

Tumor cells accumulate spontaneous and drug-induced DNA damage, but they survive because of enhanced/altered DNA repair activities [1]. The hypothesis that cancer cells are addicted to particular DNA repair pathways is supported by selective elimination of tumor cells by drugs/compounds targeting specific DNA repair mechanisms [2].
DNA double-strand breaks (DSBs), the most lethal DNA lesions, are usually repaired by BRCA1/2 -mediated homologous recombination (BRCA-HR) and DNA-PK –mediated non-homologous end-joining (D-NHEJ) in proliferating cells [3]. PARP1 may prevent accumulation of potentially lethal DNA double-strand breaks (DSBs) by playing a key role in base excision repair (BER), single-strand break (SSB) repair, alternative non-homologous end-joining (Alt-NHEJ), and/or by facilitating MRE11-mediated recruitment of RAD51 to promote stalled replication fork restart [4]. The success of the PARP inhibitor (PARPi) olaparib in BRCA1/2-deficient breast tumors has established a proof-of-concept of personalized cancer therapy utilizing synthetic lethality [5]. Moreover, we have recently demonstrated that PARPi triggers synthetic lethality also in proliferating and quiescent tumor cells deficient in BRCA-HR and DNA-PK mediated NHEJ (D-NHEJ) [6].
Unfortunately, therapeutic effect of PARPi is usually short-lived and tumor cells become unresponsive due to a variety of compensatory mechanisms [7]. In concordance, we showed that BRCA-deficient breast carcinoma cells and leukemia cells could not be completely eradicated by PARPi [6]. Therefore, more robust and rapid targeting of PARP1 is required to reduce time-dependent emergence of PARPi-resistant/refractory clones.
Most PARP inhibitors have been designed to compete with nicotinamide adenine dinucleotide (NAD) for a binding site on the PARP1 molecule [8]. This strategy resulted in the discovery of NAD-like PARP inhibitors that not only target PARPs, but, unfortunately, other enzymatic pathways involving NAD and other nucleotides as co-factors. Using such inhibitors affects multiple NAD/nucleotide-dependent enzymatic pathways, which results in secondary toxic effects proceeding from the inactivation of other pathways, while the efficiency against the PARP1 pathway is diminished. Thus, the challenge is to design inhibitors based on other activities of PARP1.
We and others reported that DSBs-dependent interaction of PARP1 with histone 4 (H4) resulted in activation of PARP1 enzymatic activity and stimulation of Alt-NHEJ [9,10]. We identified non-NAD-like inhibitor 5F02, which interfered with H4-mediated activation of PARP1 but not PARP2 and Tankyrase-1, and was effective against several types of tumors including BRCA1-deficient MDA-MB-436 cells [11]. Here we tested the hypothesis that combinations of two structurally different PARP1 inhibitors, NAD-like olaparib or talazoparib and non-NAD-like 5F02 (Figure 1A) result in more abundant elimination of BRCA1-deficient leukemia cells and reduced toxicity to normal counterparts.
Figure 1. 5F02 enhanced the effect of olaparib against BRCA1-deficient CML cells.
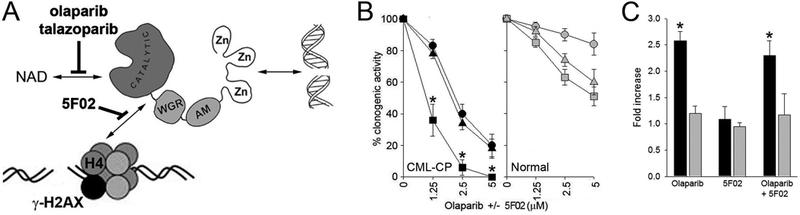
(A) Scheme illustrating anti-PARP1 activity of NAD-like (olaparib and talazoparib, both from Selleckchem) and non-NAD-like (5F02, synthesized as described in Supplemental Methods) inhibitors. (B) BCR-ABL1 –positive CML primary samples (n=3) and samples of normal hematopoietic cells (n=3) were characterized before [6]. Lin-CD34+ cells were obtained from mononuclear fractions by magnetic sorting using the EasySep negative selection human progenitor cell enrichment cocktail followed by human CD34 positive selection cocktail (StemCell Technologies) as described before [6]. Cells were incubated for 72 hrs with the indicated concentrations of olaparib (triangles), 5F02 (circles) and olaparib + 5F02 (squares) in IMDM supplemented with 10% FBS and growth factors (100 ng/ml SCF, 20 ng/ml IL-3, 100 ng/ml Flt-3 ligand, 20 ng/ml G-CSF, 20 ng/ml IL-6) followed by plating in Methocult (Stemcell Technologies). Colonies were counted after 7 days. Results represent mean % of clonogenic cells ± SD surviving the treatment (from triplicates); *p<0.005 when compared to individual treatment using the response additivity approach to study synergistic effects [15]. (C) Lin-CD34+ cells from CML-CP patient (black bars) and from healthy donor (grey bars) were incubated for 24 hrs with 2.5 μM olaparib, 2.5 μM 5F02, and olaparib + 5F02. DSBs were detected by γ-H2AX (H2AX phosphorylated on serine 139) immunofluorescence as described before [6]. Results represent relative increase of γ-H2AX immunofluorescence ± SD in comparison to normal cells (from triplicates); *p<0.05 when compared to healthy donors using unpaired two-tailed Student t test.
To test the effect of 5F02 and/or olaparib against primary tumor cells we employed BCR-ABL1-positive chronic myeloid leukemia (CML) patient cells. BCR-ABL1 oncogenic tyrosine kinase induces translational repression and degradation of BRCA1 protein which makes leukemia cells sensitive to PARPi [6,12,13]. 5F02 and olaparib exerted similar anti-leukemia effect against Lin-CD34+ peripheral blood cells from CML in chronic phase (CML-CP) (Figure 1B). However, 5F02, in contrast to olaparib, exerted very limited toxicity against normal Lin-CD34+ cells obtained from healthy donors. Moreover, the combination of 5F02 and olaparib was exceptionally effective against CML-CP cells, but showed little or no toxicity in normal cells.
As expected olaparib enhanced accumulation of DSBs (detected by γ-H2AX immunofluorescence) in Lin-CD34+ CML-CP cells when compared to normal counterparts (Figure 1C). Surprisingly, 5F02 did not induce DSBs when used alone and in combination with olaparib.
Tyrosine kinase inhibitor imatinib is a standard drug for CML treatment [14]. We reported before and also show here that a combination of imatinib + olaparib and imatinib + talazoparib exerted better anti-CML chronic phase (CML-CP) and CML accelerated phase (CML-AP) effect than the drugs used individually (Figure 2A) [6]. In addition, a similar effect was exerted by imatinib + 5F02. Interestingly, imatinib combined with olaparib or talazoaprib and 5F02 eradicated clonogenic Lin-CD34+ CML-AP and CML-AP cells in vitro, while causing minimal toxicity to normal counterparts. Moreover, the combination of imatinib + olaparib/talazoparib + 5F02 was extremely effective against CML-CP quiescent cells, but not against normal cells (Figure 2B).
Figure 2. 5F02 enhanced anti-CML effect of imatinib + talazoparib.
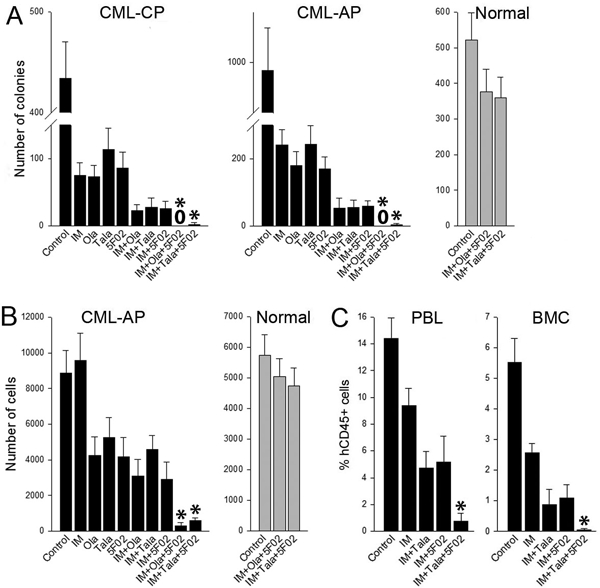
(A, B) Lin-CD34+ CML-CP/AP cells (black bars; n=3 patients of each) and normal counterparts (grey bars; n=3 healthy donors) were stained with cell trace violet (CTV) (eBioscience) and incubated for 5 days with 1μM imatinib (IM), 2.5μM 5F02, 2.5μM olaparib (Ola), 25nM talazoparib (Tala) or with indicated combinations in StemSpan®SFEM medium (Stem Cell Technologies, Vancouver, Canada) supplemented with the cocktail of growth factors (see Figure 1 legend). (A) Lin-CD34+ clonogenic cells were detected after plating in Methocult. (B) Lin-CD34+CD38-CTVmax cells were detected by flow cytometry using fluorochrome-conjugated anti-Lin (#340546), anti-CD34 (#347203, #555821) and anti-CD38 (#355790, #555460) antibodies (all from BD Pharmingen). Results represent mean number of colonies or quiescent cells ± SD from triplicates; *p<0.001 in comparison to IM+Ola, and IM+5F02 using the response additivity approach to study synergistic effects. (C) Sub-lethally total-body irradiated (600 Gy) NOD.Rag1−/−;γcnull mice expressing human IL-3, GM-CSF and SCF (NRGS) were injected with 107 Lin-CD34+ CML-CP cells and treated for 7 days with imatinib (IM) (100 mg/kg twice daily by oral gavage [6]) and indicated combinations with 5F02 (2.5 mg/kg i.p.) and/or talazoparib (Tala) (0.33 mg/kg i.v. [6]). Anti-CML effect was assessed by detection of hCD45+ cells in peripheral blood leukocytes (PBL) and bone marrow cells (BMC) at the end of treatment as described before [6]. Results represent mean ± SD percentage of hCD45+ in PBL and BMC (3–4 mice/group); *p<0.01 in comparison to IM+Tala, and IM+5F02 using the response additivity approach to study synergistic effects.
Finally, we tested the effectiveness of talazoparib + 5F02 in humanized NRGS mice bearing primary CML-CP xenograft and treated with imatinib. Talazoparib, which in mice has favorable pharmacokinetics over olaparib, and imatinib, was already examined by us in immunodeficient mice bearing CML xenotransplants [6]. Based on in vitro and in vivo pharmacokinetics (Supplemental Table 1 and Supplemental Figure 1) we administered 2.5 mg/kg of 5F02.
Imatinib reduced leukemia burden as assessed by detection of hCD45+ cells in peripheral blood and bone marrow, and addition of talazoparib or 5F02 exerted even stronger anti-leukemia effect (Figure 2C). Remarkably, combination of both PARP inhibitors with imatinib resulted in exceptionally strong synergistic therapeutic effect.
Since patients with BRCA-deficient tumors who initially respond to PARPi often develop resistance, leading to cancer relapse [7] there is an urgent need to develop novel strategies to prevent development of drug resistance, for example by killing tumor cells more rapidly and robustly. We have shown here that combination of non-NAD-like PARPi (5F02) and NAD-like PARPi (olaparib, talazoparib) exerted synergistic effect against BRCA1 and DNA-PK -deficient proliferating and quiescent leukemia stem and progenitor cells. Enhanced anti-tumor effect of 5F02 + olaparib could be also observed in BRCA2-deficient lymphomas (Supplemental Figure S2) and in BRCA1/2-deficient solid tumor cells (Supplemental Figure S3), indicating that anti-tumor synthetic lethal effect of the combination could be achieved in a variety of BRCA1/2-deficient malignant cells. Moreover, 5F02 + olaparib eliminated quiescent D-NHEJ –deficient leukemia cells. Importantly, non-NAD-like PARPi 5F02 did not enhance the toxicity of NAD-like PARPi olalarib against normal cells. Finally, the combination exerted synergistic effect in humanized immunodeficient mice bearing BRCA1/DNA-PK-deficient primary CML xenografts. Altogether, non-NAD-like PARP1i synergistically enhanced synthetic lethal effect of NAD-like PARPi in tumor cells without increasing the cytotoxic effect in normal cells.
While synthetic lethality mediated by NAD-like PARPi is usually associated with enhanced accumulation of potentially lethal DSBs, non-NAD-like PARPi 5F02 did not induce DSBs when used as a single agent and also in combination with olaparib. Thus, the mechanistic aspect of the synergistic effect of NAD-like and non-NAD-like PARPis needs to be elucidated. Nevertheless, our data suggest that combining NAD-like and allosteric non-NAD-like PARPi’s may represent a viable therapeutic strategy for enhancing tumor response to PARP inhibition and reducing the resistance that inevitably results from treatment with NAD-like PARPi’s alone.
Supplementary Material
Acknowledgements:
This work was supported by NHI/NCI 1R01 CA186238 to T.S. and Department of Deference PC160049 and the NIH R03 CA212566 to A.V.T. M.T. was supported by the Kosciuszko Foundation Scholarship for 2017/2018. B.V.L has been supported by the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement no 665735 and by the funding from Polish Ministry of Science and Higher Education funds for the implementation of international projects, 2016–2020. P.V. was supported by the Austrian Science Fund, grant F4704-B20.
Footnotes
Disclosure of interest: The authors report no conflicts of interest.
References
- 1.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C and others. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005;434:864–70. [DOI] [PubMed] [Google Scholar]
- 2.Nickoloff JA, Jones D, Lee SH, Williamson EA, Hromas R. Drugging the Cancers Addicted to DNA Repair. J Natl Cancer Inst 2017;109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell 2012;47:320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toma M, Skorski T, Sliwinski T. DNA double strand break repair - related synthetic lethality. Curr Med Chem 2018. [DOI] [PubMed] [Google Scholar]
- 5.Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med 2015;66:455–70. [DOI] [PubMed] [Google Scholar]
- 6.Nieborowska-Skorska M, Sullivan K, Dasgupta Y, Podszywalow-Bartnicka P, Hoser G, Maifrede S, Martinez E, Di Marcantonio D, Bolton-Gillespie E, Cramer-Morales K and others. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J Clin Invest 2017;127:2392–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med 2013;19:1381–8. [DOI] [PubMed] [Google Scholar]
- 8.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012;72:5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 2002;419:411–5. [DOI] [PubMed] [Google Scholar]
- 10.Thomas CJ, Kotova E, Andrake M, Adolf-Bryfogle J, Glaser R, Regnard C, Tulin AV. Kinase-mediated changes in nucleosome conformation trigger chromatin decondensation via poly(ADP-ribosyl)ation. Mol Cell 2014;53:831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas C, Ji Y, Lodhi N, Kotova E, Pinnola AD, Golovine K, Makhov P, Pechenkina K, Kolenko V, Tulin AV. Non-NAD-Like poly(ADP-Ribose) Polymerase-1 Inhibitors effectively Eliminate Cancer in vivo. EBioMedicine 2016;13:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Podszywalow-Bartnicka P, Wolczyk M, Kusio-Kobialka M, Wolanin K, Skowronek K, Nieborowska-Skorska M, Dasgupta Y, Skorski T, Piwocka K. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle 2014;13:3727–41. [DOI] [PubMed] [Google Scholar]
- 13.Dkhissi F, Aggoune D, Pontis J, Sorel N, Piccirilli N, LeCorf A, Guilhot F, Chomel JC, Ait-Si-Ali S, Turhan AG. The downregulation of BAP1 expression by BCR-ABL reduces the stability of BRCA1 in chronic myeloid leukemia. Exp Hematol 2015;43:775–80. [DOI] [PubMed] [Google Scholar]
- 14.Hehlmann R, Lauseker M, Saussele S, Pfirrmann M, Krause S, Kolb HJ, Neubauer A, Hossfeld DK, Nerl C, Gratwohl A and others. Assessment of imatinib as first-line treatment of chronic myeloid leukemia: 10-year survival results of the randomized CML study IV and impact of non-CML determinants. Leukemia 2017;31:2398–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slinker BK. The statistics of synergism. J Mol Cell Cardiol 1998;30:723–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.