D) were similar in tumor samples from all groups, as evaluated by IHC (Table 6S–9S). H&E staining on the tumor samples revealed a loss of tumor viability in those groups treated with OMTX003 and OMTX503 after day 15 of treatment (Figure 4E). Systemic signs of toxicity were found in some mice from the OMTX503-treated groups. The macroscopic toxicity observed was not translated into weight loss, as the tumor growth masked the global weight change (Figure S5B). Collectively, these results suggest that the internalization of an immunotoxin by targeting ENG in ES xenografts impaired tumor growth.
To test the efficacy of OMTX703 (cytolysin-ADC) in ES8 xenograft, an additional preclinical study was conducted. Tumor growth and Kaplan-Meier curves revealed only modest antitumor activity of OMTX703 at 30 mg/kg, as shown in Figures 5A–5C. Tumor growth was markedly impaired using the highest dose of OMTX703 (60 mg/kg, p = 0.019). Having determined an optimal dose for OMTX703, an additional experiment was conducted to assess the mechanism(s) of OMTX703 action and its potential mechanism(s) of resistance following a 2-week exposure to OMTX703 at 0, 10, 30, and 60 mg/kg; 246 proteins were assessed by reverse-phase protein lysate array (RPPA, GSE#114866). Analysis of variance (ANOVA), Pearson’s correlation as distance metric and Ward’s linkage as the clustering method using a false discovery rate (FDR) of 0.01, identified 60 proteins that discriminated between treatment groups. By principal component analysis (PCA), tumor specimens from mice treated with OMTX703 at 10 mg/kg clustered tightly with the placebo-treated group, suggesting that this dose had limited effect (Figure S6C). Surprisingly, two specimens from the OMTX703 group treated at 60 mg/kg nearly overlapped the placebo-treated cluster (Figure 5D); further scrutiny of these specimens suggested that they had acquired resistance and begun to progress (Figure S6A). RPPA analysis of the xenograft-treated specimens demonstrated two major and five minor clusters. Cluster 1 was associated with proteins significantly downregulated by OMTX703 at 30 and 60mg/kg compared to a heterogeneously-treated group of tumors from mice that received placebo control, a naked anti-endoglin Ab (OMTX003), or a subtherapeutic (i.e. 10mg/kg) dose of the OMTX703 ADC. Gene set enrichment analysis (GSEA) indicated marked suppression of proteins linked to insulin receptor signaling or downstream effectors such as PI3K and mTOR, pathways of known importance in ES (Figure S7). In contrast, the most differentially upregulated proteins resulting from OMTX703 treatment (cluster 2, Figure S8) were linked to genes associated with nerve growth factor (NGF) and its cognate receptor (TRKA). Obviously GSEA relies upon gene set enrichment and fails to reflect the site-specific protein phosphorylation states that can markedly affect protein function. Thus, the collective effect that individual phospho-proteins have upon pathway activation must be further scrutinized.
To investigate the proteomic changes associated with the heightened clinical activity of the 60 mg/kg dose, a secondary analysis was performed, which grouped the 10 mg/kg OMTX703 samples and the 10 mg/kg OMTX003 ones with the placebo-treated samples (Figure S6B). Using a FDR of 0.0001, an absolute log2 fold change of 1.5, Pearson’s correlation as distance metric and Ward’s linkage as the clustering method, 22 proteins were discriminately identified between the 3 treatment groups (Figure S6B). Notably, a protein regulator of altered metabolism (RPS6) was exclusively upregulated following OMTX703 (60 mg/kg), and a second metabolism biomarker (LDHA) was down-expressed in the 30 and 60 mg/kg-treated groups.
Efficacy study of ENG-targeting ADCs in PDX models of ES
Finally, we evaluated the efficacy of OMTX703 by using a PDX model that many believe predicts drug activity better than cell lines. Considering the in vitro data that indicate ENG expression is required to elicit an anti-proliferative effect, an initial ES PDX panel was screened to identify a suitable model to validate the therapeutic effect of OMTX703. HSJD-ES-009 was considered the best candidate because of its relatively high ENG expression as well as its high rate of successful engraftment in mice as compared to the rest of the PDX models (Figure 1G and S9A–B). Irinotecan was used as reference drug because of its potent activity as single agent in preclinical models of pediatric solid tumors (Table 10S) (24).
Criteria to quantify drug response are shown in Table 11S. Although animals did not experience weight loss during treatment (Figure 6A), OMTX703 at 60 mg/kg produced an acute toxic death rate of 50%, with 3 animals dying at days 21, 22 and 28 from treatment initiation. Regarding this fact, we proceeded to thoroughly examine the drug-induced toxicity in FFPE samples of lung, liver and kidney from the PDXs. However, by histopathological analyses, no significant differences were observed between the drug-treated and the control groups (Tables 12S–14S). Animals treated with OMTX003 experienced progressive tumor growth, as the control animals did. We found a dose-dependent antitumor response with OMTX703 at 30 mg/kg and 60 mg/kg. Interestingly, OMTX703 at 60 mg/kg induced a complete response (CR) rate of 60% at day 21 of treatment, in comparison to the reference treatment (irinotecan) which induced CR in 11% of tumors (Figure 6B and 6D).
Figure 6. OMTX703 leads to complete response and prolonged survival time in an ES PDX model.

We selected the PDX model with the highest expression of ENG as the best candidate to evaluate the efficacy of OMTX703. (A) Body weights during OMTX003, OMTX703 and irinotecan treatments. No signs of toxicity in terms of weight loss were detected. (n = 6 animals per group; means and SD) (B) Tumor volumes at the end of treatment (21 days). Irinotecan-treated group was considered as the positive control of response. (n = 6 animals per group; 8–12 tumors were evaluable in each group; individual tumor volumes are shown). * P < 0.0001 (one way ANOVA with Tukey’s multiple comparisons test); n.s. : not significant. (C) Efficacy study showing dose-dependent partial tumor growth inhibition with OMTX703. All mice showed progressive disease by the end of the study. (D) Response at the end of treatment (day 21). PD: Progressive Disease; SD: Stable Disease; PR: Partial Response; CR: Complete Response. (E) Kaplan-Meier survival curves. (F) Median Survival (MS) times: The OMTX703-treated group presented the highest median survival (60 days) compared to the reference group treated with irinotecan (46 days). * Significant as compared to control (log rank test). Upon Bonferroni correction for multiple comparisons of the median survivals of treatment groups versus control group, P value threshold < 0.0125 was considered significant.
To quantify the long-term effects of drug treatment upon survival, a second study was conducted. After cessation of treatments all groups experienced progressive tumor growth, with relapse occurring even in mice that had shown complete tumor response (Figure 6C, S9B). As with prior studies, no weight loss was observed during the survival study and weight gain was mainly due to the increase in tumor volume (Figure S9C). Median time to achieve endpoint is shown in Figures 6E–F, with significant longer median time in OMTX703 (60 mg/kg) and irinotecan-treated animals as compared to control-treated mice (60 days, p = 0.0065 for OMTX703 (60mg/kg); and 46 days, p = 0.0018 for irinotecan). Finally, IHC was performed to evaluate the possible changes on ENG and MMP14 expression amongst groups (Figure S10A–S10B, Tables 15S–16S) as well as proliferation rate by Ki67 staining (Figure S10C) and viability by H&E staining (Figure S9D). Compared to placebo-treated mice, we found a significant increase of ENG expression in mice treated with OMTX003 at 10 mg/kg (p = 0.0005-Figure S10A) and a significant increase of MMP14 expression in the groups treated with OMTX703 at 30 mg/kg (p = 0.0316), OMTX703 at 60 mg/kg (p = 0.0163) and irinotecan at 10 mg/kg (p = 0.0240) as reflected in Figure S10B.
Cell viability, measured by H&E, was not affected by the treatment in any case (Figure S9D). However, regarding the cell proliferation evaluated by Ki67 staining, we found a significant decrease in the group treated with OMTX003 10 mg/kg (p = 0.0479) and a non-significant decrease in the groups treated with OMTX703 (30 mg/kg; 60 mg/kg) and irinotecan 10 mg/kg (Figure S10C). From this result, we could hypothesize that cells with stable proliferation the ENG upregulation as a way to compensate its activity was impaired by the antibody blockade. MMP14 overexpression could be due to the strong blockade of cell proliferation, since this protein also plays an important role in cell cycle and proliferation.
Discussion
The present investigation is the first work to show the antitumor activity of ENG-targeting ADC therapies in ES. We propose that this novel approach might provide improved tumor selectivity as compared to cytotoxic agents in a subset of ES patients expressing high levels of ENG. Most cytotoxic chemotherapies used today exhibit a narrow therapeutic index dependent upon a higher proliferative rate of tumor cells compared to normal cells. The implications of ES heterogeneity in response to treatment also represent a challenge when it comes to defining upfront clinical strategies. These facts stress the need to selectively target the unique molecular features that exist in tumor cells while preventing off-target effects in normal cells. In this context, precision medicine represents an appealing yet rather challenging paradigm. Toward that end, ADCs have shown clinical promise given their ability to harness the specificity of monoclonal antibodies to more selectively deliver apoptosis-inducing molecules to epitope-bearing tumor cells (10,25–27).
Though the pathognomonic hallmarks of ES (i.e. the EWS-FLI1 fusion protein and CD99 overexpression) have been known for decades, only in the last years have ES-specific trials taken direct aim at the oncogenic fusion protein or the ETS subcomponent (28). While we eagerly await the clinical results from trials testing the first-generation EWS-FLI1 modulators, such as TK-216, the issue of innate and acquired intra-tumoral heterogeneity is likely to remain a significant hurdle that prevents—or at least substantially reduces—the likelihood that any single therapy will eradicate every tumor cell. Substantiating this concern, recent evidence from two laboratories suggests that ES cells can oscillate between high- and low-expressing states with respect to the EWS-FLI1 fusion protein. Intriguingly, low levels of EWS-FLI1 might actually result in increased migration and invasion by tumor cells, questioning the effectiveness of exclusively targeting the ES fusion protein without co-targeting of other oncogenic drivers (29,30). Though CD99 is highly expressed by ES cells, MSCs and other cell types also express it in physiological conditions (31). So far, no specific membrane receptors have been described for ES, with the exception of LINGO1. Nonetheless, it was also expressed in the brain and its expression was highly variable in ES when compared to that of CD99 (32).
The ENG receptor, as mentioned previously, is not tumor-specific and it is also expressed by normal vascular tissues. Mutations in ENG are associated with hereditary hemorrhagic telangiectasia type 1 (HHT1). HHT1 patients present massive vascular malformation (17). In cancer, the transmembrane form of ENG has been associated with poor clinical outcomes, high intratumoral microvessel densities, and an aggressive/invasive phenotype in certain tumor types (18,33,34). Interestingly, the soluble form of ENG (sENG) has been described as an antagonist of transmembrane ENG and a biomarker of hypertension, diabetes and preeclampsia (35,36). sENG has also been pointed as a circulating biomarker of poor prognosis in patients with solid tumors (37,38). However, our analysis of sENG in plasma showed no significant differences between healthy donors and ES patients, and no significant correlations with clinical parameters. To sum up, ENG presents two major roles in cancer biology: i) modulating tumor angiogenesis and ii) promoting tumor progression in certain tumor types; hence, it represents an attractive therapeutic target.
Previous works from other groups, hereby reproduced (Figure S4), have, in fact, shown that ENG-targeting with a humanized MAb impairs the ability of endothelial cells to form functional tubular structures, which are essential for tumor neo-angiogenesis (11). ENG-targeting MAbs are currently being used in clinical trials for solid tumors, mainly in combination with other anti-angiogenic agents (39–41). Expression of ENG in ES was previously described by our group in a small set of tumor samples (31). The role of ENG on ES phenotype and malignancy has already been studied in in vitro and in vivo models, where ENG was associated with increased invasiveness and metastasis formation (18). In fact, ENG is described as a “gold” receptor protein in a recent work focused on the study of the surfaceome of ES for the search of targetable membrane receptors (32). Our finding of a possible epigenetic downregulation of ENG by methylation of its promoter region in the CADO cell line is consistent with other studies in solid tumors, where the hypermethylation of ENG promoter region results in gene silencing, and was reactivated after in vitro methylation reversal assays (42,43). Moreover, the positive correlation between ENG and MMP14 observed in ES cell lines and patients’ samples, suggests that there might be an important highly regulated balance between both proteins.
A major strength of our work stems from the concurrent evaluation of ENG as a therapeutic target against ES by two academic labs, based respectively in Europe and the U.S., which focus on ES drug development. As initial proof-of-concept, we evaluated OMTX503, a humanized MAb engaging ENG linked to a non-toxic type 2 ribosome-inactivating protein nigrin-b A chain, in vitro and within murine models. The development of an anti-ENG-nigrin-b-based ADC has already shown activity towards melanoma mouse tumors, both in vitro and in vivo (44). This work showed that targeting human ENG in a xenograft model with the ADC resulted in tumor shrinkage and inhibition of neo-angiogenesis (44). It is important to mention that this MAb engaging ENG is linked to the non-toxic A chain of nigrin-b.
Though OMTX503 demonstrated moderate preclinical activity, we subsequently transitioned our research focus to ENG-targeting ADC with cytolysin, OMTX703, given its lower toxicity and broader therapeutic window. OMTX503 and OMTX703 share the same mechanism of action and both lead to cell-death after drug internalization. Our in vitro results confirmed an ENG-dependent effect for both molecules. ES cell line-derived xenografts with a high expression of ENG were used as a first in vivo approach. At this point, we found differences of response to treatment, depending on the drug administered, OMTX503 or OMTX703. We suggest that the response to treatment could be mainly influenced by the therapeutic window of each ADC.
Recognizing the potential importance of human tumor stroma in evaluating the anti-ENG ADC response, we sought to validate the xenograft results using ES PDX models that likely exhibit a wider breadth of intratumoral heterogeneity that better reflects the clinical setting. Only OMTX703 was comprehensively assessed in PDXs. Notably, the frequency of CR (60%) after 21 days of treatment in PDXs treated with OMTX703 reinforces the promising efficacy of this drug. Given that all mice relapsed in the long-term survival study, we hereby suggest the importance of drug combinations to avoid acquired drug resistance.
Taken together, the antineoplastic effects observed in our ES PDXs are highly encouraging. Though additional research is certainly required before one would propose using ENG expression in order to select patients receiving ENG-targeting therapies, the prospect of using ENG expression as a predictive biomarker of response would clearly represent a crucial step to identify ES patients most likely to benefit from anti-ENG ADC therapies and, additionally, enable tissue-based companion diagnostics. Similarly, the toxicity profiles associated to these molecules, need be to further analyzed in specific regulatory toxicological studies in additional preclinical animal models. While the current study focused primarily on the direct effect of targeting ENG in tumor cells, future research must decipher how anti-ENG ADCs affect vascular stroma and the processes responsible for endothelial cell function and angiogenesis. In that regard, the capacity for OMTX703 to jointly target the tumor and stroma raises the tantalizing possibility that OMTX703 might delay tumor growth even when ENG expression is low or absent (40,41). With the addition of more comprehensive toxicity data, ENG-targeting ADCs represent a promising therapeutic alternative that may someday help ES patients suffering from ENG-positive tumors.
Supplementary Material
Translational Relevance.
Antibody-drug conjugates represent an innovative therapeutic strategy with growing interest in the clinic. Endoglin is a mesenchymal stem cell marker, which plays an important role in the TGF-β pathway. In the current study, we demonstrate for the first time that anti-endoglin drug conjugates are a promising option for the treatment of Ewing sarcoma. Herein, we show that the expression of endoglin in Ewing sarcoma cell lines, patient-derived xenograft models, and tumor samples is heterogeneous. Hence, the screening of endoglin expression in tumor samples as a predictive marker of response to anti-endoglin antibody-drug conjugates represents a crucial step. Treatment with anti-endoglin antibody-drug conjugates, using two different drug payloads, nigrin-b A chain and cytolysin, in cell line-derived xenografts and patient-derived xenograft models, led to a drastic tumor reduction. These findings provide a strong rationale for the implementation of anti-endoglin antibody-drug conjugates in the context of precision medicine in Ewing sarcoma.
Acknowledgements:
This research has been conducted using samples from the Hospital Universitario Virgen del Rocío - Instituto de Biomedicina de Sevilla Biobank (Andalusian Public Health System Biobank and ISCIII-Red de Biobancos PT13/0010/0056). The authors thank the donors and the Hospital Universitario Virgen del Rocío - Instituto de Biomedicina de Sevilla Biobank (Andalusian Public Health System Biobank and ISCIII-Red de Biobancos PT13/0010/0056) and Xarxa de Bancs de Tumors de Catalunya (XBTC) sponsored by Pla Director d’Oncologia de Catalunya for the human specimens used in this study. The authors thank Dr. Arjan Lankester, from LUMC, for providing the plasmas and clinical data from Ewing sarcoma patients.
Funding: EDA’s lab is supported by the RETOS COLABORACIÓN project (RTC-2014-2102-1); the AECC project (GCB13131578DEÁ); ISCIII-FEDER (PI14/01466); CIBERONC (CB16/12/00361, and CB16/12/00316), and Asociación Pablo Ugarte; PPC is sponsored by the Fundación María García Estrada; AMC lab is supported by ISCIII-FEDER (CP13/00189). MJRF is supported by Consejería de Salud de la Junta de Andalucía (ECAIF2-0176-2013) JL’s lab is supported by the National Institutes of Health (R01 CA180279) and the University of Texas MD Anderson Cancer Center’s Support Grant CA016672.
Footnotes
Conflict of interest: SD, CF, MF, LS are Oncomatryx employees. JL serves as an external advisor to Oncomatryx, who partially supported his lab's endoglin-related research. All the other authors declare no potential conflicts of interest.
References
- 1.Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop 2016;7(9):527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zucman J, Delattre O, Desmaze C, Plougastel B, Joubert I, Melot T, et al. Cloning and characterization of the Ewing’s sarcoma and peripheral neuroepithelioma t(11;22) translocation breakpoints. Genes Chromosomes Cancer 1992;5(4):271–7. [DOI] [PubMed] [Google Scholar]
- 3.Sheffield NC, Pierron G, Klughammer J, Datlinger P, Schonegger A, Schuster M, et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat Med 2017;23(3):386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huertas-Martinez J, Court F, Rello-Varona S, Herrero-Martin D, Almacellas-Rabaiget O, Sainz-Jaspeado M, et al. DNA methylation profiling identifies PTRF/Cavin-1 as a novel tumor suppressor in Ewing sarcoma when co-expressed with caveolin-1. Cancer letters 2017;386:196–207. [DOI] [PubMed] [Google Scholar]
- 5.Franzetti GA, Laud-Duval K, van der Ent W, Brisac A, Irondelle M, Aubert S, et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornaz-Buros S, Riggi N, DeVito C, Sarre A, Letovanec I, Provero P, et al. Targeting cancer stem-like cells as an approach to defeating cellular heterogeneity in Ewing sarcoma. Cancer Res 2014;74(22):6610–22. [DOI] [PubMed] [Google Scholar]
- 7.Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov 2006;5(2):147–59. [DOI] [PubMed] [Google Scholar]
- 8.Citores L, Ferreras JM, Munoz R, Benitez J, Jimenez P, Girbes T. Targeting cancer cells with transferrin conjugates containing the non-toxic type 2 ribosome-inactivating proteins nigrin b or ebulin l. Cancer Lett 2002;184(1):29–35. [DOI] [PubMed] [Google Scholar]
- 9.Munoz R, Arias Y, Ferreras JM, Rojo MA, Gayoso MJ, Nocito M, et al. Targeting a marker of the tumour neovasculature using a novel anti-human CD105-immunotoxin containing the non-toxic type 2 ribosome-inactivating protein nigrin b. Cancer Lett 2007;256(1):73–80. [DOI] [PubMed] [Google Scholar]
- 10.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nature reviews Drug discovery 2017;16(5):315–37. [DOI] [PubMed] [Google Scholar]
- 11.Rosen LS, Gordon MS, Robert F, Matei DE. Endoglin for targeted cancer treatment. Current oncology reports 2014;16(2):365. [DOI] [PubMed] [Google Scholar]
- 12.Lastres P, Letamendia A, Zhang H, Rius C, Almendro N, Raab U, et al. Endoglin modulates cellular responses to TGF-beta 1. J Cell Biol 1996;133(5):1109–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen CZ, Li M, de Graaf D, Monti S, Gottgens B, Sanchez MJ, et al. Identification of endoglin as a functional marker that defines long-term repopulating hematopoietic stem cells. Proc Natl Acad Sci U S A 2002;99(24):15468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawinkels LJ, Kuiper P, Wiercinska E, Verspaget HW, Liu Z, Pardali E, et al. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer research 2010;70(10):4141–50. [DOI] [PubMed] [Google Scholar]
- 15.Lopez-Novoa JM, Bernabeu C. The physiological role of endoglin in the cardiovascular system. Am J Physiol Heart Circ Physiol 2010;299(4):H959–74. [DOI] [PubMed] [Google Scholar]
- 16.Arthur HM, Ure J, Smith AJ, Renforth G, Wilson DI, Torsney E, et al. Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol 2000;217(1):42–53. [DOI] [PubMed] [Google Scholar]
- 17.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nature genetics 1994;8(4):345–51. [DOI] [PubMed] [Google Scholar]
- 18.Pardali E, van der Schaft DW, Wiercinska E, Gorter A, Hogendoorn PC, Griffioen AW, et al. Critical role of endoglin in tumor cell plasticity of Ewing sarcoma and melanoma. Oncogene 2011;30(3):334–45. [DOI] [PubMed] [Google Scholar]
- 19.van der Schaft DW, Hillen F, Pauwels P, Kirschmann DA, Castermans K, Egbrink MG, et al. Tumor cell plasticity in Ewing sarcoma, an alternative circulatory system stimulated by hypoxia. Cancer Res 2005;65(24):11520–8. 10.1158/0008-5472.CAN-05-2468. [DOI] [PubMed] [Google Scholar]
- 20.Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer 2003;3(6):411–21. [DOI] [PubMed] [Google Scholar]
- 21.Volkel T, Holig P, Merdan T, Muller R, Kontermann RE. Targeting of immunoliposomes to endothelial cells using a single-chain Fv fragment directed against human endoglin (CD105). Biochimica et biophysica acta 2004;1663(1–2):158–66. [DOI] [PubMed] [Google Scholar]
- 22.Fracasso G, Bellisola G, Castelletti D, Tridente G, Colombatti M. Immunotoxins and other conjugates: preparation and general characteristics. Mini reviews in medicinal chemistry 2004;4(5):545–62. [DOI] [PubMed] [Google Scholar]
- 23.Thorpe PE, Wallace PM, Knowles PP, Relf MG, Brown AN, Watson GJ, et al. New coupling agents for the synthesis of immunotoxins containing a hindered disulfide bond with improved stability in vivo. Cancer research 1987;47(22):5924–31. [PubMed] [Google Scholar]
- 24.Stewart E, Goshorn R, Bradley C, Griffiths LM, Benavente C, Twarog NR, et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell reports 2014;9(3):829–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krop IE, Kim SB, Gonzalez-Martin A, LoRusso PM, Ferrero JM, Smitt M, et al. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. The Lancet Oncology 2014;15(7):689–99. [DOI] [PubMed] [Google Scholar]
- 26.Gan HK, van den Bent M, Lassman AB, Reardon DA, Scott AM. Antibody-drug conjugates in glioblastoma therapy: the right drugs to the right cells. Nature reviews Clinical oncology 2017. [DOI] [PubMed] [Google Scholar]
- 27.Lambert JM, Morris CQ. Antibody-Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Advances in therapy 2017;34(5):1015–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ordonez JL, Osuna D, Herrero D, de Alava E, Madoz-Gurpide J. Advances in Ewing’s sarcoma research: where are we now and what lies ahead? Cancer research 2009;69(18):7140–50. [DOI] [PubMed] [Google Scholar]
- 29.Franzetti GA, Laud-Duval K, van der Ent W, Brisac A, Irondelle M, Aubert S, et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017;36(25):3505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barber-Rotenberg JS, Selvanathan SP, Kong Y, Erkizan HV, Snyder TM, Hong SP, et al. Single enantiomer of YK-4–279 demonstrates specificity in targeting the oncogene EWS-FLI1. Oncotarget 2012;3(2):172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amaral AT, Manara MC, Berghuis D, Ordonez JL, Biscuola M, Lopez-Garcia MA, et al. Characterization of human mesenchymal stem cells from ewing sarcoma patients. Pathogenetic implications. PloS one 2014;9(2):e85814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Town J, Pais H, Harrison S, Stead LF, Bataille C, Bunjobpol W, et al. Exploring the surfaceome of Ewing sarcoma identifies a new and unique therapeutic target. Proceedings of the National Academy of Sciences of the United States of America 2016;113(13):3603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dallas NA, Samuel S, Xia L, Fan F, Gray MJ, Lim SJ, et al. Endoglin (CD105): a marker of tumor vasculature and potential target for therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2008;14(7):1931–7. [DOI] [PubMed] [Google Scholar]
- 34.Uneda S, Toi H, Tsujie T, Tsujie M, Harada N, Tsai H, et al. Anti-endoglin monoclonal antibodies are effective for suppressing metastasis and the primary tumors by targeting tumor vasculature. International journal of cancer Journal international du cancer 2009;125(6):1446–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blazquez-Medela AM, Garcia-Ortiz L, Gomez-Marcos MA, Recio-Rodriguez JI, Sanchez-Rodriguez A, Lopez-Novoa JM, et al. Increased plasma soluble endoglin levels as an indicator of cardiovascular alterations in hypertensive and diabetic patients. BMC medicine 2010;8:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Young BC, Levine RJ, Karumanchi SA. Pathogenesis of preeclampsia. Annual review of pathology 2010;5:173–92. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi N, Kawanishi-Tabata R, Haba A, Tabata M, Haruta Y, Tsai H, et al. Association of serum endoglin with metastasis in patients with colorectal, breast, and other solid tumors, and suppressive effect of chemotherapy on the serum endoglin. Clinical cancer research : an official journal of the American Association for Cancer Research 2001;7(3):524–32. [PubMed] [Google Scholar]
- 38.Vo MN, Evans M, Leitzel K, Ali SM, Wilson M, Demers L, et al. Elevated plasma endoglin (CD105) predicts decreased response and survival in a metastatic breast cancer trial of hormone therapy. Breast cancer research and treatment 2010;119(3):767–71. [DOI] [PubMed] [Google Scholar]
- 39.Rosen LS, Hurwitz HI, Wong MK, Goldman J, Mendelson DS, Figg WD, et al. A phase I first-in-human study of TRC105 (Anti-Endoglin Antibody) in patients with advanced cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2012;18(17):4820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon MS, Robert F, Matei D, Mendelson DS, Goldman JW, Chiorean EG, et al. An open-label phase Ib dose-escalation study of TRC105 (anti-endoglin antibody) with bevacizumab in patients with advanced cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20(23):5918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Tian H, Blobe GC, Theuer CP, Hurwitz HI, Nixon AB. Effects of the combination of TRC105 and bevacizumab on endothelial cell biology. Investigational new drugs 2014;32(5):851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Leary K, Shia A, Cavicchioli F, Haley V, Comino A, Merlano M, et al. Identification of Endoglin as an epigenetically regulated tumour-suppressor gene in lung cancer. British journal of cancer 2015;113(6):970–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong VC, Chan PL, Bernabeu C, Law S, Wang LD, Li JL, et al. Identification of an invasion and tumor-suppressing gene, Endoglin (ENG), silenced by both epigenetic inactivation and allelic loss in esophageal squamous cell carcinoma. International journal of cancer Journal international du cancer 2008;123(12):2816–23. [DOI] [PubMed] [Google Scholar]
- 44.Munoz R, Arias Y, Ferreras JM, Jimenez P, Langa C, Rojo MA, et al. In vitro and in vivo effects of an anti-mouse endoglin (CD105)-immunotoxin on the early stages of mouse B16MEL4A5 melanoma tumours. Cancer immunology, immunotherapy : CII 2013;62(3):541–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.