

Abstract
Presynaptic α2δ subunits of voltage-gated calcium channels regulate channel abundance and are involved in glutamatergic synapse formation. However, little is known about the specific functions of the individual α2δ isoforms and their role in GABAergic synapses. Using primary neuronal cultures of embryonic mice of both sexes, we here report that presynaptic overexpression of α2δ-2 in GABAergic synapses strongly increases clustering of postsynaptic GABAARs. Strikingly, presynaptic α2δ-2 exerts the same effect in glutamatergic synapses, leading to a mismatched localization of GABAARs. This mismatching is caused by an aberrant wiring of glutamatergic presynaptic boutons with GABAergic postsynaptic positions. The trans-synaptic effect of α2δ-2 is independent of the prototypical cell-adhesion molecules α-neurexins (α-Nrxns); however, α-Nrxns together with α2δ-2 can modulate postsynaptic GABAAR abundance. Finally, exclusion of the alternatively spliced exon 23 of α2δ-2 is essential for the trans-synaptic mechanism. The novel function of α2δ-2 identified here may explain how abnormal α2δ subunit expression can cause excitatory–inhibitory imbalance often associated with neuropsychiatric disorders.
SIGNIFICANCE STATEMENT Voltage-gated calcium channels regulate important neuronal functions such as synaptic transmission. α2δ subunits modulate calcium channels and are emerging as regulators of brain connectivity. However, little is known about how individual α2δ subunits contribute to synapse specificity. Here, we show that presynaptic expression of a single α2δ variant can modulate synaptic connectivity and the localization of inhibitory postsynaptic receptors. Our findings provide basic insights into the development of specific synaptic connections between nerve cells and contribute to our understanding of normal nerve cell functions. Furthermore, the identified mechanism may explain how an altered expression of calcium channel subunits can result in aberrant neuronal wiring often associated with neuropsychiatric disorders such as autism or schizophrenia.
Keywords: auxiliary subunits, cacna2d, cultured hippocampal neurons, imaging, immunocytochemistry, voltage-gated calcium channels
Introduction
Auxiliary α2δ subunits are traditionally envisioned as potent modulators of voltage-gated calcium channels (Felix et al., 1997; Klugbauer et al., 1999). Recently, synaptic functions of all four α2δ subunit isoforms have been suggested, which may be partly or entirely independent of the channel complex: α2δ-1 has been identified as postsynaptic receptor for glia-secreted thrombospondins, which mediate excitatory synaptogenesis (Eroglu et al., 2009; Risher et al., 2018). In mice, loss of α2δ-2 affected morphology and function of cerebellar Purkinje neurons (Barclay et al., 2001; Brodbeck et al., 2002), as well as structure and function of auditory hair cell synapses (Fell et al., 2016). Additionally, α2δ-2 has been identified as a suppressor of axonal regeneration in the adult CNS (Tedeschi et al., 2016). The invertebrate homologs of α2δ-3 are crucial for the presynaptic development of motoneurons (Kurshan et al., 2009; Caylor et al., 2013) and, in mice, loss of α2δ-3 results in aberrant synapse formation of auditory nerve fibers (Pirone et al., 2014). Finally, α2δ-4 is required for the organization of rod and cone photoreceptor synapses (Wang et al., 2017; Kerov et al., 2018). The overall importance of α2δ subunits is emphasized by their involvement in various neurological disorders: CACNA2D1 and CACNA2D2, the genes encoding α2δ-1 and α2δ-2, have been linked to epilepsy (Chioza et al., 2009; Edvardson et al., 2013; Pippucci et al., 2013; Vergult et al., 2015; Butler et al., 2018); CACNA2D3 is a potential risk gene for autism spectrum disorders (Iossifov et al., 2012; De Rubeis et al., 2014); and all three genes may be associated with schizophrenia (Purcell et al., 2014; Moons et al., 2016). Mutations in CACNA2D4 can cause retinal dysfunction in humans (Ba-Abbad et al., 2016) and a partial deletion of CACNA2D4 was identified in patients with late-onset bipolar disorder (Van Den Bossche et al., 2012).
Despite the increasing knowledge, it is still largely elusive how the individual α2δ isoforms contribute to the formation and function of central synapses. The fact that α2δ subunits constitute large extracellular and highly glycosylated proteins (Davies et al., 2007; Bauer et al., 2010) puts them in an ideal position for extracellular functions, as previously suggested (Fell et al., 2016; Wang et al., 2017). Indeed, a recent study provided evidence for an interaction of α2δ-1 with NMDARs in dorsal horn neurons (Chen et al., 2018) and presynaptic α-neurexins (α-Nrxns) are potential candidates for mediating synaptic functions of α2δ subunits (Tong et al., 2017; Brockhaus et al., 2018). However, the role of α2δ subunits has until now only been addressed in excitatory glutamatergic synapses. This is surprising because α2δ subunits are strongly expressed in GABAergic neurons (Cole et al., 2005; Schlick et al., 2010) and aberrant GABAergic signaling is primarily implicated in the etiology of the abovementioned neurological disorders (e.g., epilepsy; Bonansco and Fuenzalida, 2016). Finally, the brain robustly expresses α2δ-1, α2δ-2, and α2δ-3 (Schlick et al., 2010); however, whether and how the individual isoforms contribute to specific functions in neurons simultaneously expressing all isoforms is elusive.
Therefore, to address α2δ subunit-mediated synaptic functions, we analyzed the consequences of presynaptic α2δ subunit expression in glutamatergic and GABAergic synapses. We show that presynaptic overexpression of α2δ-2 specifically induces the formation of mismatched glutamatergic synapses by trans-synaptically recruiting postsynaptic GABAARs. Moreover, α2δ-2 recruits postsynaptic GABAARs also in GABAergic synapses and thus independently of the presynaptic neurotransmitter identity. Most importantly, the mismatched synapse formation is explained by an altered wiring of glutamatergic axons to GABAergic postsynaptic positions. Interestingly, the α2δ-2-induced GABAAR clustering is further upregulated in glutamatergic neurons lacking all three α-Nrxns. Therefore, α-Nrxns can modulate the presynaptic effect of α2δ-2; however, they are not required for the trans-synaptic role of α2δ-2 in recruiting GABAARs. Together, our findings prove that presynaptic α2δ-2 acts trans-synaptically on postsynaptic GABAAR. The fact that increased α2δ-2 expression triggers aberrant axonal wiring is particularly interesting in light of neurological disorders associated with axonal wiring defects and altered excitatory–inhibitory balance (Huang and Hsueh, 2015; Lee et al., 2017).
Materials and Methods
Breeding and genotyping procedures
Animals.
Animal procedures for WT BALB/c and α2δ mutant mice were performed at the Medical University Innsbruck in compliance with government regulations and approved by the Austrian Federal Ministry of Science, Research and Economy (license numbers BMWFW-66.011/0113-WF/V/3b/2014 and BMWFW-66.011/0114-WF/V/3b/2014). Regular reports including the mouse numbers used for this project were given to the Austrian Federal Ministry of Science, Research and Economy (bmwfw). Animal experiments at the University of Münster [α-Nrxn triple knock-out (TKO) and control mice] were performed in accordance with government regulations for animal welfare and approved by the Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV, NRW, Germany, license numbers 84-02.05.20.11.209 and 84-02.04.2015.A423). Mice were maintained at the central animal facilities in Innsbruck and Münster under standard housing conditions with food and water available ad libitum on a 12 h light/dark cycle.
Breeding and genotyping of mutant mice.
WT and α2δ-3−/− mice used for qRT-PCR and LacZ reporter expression were obtained from breeding double heterozygous α2δ-1+/−, α2δ-3+/− mice having a mixed 129J × C57BL/6 background. The α2δ-1 knock-out mouse strain (α2δ-1−/−) was previously generated and characterized (Fuller-Bicer et al., 2009; Patel et al., 2013; Mastrolia et al., 2017). Genotyping for the Cacna2d1 gene was done as published previously (Fuller-Bicer et al., 2009) with some modifications by use of standard PCR conditions (annealing at 52°C for 30 s). Primers were as follows: WT-F1: 5′-GAGCTTTCTTTCTTCTGATTCCAC-3′, mutant-F2: 5′-CTGCACGAGACTAGTGAGACG-3′, R: 5′-ACATTCTCAAGACTGTAGGCAGAG-3′. Expected band sizes were 346 bp for WT (α2δ-1+/+) and 635 bp for knock-out (α2δ-1−/−) animals, respectively, and heterozygous mice showed both bands. The α2δ-3 knock-out mice (α2δ-3−/−) generated by Deltagen (B6.129P2-Cacna2d3tm1Dgen; Neely et al., 2010) were originally purchased from The Jackson Laboratory and provided by Jutta Engel (Saarland University, Germany). Knock-out was obtained by targeted insertion of a bacterial LacZ cassette into the Cacna2d3 gene such that the endogenous promoter drives the expression of β-galactosidase. The following primers were used for detecting the WT allele (F1-R, 183 bp fragment) and the knock-out allele (F2-R, 331 bp fragment): F1: 5′-TAGAAAAGATGCACTGGTCACCAGG-3′, F2: 5′-GGGCCAGCTCATTCCTCCCACTCAT-3′, R: 5′-GCAGAAGGCACATTGCCATACTCAC-3′ by use of standard PCR conditions (annealing at 63°C for 30 s). α-Nrxn TKO mice were generated as described previously (Missler et al., 2003; Dudanova et al., 2007). Genotyping of α-Nrxn-deficient mice was performed at the same time of neuronal preparation and as described previously (Missler et al., 2003).
Cell culture and transfection procedures
Lentiviral production.
Lentiviruses were produced by transient transfection of confluent tsa201 cells with the lentiviral expression vectors containing pHR-βA-eGFP, pHR-βA-mcherry, or pHR-βA-eGFP*α2δ-2 in combination with psPAX2 (packaging plasmid) and the pVSV (envelope plamid) using Metafectene (Biontex Laboratories). The following day, medium was changed to neuronal plating medium (NPM; consisting of MEM, 10% horse serum, 0.2% glucose, and 1 mm sodium pyruvate) and, after 24 and 48 h, supernatants containing the viruses were harvested, sterile filtered (0.20 μm), aliquoted, and stored at −20°C. Cultured hippocampal and striatal neurons were infected immediately after plating with the lentiviral medium supernatant diluted 1:4 in NPM and incubated for 4 h in a humidified incubator (95% air and 5% CO2) at 37°C. More details for infection procedures are given in the individual sections below.
Primary cultures of hippocampal neurons for fluorescence imaging.
Low-density cultures of hippocampal neurons were obtained from 16.5- to 18-d-old embryonic BALB/c mice of either sex as described previously (Obermair et al., 2004; Kaech and Banker, 2006; Stanika et al., 2016; Folci et al., 2018). In brief, hippocampi were dissected in cold Hank's balanced salt solution (HBSS) following dissociation by 2.5% trypsin-EDTA treatment and trituration. Dissociated neurons were plated at a density of ∼3500 or 7000 cells/cm2 on 18 mm glass coverslips (#1.5; GML) coated with poly-l-lysine (Sigma-Aldrich) in 60 mm culture dishes. After allowing the neurons to attach for 3–4 h, coverslips were transferred neuron-side down into a 60 mm culture dish containing a glial feeder layer. Maintenance of neurons and glia was done in serum-free neurobasal medium supplemented with Glutamax and B-27 (NBKO, all ingredients from Thermo Fisher Scientific). Three days after plating, Ara-C (5 μm) was added to stop glial proliferation and 1/3 of the medium was replaced weekly with fresh maintenance medium. Plasmids were introduced into neurons at 6 days in vitro (DIV) with Lipofectamine 2000-mediated transfection (Thermo Fisher Scientific) as described previously (Obermair et al., 2004). For cotransfection of pβA-eGFP plus pβA-α2δ or mCherry plus pβA-α2δ, 1.5 μg of total DNA was used at a molar ratio of 0.7:1. Control neurons from the same culture preparations were transfected with 1–2 μg of pβA-eGFP or mCherry, respectively. Labeling of cells for superresolution gated stimulated emission depletion (gSTED) microscopy (see Fig. 11A–C) was done by introducing mCherry or mCherry plus α2δ-2 at 6 DIV and pβA-eGFP at 7 DIV with lipofection. Cells were processed for immunostaining experiments between 20 and 30 DIV.
Figure 11.

Presynaptic α2δ-2 induces the aberrant wiring of glutamatergic axons to dendritic shafts. A, D, To analyze the position of α2δ-2-induced mismatched synapses on postsynaptic dendrites of excitatory (A, hippocampal neurons) and inhibitory neurons (D, MSNs), presynaptic (control or α2δ-2) and postsynaptic neurons were labeled with mCherry (red) and eGFP (green), respectively (for details, see Materials and Methods). The magnified insets in A and D demonstrate the close association of presynaptic axonal varicosities (mCherry) with postsynaptic dendritic spines (eGFP), as expected for excitatory spine synapses. Using gSTED microscopy, the distance from the dendritic shaft to the contact zone of each synaptic bouton was measured (A, arrows and lines in the magnified inset). B, E, gSTED microscopy confirms the preferential location of excitatory synapses on dendritic spines of both glutamatergic or GABAergic neurons (white arrowheads and sketch in B) and, most importantly, suggests an aberrant wiring of putative glutamatergic axons expressing α2δ-2 with postsynaptic sites along dendritic shafts (blue arrowheads and sketch in B), as typically observed for GABAergic synapses. C, Corresponding postsynaptic location of each contacting bouton was categorized as spine (green), shaft (gray), or as unclear (white; top) and the distance from the dendritic shaft to the contact point was measured (bottom). Although the vast majority of synaptic contact points in the control condition are located on dendritic spines (80%), presynaptic expression of α2δ-2 shifted the preferential contact points to dendritic shafts (65%, top). This is further confirmed by a significantly decreased contact point to shaft distance of α2δ-2-expressing compared with control synapses. Values for individual boutons (dots) and means (lines) ± SEM are shown. Dot colors (bottom) show the respective categorized synapse position (spine, green; shaft, gray; and unclear, white) presented in the top. Data from two independent culture preparations and 118 (control) and 75 (+α2δ-2) boutons were analyzed. Statistics: C, top: χ2 test: χ2(2) = 63.4, ***p < 0.001; bottom: Mann–Whitney U test: ***p < 0.001. E, F, gSTED microscopy (E) and high-resolution fluorescence microscopy (F) revealed that, similar to hippocampal neurons, presynaptic expression of α2δ-2 in cortical neurons shifted the preferential contact points to dendritic shafts of GABAergic MSNs, as indicated by gephyrin labeling and the dendritic position (blue arrowheads and line scan in F). Representative micrographs of two independent cultures are shown. Scale bars, 50 μm (overview), 3 μm (insets, A, D), and 2 μm (B, E, F).
For α-Nrxn TKO, WT and mutant mice of either sex were used for neuronal cultures derived from timed pregnant dams at embryonic day 17 (E17). Dissociated primary neurons were prepared in HBSS from single hippocampi as described previously (Neupert et al., 2015). Briefly, cell suspensions obtained after 0.25% trypsin treatment and trituration were plated onto 18 mm glass coverslips (Menzel-Glaeser) coated with poly-l-lysine (Sigma-Aldrich) at a density of 55,000 cells/coverslip. After 4 h at 37°C in plating medium (MEM, 10% horse serum, 0.6% glucose, and 1 mm sodium pyruvate), coverslips were inverted onto a 70–80% confluent monolayer of astrocytes grown in 12-well plates (Falcon) and incubated in Neurobasal medium supplemented with B27, 0.5 mm glutamine, and 12.5 μm glutamate. After 3 d, media were refreshed with Neurobasal medium supplemented with B27, 0.5 mm glutamine, and 5 μm AraC. Cultures were maintained at 37°C in a humidified incubator with an atmosphere of 95% air and 5% CO2. Neurons were transfected at 14 DIV using lipofectamine (Thermo Fisher Scientific) and experiments were performed between 20 and 25 DIV.
Primary cocultures of WT cortical and striatal neurons.
Because the inclusion of glutamatergic neurons is required for the proper development of GABAergic medium spiny neurons (MSNs) in culture (Segal et al., 2003), we modified the protocol for hippocampal neurons and adapted the striatal–cortical coculture published previously (Penrod et al., 2011). Cocultures of cortical neurons and striatal MSNs were prepared from 16.5- to 18-d-old embryonic BALB/c mice of either sex as summarized in Figure 7. Briefly, fetuses were removed from the uterus, decapitated, and brains were dissected in cold HBSS. After separating the cerebral hemispheres and stripping away the meninges, hemispheres were placed medial surface up, showing the hippocampus as a clearly visible structure. A region of the prefrontal cortex was dissected, minced, and transferred to a 15 ml Falcon tube containing HBSS (see Fig. 7A). The remaining cortical tissue was peeled along the line of the hippocampus to reveal the striatum, which was scooped out using small curved scissors and transferred to a separate tube. Striatal and cortical tissue of at least 2 hemispheres was separately collected and dissociated by 2.5% trypsin-EDTA treatment and trituration as described for hippocampal neurons (see above). Plasmid DNA–lipid complexes were prepared according to the Lipofectamine 2000-mediated transfection protocol (Thermo Fisher Scientific) and ∼2.4 × 105 striatal neurons were transfected for 20 min in a 37°C water bath keeping the total volume to 1 ml with NBKO. Subsequently, the cell suspension was directly seeded on poly-l-lysine-coated glass coverslips within a 60 mm culture dish containing 4 ml of prewarmed NPM and striatal neurons were allowed to attach at 37°C. For the entire transfection procedure, dissociated cortical neurons were maintained in HBSS in a 15 ml tube at 37°C and occasionally swirled. After 2 h, transfection of striatal neurons was stopped by replacing the transfection-plating solution with 5 ml of fresh, prewarmed NPM and untransfected cortical neurons were seeded onto striatal neurons in a ratio of 2 (cortical neurons) to 3 (MSNs) at a total density of ∼14,000 cells/cm2. For the immunostaining experiments and electrophysiological recordings shown in Figure 7, striatal neurons were plated at a density of 4000 or 7000 cells/cm2, respectively, and infected with a lentiviral pHR-βA-eGFP construct. Following attachment of MSNs and viral infection for 4 h, untransfected cortical neurons were plated at a density of 2800 cells/cm2. Cortical cells were allowed to attach for 3–4 h and coverslips were transferred neuron-side down into a 60 mm culture dish containing a glial feeder layer. In cocultures for superresolution gSTED microscopy (see Fig. 11E), striatal and cortical neurons were labeled with eGFP and mCherry, respectively. For this purpose, striatal neurons were plated at a density of 7000 cells/cm2 on poly-l-lysine-coated glass coverslips in a 60 mm culture dish containing 4 ml of prewarmed NPM. MSNs were virally infected using a lentiviral pHR-βA-eGFP construct and allowed to attach for 3–4 h. In the meantime, ∼2.0 × 105 cortical neurons were transfected at 37°C in a water bath, keeping the total volume to 1 ml with NBKO. To this end, mCherry or mCherry plus α2δ-2 were introduced using the Lipofectamine 2000-mediated transfection protocol (Thermo Fisher Scientific). Total DNA amounts and molar ratios were the same as described above for hippocampal neurons. Infection of striatal neurons was stopped by replacing the virus-plating solution with 4 ml of fresh, prewarmed NPM. The cell suspension containing the cortical neurons and transfection solution was centrifuged for 5 min at 1000 rpm at 4°C. Finally, cortical neurons were resuspended in 1 ml of NBKO, seeded onto striatal neurons, and allowed to attach for 3–4 h before coverslips were transferred neuron-side down into a 60 mm culture dish containing a glial feeder layer. Ara-C treatment and maintenance of neurons and glia were as described above. Cells were processed for immunostaining and patch-clamp experiments at 21–27 and 13–14 DIV, respectively.
Figure 7.

Presynaptic and postsynaptic differentiation of GABAergic MSNs. A, Schematic illustration of the coculture procedure: Cerebral hemispheres were dissected and stripped of meninges (1). Parts of the prefrontal cortex and striatum were dissected as shown (2). Striatal and cortical tissue was collected separately and dissociated using trypsin-EDTA and trituration (3). Cortical neurons were plated on poly-l-lysine-coated coverslips while striatal neurons were transfected with soluble eGFP (4). Subsequently, striatal neurons were plated onto cortical neurons in a ratio of 3:2 (5) and maintained above a glial feeder layer (6). B, Lentiviral infection or lipofection with soluble eGFP allowed discriminating MSNs from cortical neurons. C–E, Double immunofluorescence of striatal–cortical cocultures (24–26 DIV) with presynaptic and postsynaptic markers for excitatory and inhibitory synapses. Neuronal morphology is outlined by eGFP. C, GABAergic synapses of transfected eGFP-positive MSNs showed immunoreactivity for vGAT, whereas vGLUT1 was absent. D, Axons of cortical neurons formed excitatory synapses on MSN spine heads (vGLUT1, white arrowheads), whereas GABAergic synapses were located on the dendritic shaft (vGAT, blue arrowhead). E, Labeling of PSD-95 in spine heads opposite synapsin-positive presynaptic terminals further indicated the presence of functional excitatory synapses on MSNs. F, Patch-clamp analysis of mIPSCs and mEPSCs in 14 DIV neurons confirmed the functionality of GABAergic and glutamatergic synapses. Representative micrographs of two independent cultures are shown. Scale bars, 50 μm (B), 3 μm (overview), and 1 μm (magnified selections; C–E).
Neuronal cultures for paired recordings.
Culture protocols were modified to enable electrophysiological recordings from pairs of isolated neurons. Poly-l-lysine solution (Sigma-Aldrich) was sprayed on glass coverslips, producing uniformly distributed single dots of neuronal substrate. Hippocampal neurons were seeded at a density of 880 cells/cm2 and infected with pHR-βA-eGFP*α2δ-2. Cells were processed for electrophysiological experiments at 13–18 DIV.
Molecular biology
TaqMan qRT-PCR gene expression analysis.
For expression analysis of α2δ subunits (α2δ-1 to α2δ-4), RNA was isolated from 3 individual preparations of mono-cultured MSNs and adult male mouse striatum (7–8 weeks) as described previously (Schlick et al., 2010). Briefly, striatal neurons were prepared from 16.5- to 18-d-old embryonic BALB/c mice of either sex and plated on poly-l-lysine-coated glass coverslips in four 60 mm culture dishes at a density of 10,500 cells/cm2. After 24–25 d in culture, neurons were harvested for subsequent RNA extraction by trypsin treatment and homogenized with QiaShredder columns (Qiagen). For RNA isolation from brain tissue, WT mice obtained from double-heterozygous α2δ-1+/−, α2δ-3+/− breedings were killed by CO2 exposure, decapitated, and the striatum was dissected in cold HBSS and stored at −80°C in RNAlater RNA Stabilization Reagent (Qiagen) until further use. Tissue samples were disrupted using a Sonicator (UP200S; Hieschler) and QiaShredder columns (Qiagen). After homogenization, total RNA was immediately isolated with the RNeasy Protect Mini Kit following the manufacturer's instructions (Qiagen) and concentrations were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Subsequently, 1 μg (striatum) or 5.7 μl (∼50 ng, neurons) of RNA was transcribed with Superscript II reverse transcriptase (Thermo Fisher Scientific) and random hexamers (Promega). qRT-PCR (50 cycles) was performed in duplicates using a maximum amount of 20 ng RNA equivalent of cDNA per sample and the following TaqMan gene expression assays (Thermo Fisher Scientific): α2δ-1 (ID: Mm00486607_m1), α2δ-2 (ID: Mm00457825_m1), α2δ-3 (ID: Mm00486613_m1), α2δ-4 (ID: Mm01190105_m1), and Hprt1 (ID: Mm00446968_m1) as an endogenous control. The absolute number of α2δ transcripts was calculated by applying standard curves generated from PCR products of known concentrations (Schlick et al., 2010). To compare the relative expression of distinct α2δ subunits in different preparations, data were normalized to the preparation with the highest Hprt1 expression.
Expression vectors and cloning procedures.
To facilitate neuronal expression, all constructs were cloned into a eukaryotic expression plasmid containing a neuronal chicken β-actin promoter, pβA. Cloning of all constructs was confirmed by sequencing (Eurofins Genomics).
For pβA-α2δ-1: Mouse α2δ-1 was cloned from genomic cDNA derived from mouse cerebellum. Primer sequences were selected according to GenBank NM-001110844. Briefly, the cDNA of α2δ-1 was amplified by PCR in three fragments. The forward primer used for amplifying fragment 1 introduced a NotI site and the Kozak sequence (CCTACC) upstream of the starting codon and the reverse primer used for amplifying fragment 3 introduced a KpnI and a SalI site after the stop codon. Fragment 2 (nt 1442–2564) was MfeI/BamHI digested and fragment 3 (nt 2335–3276) was KpnI/BamHI digested and co-ligated in the corresponding MfeI/KpnI sites of the pβA vector, yielding an intermediate construct. Fragment 1 (nt 1–1575) was NotI/MfeI digested and co-ligated with the SalI/MfeI-digested intermediate construct containing fragments 2 and 3 and the NotI/SalI-digested pβA vector, yielding pβA-α2δ-1 (GenBank accession number MK327276).
For pβA-2HA-α2δ-1: The putative signal peptide (aa1–24) was predicted using Signal P (SignalP 4.0: discriminating signal peptides from transmembrane regions (Petersen et al., 2011). A double hemagglutinin tag (2HA) followed by a TEV cleavage site was introduced between the third and fourth amino acids after the predicted signal peptide cleavage site of mouse α2δ-1; that is, residue F27. Introduction of this sequence did not alter the predicted cleavage site. Briefly, the cDNA sequence of α2δ-1 (nt 1–516) was PCR amplified with overlapping primers introducing the double HA tag and the TEV cleavage site in separate PCRs using pβA-α2δ-1 as a template. The two separate PCR products were then used as templates for a final PCR with flanking primers to connect the nucleotide sequences. The resulting fragment was then NotI/BglII digested and ligated into the corresponding sites of pβA-α2δ-1, yielding pβA-2HA-α2δ-1.
For pβA-α2δ-2 (v1): Mouse α2δ-2 was cloned from genomic cDNA from mouse brain. Primer sequences were selected according to GenBank NM-001174049. The cDNA of α2δ-2 was amplified by PCR in four fragments. The forward primer used for amplifying fragment 1 introduced a HindIII site and the Kozak sequence (CCTACC). Fragment 1 was isolated from cerebellum, whereas the other three fragments were isolated from hippocampus. Fragment 1 (nt 1–686) and Fragment 2 (nt 323–1294) were HindIII/BamHI and BamHI/EcoRI digested, respectively, and co-ligated in the corresponding HindIII/EcoRI sites of the pBS (Bluescript) vector, yielding the intermediate construct pBS-α2δ-2-part1. Fragment 3 (nt 1137–2359) was EcoRI/PmlI digested and ligated into the corresponding sites of the pSPORT vector, yielding the intermediate construct pSPORT-α2δ-2-part2. Fragment 4 (nt 2226–3444) was BmtI/XbaI digested and ligated into the corresponding sites of pSPORT-part2, yielding the intermediate construct pSPORT-α2δ-2-part3. pSPORT-α2δ-2-part3 (v1) was EcoRI/XbaI digested and the band containing fragments 3–4 (bp 1137–3444) was ligated into pBS-α2δ-2-part1, yielding pBS-α2δ-2. This construct was then HindIII/XbaI digested and the cDNA of α2δ-2 was ligated into the pβA vector, yielding pβA-α2δ-2 (v1) (GenBank accession number MK327277).
For pβA-2HA-α2δ-2 (v1): A putative signal peptide was not reliably predicted using Signal P (SignalP 4.0: discriminating signal peptides from transmembrane regions; Petersen et al., 2011); however, the highest prediction showed that the signal peptide comprises residues 1–64. The 2HA tag followed by a thrombin cleavage site was therefore introduced after the predicted signal peptide cleavage site of mouse α2δ-2; that is, residue A64. Introduction of this sequence did not alter the predicted cleavage site. Briefly the cDNA sequence of α2δ-2 (nt 1–761) was PCR amplified with overlapping primers introducing the double HA tag and the thrombin cleavage site in separate PCRs using pβA-α2δ-2 as a template. The two separate PCR products were then used as templates for a final PCR with flanking primers to connect the nucleotide sequences. The resulting fragment was then HindIII/AflII digested and ligated into α2δ-2 (v1).
For pβA-α2δ-2 (v2): An alternative splice variant was isolated from cerebellum, corresponding to GenBank NM-020263. Fragment 3 contained an additional 21 nt at position nt 1992 and was cloned into pSPORT together with fragment 4, yielding pSPORT-α2δ-2-part3 (v2). pSPORT-α2δ-2-part3 (v2) was digested with ClaI/BglII and cloned into the corresponding sites of pβA-α2δ-2 (v1), yielding pβA-α2δ-2 (v2) (GenBank accession number MK327278).
For pβA-α2δ-2 (v3): An alternative splice variant was isolated from hippocampus, corresponding to GenBank NM-001174048. Fragment 4 contained an additional 3 nt at position nt 2598 (resulting in an additional Q residue) and an additional 6 nt at position 3219 nt (translated in CPA instead of S) and was cloned into pSPORT together with fragment 3, yielding pSPORT-α2δ-2-part3 (v3). pSPORT-α2δ-2-part3 (v3) was digested with ClaI/XbaI and cloned into the corresponding sites of pβA-α2δ-2 (v1), yielding pβA-α2δ-2 (v3) (GenBank accession number MK327279).
For pβA-α2δ-3: Mouse α2δ-3 was cloned from genomic cDNA from mouse hippocampus. Primer sequences were selected according to GenBank NM-009785. Briefly, the cDNA of α2δ-3 was amplified by PCR in four fragments. The forward primer used for amplifying fragment 1 introduced a NotI site and the Kozak sequence (CCTACC) upstream of the starting codon. Fragment 3 (nt 1520–2817) was then SacI/PstI digested and fragment 4 (nt 2727–3276) was DraI/PstI digested and co-ligated in the corresponding SacI/SmaI sites of the pSPORT vector, yielding an intermediate construct. Fragment1 (nt 1–653) was then NotI/BamHI digested and fragment2 (535–1636) was SacI/BamHI digested and co-ligated with the SacI/NotI digested intermediate construct containing fragments 3 and 4, yielding pSPORT-α2δ-3. The cloned cDNA of α2δ-3 was then NotI/RsrII digested and ligated into the corresponding sites of the pβA vector, yielding pβA-α2δ-3 (GenBank accession number MK327280).
For pβA-2HA-α2δ-3: The putative signal peptide (aa 1–28) was predicted using Signal P (SignalP 4.0: discriminating signal peptides from transmembrane regions; Petersen et al., 2011). The 2HA tag followed by a thrombin cleavage site was therefore introduced after the predicted signal peptide cleavage site of mouse α2δ-3; that is, residue D28. Introduction of this sequence did not alter the predicted cleavage site. Briefly, the cDNA sequence of α2δ-3 (nt 1–653) was PCR amplified with overlapping primers introducing the double HA tag and the thrombin cleavage site in separate PCRs using pβA-α2δ-3 as a template. The two separate PCR products were then used as templates for a final PCR with flanking primers to connect the nucleotide sequences. The resulting fragment was then NotI/BsrGI digested and ligated into the corresponding sites of pβA-α2δ-3, yielding pβA-2HA-α2δ-3.
For pHR-pβA-eGFP*α2δ-2 (v1): To remove the HA tag from pβA-eGFP*2HA-α2δ-2, pβA-eGFP*2HA-α2δ-2 was XhoI/RsrII digested to isolate part of the promoter and the eGFP coding sequence. The obtained fragment was then inserted in the corresponding sites of pβA-α2δ-2, yielding pβA-eGFP*α2δ-2. For generating the viral vector pHR-pβA-eGFP*α2δ-2, the eGFP*α2δ-2 coding sequence was introduced with HindIII/XbaI into a custom-built pENTR vector and inserted into a custom-built destination vector, pHR-βA-DEST, using the LR Clonase II enzyme mixture (GATEWAY; Invitrogen), yielding pHR-pβA-eGFP*α2δ-2 (v1).
For pβA-eGFP*2HA-α2δ-2 (v1): The eGFP coding sequence, followed by a stop codon and the Kozak sequence (CCTACC), were introduced by PCR between the Kozak sequence and the coding sequence of 2HA-α2δ-2. The resulting fragment was then HindIII/PpuMI digested and ligated into the corresponding sites of pβA-2HA-α2δ-2, yielding pβA-eGFP*2HA-α2δ-2. This construct was then transfected in hippocampal cultures with lipofectamine to insure that both the eGFP and HA signals were present in transfected neurons.
For pHR-pβA-eGFP: The cloning procedure to generate this plasmid was described previously (Subramanyam et al., 2009).
For pHR-pβA-mcherry: The coding sequence of mcherry was inserted into a custom-built destination vector, pHR-βA-DEST, using LR Clonase II enzyme mixture (GATEWAY; Invitrogen) as described previously (Etemad et al., 2014).
Immunocytochemistry and microscopy
Antibodies.
Information on primary and secondary antibodies is summarized in Table 1.
Table 1.
List of primary and secondary antibodies
| Species | Dilution | Source | |
|---|---|---|---|
| Primary antibodies | |||
| Anti-HA | Rat, monoclonal, clone 3F10 | 1:100 (LIVE/A594) | Roche (catalog #11867423001, RRID:AB_390918) |
| Anti-GABAARβ2/3 | Mouse, monoclonal, clone bd17 | 1:500 (A594) | Millipore (catalog #MAB341, RRID:AB_2109419) |
| 1:250 (A350/OG488) | |||
| Anti-GABAARα1N | Rabbit, polyclonal | 1:2000 (A594) | (Hörtnagl et al., 2013) |
| Anti-GABAARα2C | Rabbit, polyclonal | 1:1000 (A594) | (Hörtnagl et al., 2013) |
| Anti-GABAARα3N | Rabbit, polyclonal | 1:500 (A594) | (Hörtnagl et al., 2013) |
| Anti-GABAARα4N | Rabbit, polyclonal | 1:500 (A594) | (Hörtnagl et al., 2013) |
| Anti-GABAARβ2L | Rabbit, polyclonal | 1:500 (A594) | (Hörtnagl et al., 2013) |
| Anti-GABAARβ3L | Rabbit, polyclonal | 1:2000 (A594) | (Hörtnagl et al., 2013) |
| Anti-GABAARγ2L | Rabbit, polyclonal | 1:500 (A594) | (Hörtnagl et al., 2013) |
| Anti-GABAARδN | Rabbit, polyclonal | 1:100 (A594) | (Hörtnagl et al., 2013) |
| Anti-gephyrin | Mouse, monoclonal, clone mab7a | 1:2000 (A594) | Synaptic Systems (catalog #147 021, RRID:AB_2232546) |
| 1:1000 (A350/STAR440) | |||
| Anti-GLUR1 | Rabbit, polyclonal | 1:1000 (A594) | Upstate (catalog #06-306) |
| Anti-PSD-95 | Mouse, monoclonal, clone 6G6–1C9 | 1:1000 (A594) | Thermo Fisher Scientific (catalog #MA1-045, RRID:AB_325399) |
| Anti-synapsin1/2 | Rabbit, polyclonal | 1:2000 (A350) | Synaptic Systems (catalog #106 002, RRID:AB_887804) |
| Anti-synapsin1 | Mouse, monoclonal, clone 46.1 | 1:500 (A350) | Synaptic Systems (catalog #106 011, RRID:AB_2619772) |
| Anti-vGLUT1 | Rabbit, polyclonal | 1:2000 (A350) | Synaptic Systems (catalog #135 002, RRID:AB_2315546) |
| Anti-vGLUT1 | Mouse, monoclonal, clone 317G6 | 1:500 (A594) | Synaptic Systems (catalog #135 511, RRID:AB_887879) |
| 1:250 (A350) | |||
| Anti-vGAT | Rabbit, polyclonal | 1:500 (A350) | Synaptic Systems (catalog #131 002, RRID:AB_887871) |
| 1:250 (STAR440) | |||
| Secondary antibodies | |||
| Alexa Fluor 350 | Goat anti-mouse | 1:500 | Thermo Fisher Scientific (catalog #A-21049, RRID:AB_2535717) |
| Goat anti-rabbit | 1:500 | Thermo Fisher Scientific (catalog #A-21068, RRID:AB_2535729) | |
| Alexa Fluor 594 | Goat anti-mouse | 1:4000 | Thermo Fisher Scientific (catalog #A-11032, RRID:AB_2534091) |
| Goat anti-rabbit | 1:4000 | Thermo Fisher Scientific (catalog #A-11037, RRID:AB_2534095) | |
| Goat anti-rat | 1:4000 | Thermo Fisher Scientific (catalog #A-11007, RRID:AB_10561522) | |
| Oregon Green 488 | Goat anti-mouse | 1:500 | Thermo Fisher Scientific (catalog #O-11033, RRID:AB_2539797) |
| STAR 440 | Goat anti-mouse | 1:500 | Abberior (catalog #2-0002-003-7, RRID:AB_2756514) |
High-resolution fluorescence microscopy.
Immunolabeling of permeabilized or live-cell-stained neurons was performed as described previously (Obermair et al., 2004, 2010; Stanika et al., 2016; Folci et al., 2018) and information on antibodies is given in Table 1. For PSD-95 labeling of eGFP-transfected neurons, cultures were fixed in 4% paraformaldehyde/4% sucrose (pF) in PBS for 20 min at room temperature, rinsed in PBS followed by a second fixation with absolute methanol for 10 min at −20°C. For all other antibody combinations, neurons were fixed with pF only. Subsequent to fixation and washing, cultures were incubated for 30 min in 5% normal goat serum in PBS containing 0.2% bovine serum albumin (BSA) and 0.2% Triton X-100 (PBS/BSA/Triton), enabling membrane permeabilization. Primary antibodies were applied overnight in PBS/BSA/Triton at 4°C and detected by fluorochrome-conjugated secondary antibodies incubated for 1 h at room temperature. For surface staining of HA-tagged α2δ constructs, transfected neurons were incubated with rat-anti-HA antibody for 20 min at 37°C following rinsing in warm HBSS and fixation with pF for 10 min at room temperature. Subsequent washing and blocking steps were done with PBS and PBS/BSA, respectively, followed by 1 h incubation with fluorochrome-conjugated secondary goat-anti-rat Alexa Fluor 594 antibody. After rinsing and postfixation of cells in pF for 5 min, neurons were permeabilized by blocking with PBS/BSA/Triton. Primary mouse-anti-synapsin antibody was applied overnight at 4°C and detected with goat-anti-mouse Alexa Fluor 350 antibody. Coverslips were either mounted in p-phenylenediamine glycerol (Flucher et al., 1993) or DAKO fluorescence mounting medium (Agilent Technologies) for α-Nrxn knock-out and control cultures. Hippocampal cultures were mainly viewed with an Axio Imager microscope (Carl Zeiss) using a 63× 1.4 numerical aperture (NA) oil-immersion objective lens and 14-bit grayscale images were recorded with a cooled CCD camera (SPOT; Diagnostic Instruments) using MetaMorph software (Molecular Devices). α-Nrxn knock-out cultures and MSNs were observed with a BX53 microscope (Olympus) using a 60× 1.42 NA oil-immersion objective lens and 14-bit grayscale images were recorded with a cooled CCD camera (XM10; Olympus) using cellSens Dimension software (Olympus). To analyze presynaptic and postsynaptic differentiation cover glasses were systematically screened for contact sites between axons of presynaptic neurons [transfected with the fluorescent marker alone (eGFP or mCherry only) or cotransfected with the marker and target construct (eGFP or mCherry plus α2δ)] and dendrites or somas of nontransfected postsynaptic neurons. Images of randomly selected well differentiated cells were acquired with the same exposure and gain settings for all conditions within an experiment. These settings were initially adjusted to facilitate optimal visualization of peripheral cell compartments (axons and presynaptic boutons) also in weakly expressing neurons. Therefore, overly saturated neurons (based on eGFP and mCherry levels) were excluded from analysis and only cells with medium to low eGFP or mCherry expression were selected for further analysis. Images were analyzed with MetaMorph software (Molecular Devices) as described below. Figures were assembled in Adobe Photoshop CS6 and linear adjustments were done to correct black level and contrast.
gSTED microscopy.
For gSTED microscopy of mCherry-transfected permeabilized hippocampal and striatal MSNs (see Fig. 3), immunolabeling was done as described for high-resolution fluorescence microscopy with some modifications. Briefly, mouse-anti-GABAARβ2/3 antibody was used at higher concentrations (1:250) and detected with secondary goat-anti-mouse Oregon Green (OG) 488 (1:500). Coverslips were mounted in DABCO/glycerol to retard photobleaching (2% DABCO, 90% glycerol, buffered in Tris-HCl, pH 8.5; DABCO and glycerol were purchased from Carl Roth). Samples were visualized using a TCS SP8 gSTED microscope equipped with an HCXPL APO 100× 1.4 NA oil-immersion objective (Leica Microsystems) and images were scanned at a pixel size of 15 nm. For hippocampal neurons, individual fluorophores were recorded in the following sequence (excitation/detection range wavelength in nm): mCherry (575/600–690; gate 0.20–6 ns, in confocal mode) and OG 488 (mouse-anti-GABAARβ2/3; 502/512–581; gate 1–6 ns, STED laser at 592 nm 100%). For MSNs, individual fluorophores were recorded in the following sequence (excitation/detection range wavelength in nm): mCherry (mCherry only or mCherry + α2δ-2; 575/600–690; gate 0.20–6 ns, in confocal mode) and OG 488 (mouse-anti-GABAARβ2/3; 502/510–582; gate 1–6.2 ns, STED laser at 592 nm 100%). To study the localization of excitatory synapses on postsynaptic dendrites of excitatory neurons (see Fig. 11B), hippocampal neurons labeled with mCherry and eGFP, respectively, were fixed in 4% pF in PBS for 20 min at room temperature, washed in PBS, and mounted in DABCO/glycerol. Fluorophores were recorded in the following sequence (excitation/detection range wavelength in nm): mCherry (presynaptic axon: mCherry only or mCherry + α2δ-2; 584/600–688; gate 0.20–6 ns in confocal mode) and eGFP (postsynaptic dendrite: eGFP only, 490/508–584 or 520–584; gate 1–6.2 ns, STED laser at 592 nm). To enable clear visualization of dendritic structures, STED laser intensity was adjusted according to the eGFP signal. To study the localization of excitatory synapses on postsynaptic dendrites of inhibitory GABAergic neurons (see Fig. 11E), cortical neurons and MSNs were labeled with mCherry and eGFP, respectively, as described above. Primary mouse-anti-gephyrin (1:1000) was applied overnight in PBS/BSA/Triton at 4°C and detected with secondary goat-anti-mouse STAR440 (1:500). Fluorophores were recorded in the following sequence (excitation/detection range wavelength in nm): mCherry (presynaptic axon: mCherry only or mCherry + α2δ-2; 580/600–690; gate 0.5–6 ns in confocal mode), eGFP (postsynaptic dendrite: eGFP only, 470/508–580; gate 1–6.0 ns, STED laser at 592 nm), and STAR440 (mouse-anti-gephyrin; 470/480–570; gate 1–6.0 ns, STED laser at 592 nm). STED laser intensity was adjusted according to the eGFP signal to enable clear visualization of dendritic structures and to reduce cross-activation during the detection of STAR440. Channel dye separation was performed to further split the eGFP and STAR440 signals. Images were recorded with LAS AF software (Leica Microsystems), analyzed using MetaMorph software (Molecular Devices), and figures were assembled in Adobe Photoshop CS6 and linear adjustments were done to correct black level and contrast.
Figure 3.
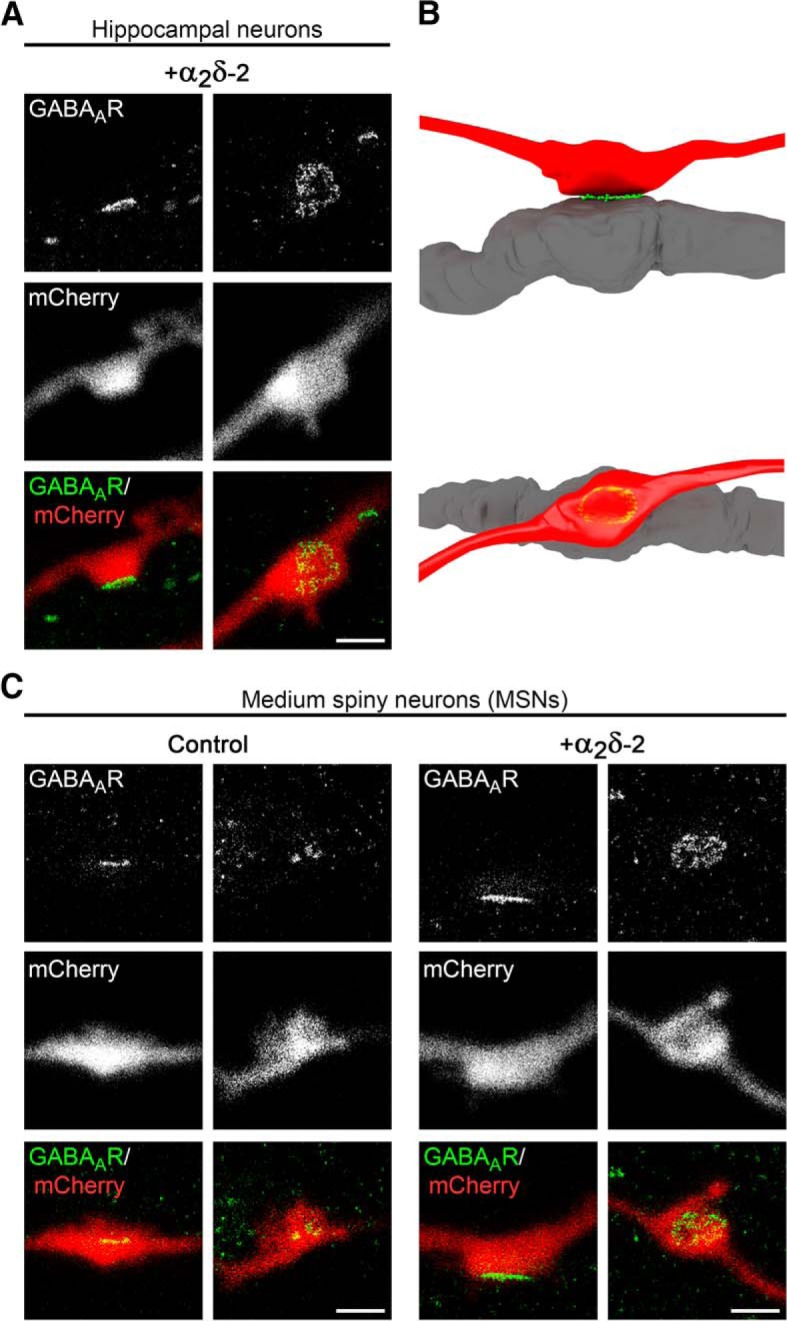
GABAAR clusters are confined to the postsynaptic membrane. gSTED micrographs of cultured hippocampal (A) or MSNs (C) transfected with α2δ-2 and mCherry or mCherry only (control; 20–30 DIV). Transfected neurons (red, detected in confocal mode) were immunolabeled for the GABAAR (green, detected in gSTED mode). In all conditions, postsynaptic GABAAR clusters are closely opposed to mCherry-positive presynaptic boutons. B, 3D model showing that the GABAAR staining pattern depends on the orientation of the imaged synapse, which applies both for hippocampal as well as MSNs. Scale bar, 1 μm.
LacZ reporter expression.
Using the LacZ reporter function of the mutated Cacna2d3 gene (Cacna2d3tm1Dgen; (Neely et al., 2010), β-galactosidase activity was analyzed in male α2δ-3−/− knock-out mice. Eight-week-old mice were killed by CO2 asphyxiation, decapitated, and brains were removed from the skull. Subsequently, hemispheres were separated with a sagittal cut along the midline, placed medial side down on a flat piece of thin acryl glass, and frozen in 2-methylbutan (Carl Roth) cooled to −80°C. Native samples were stored at −80°C until further processing and transferred to −20°C 1 d before sectioning. Twenty-micrometer-thick sagittal sections of one hemisphere were obtained with a cryotome (NX50; Histocom), collected on polylysine-coated glass slides (Lactan), and stored at −20°C until further use. Sections were air dried at room temperature for 15 min, fixed with freshly prepared cold 4% pF and 0.2% glutaraldehyde (Sigma-Aldrich) for 10 min, and washed repeatedly in PBS for 30 min. X-Gal staining reaction was done with X-Gal (Boehringer Mannheim; 4% in N,N-dimethylformamide) diluted 1:40 in prewarmed X-Gal buffer (5 mm K3Fe(CN)6, 5 mm K4Fe(CN)6 · 3H2O, 2 mm MgCl2 in PBS; Sigma-Aldrich) at 37°C in a humidified incubator. Reaction was stopped after 24 h by several washes with PBS and rinsing in Milli-Q water (Millipore) at room temperature. Sections were counterstained with 0.2% eosin (Carl Roth) in 0.1 m CH3COOH in Milli-Q, dehydrated by a graded series of ethanol, followed by washing in xylene (Carl Roth) and mounting with Eukitt (Christine Gröpl, Tulln, Austria). Representative images were recorded with a BX53 microscope (Olympus) equipped with a SC100 color camera (Olympus). Eight-bit panorama photographs were created by scanning specimens with a 4× 0.16 NA objective and using the manual multiple image alignment (MIA) function in cellSens Dimension software (Olympus).
Image analysis and quantification
Colocalization of synaptic proteins.
The synaptic localization of HA-tagged α2δ subunits, presynaptic (synapsin, vGAT, vGLUT1) and postsynaptic proteins (GABAAR subtypes, gephyrin, GLUR1) were analyzed with the “line scan” function in MetaMorph software (Molecular Devices) (Di Biase et al., 2009). Average fluorescence intensities of green (A488), blue (A350), and red (A594) signals were measured along a line of 3 or 8 μm length, background subtracted, and plotted in Microsoft Excel.
Quantification of fluorescent clusters in single boutons.
To analyze the effects of presynaptic expression of α2δ subunits in GABAergic synapses of cultured MSNs, 14-bit grayscale images from triple-fluorescence labeling were acquired from the eGFP (green), GABAARβ2/3 (red), and vGAT (blue) channels. Images were analyzed with a custom programmed and semiautomated MetaMorph journal (Molecular Devices) as follows. Corresponding eGFP and vGAT images were superimposed, eGFP/vGAT-positive varicosities (putative GABAergic synapses) were randomly chosen as regions of interest (ROIs), and a region for background subtraction was selected. Axonal varicosities were defined as prominent swellings with higher fluorescence signal compared with the adjacent axonal shaft. Subsequently, eGFP, GABAAR, and vGAT grayscale images were thresholded to include the entire area of individual varicosities (eGFP) and synaptic clusters (GABAAR, vGAT). Selected ROIs were then transferred to the eGFP image and, by applying the “shrink region to fit” tool, automatic boundaries were drawn according to the threshold enabling only colocalized clusters to be analyzed. ROIs and background regions were further transferred to the thresholded GABAAR and vGAT channel images and ROI staining intensities were recorded. Therefore, the following parameters could be measured for the individual synapses in each of the channels in a blinded manner: eGFP threshold area as a measure for bouton size and average and integrated fluorescence intensities providing information on the size and intensity of clusters. To address the effects of presynaptic expression of α2δ subunits in glutamatergic synapses of α-Nrxn TKO neurons, 14-bit grayscale images from triple-fluorescence labeling were acquired from the eGFP (green), GABAARβ2/3 (red), and vGLUT1 (blue) channels. Images were analyzed with a custom programmed and semiautomated journal as described above. Corresponding eGFP and vGLUT1 images were superimposed and eGFP/vGLUT1-positive varicosities (putative glutamatergic synapses) were randomly selected as ROIs. To analyze the effects of presynaptic expression of α2δ subunits in glutamatergic synapses of cultured hippocampal WT neurons, 14-bit grayscale images from triple-fluorescence labeling were acquired from the eGFP (green), GLUR1 (red), and GABAARβ2/3/gephyrin (blue) channel. Images were analyzed in the same manner as mentioned above but without the use of a semiautomatic MetaMorph journal. For all experiments, background subtractions for individual channels and further data organization and analyses were done with Microsoft Excel. Mean values for individual cells were calculated from individual boutons and normalized to the control (eGFP only) or α2δ-2 condition within each culture preparation. Cumulative frequency distribution of GABAARβ2/3 fluorescence intensities of single boutons was done to visualize the effect of α2δ subunits on the whole GABAAR population. Two to four independent culture preparations were analyzed and details are given in the respective figure legends.
Quantification of gephyrin content in mismatched and neighboring synapses.
To compare gephyrin content of α2δ-2-induced mismatched synapses with endogenous clusters, 14-bit grayscale images from triple-fluorescence labelings were acquired from the eGFP (green), GLUR1 (red), and gephyrin (blue) channel and analyzed with MetaMorph software (Molecular Devices). First, corresponding eGFP, GLUR1, and gephyrin images were superimposed and eGFP-positive varicosities expressing α2δ-2 (putative mismatched synapses) were selected as ROIs and a region was selected for background subtraction. eGFP, GLUR1, and gephyrin grayscale images were thresholded to include the entire area of individual varicosities (eGFP) and synaptic clusters (GLUR1, gephyrin). Regions were transferred to the eGFP image, shrunk to fit the threshold area, and further transferred to the thresholded gephyrin channel images. After measuring the integrated gephyrin intensity of eGFP-positive mismatched synapses, ROIs of 10 untransfected neighboring WT synapses located on the same dendrite were selected and regions were shrunk according to the gephyrin threshold. The GLUR1 staining served as orientation to outline dendritic morphology of untransfected neurons. Per image, one mismatched gephyrin cluster induced by α2δ-2 was compared with the mean of 10 endogenous clusters. Dots in graph represent values for individual boutons (mismatched synapses) and means of 10 endogenous clusters measured per image. Values were normalized to endogenous gephyrin intensities of neighboring synapses within each culture preparation. Three independent culture preparations were done and eight to 10 images per preparation were analyzed.
Analysis of synapse localization in gSTED images.
Scans of mCherry-labeled boutons (control or α2δ-2) contacting eGFP-labeled postsynaptic dendrites were acquired from the eGFP (green, gSTED mode) and mCherry (red, confocal mode) channels. Axonal varicosities were visible as prominent swellings with higher fluorescence signal compared with the adjacent axonal shaft. Initially, linear contrast adjustments were done with LAS AF software (Leica Microsystems), thus enabling clear visualization of postsynaptic dendritic spines and the potential overlap of fluorescence signals, which was used to define the contact zone of putative synapses. Superimposed images were exported into a TIFF format and further analyzed using MetaMorph software (Molecular Devices). To determine the position of synapses on postsynaptic elements, two types of analysis were used. First, the postsynaptic contact sites of presynaptic boutons were manually classified as dendritic shafts, dendritic spines, or unclear localizations. Second, the distances from the postsynaptic dendritic shafts to the contact points with the presynaptic boutons were calculated as follows. First, the distances between the center of the dendritic shafts and the shaft surfaces were determined and measured. Second, the distances between the center of dendritic shafts and the contact zone (presynaptic bouton with postsynaptic dendrite) were determined and measured. Third, the actual distance from the dendritic shaft surface to the contact zone was determined by subtracting the individual center surface distances from the center contact zone distances. Dots in graph (Fig. 11C) represent values for individual boutons and means (lines) ± SEM are shown. Two independent culture preparations were analyzed, of which one experiment was performed in a blinded manner.
Electrophysiology
Induced (evoked) AMPAR-mediated synaptic responses were recorded from paired hippocampal neurons (holding potential −70mV) using the whole-cell patch-clamp technique. Neurons were stimulated with 10 ms depolarization pulse from a holding potential of −70 to 60 mV. Patch pipettes were pulled from borosilicate glass (Harvard Apparatus), fire-polished (Microforge MF-830; Narishige), and had resistances of 2.5–4 MΩ when filled with the following (in mm): 130 K-gluconate, 1 MgCl2, 10 HEPES, 5 EGTA, 4 Mg-ATP, and 0.3 Na-GTP, pH 7.2 with KOH. The bath solution contained the following (in mm): 137 NaCl, 3 KCl, 10 HEPES, 2 MgCl2, 1.8 CaCl2, 0.05 DL-AP5, and 10 glucose, pH 7.4 with NaOH. Currents were recorded with an EPC 10 amplifier controlled by PatchMaster software (HEKA Elektronik Dr. Schulze). To test synaptic plasticity (or paired-pulse plasticity), we applied a pair of pulses with varying interpulse intervals (10, 25, 50, 100, 250, or 500 ms) and calculated the paired-pulse amplitude ratio (P2/P1).
To test spontaneous activity of mixed cortical-MSN cultures, TTX (1 μm) and bicuculline (10 μm) or CNQX (10 μm) were added to bath solution and miniature EPSCs (mEPSCs; holding potential −70 mV) or miniature IPSCs (mIPSCs; holding potential 0 mV), respectively, were recorded.
Sequence alignment and homology modeling
Information on α2δ-2 splice sites was obtained from the Uniprot (Q6PHS9) and Ensemble (ENSMUSG00000010066) databases. Protein sequences of the distinct variants were aligned with CLUSTAL O (1.2.4) multiple sequence alignment tool. The high-resolution structure of α2δ-1 (PDB code: 5GJV; Wu et al., 2016) was used as a template to model the structure of α2δ-2 splice variants. The pdb files were generated with the Swiss-Model server (http://www.expasy.org/swissmod/SWISS-MODEL.html) and models were exported and further analyzed using UCSF Chimera (http://www.rbvi.ucsf.edu/chimera).
Experimental design and statistical analysis
According to the RRR principle, the number of mice used was kept to the minimum necessary for a statistical representative analysis, which was comparable to numbers reported in previous studies. Where indicated and possible, investigators were partially blinded during experiments and analyses. Moreover, analysis of presynaptic and postsynaptic proteins was done in a blinded manner as described above. Two to four independent culture preparations were analyzed per experiment and details on cell or bouton numbers are given in the respective figure legends. The graphs show values of individual cells/boutons (dots) and means (line) ± SEM. The numbers (n) used to calculate SEMs were either given by number of animals used (qRT-PCR) or the number of analyzed cells/boutons. Before statistical analysis, the distribution of datasets was evaluated with histograms. Whenever datasets were strongly skewed or normality was experimentally excluded (e.g., a large proportion of “0” values), raw data were log10 transformed and data distribution was reevaluated by histograms. For normally distributed raw or log10-transformed datasets, unpaired t test or one-way ANOVA with Holm–Sidak post hoc test was used. Alternatively, when log10 transformation did not result in normally distributed data, the Mann–Whitney U test or Kruskal–Wallis ANOVA with Dunn's post hoc analysis was applied. Significance levels (p-values) of statistical tests and post hoc analysis are presented in the respective figure legends. The model in Figure 3 was generated with Maya software (version 2018; Autodesk). Data, graphs and figures were organized, analyzed, and assembled using Microsoft Excel, GraphPad Prism 6, SigmaPlot (Systat Software), and Adobe Photoshop CS6.
Results
Presynaptic overexpression of α2δ-2 induces the formation of mismatched synapses in excitatory hippocampal neurons
To date, the synaptic functions of α2δ subunits have been identified in cells and tissues primarily expressing a single dominant α2δ isoform (Eroglu et al., 2009; Pirone et al., 2014; Fell et al., 2016; Wang et al., 2017). To study the involvement of the individual α2δ isoforms in glutamatergic synapse formation and differentiation, we homologously overexpressed mouse α2δ-1, α2δ-2, and α2δ-3 cDNAs in WT mouse hippocampal cultures together with soluble eGFP. To analyze the consequences on synapse differentiation, transfected neurons were immunolabeled against the vesicular glutamate transporter (vGLUT1), a marker for presynaptic excitatory synapses, and the postsynaptic GABAARβ2/3 subunit (GABAAR), which is typically absent in glutamatergic synapses. Immunofluorescence analysis revealed that, as expected, in control hippocampal neurons (eGFP only) axonal varicosities were positive for presynaptic vGLUT1 and negative for postsynaptic GABAAR labeling (Fig. 1A,B, first column). Surprisingly, presynaptic expression of α2δ-2 induced a robust localization of GABAAR clusters opposite vGLUT1-positive presynaptic boutons (Fig. 1B, blue arrowheads). These apparently mismatched synapses are atypical because they are formed between excitatory nerve terminals and inhibitory postsynaptic receptors. Importantly, mismatched postsynaptic receptor localization was not observed opposite terminals expressing α2δ-1 or α2δ-3.
Figure 1.

Presynaptic expression of α2δ-2 in cultured hippocampal neurons induces the formation of mismatched synapses. Representative immunofluorescence micrographs of cultured hippocampal neurons cotransfected with distinct α2δ subunits and soluble eGFP. Putative presynaptic en passent boutons (arrowheads) were identified as eGFP-filled axonal varicosities along dendrites of untransfected neurons (axons are outlined with dashed lines). A, B, Double labeling of transfected neurons (20–30 DIV) for vGLUT1 and the GABAAR β2/3 subunit. Colocalization of fluorescence signals was analyzed using line scans. A, In control boutons (eGFP only), potential glutamatergic synapses are positive for presynaptic vGLUT1 but negative for postsynaptic GABAAR (summarized in sketch). B, Presynaptic expression of α2δ-2 (blue arrowheads) induces postsynaptic GABAAR localization opposite transfected vGLUT1 positive nerve terminals. In contrast, postsynaptic GABAARs are not expressed opposite glutamatergic synapses expressing α2δ-1, α2δ-3, or eGFP only (control). C, D, Double labeling of transfected neurons (20–30 DIV) for vGAT and postsynaptic GABAAR. Colocalization of fluorescence signals was analyzed using line scans. C, In control boutons (eGFP only), potential glutamatergic synapses are negative for presynaptic vGAT and postsynaptic GABAAR (summarized in sketch). D, Transfected axonal varicosities were negative for vGAT in all conditions. Note the specific GABAAR labeling opposite vGAT-negative nerve terminals expressing α2δ-2 (blue arrowheads), which is in contrast to the colocalized fluorescence signals of vGAT and GABAAR in untransfected GABAergic neighboring synapses (asterisks). Representative images of two independent culture preparations are shown. Scale bars, 10 μm (A,C) and 3 μm (B,D).
Excitatory and inhibitory neurotransmitter transporters can coexist in glutamatergic hippocampal and cerebellar mossy fiber terminals, possibly leading to the corelease of glutamate and GABA and the localization of postsynaptic GABAARs in spines (Bergersen et al., 2003; Münster-Wandowski et al., 2016). Therefore, we next tested whether vesicular GABA transporters (vGATs) are present in synapses overexpressing presynaptic α2δ subunits. As expected and in contrast to neighboring untransfected GABAergic synapses (Fig. 1D, asterisks), presynaptic vGAT and postsynaptic GABAARs were not found in putative excitatory synapses expressing α2δ-1, α2δ-3, or eGFP only (Fig. 1C,D). Most importantly, presynaptic vGAT labeling was also absent in synapses expressing presynaptic α2δ-2, which again showed strong postsynaptic GABAAR clustering (Fig. 1D, +α2δ-2, blue arrowheads). Therefore, the mismatched localization of postsynaptic GABAARs cannot be explained by the existence of inhibitory neurotransmitters within glutamatergic nerve terminals.
To further characterize our observation, we next quantitatively assessed the postsynaptic receptor composition of mismatched glutamatergic synapses by double labeling transfected hippocampal neurons with markers for excitatory and inhibitory synapses (Fig. 2A–C). Immunofluorescence analysis revealed that, in control hippocampal neurons (eGFP only) postsynaptic AMPARs (GLUR1), but not GABAARs, were expressed opposite transfected presynaptic terminals, as expected for putative glutamatergic synapses (Fig. 2A, control). Consistent with our initial observations (Fig. 1), postsynaptic GABAAR fluorescent intensities were strongly and significantly increased opposite α2δ-2-expressing presynaptic terminals (Fig. 2A,D). The elevated GABAA receptor abundance (Fig. 2A) was accompanied by a 76% reduction of GLUR1 clustering compared with control (Fig. 2E). Surprisingly, presynaptic expression of α2δ-3, which did not alter postsynaptic GABAAR clustering, also caused a significant reduction of GLUR1 receptors (66% reduction vs control). Finally, we also observed a slight but not significant tendency for a reduced GLUR1 receptor expression opposite α2δ-1-overexpressing boutons (Fig. 2E). We next tested whether gephyrin, a key organizer of GABAergic synapses that anchors, clusters, and stabilizes GABAAR and other postsynaptic elements of inhibitory synapses (Choii and Ko, 2015), was also recruited to mismatched GABAAR-positive synapses. Indeed, we found that the strong increase in GABAAR expression opposite α2δ-2-expressing terminals was accompanied by similarly increased gephyrin abundance, whereas it was missing from control and α2δ-1- or α2δ-3-expressing synapses (Fig. 2B,F).
Figure 2.

Presynaptic expression of α2δ-2 alters the postsynaptic composition of glutamatergic synapses. A, B, Representative immunofluoresence micrographs of cultured hippocampal neurons (24–30 DIV) cotransfected with distinct α2δ subunits and soluble eGFP (control condition with eGFP alone is summarized in sketch, C). Colocalization of fluorescence signals was analyzed using line scans. A, Colabeling of the AMPA receptor subtype GLUR1 and the GABAAR. Presynaptic expression of α2δ-2 (blue arrowheads) induces postsynaptic GABAAR localization with a concomitant reduction of the GLUR1 fluorescence signal. In contrast, postsynaptic GABAARs are not expressed opposite putative glutamatergic synapses expressing α2δ-1, α2δ-3 or eGFP only (control). B, Colabeling of GLUR1 and gephyrin, the scaffolding protein of GABAergic synapses. Presynaptic expression of α2δ-2 (blue arrowheads) induces postsynaptic gephyrin localization with a concomitant reduction of the GLUR1 fluorescence signal. In contrast, postsynaptic gephyrin immunoreactivity is absent opposite putative glutamatergic synapses expressing α2δ-1, α2δ-3, or eGFP only (control). D–F, Quantitative analysis of GABAAR (D), GLUR1 (E), and gephyrin (F) fluorescence intensities. Values were normalized to α2δ-2 (D, F) or eGFP only (control, E) fluorescence intensities within each culture preparation. Note the significant increase of postsynaptic GABAAR (D) and gephyrin (F) clusters opposite presynaptic boutons expressing α2δ-2. In contrast, integrated intensity of GLUR1 was significantly reduced opposite axonal varicosities transfected with α2δ-2 or α2δ-3. Values for individual cells (dots) and means (line) ± SEM are shown. Data are shown from three independent culture preparations; 11–25 (D), 22–30 (E), and 11–28 (F) cells were analyzed in each condition. Statistics: D, Kruskal–Wallis ANOVA with Dunn's post hoc analysis: H(4) = 51.6, p < 0.0001, post hoc: ****p < 0.0001 between α2δ-2 and all other conditions; E, ANOVA on log10-transformed data with Holm–Sidak post hoc analysis: F(3,100) = 18.6, p < 0.001, post hoc: ***p < 0.001 between control/α2δ-1 and α2δ-2/α2δ-3; F, Kruskal–Wallis ANOVA with Dunn's post hoc analysis: H(4) = 64.6, p < 0.0001, post hoc: ****p < 0.0001 between α2δ-2 and all other conditions. Asterisks in graphs indicate the significant difference compared with control. Scale bars, 3 μm.
Presynaptic α2δ-2 recruits synaptic GABAAR subtypes
Because immunofluorescence analysis suggested an effect on postsynaptic GABAAR clusters opposite presynaptic boutons, we next used superresolution gSTED microscopy to verify their subsynaptic localization (Fig. 3A). Postsynaptic GABAAR clusters appear closely opposed to mCherry-positive, α2δ-2-overexpressing presynaptic boutons: depending on the orientation of the imaged synapses, the GABAAR-labeling pattern is either visible as a thin line (side view; Fig. 3A, first column and Fig. 3B, top) or as a ring-like structure (planar view; Fig. 3A, second column and Fig. 3B, bottom). These labeling patterns support a confined localization of α2δ-2-recruited GABAARs in the postsynaptic membrane.
GABAARs are pentameric ligand-gated chloride channels consisting typically of α, β, γ, and δ subunits (Hörtnagl et al., 2013). Because the antibody used in our colocalization experiments (Figs. 1, 2) detects only the GABAAR β2 and 3 subunits, we next analyzed whether and which additional GABAAR subtypes can be recruited by presynaptic α2δ-2. Immunofluorescence analysis revealed robust labeling of the typically synaptic GABAAR subunits α1, α2, β3, and γ2 opposite α2δ-2 transfected vGLUT1-positive nerve terminals (Fig. 4A). In contrast, the mainly extrasynaptic subunits α3, α4, β2, and δ (Hörtnagl et al., 2013) were either not detectable in these α2δ-2-expressing synapses or diffusely and extrasynaptically located (Fig. 4C). Importantly, mismatched postsynaptic receptor localization of all distinct subtypes was neither observed opposite terminals expressing eGFP only (control) nor those expressing α2δ-1 or α2δ-3 (Fig. 4B,D, examples for α1 and α3 subunits are shown). Because there was no apparent difference between postsynaptic α1, α2, β3, γ2, and β2/3 labeling, we continued using the commercially available and extensively characterized β2/3 antibody for all subsequent experiments (Goffin et al., 2010; Arama et al., 2015; Stefanits et al., 2018).
Figure 4.

Presynaptic expression of α2δ-2 induces the recruitment of synaptic GABAAR subtypes. Representative immunofluorescence micrographs of cultured hippocampal neurons cotransfected with distinct α2δ subunits and soluble eGFP. Transfected permeabilized neurons (20–30 DIV) were double stained for vGLUT1 and different postsynaptic GABAAR subtypes. Colocalization of fluorescence signals within eGFP-filled axonal varicosities (arrowheads, axons are outlined with dashed lines) was analyzed using line scans. A, B, Immunofluorescence analysis identified intensely fluorescent clusters of the GABAAR subunits α1, α2, β3, and γ2 opposite α2δ-2 expressing glutamatergic (vGLUT1-positive) nerve terminals (A). In contrast, these postsynaptic GABAAR subtypes are absent opposite putative glutamatergic synapses expressing eGFP only (control), α2δ-1, or α2δ-3 (B, micrographs depict examples for α1). C, D, Labeling of the GABAAR subunits α3, α4, β2, and δ displayed weak and mainly extrasynaptic immunoreactivity in all conditions. Note that all GABAAR subtypes presented for α2δ-2 were also analyzed in hippocampal neurons expressing eGFP only, α2δ-1, and α2δ-3. Representative images of two independent cultures are shown. Scale bars, 3 μm.
Potential mechanisms explaining an α2δ-2-induced mismatched synapse formation
So far, our results show that the specific expression of a single α2δ subunit isoform, α2δ-2, in presynaptic glutamatergic terminals triggers a mismatched localization of postsynaptic GABAARs, which was accompanied by a strong reduction of postsynaptic AMPARs. The underlying mechanism could be explained by three hypotheses (Fig. 5): First, it is known that an elevated expression of α2δ subunits increases presynaptic calcium channel abundance and current densities (Hoppa et al., 2012; Geisler et al., 2015) and thus likely enhances synaptic transmission (Zhou and Luo, 2015; Chen et al., 2018). Therefore, postsynaptic GABAARs could be recruited opposite these glutamatergic boutons in an attempt to suppress excessive glutamatergic excitation (Shrivastava et al., 2011). If this hypothesis applies, then GABAAR abundance at inhibitory synapses should not change upon α2δ-2 overexpression in GABAergic presynaptic terminals (Fig. 5A). Second, presynaptic α2δ-2, an extracellular protein extending far into the synaptic cleft, may contribute to the anchoring of postsynaptic GABAARs by a trans-synaptic mechanism. Such a specific role of α2δ-2 should be independent of the synapse type and thus GABAAR abundance should be affected when α2δ-2 is overexpressed in both glutamatergic and GABAergic synapses (Fig. 5B). Independent of the two hypotheses, the observed reduction in GLUR1 labeling opposite α2δ-2-expressing glutamatergic terminals (Fig. 2) could be explained by GABAARs competing for AMPAR slots within dendritic spines. Alternatively, however, presynaptic α2δ-2 could also trans-synaptically induce an aberrant axonal wiring by guiding glutamatergic axons to GABAergic postsynaptic locations along dendritic shafts (Fig. 5C). In this scenario, the reduced GLUR1 expression would be a secondary effect because no GLUR1 receptors are to be expected in postsynaptic GABAergic synapses. To distinguish between these hypotheses, we next studied the consequence of presynaptic α2δ-2-overexpression on the molecular composition of GABAergic synapses.
Figure 5.

Potential mechanisms explaining the observed mismatched synapse formation of glutamatergic nerve terminals expressing α2δ-2. A, Compensatory mechanism. Elevated expression of α2δ subunits increases presynaptic calcium channel abundance and current densities and thus glutamate release. Therefore, GABAARs could be recruited to the dendritic spine in an attempt to compensate for excessive excitatory synaptic activity. If this is the case, then GABAAR abundance at inhibitory synapses should not change upon α2δ-2 overexpression in GABAergic presynaptic terminals. B, α2δ-2 may be involved in trans-synaptically anchoring postsynaptic GABAARs. In this scenario, GABAAR abundance should be increased when α2δ-2 is overexpressed in both glutamatergic and GABAergic synapses. C, Trans-synaptic function of α2δ-2 could also induce aberrant axonal wiring by guiding glutamatergic axons to GABAergic postsynaptic locations positioned along dendritic shafts. This is in contrast to the normal situation in which glutamatergic synapses are generally formed on dendritic spines.
MSNs express three presynaptic α2δ isoforms
The cultures of hippocampal neurons used for the analysis of experiments presented in Figures 1, 2, 3, and 4 contain only 5–10% GABAergic interneurons (Obermair et al., 2003). Consequentially transfected GABAergic neurons are rarely observed. Therefore, to quantitatively address the role of α2δ subunits in GABAergic synapses, we established cultures of striatal neurons, which consist of ∼95% inhibitory MSNs. Quantitative RT-PCR revealed that differentiated monocultured MSNs (24–25 DIV) expressed three neuronal α2δ isoforms (α2δ-1, α2δ-2, and α2δ-3) similar to the adult mouse striatum (Fig. 6A). Whereas α2δ-2 and α2δ-3 expression levels were similar in cultured neurons, the higher abundance of α2δ-3 transcripts in 8-week-old striatal tissue was consistent with previous studies (Cole et al., 2005). The strong expression of α2δ-3 was confirmed on cryosections of a mouse line carrying a lacZ reporter gene in the Cacna2d3 locus using β-galactosidase histochemistry (Fig. 6B). The α2δ-4 isoform, which is predominantly present in the retina, was hardly detectable and thus not considered for the following experiments. For studying presynaptic α2δ subunit expression in differentiated GABAergic neurons, we adapted a coculture system of GABAergic MSNs with glutamatergic cortical neurons (Penrod et al., 2011) (Fig. 7A). Lentiviral infection of MSNs with soluble eGFP just before starting the coculture allowed discriminating them from excitatory cortical neurons (Fig. 7B) based on their eGFP fluorescence. Immunolabeling of presynaptic and postsynaptic markers for excitatory (vGLUT1, PSD-95) and inhibitory (vGAT) synapses demonstrated that glutamatergic and GABAergic synapses formed properly on dendritic spines and shafts, respectively (Fig. 7C–E). Moreover, patch-clamp recordings of spontaneous mEPSCs and mIPSCs in MSNs further confirmed the functionality of glutamatergic and GABAergic synapses in 14 DIV cocultures (Fig. 7F). Finally, live-cell labeling of MSNs cotransfected with HA-tagged α2δ subunits and soluble eGFP revealed that all α2δ isoforms are expressed on the surface of presynaptic axonal varicosities and thus can in theory contribute to synaptic functions in GABAergic neurons (Fig. 6C).
Figure 6.

Striatum and cultured MSNs express three neuronal α2δ subunits. A, Absolute qRT-PCR analysis revealed a stable expression of α2δ-1, α2δ-2, and α2δ-3 in adult mouse striatum and monocultured MSNs (24–25 DIV). Although α2δ-3 was the dominant isoform in striatal tissue, mRNA levels for α2δ-2 and α2δ-3 were similarly abundant in cultured MSNs. Error bars indicate mean ± SEM. Data from three independent culture/tissue preparations are shown. B, β-galactosidase staining of sagittal cryosections of α2δ-3 knock-out mice carrying a LacZ cassette revealed intense labeling of striatum (Str), hippocampus (Hc), and olfactory tubercle (Ot). Lower expression was detected in the cortex (Cx), thalamus (Th), olfactory bulb (Ob), and parts of the cerebellum (Cb). C, Schematic representation of the epitope-tagged α2δ subunits depicting the position of the extracellular 2HA tag inserted downstream of the signal peptide (SP), cache domains (C1–C4), and VWA (van Willebrand factor type A). Cultured MSNs were transfected with HA-tagged α2δ subunits together with soluble eGFP and live labeled with an antibody against the HA epitope at 24 DIV. All α2δ isoforms are expressed at the surface of presynaptic boutons, which is also shown by line scan analysis of α2δ-1, α2δ-2, and α2δ-3 (red) in relation to synapsin (blue) and eGFP (green). The sketch summarizes the observed labeling patterns. Representative images of three (B) and one (C) independent preparation(s) are shown. Statistics: A, ANOVA on log10-transformed data with Holm–Sidak post hoc analysis: cultured MSNs: F(3,8) = 460, p < 0.001; post hoc: p < 0.001 between all α2δ subunits except α2δ-2 vs α2δ-3 (p = 0.34); striatum: F(3,8) = 891, p < 0.001; post hoc: p < 0.001 between all α2δ subunits except α2δ-1 vs α2δ-2 (p = 0.30). Scale bars, 2 mm (B) and 1 μm (C).
Presynaptic overexpression of α2δ-2 upregulates postsynaptic GABAARs in inhibitory synapses
To distinguish between our hypotheses (Fig. 5), we coexpressed individual α2δ isoforms together with soluble eGFP in differentiated GABAergic MSNs. As expected, immunofluorescence analysis revealed that, in control MSNs (eGFP only), axonal varicosities were typically positive for presynaptic vGAT and postsynaptic GABAAR labeling (Fig. 8A,B). Postsynaptic GABAARs were similarly localized opposite vGAT-positive terminals expressing the individual α2δ isoforms (Fig. 8B). However, quantitative analysis of GABAAR fluorescence intensities of individual cells revealed that GABAAR labeling was strongly (∼2-fold) and significantly increased opposite boutons expressing the α2δ-2 isoform (Fig. 8C). Furthermore, presynaptic expression of α2δ-3 caused a 20% decrease of GABAAR labeling intensity compared with control (Fig. 8C). Plotting the cumulative frequency distribution of GABAAR fluorescence intensities of the single synapses demonstrates that presynaptic α2δ subunit isoforms differentially regulate postsynaptic GABAAR abundance in MSNs (Fig. 8D). GABAAR clusters were increased in size and intensity in the majority of synapses overexpressing α2δ-2 (blue line), causing a shift of the entire population to the right toward higher fluorescence intensities compared with control (eGFP only, green line). In contrast, ∼24% and ∼40% of all boutons expressing α2δ-1 (yellow line) and α2δ-3 (magenta line), respectively, lacked postsynaptic GABAAR labeling and the remaining population was left shifted toward smaller clusters. The slightly but significantly reduced GABAAR clustering opposite α2δ-3-overexpressing boutons (Fig. 8C) was associated with smaller presynaptic boutons (27% smaller than control; Fig. 8E) and decreased presynaptic vGAT content (46% lower than control; Fig. 8F). Conversely, presynaptic expression of α2δ-2, which strongly increased postsynaptic GABAAR clustering, affected neither bouton size nor vGAT labeling intensity.
Figure 8.

Presynaptic expression of α2δ-2 induces upregulation of postsynaptic GABAARs in MSNs. A, B, Representative immunofluorescence micrographs of cultured MSNs cotransfected with distinct α2δ subunits and soluble eGFP. Transfected neurons (21–28 DIV) were immunolabeled for vGAT and the GABAAR. Colocalization of fluorescence signals within eGFP-filled axonal varicosities (arrowheads, axons are outlined with dashed lines) was analyzed using line scans. A, GABAergic synapses transfected with eGFP only (control) show matched presynaptic vGAT and postsynaptic GABAAR immunoreactivity (summarized in sketch). B, Similar to control, postsynaptic GABAARs were localized opposite vGAT-positive presynaptic terminals expressing individual α2δ isoforms (see also colocalization in line scans). Most importantly, GABAAR clusters were larger and more intense opposite synaptic boutons expressing α2δ-2 (blue arrowheads). C–F, Quantitative analysis of GABAAR fluorescence intensity (C), cumulative frequency distribution of GABAAR fluorescence intensity (D), bouton size (E), and vGAT fluorescence intensity (F). Values for individual cells (dots) and means (lines) ± SEM are shown. Values were normalized to the control within each culture preparation. Data from four independent culture preparations and 34–42 cells were analyzed in each condition. Statistics: ANOVA on log10-transformed data with Holm–Sidak post hoc analysis: C, F(3,152) = 17.6, p < 0.001; post hoc: ***p < 0.001 between α2δ-2 and control, ***p < 0.001 between α2δ-2 and α2δ-1/α2δ-3, **p = 0.004 between control and α2δ-3, p = 0.2 between control and α2δ-1; E, F(3,152) = 5.1, p < 0.01; post hoc: **p < 0.01 between α2δ-3 and control/α2δ-2, p = 0.08 between α2δ-3 and α2δ-1; F, F(3,152) = 8.0, p < 0.001; post hoc: ***p < 0.001 between α2δ-3 and control, **p < 0.01 between α2δ-3 and α2δ-1/α2δ-2. Asterisks in graphs indicate the significant difference compared with control. Scale bars, 10 μm (A) and 3 μm (B).
Similar to mismatched glutamatergic synapses (see above), we used superresolution gSTED microscopy to visualize the subsynaptic localization of postsynaptic GABAAR clusters in cultured MSNs (Fig. 3C). MSNs were cotransfected with α2δ-2 and soluble mCherry or mCherry only (control). In both conditions, postsynaptic GABAAR clusters appeared closely opposed to mCherry-positive presynaptic boutons, thus confirming their localization in the postsynaptic membrane. Moreover, similar to hippocampal neurons, the GABAAR pattern was either visible as a thin line (side view; Fig. 3C, first and third column and Fig. 3B, top) or a ring-like structure (planar view; Fig. 3C, second and fourth column and Fig. 3B, bottom). Importantly, gSTED imaging further confirmed that presynaptic expression of α2δ-2 increased GABAAR density at the corresponding postsynaptic side.
α-Nrxns modulate postsynaptic GABAAR abundance set by presynaptic α2δ-2
Thus far, our results revealed that presynaptic α2δ-2 strongly induced postsynaptic GABAA receptor clustering in both glutamatergic and GABAergic synapses, suggesting that α2δ-2 acts trans-synaptically according to our hypothesis outlined in Figure 5. This means that α2δ-2 could either directly interact with postsynaptic receptors or may act via other synaptic organizer molecules such as α-Nrxns. Presynaptic α-Nrxns are synaptic cell adhesion molecules whose general importance in a variety of synaptic functions is well established (Reissner et al., 2013; Südhof, 2017). Several lines of evidence implicate α-Nrxns as interesting candidates for mediating a possible indirect interaction between α2δ-2 and GABAARs. First, α-Nrxns have been linked functionally to high-voltage-dependent Ca2+channels, which has strong effects on presynaptic neurotransmitter release (Missler et al., 2003; Chen et al., 2017). Second, α-Nrxns have been implicated as organizing molecules at both excitatory glutamatergic and inhibitory GABAergic synapses by promoting clustering of postsynaptic AMPARs (Heine et al., 2008; Aoto et al., 2013, 2015) and GABAARs (Kang et al., 2008). Third, the presynaptic complex of α-Nrxn and its ligand neurexophilin-1 is characteristic for inhibitory interneurons and can induce postsynaptic GABAAR recruitment when ectopically expressed at excitatory synapses (Born et al., 2014). Fourth, neurexin-1α has recently been shown to modulate P/Q-type (Brockhaus et al., 2018) and N-type (Tong et al., 2017) calcium channels by cooperating with α2δ subunits. Therefore, to test the possibility that upregulation of postsynaptic GABAARs depends on α-Nrxns, we expressed α2δ-2 in glutamatergic synapses lacking all three α-Nrxn isoforms (Missler et al., 2003). We transfected eGFP alone or in combination with α2δ-2 or α2δ-3 in hippocampal cultures from WT and α-Nrxn TKO mice. Immunofluorescence analysis revealed that, as expected, in control hippocampal neurons, axonal varicosities were typically positive for presynaptic vGLUT1 but negative for postsynaptic GABAAR labeling (Fig. 9A,C, WT control). Consistent with our previous observations, presynaptic expression of α2δ-2 induced the formation of mismatched synapses (Fig. 9A,C, WT+α2δ-2), whereas expression of α2δ-3 had no effect (p = 0.64; Fig. 9A,C, WT+α2δ-3). Strikingly, expression of presynaptic α2δ-2 in α-Nrxn TKO neurons induced postsynaptic GABAAR clustering opposite glutamatergic boutons more strongly than in WT (Fig. 9A,C, α-Nrxn TKO+α2δ-2). Quantitative analysis revealed that this effect was three times higher compared with the WT+α2δ-2 condition, suggesting that the propensity of α2δ-2 in inducing mismatched synapses was enhanced in the absence of α-Nrxns. In fact, increased GABAAR abundance was already visible in nontransfected α-Nrxn TKO synapses in which the presence of endogenous α2δ-2 may be responsible for a significantly increased baseline level of mismatched GABAARs (p = 0.023; Fig. 9A,C). This is an important result because it suggests that no overexpression of α2δ-2 is needed to induce postsynaptic GABAAR clustering, but an imbalance between α2δ-2 and α-Nrxns. These differential effects can be best compared by blotting the cumulative frequency distribution of GABAAR fluorescence intensities of all individual synapses analyzed (Fig. 9D). Compared with WT (control, green line), the entire population of synapses was right-shifted toward larger GABAAR clusters in α-Nrxn TKO neurons (dashed green line). Presynaptic expression of transfected α2δ-2 strongly shifted the distributions toward larger GABAAR clusters in WT (+α2δ-2, blue line) and α-Nrxn TKO neurons (TKO+α2δ-2, dashed blue line). Presynaptic expression of α2δ-3 had no effect in WT neurons, which, due to the lack of mismatched synapses, basically displayed only background GABAAR intensity (+α2δ-3, magenta line). Interestingly, overexpression of α2δ-3 in α-Nrxn TKO neurons reduced the elevated GABAAR clusters to control levels (TKO+α2δ-3, dashed magenta line). This possibly indicates that overexpressed, abundant α2δ-3 blocked the effect by endogenous α2δ-2 on baseline GABAAR intensity in TKO synapses. Together, our data suggest that presynaptic α2δ-2 potently induced mismatched synapse formation with GABAAR clustering in the absence of α-Nrxns. This demonstrates that the trans-synaptic role of α2δ-2 in recruiting postsynaptic GABAARs is independent of α-Nrxns. However, the fact that GABAAR abundance was already increased in α-Nrxn TKO synapses without overexpression of α2δ-2 and that the abundance could be differentially modulated by presynaptic α2δ-2 and α2δ-3 suggests a cooperative and trans-synaptic activity of α2δ subunits and α-Nrxns in fine-tuning postsynaptic GABAAR levels.
Figure 9.

α-Nrxn modulates the effect of α2δ-2 on GABAARs. Representative immunofluorescence micrographs of cultured hippocampal neurons (20–25 DIV) prepared from WT or α-Nrxn TKO mice. A, B, In control boutons (eGFP only), potential glutamatergic synapses are positive for presynaptic vGLUT1 but negative for postsynaptic GABAAR staining (summarized in sketch, B). Consistent with our initial observation (see Fig. 1), presynaptic expression of α2δ-2 (blue arrowheads) induces postsynaptic GABAAR localization opposite vGLUT1-positive nerve terminals in WT neurons. Most importantly, in α-Nrxn TKO synapses, the expression of α2δ-2 strongly induced postsynaptic GABAAR localization (blue asterisks). C, Postsynaptic GABAARs clusters (integrated intensities) are significantly increased opposite glutamatergic nerve terminals expressing α2δ-2 compared with control (eGFP only). This effect is much stronger in α-Nrxn TKO synapses compared with WT neurons (3-fold higher GABAAR fluorescence intensity). D, Cumulative frequency distribution further reveals that the vast majority of glutamatergic boutons are positive for postsynaptic GABAARs in WT+α2δ-2 (88%) and TKO+α2δ-2 (97%) and that the population is shifted toward larger and more intense clusters. However, this analysis also demonstrates that mismatched GABAARs already form opposite α-Nrxn TKO synapses, although at a lower overall intensity. Values for individual cells (dots) and means (lines) ± SEM are shown. Values were normalized to the WT control within each culture preparation. Data from two independent culture preparations and 12–19 cells were analyzed within each condition. Statistics: C, ANOVA on log10-transformed data with Holm–Sidak post hoc analysis: genotype: F(1,86) = 5.3, p = 0.023; transfection: F(2,86) = 49.0, p < 0.001; genotype × transfection: F(2,86) = 3.7, p = 0.03; post hoc: *p < 0.05, ***p < 0.001. Asterisks in graphs indicate the significant difference compared with control. Scale bar, 3 μm.
α2δ-2 induces the aberrant wiring of glutamatergic axons to GABAergic postsynaptic positions
Our experiments described above demonstrate that presynaptic α2δ-2 induces the recruitment of postsynaptic GABAARs opposite glutamatergic and GABAergic presynaptic boutons. Moreover, this effect of α2δ-2 is independent of the presynaptic neurotransmitter identity, suggesting a trans-synaptic activity in recruiting/anchoring postsynaptic GABAARs. As hypothesized (Fig. 5), such a trans-synaptic role of α2δ-2 could result in two types of mismatched synapses: the GABAARs could either be recruited to postsynaptic compartments of dendritic spine synapses with a concomitant reduction of the resident glutamate receptors or α2δ-2 could induce an altered wiring of glutamatergic axons to GABAergic postsynaptic locations along the dendritic shaft.
To distinguish between these two possibilities, we focused more closely on the position of the mismatched synapses along the postsynaptic neurons. First, it is important to emphasize that α2δ-2 induced the upregulation of postsynaptic GABAergic components only at the sites contacted by the presynaptic axon without affecting the neighboring synapses. This is best visualized by the observation that gephyrin fluorescence intensity was 4.5 times higher in α2δ-2-induced mismatched synapses (Fig. 10A–C, asterisks in A and B) compared with neighboring endogenous GABAergic synapses situated on the same dendrite, which are formed by axons from untransfected neurons (Fig. 10A–C, arrowheads in A and B). This finding further supports a local and synapse-specific effect of presynaptic α2δ-2 rather than a global alteration in the expression of GABAergic postsynaptic components. Importantly, α2δ-2-overexpressing glutamatergic axons preferentially formed synapses at typical GABAergic postsynaptic locations, namely linearly aligned along the dendritic shaft (Fig. 10D, right column, blue arrowheads), in contrast to control axons, which preferably contacted dendritic spines (Fig. 10D, left column, white arrowheads). Therefore, to determine the exact position of mismatched synapses on postsynaptic dendrites, we established hippocampal cultures in which presynaptic (control or α2δ-2) and postsynaptic neurons were transfected with mCherry (red) and eGFP (green), respectively (Fig. 11A–C). gSTED microscopy allowed precise visualization of the contact point between the presynaptic mCherry-positive axon and the postsynaptic eGFP-positive dendrite (Fig. 11A,B). As expected for glutamatergic excitatory synapses, the vast majority of axonal varicosities was located on dendritic spines in the control condition (80% spine, 13% shaft, 7% unclear; Fig. 11B,C, top). In contrast, presynaptic expression of α2δ-2 significantly shifted the preferential contact points to dendritic shafts (24% spine, 65% shaft, 11% unclear; χ2 test: p < 0.001). This finding was further corroborated by a highly significant reduction of the mean distance from the contacting presynaptic boutons to the postsynaptic dendritic shafts in α2δ-2-transfected compared with control axons (mean distance ± SEM: control: 0.92 ± 0.05 μm; α2δ-2: 0.54 ± 0.05 μm; Mann–Whitney U test: p < 0.001; Fig. 11C, bottom). Importantly, applying the same methodological approach for the coculture of GABAergic MSNs and glutamatergic cortical neurons (Fig. 11D, also see Fig. 7) revealed that axonal varicosities from control cortical neurons generally contacted dendritic spines of postsynaptic inhibitory MSNs (total of 65 synapses: 83% spine, 1.5% shaft, 15.5% unclear: Fig. 11E,F, top). In contrast, the presynaptic expression of α2δ-2 shifted the preferential contact points of cortical boutons to dendritic shafts of MSNs (total of 51 synapses: 19.6% spine, 72.6% shaft, 7.8% unclear; Fig. 11E,F, bottom, gephyrin labeling and position at dendritic shaft). These findings demonstrate that the α2δ-2-induced aberrant wiring is independent of the identity of the postsynaptic neuron.
Figure 10.

Mismatched synapses show characteristics of GABAergic shaft synapses. A, D, Representative immunofluorescence micrographs of cultured hippocampal neurons cotransfected with α2δ-2 and soluble eGFP. Colabeling of transfected neurons (24 DIV) for postsynaptic GLUR1 and gephyrin is shown. A, Presynaptic expression of α2δ-2 induces a strong upregulation of postsynaptic gephyrin (asterisk) compared with neighboring endogenous GABAergic synapses situated on the same dendrite (arrowheads). Although GLUR1 expression was reduced in mismatched synapses (*; see Fig. 2), it was absent in endogenous GABAergic synapses (arrowheads, see sketch in B). C, Quantitative analysis of gephyrin fluorescence intensities in GABAergic neighboring and mismatched synapses. Dots represent values for individual boutons (mismatched synapses) and means of 10 endogenous clusters measured per image. Results were normalized to endogenous gephyrin intensities of neighboring synapses within each culture preparation. Data from three independent culture preparations are shown. Statistics: t test on log10-transformed data: t(50) = 9.5; ***p < 0.001. D, Left, Control boutons transfected with eGFP only form glutamatergic synapses on dendritic spines (gephyrin negative, GLUR1 positive; white arrowheads). Right, Expression of α2δ-2 in putative glutamatergic boutons induces the formation of mismatched synapses (gephyrin-positive, GLUR1-negative) along the shaft of the dendrite (blue arrowheads). Dendritic morphology of untransfected neurons was outlined according to GLUR1 labeling. Scale bars, 5 μm (A) and 4 μm (B).
Together, our results provide strong evidence that overexpression of presynaptic α2δ-2 induces the aberrant wiring of glutamatergic axons to postsynaptic GABAergic locations, resulting in a mismatch of presynaptic excitatory neurotransmitters and postsynaptic inhibitory receptors.
Reduced synaptic transmission in aberrantly wired synapses
Excitatory and inhibitory synapses require matched presynaptic and postsynaptic specializations to enable efficient and synchronous neurotransmission. To assess the functional consequences of α2δ-2 overexpression in presynaptic boutons of aberrantly wired glutamatergic synapses, we performed whole-cell patch-clamp recordings on synaptically connected pairs of isolated neurons grown on poly-l-lysine dots (Fig. 12A). We first investigated whether AMPAR-mediated evoked EPSCs (eEPSCs) between individual pairs of hippocampal neurons were altered by the lentiviral overexpression of α2δ-2 in the presynaptic neuron. Isolated synaptic currents were measured in the postsynaptic neuron in response to an action potential induced in the presynaptic neuron. The presynaptic action potential followed a given waveform recorded from WT hippocampal neurons (Fig. 12B). This experimental setup allowed to directly compare synaptic transmission in glutamatergic control synapses (presynaptic cell = control, postsynaptic cell = α2δ-2 overexpressing) and mismatched synapses (presynaptic cell = α2δ-2 overexpressing, postsynaptic cell = control). In each of seven neuronal pairs recorded, the eEPSC amplitude was lower in the mismatched compared with the control pair (on average reduced by 48%) (Fig. 12C). To discriminate whether the eEPSC reduction is the result of the reduced postsynaptic AMPA receptor expression associated with the aberrantly wired axons or if presynaptic α2δ-2 reduces release probability, we next measured synaptic paired-pulse response ratios (PPRs) (Katz and Miledi, 1968; Zucker and Regehr, 2002). eEPSCs were recorded in the postsynaptic neuron in response to two action potentials elicited in the presynaptic neuron at different interstimulus intervals. As previously reported for hippocampal pyramidal neurons, calculated PPRs displayed facilitation (Fig. 12D) with a decline in the magnitude at increasing intervals (Dobrunz et al., 1997). Overexpression of α2δ-2 in presynaptic neurons slightly increased facilitation at 25 and 50 ms compared with the initial 10 ms interval. The strong reduction of eEPSC amplitudes is thus consistent with a reduced number of functional synapses caused by the aberrant wiring of glutamatergic α2δ-2-expressing boutons with GABAergic postsynaptic sites. Conversely, the only moderately altered paired-pulse facilitation (PPF) indicates that the decreased amplitude of eEPSCs was not caused by a reduced presynaptic release probability. The observed slightly increased PPF may reflect an increased abundance of presynaptic calcium channels caused by the overexpression of α2δ subunits, as has been previously demonstrated (Hoppa et al., 2012).
Figure 12.
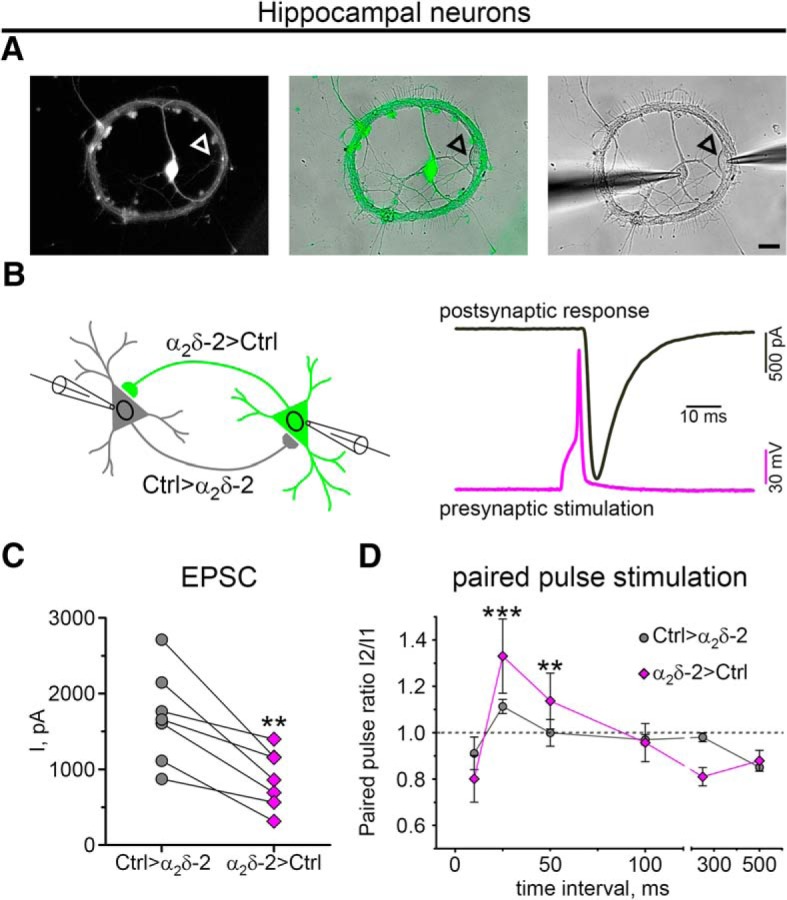
Presynaptic expression of α2δ-2 affects synaptic transmission in aberrantly wired synapses. A–C, Experimental paradigm to study the functional consequences of presynaptic α2δ-2 overexpression in mismatched glutamatergic synapses of cultured hippocampal neurons. A, Neurons were cultured in pairs of two synaptically connected cells in which one represents an untransfected control neuron (arrowhead) and the other an α2δ-2-overexpressing neuron (eGFP labeled). B, Whole-cell patch-clamp recordings of excitatory synaptic transmission (13–18 DIV). An action potential in neuron 1 elicited by a given AP waveform recorded from WT hippocampal neurons caused eEPSCs in neuron 2. Synaptic transmission was analyzed in both directions of synaptically connected pairs. Therefore, putative glutamatergic spine synapses (control innervating α2δ-2-overexpressing neuron) and putative mismatched synapses (α2δ-2 overexpressing innervating control neuron) could be directly compared within the same pair. C, D, Quantitative analysis of mean eEPSC peak amplitudes (C) and paired-pulse response ratios (PPRs) as a measure for synaptic plasticity (D). C, eEPSCs were strongly reduced (48%) in mismatched synapses. D, Dashed line shows the boundary between paired-pulse depression (PPR < 1) and facilitation (PPR > 1). Note the slight significant increase in facilitation in mismatched synapses at 25 and 50 ms compared with the initial 10 ms interval. Values for individual pairs (dots) and means (lines) ± SEM are shown. Data from three independent culture preparations from seven (C) and six (D) pairs were analyzed. Statistics: C, paired t test: t(6) = 4.6; **p < 0.01; D, two-way repeated-measures ANOVA: condition: F(1,45) = 0.02, p = 0.9; interval: F(5,45) = 11.1, p < 0.001; condition × interval: F(5,45) = 2.7, p = 0.03; Holm–Sidak post hoc analysis: **p = 0.004, ***p < 0.001 compared with 10 ms within α2δ-2 to control. Scale bar, 20 μm.
Lack of exon 23 in α2δ-2 splice variants mediates the trans-synaptic effect on GABAARs
Our results revealed a specific trans-synaptic function of only one of the three expressed α2δ isoforms, namely α2δ-2. Sequence comparisons of the previously known alternatively spliced regions (Hobom et al., 2000) identified three potential splice sites in α2δ-2 (Fig. 13A, scheme): inclusion or absence of exon 23 and two alternative splice sites at exons 30 and 38. Using homology modeling, we explored the potential implication of these sequence differences on the structural fold of α2δ-2 (Fig. 14) based on the recently suggested α2δ-1 cryo-EM structure (Wu et al., 2016). Inclusion of exon 23 resulted in the formation of an extra loop that might disrupt an α-helix (Figs. 13B, 14B), whereas alternative splicing of exons 30 and 38 predicted only small changes in the secondary structure (Fig. 14C,D). Therefore, to test whether alternative splicing at these sites is involved in the trans-synaptic function of α2δ-2, we cloned and compared three splicing variants (α2δ-2-v1, α2δ-2-v2, and α2δ-2-v3, α2δ-2-v1 = original construct; Fig. 13A). To determine the potential of the splice variants in inducing mismatched synapses, we transfected WT hippocampal neurons with the three distinct α2δ-2 variants together with soluble eGFP. Strikingly, only the splice variants lacking exon 23 (α2δ-2-v1 and α2δ-2-v3) induced the clustering of postsynaptic GABAARs opposite transfected glutamatergic nerve terminals (Fig. 13C, ΔExon 23). The fact that α2δ-2-v1 and α2δ-2-v3 both induced mismatched synapse formation excludes a major role of exons 30 and 38, which differ in α2δ-2-v1 and α2δ-2-v3. In contrast, α2δ-2-v2, which contains the exon 23 insert, failed completely to induce mismatched synapse formation (Fig. 13C, +Exon 23), indicating that the trans-synaptic function of α2δ-2 on GABAARs is transcriptionally regulated. Together, our results have uncovered a highly specific role of the α2δ-2 subunit of voltage-gated calcium channels in clustering postsynaptic GABAARs, an important process that is further regulated by the presence of α-Nrxns and by alternative splicing.
Figure 13.

Lack of exon 23 in α2δ-2 splice variants mediates the trans-synaptic effect on GABAARs. A, Schematic overview illustrating the approximate positions of three alternatively spliced regions of α2δ-2. Sequence alignment between α2δ-2-v1, the original construct used in this study, and two additional α2δ-2 variants (v2, v3) reveals alternative splicing of exons 23, 30, and 38. B, Using homology modeling, we tested the potential consequences of alternative splicing on the structure prediction based on the high-resolution structure of α2δ-1 (PDB code: 5GJV; Wu et al., 2016; see Fig. 14). Inclusion of exon 23 in α2δ-2-v2 suggested the formation of an extra loop (arrowhead) leading to the disruption of an α-helix present in α2δ-2-v1 and α2δ-2-v3. C, Representative immunofluorescence micrographs of cultured hippocampal neurons (21–26 DIV) cotransfected with distinct α2δ-2 splice variants and soluble eGFP. Presynaptic expression of either α2δ-2-v1 (original construct) or α2δ-2-v3, which both lack exon 23, robustly induces a mismatched GABAAR localization opposite vGLUT1-positive nerve terminals (blue arrowheads). In contrast, presynaptic expression α2δ-2-v2, which contains exon 23, failed to induce mismatched synapse formation (white arrowheads, line scans). Asterisks mark endogenous GABAAR clusters from untransfected neighboring synapses not colocalizing with presynaptic vGLUT1. Representative images of two independent cultures are shown. Scale bar, 3 μm.
Figure 14.

Splicing sites of α2δ-2 and implications on secondary structure. A, Using homology modeling, we tested the potential consequences of alternative splicing on the structure prediction of three distinct α2δ-2 splice variants (v1–v3) based on the high-resolution structure of α2δ-1 (PDB code: 5GJV; Wu et al., 2016). Although some of the longer loops generally seemed to be quite flexible and differently orientated in all three models (gray arrows), alternative splicing of exons 23, 30, and 38 resulted in differences between secondary structure elements in distinct variants (boxed regions in B–D). B, Higher magnification showing that the inclusion of exon 23 in α2δ-2-v2 suggests the formation of an extra loop (arrowhead and green selection in overlay), leading to the disruption of an α-helix present in α2δ-2-v1 and α2δ-2-v3 (see also Fig. 13). C, Structure modeling further proposes that the insertion of three base pairs in exon 30 of α2δ-2-v3 causes the formation of a short α-helix in the depicted loop, which is absent in α2δ-2-v1 and α2δ-2-v2. D, Moreover, models implicate that alternative splicing of exon 38 affects the length of a β sheet at this position, which is longer in α2δ-2-v1 and α2δ-2-v2.
Discussion
The experiments described here reveal that presynaptic expression of a single splice variant of α2δ-2 trans-synaptically regulates postsynaptic GABAAR abundance. Therefore, presynaptic α2δ-2 modulates postsynaptic GABAAR clustering independently of the presynaptic neurotransmitter, induces an aberrant wiring of glutamatergic axons with postsynaptic GABAergic sites of both glutamatergic and GABAergic neurons, and alters synaptic transmission in mismatched synaptic pairs. These results demonstrate for the first time that presynaptic α2δ subunits are able to trans-synaptically regulate synapse specification and axonal wiring.
α2δ-2 induces the formation of mismatched synapses via a trans-synaptic mechanism
Excitatory and inhibitory synapses are characterized by their distinct molecular composition and subcellular localization in neurons. The proper alignment of presynaptic and postsynaptic specializations is a hallmark of synapse formation and a prerequisite for efficient and synchronous excitatory and inhibitory synaptic transmission (Lardi-Studler and Fritschy, 2007). Only a small number of studies have reported mismatched synapses in vitro. In micro-island cultures of autaptic hippocampal neurons, the vast majority of GABAARs and gephyrin clusters are located opposite glutamatergic terminals, suggesting that postsynaptic compartments can be recruited without the proper presynaptic input (Rao et al., 2000; Christie et al., 2002). Moreover, in low-density cultures of hippocampal and cerebellar granule neurons, mismatches of presynaptic and postsynaptic proteins can form during early developmental stages, but are essentially absent in differentiated cultures (Brünig et al., 2002; Anderson et al., 2004; Studler et al., 2005). In contrast to these studies, the mismatched synapses reported in our study are specifically induced by the presynaptic expression of α2δ-2 and are independent from neurotransmitter input, suggesting that α2δ-2 operates via a different mechanism.
Mixed synapses with glutamatergic and GABAergic properties have also been detected in different brain areas, indicating a functional role in vivo. For example, postsynaptic GABAARs are present opposite presynaptic glutamatergic mossy fiber axon terminals in the cerebellar cortex and hippocampus (Nusser et al., 1996a,b; Bergersen et al., 2003). Physiologically, these receptors may be involved in fine-tuning excitatory synaptic transmission in response to GABA spillover from surrounding synapses (Isaacson et al., 1993). Although we cannot completely rule out GABA spillover from adjacent neurons, it is important to note that the α2δ-2-induced mismatched synapses do not express presynaptic vGAT. Therefore, the recruitment of postsynaptic GABAARs is entirely independent of presynaptic GABA release. These findings are consistent with a recent report showing that, even in the absence of presynaptic glutamate release, dendritic spines are formed and AMPARs are present in glutamatergic synapses (Sigler et al., 2017).
Generally, excitatory and inhibitory synapses are mostly targeted to different subcellular regions of the postsynaptic neuron (Lardi-Studler and Fritschy, 2007). Therefore, glutamatergic boutons predominantly contact postsynaptic dendritic spines (Peters, 2002), whereas GABAergic nerve terminals mostly connect to the shaft of the dendrite (Markram et al., 2004). Nevertheless, converging lines of evidence demonstrate morphologically mixed synapse types in different parts of the brain. In the adult neocortex, for example, two distinct types of GABAergic synapses are positioned either on the shaft or spine of the dendrite, which are suggested to have different roles in shaping dendritic activity (Chen et al., 2012). It is hypothesized that these constellations facilitate the shunting of excitatory currents, thereby balancing excitatory and inhibitory neurotransmission (Nusser et al., 1996a; Root et al., 2014). Along these lines, we proposed that the underlying mechanism of α2δ-2-induced mismatched synapses could be explained by a compensatory upregulation of postsynaptic GABAARs due to increased excitatory transmission upon α2δ overexpression (Zhou and Luo, 2015; Chen et al., 2018). However, several of our observations argue against this theory. First, using distinct experimental paradigms, we show here that α2δ-1 and α2δ-3, which both enhance excitatory synaptic transmission and increase presynaptic release probability (Hoppa et al., 2012), do not induce postsynaptic clustering of synaptic GABAAR subtypes. Second, the effect of α2δ-2 is independent of the presynaptic neurotransmitter identity, thus providing compelling evidence for a trans-synaptic function. Third, solely α2δ-2 splice variants lacking exon 23 specifically induced the recruitment of postsynaptic GABAARs opposite glutamatergic nerve terminals. Fourth, the effect of α2δ-2 is restricted to transfected synapses, emphasizing a synapse-specific role rather than a global alteration of GABAergic postsynaptic components.
Presynaptic α2δ subunits differentially modulate postsynaptic GABAAR abundance via distinct mechanisms
We found diverging effects of presynaptic α2δ-2 and α2δ-3 on the postsynaptic GABAAR abundance of GABAergic MSNs because α2δ-2 strongly increased and α2δ-3 reduced postsynaptic GABAAR levels (Fig. 8). In contrast to presynaptic α2δ-2 expression, which modulated the postsynaptic composition without apparently affecting the presynaptic side, the effect of α2δ-3 was associated with smaller presynaptic boutons and decreased presynaptic vGAT content. Smaller presynaptic boutons may also explain the reduced GLUR1 content opposite α2δ-3-expressing synapses (Fig. 2). Together, these data suggest that presynaptic α2δ subunits regulate presynaptic differentiation and postsynaptic GABAAR abundance by two independent mechanisms. Although expression of α2δ-3 induces smaller synapses associated with a reduced postsynaptic receptor density, presynaptic α2δ-2 modulates postsynaptic GABAAR abundance but does not affect presynaptic bouton size. Altered expression of mammalian α2δ-3 (Pirone et al., 2014) and its invertebrate homologs (Kurshan et al., 2009; Caylor et al., 2013) has previously been shown to affect the size and morphology of presynaptic boutons of auditory nerve fibers and motoneurons, respectively.
Evidence for an α2δ-2-mediated trans-synaptic signaling cascade
To date, surprisingly little is known on the specific signals imperative for the proper alignment of inhibitory synapses and the targeting of GABAARs and gephyrin to the postsynaptic compartment. In vitro cultured neurons provide an excellent and reduced model system allowing us to address basic cell biological mechanisms. Most importantly, they are less prone to potential compensatory mechanisms compared with the in vivo situation (Dong et al., 2007; Brown et al., 2016; Chen et al., 2017). Likewise, overexpression of proteins in their native environment provides a powerful experimental tool but needs to be carefully discussed, particularly in relation to the specific physiological roles. Therefore, it is conceivable that an aberrantly high accumulation of presynaptic α2δ-2 proteins may induce artificial synaptic connections. Nevertheless, our study provides several lines of evidence supporting a presynaptic role of α2δ-2 in the trans-synaptic alignment of synapses and receptor clustering. First, we observed that presynaptic expression of a single splice variant of α2δ-2 trans-synaptically regulates postsynaptic GABAAR abundance. Second, overexpression of α2δ-2 specifically induces an aberrant wiring of glutamatergic axons to postsynaptic GABAergic sites independently of the postsynaptic neuron type (Fig. 11). Third, whereas mismatched synapses lack glutamate receptors, they contain different synaptic GABAAR subtypes and gephyrin. Importantly, the strongly reduced GLUR1 expression (Fig. 2) in the mismatched and aberrantly wired synapses is a secondary effect because no AMPARs are to be expected in postsynaptic GABAergic synapses (Fig. 5C, model). Fourth, gephyrin abundance is higher in α2δ-2-induced mismatched synapses compared with untransfected endogenous GABAergic synapses. Considering that gephyrin immobilizes synaptic GABAAR clusters in hippocampal neurons (Jacob et al., 2005), we provide strong evidence that gephyrin recruitment occurs secondary to synaptic GABAAR clustering.
However, it is still unknown whether the interaction with postsynaptic receptors is mediated by a direct trans-synaptic action of α2δ-2 or if it may be indirectly mediated by the interaction with other primary synaptic organizers (see also discussion in Fell et al., 2016). Several studies implicate the trans-synaptic cell adhesion molecules α-Nrxns as the most interesting candidates for mediating a possible indirect interaction between α2δ-2 and GABAARs (Missler et al., 2003; Kang et al., 2008; Born et al., 2014; Tong et al., 2017). Here, we demonstrate that presynaptic α2δ-2 also potently induces mismatched synapse formation in the absence of α-Nrxns. Although this finding supports an α2δ-2-mediated trans-synaptic effect and possibly a direct trans-synaptic interaction of α2δ-2 with postsynaptic GABAARs, it does not exclude the potential involvement of other cell adhesion molecules not addressed in our study (Graf et al., 2004; Lardi-Studler and Fritschy, 2007).
The effect of overexpressed α2δ-2 was more pronounced in α-Nrxn TKO synapses compared with WT control synapses. We observed that the baseline expression of postsynaptic GABAARs in TKO was already significantly increased, which shows that no overexpression of α2δ-2 is needed for inducing postsynaptic GABAAR clustering. This finding may indicate that the presence of α-Nrxns negatively influences GABAAR recruitment by α2δ-2. Alternatively, the loss of α-Nrxns could be either overproportionally compensated by the still present β-neurexins (Missler et al., 2003) or shift the modulation of postsynaptic receptor abundance to the neuroligin-2/Slitrk3 or the neuroligin-2/MDGA1 complexes (Pettem et al., 2013; Li et al., 2017). Independently of the detailed underlying mechanism, our findings support a cooperative action of α2δ subunits and neurexins in fine-tuning synaptic functions (Tong et al., 2017; Brockhaus et al., 2018).
Future implications and potential relevance to neurological disorders
One important finding presented here is that only α2δ-2 splice variants lacking exon 23 induced a mismatched localization of postsynaptic GABAARs. This is interesting for several reasons. First, the presynaptic effect of α2δ-2 lacking a single spliced exon is highly specific and thus argues against a simple unspecific effect caused by homologous overexpression of α2δ isoforms in our experimental paradigm. Second, in light of discussing potential interaction mechanisms (see above), it is tempting to speculate that α2δ-2 interacts with its partners via the region surrounding the lacking exon 23, which will be tested in future experiments. Third, our findings show that the trans-synaptic function of α2δ-2 may be independent of the calcium channel complex. This is further supported by a previous study showing that heterologous coexpression of different α2δ-2 splice variants with various α1 subunits caused similar effects on calcium channel current densities and activation/inactivation kinetics (Hobom et al., 2000). Fourth, and most importantly, alternative splicing has also been described for mouse and human genes encoding for α2δ-1 and α2δ-4 (Lana et al., 2014; Bacchi et al., 2015). This is particularly interesting considering the previously identified associations between α2δ subunits with neurological disorders (Chioza et al., 2009; Edvardson et al., 2013; Pippucci et al., 2013; Vergult et al., 2015; Butler et al., 2018; Valence et al., 2018). Two distinct splice site mutations in CACNA2D2, for example, lead to the truncation of the protein, causing epilepsy, dyskinesia, and cerebellar atrophy in one patient (Pippucci et al., 2013), whereas the other mutation is associated with congenital ataxia (Valence et al., 2018). These clinically observed phenotypes are similar to the previously described phenotype of Cacna2d2 mutant or null mouse models (Barclay et al., 2001; Brodbeck et al., 2002; Ivanov et al., 2004) and may be caused by the loss of functional GABAergic synapses and the consequential increased excitatory activity. Moreover, α2δ subunits are potential risk genes for autism spectrum disorders (Iossifov et al., 2012; De Rubeis et al., 2014) and schizophrenia (Purcell et al., 2014; Moons et al., 2016), diseases classically associated with altered neuronal connectivity (Belmonte et al., 2004; Fornito and Bullmore, 2015). The findings presented here provide a possible mechanistic explanation for how altered α2δ subunit expression may be linked to neurological disorders. However, for the future understanding of the precise underlying disease mechanisms, it will be necessary to reveal whether all α2δ subunit isoforms can act as similarly specific trans-synaptic organizers in distinct synapses. Finally, our findings allow hypothesizing that presynaptic α2δ subunits are critical determinants of synapse specificity and connectivity. Therefore, alterations in the balance of presynaptic α2δ isoforms may underlie synaptic plasticity and, conversely, pathological axonal wiring observed in neurological disorders.
Footnotes
This work was supported by the Austrian Science Fund (Grants SFB F4415 and DOC 30-B30 to G.J.O.) and the Deutsche Forschungsgemeinschaft (Grant SFB1348-TPA03 to M.M.). This work is part of the doctoral theses of S.G. and C.L.S. We thank Ariane Benedetti, Martin Heitz, and Cornelia Ablinger for technical support; Martin Offterdinger from the Biooptics facility of the Medical University Innsbruck for help with superresolution microscopy; Hermann Dietrich, Anja Beierfuß, and their team for animal care; Arnold Schwartz and Jutta Engel for providing α2δ-1 and α2δ-3 knock-out mice, respectively; Bernhard Flucher for critical discussions; Günther Sperk for providing the rabbit GABAAR antibodies; Stefan Geley for help with cloning the mCherry construct; and Sebastian Honke for help with 3D modeling.
The authors declare no competing financial interests.
References
- Anderson TR, Shah PA, Benson DL (2004) Maturation of glutamatergic and GABAergic synapse composition in hippocampal neurons. Neuropharmacology 47:694–705. 10.1016/j.neuropharm.2004.07.023 [DOI] [PubMed] [Google Scholar]
- Aoto J, Martinelli DC, Malenka RC, Tabuchi K, Südhof TC (2013) Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell 154:75–88. 10.1016/j.cell.2013.05.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Földy C, Ilcus SM, Tabuchi K, Südhof TC (2015) Distinct circuit-dependent functions of presynaptic neurexin-3 at GABAergic and glutamatergic synapses. Nat Neurosci 18:997–1007. 10.1038/nn.4037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arama J, Abitbol K, Goffin D, Fuchs C, Sihra TS, Thomson AM, Jovanovic JN (2015) GABAA receptor activity shapes the formation of inhibitory synapses between developing medium spiny neurons. Front Cell Neurosci 9:290. 10.3389/fncel.2015.00290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba-Abbad R, Arno G, Carss K, Stirrups K, Penkett CJ, Moore AT, Michaelides M, Raymond FL, Webster AR, Holder GE (2016) Mutations in CACNA2D4 cause distinctive retinal dysfunction in humans. Ophthalmology 123:668–671.e2. 10.1016/j.ophtha.2015.09.045 [DOI] [PubMed] [Google Scholar]
- Bacchi N, Messina A, Burtscher V, Dassi E, Provenzano G, Bozzi Y, Demontis GC, Koschak A, Denti MA, Casarosa S (2015) A new splicing isoform of Cacna2d4 mimicking the effects of c.2451insC mutation in the retina: novel molecular and electrophysiological insights. Invest Ophthalmol Vis Sci 56:4846–4856. 10.1167/iovs.15-16410 [DOI] [PubMed] [Google Scholar]
- Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J, Canti C, Meir A, Page KM, Kusumi K, Perez-Reyes E, Lander ES, Frankel WN, Gardiner RM, Dolphin AC, Rees M (2001) Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci 21:6095–6104. 10.1523/JNEUROSCI.21-16-06095.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CS, Tran-Van-Minh A, Kadurin I, Dolphin AC (2010) A new look at calcium channel α2δ subunits. Curr Opin Neurobiol 20:563–571. 10.1016/j.conb.2010.05.007 [DOI] [PubMed] [Google Scholar]
- Belmonte MK, Allen G, Beckel-Mitchener A, Boulanger LM, Carper RA, Webb SJ (2004) Autism and abnormal development of brain connectivity. J Neurosci 24:9228–9231. 10.1523/JNEUROSCI.3340-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergersen L, Ruiz A, Bjaalie JG, Kullmann DM, Gundersen V (2003) GABA and GABAA receptors at hippocampal mossy fibre synapses. Eur J Neurosci 18:931–941. 10.1046/j.1460-9568.2003.02828.x [DOI] [PubMed] [Google Scholar]
- Bonansco C, Fuenzalida M (2016) Plasticity of hippocampal excitatory–inhibitory balance: missing the synaptic control in the epileptic brain. Neural Plast 2016:8607038. 10.1155/2016/8607038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born G, Breuer D, Wang S, Rohlmann A, Coulon P, Vakili P, Reissner C, Kiefer F, Heine M, Pape HC, Missler M (2014) Modulation of synaptic function through the α-neurexin-specific ligand neurexophilin-1. Proc Natl Acad Sci U S A 111:E1274–E1283. 10.1073/pnas.1312112111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhaus J, Schreitmüller M, Repetto D, Klatt O, Reissner C, Elmslie K, Heine M, Missler M (2018) α-neurexins together with α2δ-1 auxiliary subunits regulate ca2+ influx through caV2.1 channels. J Neurosci 38:8277–8294. 10.1523/JNEUROSCI.0511-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbeck J, Davies A, Courtney JM, Meir A, Balaguero N, Canti C, Moss FJ, Page KM, Pratt WS, Hunt SP, Barclay J, Rees M, Dolphin AC (2002) The ducky mutation in Cacna2d2 results in altered purkinje cell morphology and is associated with the expression of a truncated alpha 2 delta-2 protein with abnormal function. J Biol Chem 277:7684–7693. 10.1074/jbc.M109404200 [DOI] [PubMed] [Google Scholar]
- Brown LE, Nicholson MW, Arama JE, Mercer A, Thomson AM, Jovanovic JN (2016) γ-aminobutyric acid type A (GABAA) receptor subunits play a direct structural role in synaptic contact formation via their N-terminal extracellular domains. J Biol Chem 291:13926–13942. 10.1074/jbc.M116.714790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünig I, Suter A, Knuesel I, Lüscher B, Fritschy JM (2002) GABAergic terminals are required for postsynaptic clustering of dystrophin but not of GABA(A) receptors and gephyrin. J Neurosci 22:4805–4813. 10.1523/JNEUROSCI.22-12-04805.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler KM, Holt PJ, Milla SS, da Silva C, Alexander JJ, Escayg A (2018) Epileptic encephalopathy and cerebellar atrophy resulting from compound heterozygous CACNA2D2 variants. Case Rep Genet 2018:6308283. 10.1155/2018/6308283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caylor RC, Jin Y, Ackley BD (2013) The caenorhabditis elegans voltage-gated calcium channel subunits UNC-2 and UNC-36 and the calcium-dependent kinase UNC-43/CaMKII regulate neuromuscular junction morphology. Neural Dev 8:10. 10.1186/1749-8104-8-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Villa KL, Cha JW, So PT, Kubota Y, Nedivi E (2012) Clustered dynamics of inhibitory synapses and dendritic spines in the adult neocortex. Neuron 74:361–373. 10.1016/j.neuron.2012.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li L, Chen SR, Chen H, Xie JD, Sirrieh RE, MacLean DM, Zhang Y, Zhou MH, Jayaraman V, Pan HL (2018) The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep 22:2307–2321. 10.1016/j.celrep.2018.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Jiang M, Zhang B, Gokce O, Südhof TC (2017) Conditional deletion of all neurexins defines diversity of essential synaptic organizer functions for neurexins. Neuron 94:611–625.e4. 10.1016/j.neuron.2017.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chioza BA, et al. (2009) Genome wide high density SNP-based linkage analysis of childhood absence epilepsy identifies a susceptibility locus on chromosome 3p23–p14. Epilepsy Res 87:247–255. 10.1016/j.eplepsyres.2009.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choii G, Ko J (2015) Gephyrin: a central GABAergic synapse organizer. Exp Mol Med 47:e158–e158. 10.1038/emm.2015.5 [DOI] [PubMed] [Google Scholar]
- Christie SB, Miralles CP, De Blas AL (2002) GABAergic innervation organizes synaptic and extrasynaptic GABAA receptor clustering in cultured hippocampal neurons. J Neurosci 22:684–697. 10.1523/JNEUROSCI.22-03-00684.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA, Gu G (2005) Differential distribution of voltage-gated calcium channel alpha-2 delta (alpha2delta) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 491:246–269. 10.1002/cne.20693 [DOI] [PubMed] [Google Scholar]
- Davies A, Hendrich J, Van Minh AT, Wratten J, Douglas L, Dolphin AC (2007) Functional biology of the alpha(2)delta subunits of voltage-gated calcium channels. Trends Pharmacol Sci 28:220–228. 10.1016/j.tips.2007.03.005 [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515:209–215. 10.1038/nature13772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Biase V, Flucher BE, Obermair GJ (2009) Resolving sub-synaptic compartments with double immunofluorescence labeling in hippocampal neurons. J Neurosci Methods 176:78–84. 10.1016/j.jneumeth.2008.08.025 [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Huang EP, Stevens CF (1997) Very short-term plasticity in hippocampal synapses. Proc Natl Acad Sci U S A 94:14843–14847. 10.1073/pnas.94.26.14843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong N, Qi J, Chen G (2007) Molecular reconstitution of functional GABAergic synapses with expression of neuroligin-2 and GABAA receptors. Mol Cell Neurosci 35:14–23. 10.1016/j.mcn.2007.01.013 [DOI] [PubMed] [Google Scholar]
- Dudanova I, Tabuchi K, Rohlmann A, Südhof TC, Missler M (2007) Deletion of α-neurexins does not cause a major impairment of axonal pathfinding or synapse formation. J Comp Neurol 502:261–274. 10.1002/cne.21305 [DOI] [PubMed] [Google Scholar]
- Edvardson S, Oz S, Abulhijaa FA, Taher FB, Shaag A, Zenvirt S, Dascal N, Elpeleg O (2013) Early infantile epileptic encephalopathy associated with a high voltage gated calcium channelopathy. J Med Genet 50:118–123. 10.1136/jmedgenet-2012-101223 [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Özkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA (2009) Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139:380–392. 10.1016/j.cell.2009.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etemad S, Obermair GJ, Bindreither D, Benedetti A, Stanika R, Di Biase V, Burtscher V, Koschak A, Kofler R, Geley S, Wille A, Lusser A, Flockerzi V, Flucher BE (2014) Differential neuronal targeting of a new and two known calcium channel β4 subunit splice variants correlates with their regulation of gene expression. J Neurosci 34:1446–1461. 10.1523/JNEUROSCI.3935-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix R, Gurnett CA, De Waard M, Campbell KP (1997) Dissection of functional domains of the voltage-dependent Ca2+ channel alpha2delta subunit. J Neurosci 17:6884–6891. 10.1523/JNEUROSCI.17-18-06884.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell B, Eckrich S, Blum K, Eckrich T, Hecker D, Obermair GJ, Münkner S, Flockerzi V, Schick B, Engel J (2016) α2δ2 controls the function and trans-synaptic coupling of Cav1.3 channels in mouse inner hair cells and is essential for normal hearing. J Neurosci 36:11024–11036. 10.1523/JNEUROSCI.3468-14.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher BE, Andrews SB, Fleischer S, Marks AR, Caswell A, Powell JA (1993) Triad formation: organization and function of the sarcoplasmic reticulum calcium release channel and triadin in normal and dysgenic muscle in vitro. J Cell Biol 123:1161–1174. 10.1083/jcb.123.5.1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folci A, Steinberger A, Lee B, Stanika R, Scheruebel S, Campiglio M, Ramprecht C, Pelzmann B, Hell JW, Obermair GJ, Heine M, Di Biase V (2018) Molecular mimicking of C-terminal phosphorylation tunes the surface dynamics of caV1.2 calcium channels in hippocampal neurons. J Biol Chem 293:1040–1053. 10.1074/jbc.M117.799585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornito A, Bullmore ET (2015) Reconciling abnormalities of brain network structure and function in schizophrenia. Curr Opin Neurobiol 30:44–50. 10.1016/j.conb.2014.08.006 [DOI] [PubMed] [Google Scholar]
- Fuller-Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N, Muth JN, Mikala G, Petrashevskaya NN, Jordan MA, Zhang SP, Qin N, Flores CM, Isaacsohn I, Varadi M, Mori Y, Jones WK, Schwartz A (2009) Targeted disruption of the voltage-dependent calcium channel α2/δ-1-subunit. Am J Physiol Heart Circ Physiol 297:H117–H124. 10.1152/ajpheart.00122.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Schöpf CL, Obermair GJ (2015) Emerging evidence for specific neuronal functions of auxiliary calcium channel alpha(2)delta subunits. Gen Physiol Biophys 34:105–118. 10.4149/gpb_2014037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin D, Ali AB, Rampersaud N, Harkavyi A, Fuchs C, Whitton PS, Nairn AC, Jovanovic JN (2010) Dopamine-dependent tuning of striatal inhibitory synaptogenesis. J Neurosci 30:2935–2950. 10.1523/JNEUROSCI.4411-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM (2004) Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119:1013–1026. 10.1016/j.cell.2004.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine M, Thoumine O, Mondin M, Tessier B, Giannone G, Choquet D (2008) Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc Natl Acad Sci U S A 105:20947–20952. 10.1073/pnas.0804007106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobom M, Dai S, Marais E, Lacinova L, Hofmann F, Klugbauer N (2000) Neuronal distribution and functional characterization of the calcium channel α2δ-2 subunit. Eur J Neurosci 12:1217–1226. 10.1046/j.1460-9568.2000.01009.x [DOI] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA (2012) α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486:122–125. 10.1038/nature11033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hörtnagl H, Tasan RO, Wieselthaler A, Kirchmair E, Sieghart W, Sperk G (2013) Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience 236:345–372. 10.1016/j.neuroscience.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TN, Hsueh YP (2015) Brain-specific transcriptional regulator T-brain-1 controls brain wiring and neuronal activity in autism spectrum disorders. Front Neurosci 9:406. 10.3389/fnins.2015.00406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, et al. (2012) De novo gene disruptions in children on the autistic spectrum. Neuron 74:285–299. 10.1016/j.neuron.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Solís JM, Nicoll RA (1993) Local and diffuse synaptic actions of GABA in the hippocampus. Neuron 10:165–175. 10.1016/0896-6273(93)90308-E [DOI] [PubMed] [Google Scholar]
- Ivanov SV, Ward JM, Tessarollo L, McAreavey D, Sachdev V, Fananapazir L, Banks MK, Morris N, Djurickovic D, Devor-Henneman DE, Wei MH, Alvord GW, Gao B, Richardson JA, Minna JD, Rogawski MA, Lerman MI (2004) Cerebellar ataxia, seizures, premature death, and cardiac abnormalities in mice with targeted disruption of the Cacna2d2 gene. Am J Pathol 165:1007–1018. 10.1016/S0002-9440(10)63362-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Bogdanov YD, Magnus C, Saliba RS, Kittler JT, Haydon PG, Moss SJ (2005) Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J Neurosci 25:10469–10478. 10.1523/JNEUROSCI.2267-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G (2006) Culturing hippocampal neurons. Nat Protoc 1:2406–2415. 10.1038/nprot.2006.356 [DOI] [PubMed] [Google Scholar]
- Kang Y, Zhang X, Dobie F, Wu H, Craig AM (2008) Induction of GABAergic postsynaptic differentiation by α-neurexins. J Biol Chem 283:2323–2334. 10.1074/jbc.M703957200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R (1968) The role of calcium in neuromuscular facilitation. J Physiol 195:481–492. 10.1113/jphysiol.1968.sp008469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerov V, Laird JG, Joiner ML, Knecht S, Soh D, Hagen J, Gardner SH, Gutierrez W, Yoshimatsu T, Bhattarai S, Puthussery T, Artemyev NO, Drack AV, Wong RO, Baker SA, Lee A (2018) α2δ-4 is required for the molecular and structural organization of rod and cone photoreceptor synapses. J Neurosci 38:6145–6160. 10.1523/JNEUROSCI.3818-16.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Lacinová L, Marais E, Hobom M, Hofmann F (1999) Molecular diversity of the calcium channel alpha2delta subunit. J Neurosci 19:684–691. 10.1523/JNEUROSCI.19-02-00684.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurshan PT, Oztan A, Schwarz TL (2009) Presynaptic alpha2delta-3 is required for synaptic morphogenesis independent of its Ca2+-channel functions. Nat Neurosci 12:1415–1423. 10.1038/nn.2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana B, Schlick B, Martin S, Pratt WS, Page KM, Goncalves L, Rahman W, Dickenson AH, Bauer CS, Dolphin AC (2014) Differential upregulation in DRG neurons of an alpha2delta-1 splice variant with a lower affinity for gabapentin after peripheral sensory nerve injury. Pain 155:522–533. 10.1016/j.pain.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardi-Studler B, Fritschy JM (2007) Matching of pre- and postsynaptic specializations during synaptogenesis. Neuroscientist 13:115–126. 10.1177/1073858406296803 [DOI] [PubMed] [Google Scholar]
- Lee E, Lee J, Kim E (2017) Excitation/Inhibition imbalance in animal models of autism spectrum disorders. Biol Psychiatry 81:838–847. 10.1016/j.biopsych.2016.05.011 [DOI] [PubMed] [Google Scholar]
- Li J, Han W, Pelkey KA, Duan J, Mao X, Wang YX, Craig MT, Dong L, Petralia RS, McBain CJ, Lu W (2017) Molecular dissection of Neuroligin 2 and Slitrk3 reveals an essential framework for GABAergic synapse development. Neuron 96:808–826.e8. 10.1016/j.neuron.2017.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C (2004) Interneurons of the neocortical inhibitory system. Nat Rev Neurosci 5:793–807. 10.1038/nrn1519 [DOI] [PubMed] [Google Scholar]
- Mastrolia V, Flucher SM, Obermair GJ, Drach M, Hofer H, Renström E, Schwartz A, Striessnig J, Flucher BE, Tuluc P (2017) Loss of α2δ-1 calcium channel subunit function increases the susceptibility for diabetes. Diabetes 66:897–907. 10.2337/db16-0336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC (2003) α-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423:939–948. 10.1038/nature01755 [DOI] [PubMed] [Google Scholar]
- Moons T, De Hert M, Gellens E, Gielen L, Sweers K, Jacqmaert S, van Winkel R, Vandekerckhove P, Claes S (2016) Genetic evaluation of schizophrenia using the Illumina HumanExome chip. PLoS One 11:e0150464. 10.1371/journal.pone.0150464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münster-Wandowski A, Zander JF, Richter K, Ahnert-Hilger G (2016) Co-existence of functionally different vesicular neurotransmitter transporters. Front Synaptic Neurosci 8:4. 10.3389/fnsyn.2016.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely GG, et al. (2010) A genome-wide drosophila screen for heat nociception identifies α2δ3 as an evolutionarily conserved pain gene. Cell 143:628–638. 10.1016/j.cell.2010.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert C, Schneider R, Klatt O, Reissner C, Repetto D, Biermann B, Niesmann K, Missler M, Heine M (2015) Regulated dynamic trafficking of neurexins inside and outside of synaptic terminals. J Neurosci 35:13629–13647. 10.1523/JNEUROSCI.4041-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Benke D, Fritschy JM, Somogyi P (1996a) Differential synaptic localization of two major gamma-aminobutyric acid type A receptor alpha subunits on hippocampal pyramidal cells. Proc Natl Acad Sci U S A 93:11939–11944. 10.1073/pnas.93.21.11939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Stephenson FA, Somogyi P (1996b) The alpha 6 subunit of the GABAA receptor is concentrated in both inhibitory and excitatory synapses on cerebellar granule cells. J Neurosci 16:103–114. 10.1523/JNEUROSCI.16-01-00103.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermair GJ, Kaufmann WA, Knaus HG, Flucher BE (2003) The small conductance Ca2+-activated K+ channel SK3 is localized in nerve terminals of excitatory synapses of cultured mouse hippocampal neurons. Eur J Neurosci 17:721–731. 10.1046/j.1460-9568.2003.02488.x [DOI] [PubMed] [Google Scholar]
- Obermair GJ, Szabo Z, Bourinet E, Flucher BE (2004) Differential targeting of the L-type Ca2+ channel alpha 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur J Neurosci 19:2109–2122. 10.1111/j.0953-816X.2004.03272.x [DOI] [PubMed] [Google Scholar]
- Obermair GJ, Schlick B, Di Biase V, Subramanyam P, Gebhart M, Baumgartner S, Flucher BE (2010) Reciprocal interactions regulate targeting of calcium channel beta subunits and membrane expression of alpha1 subunits in cultured hippocampal neurons. J Biol Chem 285:5776–5791. 10.1074/jbc.M109.044271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Bauer CS, Nieto-Rostro M, Margas W, Ferron L, Chaggar K, Crews K, Ramirez JD, Bennett DL, Schwartz A, Dickenson AH, Dolphin AC (2013) α2δ-1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. J Neurosci 33:16412–16426. 10.1523/JNEUROSCI.1026-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penrod RD, Kourrich S, Kearney E, Thomas MJ, Lanier LM (2011) An embryonic culture system for the investigation of striatal medium spiny neuron dendritic spine development and plasticity. J Neurosci Methods 200:1–13. 10.1016/j.jneumeth.2011.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A. (2002) Examining neocortical circuits: some background and facts. J Neurocytol 31:183–193. 10.1023/A:1024157522651 [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- Pettem KL, Yokomaku D, Takahashi H, Ge Y, Craig AM (2013) Interaction between autism-linked MDGAs and neuroligins suppresses inhibitory synapse development. J Cell Biol 200:321–336. 10.1083/jcb.201206028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippucci T, Parmeggiani A, Palombo F, Maresca A, Angius A, Crisponi L, Cucca F, Liguori R, Valentino ML, Seri M, Carelli V (2013) A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS One 8:e82154. 10.1371/journal.pone.0082154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirone A, Kurt S, Zuccotti A, Rüttiger L, Pilz P, Brown DH, Franz C, Schweizer M, Rust MB, Rübsamen R, Friauf E, Knipper M, Engel J (2014) α2δ-3 is essential for normal structure and function of auditory nerve synapses and is a novel candidate for auditory processing disorders. J Neurosci 34:434–445. 10.1523/JNEUROSCI.3085-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kähler A, Duncan L, Stahl E, Genovese G, Fernández E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PK, Banks E, Shakir K, et al. (2014) A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506:185–190. 10.1038/nature12975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Cha EM, Craig AM (2000) Mismatched appositions of presynaptic and postsynaptic components in isolated hippocampal neurons. J Neurosci 20:8344–8353. 10.1523/JNEUROSCI.20-22-08344.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner C, Runkel F, Missler M (2013) Neurexins. Genome Biol 14:213. 10.1186/gb-2013-14-9-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher WC, Kim N, Koh S, Choi JE, Mitev P, Spence EF, Pilaz LJ, Wang D, Feng G, Silver DL, Soderling SH, Yin HH, Eroglu C (2018) Thrombospondin receptor α2δ-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J Cell Biol 217:3747–3765. 10.1083/jcb.201802057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DH, Mejias-Aponte CA, Zhang S, Wang HL, Hoffman AF, Lupica CR, Morales M (2014) Single rodent mesohabenular axons release glutamate and GABA. Nat Neurosci 17:1543–1551. 10.1038/nn.3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlick B, Flucher BE, Obermair GJ (2010) Voltage-activated calcium channel expression profiles in mouse brain and cultured hippocampal neurons. Neuroscience 167:786–798. 10.1016/j.neuroscience.2010.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M, Greenberger V, Korkotian E (2003) Formation of dendritic spines in cultured striatal neurons depends on excitatory afferent activity. Eur J Neurosci 17:2573–2585. 10.1046/j.1460-9568.2003.02696.x [DOI] [PubMed] [Google Scholar]
- Shrivastava AN, Triller A, Sieghart W (2011) GABAA receptors: post-synaptic co-localization and cross-talk with other receptors. Front Cell Neurosci 5:7. 10.3389/fncel.2011.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigler A, Oh WC, Imig C, Altas B, Kawabe H, Cooper BH, Kwon HB, Rhee JS, Brose N (2017) Formation and maintenance of functional spines in the absence of presynaptic glutamate release. Neuron 94:304–311.e4. 10.1016/j.neuron.2017.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanika R, Campiglio M, Pinggera A, Lee A, Striessnig J, Flucher BE, Obermair GJ (2016) Splice variants of the CaV1.3 L-type calcium channel regulate dendritic spine morphology. Sci Rep 6:34528. 10.1038/srep34528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanits H, Milenkovic I, Mahr N, Pataraia E, Hainfellner JA, Kovacs GG, Sieghart W, Yilmazer-Hanke D, Czech T (2018) GABA A receptor subunits in the human amygdala and hippocampus: immunohistochemical distribution of 7 subunits. J Comp Neurol 526:324–348. 10.1002/cne.24337 [DOI] [PubMed] [Google Scholar]
- Studler B, Sidler C, Fritschy JM (2005) Differential regulation of GABAA receptor and gephyrin postsynaptic clustering in immature hippocampal neuronal cultures. J Comp Neurol 484:344–355. 10.1002/cne.20472 [DOI] [PubMed] [Google Scholar]
- Subramanyam P, Obermair GJ, Baumgartner S, Gebhart M, Striessnig J, Kaufmann WA, Geley S, Flucher BE (2009) Activity and calcium regulate nuclear targeting of the calcium channel beta4b subunit in nerve and muscle cells. Channels (Austin) 3:343–355. 10.4161/chan.3.5.9696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. (2017) Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171:745–769. 10.1016/j.cell.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, Dupraz S, Laskowski CJ, Xue J, Ulas T, Beyer M, Schultze JL, Bradke F (2016) The calcium channel subunit Alpha2delta2 suppresses axon regeneration in the adult CNS. Neuron 92:419–434. 10.1016/j.neuron.2016.09.026 [DOI] [PubMed] [Google Scholar]
- Tong XJ, López-Soto EJ, Li L, Liu H, Nedelcu D, Lipscombe D, Hu Z, Kaplan JM (2017) Retrograde synaptic inhibition is mediated by α-neurexin binding to the α2δ subunits of N-type calcium channels. Neuron 95:326–340.e5. 10.1016/j.neuron.2017.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valence S, Cochet E, Rougeot C, Garel C, Chantot-Bastaraud S, Lainey E, Afenjar A, Barthez MA, Bednarek N, Doummar D, Faivre L, Goizet C, Haye D, Heron B, Kemlin I, Lacombe D, Milh M, Moutard ML, Riant F, Robin S, et al. (2018) Exome sequencing in congenital ataxia identifies two new candidate genes and highlights a pathophysiological link between some congenital ataxias and early infantile epileptic encephalopathies. Genet Med. Advance online publication. Retrieved July 12, 2018. doi: 10.1038/s41436-018-0089-2. [DOI] [PubMed] [Google Scholar]
- Van Den Bossche MJ, Strazisar M, De Bruyne S, Bervoets C, Lenaerts A-S, De Zutter S, Nordin A, Norrback K-F, Goossens D, De Rijk P, Green EK, Grozeva D, Mendlewicz J, Craddock N, Sabbe BG, Adolfsson R, Souery D, Del-Favero J (2012) Identification of a CACNA2D4 deletion in late onset bipolar disorder patients and implications for the involvement of voltage-dependent calcium channels in psychiatric disorders. Am J Med Genet Part B Neuropsychiatr Genet 159B:465–475. 10.1002/ajmg.b.32053 [DOI] [PubMed] [Google Scholar]
- Vergult S, Dheedene A, Meurs A, Faes F, Isidor B, Janssens S, Gautier A, Le Caignec C, Menten B (2015) Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. Eur J Hum Genet 23:628–632. 10.1038/ejhg.2014.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Fehlhaber KE, Sarria I, Cao Y, Ingram NT, Guerrero-Given D, Throesch B, Baldwin K, Kamasawa N, Ohtsuka T, Sampath AP, Martemyanov KA (2017) The auxiliary calcium channel subunit α2δ4 is required for axonal elaboration, synaptic transmission, and wiring of rod photoreceptors. Neuron 93:1359–1374.e6. 10.1016/j.neuron.2017.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q, Yan N (2016) Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 A resolution. Nature 537:191–196. 10.1038/nature19321 [DOI] [PubMed] [Google Scholar]
- Zhou C, Luo ZD (2015) Nerve injury-induced calcium channel alpha-2-delta-1 protein dysregulation leads to increased pre-synaptic excitatory input into deep dorsal horn neurons and neuropathic allodynia. Eur J Pain 19:1267–1276. 10.1002/ejp.656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]