

Abstract
Tuberous sclerosis complex (TSC) and focal cortical dysplasia (FCD) are focal malformations of cortical development (FMCDs) that are highly associated with intractable epilepsy. TSC and FCD are mTORopathies caused by a spectrum of pathogenic variants in the mechanistic target of rapamycin (mTOR) pathway genes leading to differential activation of mTOR signaling. However, whether the degree of mTOR hyperactivity influences disease severity remains unclear. Here, we examined the effects of differential mTOR hyperactivity levels on epilepsy and associated neuropathology in a mouse model of TSC and FCD. Constitutively active Rheb (RhebCA), the canonical activator of mTOR complex 1 (mTORC1), was expressed in mouse embryos of either sex via in utero electroporation at low, intermediate, and high concentrations to induce different mTORC1 activity levels in developing cortical neurons. We found that RhebCA expression induced mTORC1 hyperactivation and increased neuronal soma size and misplacement in a dose-dependent manner. No seizures were detected in the low RhebCA mice, whereas the intermediate and high RhebCA mice displayed spontaneous, recurrent seizures that significantly increased with higher RhebCA concentrations. Seizures were associated with a global increase in microglial activation that was notably higher in the regions containing RhebCA-expressing neurons. These data demonstrate that neuronal mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in experimental TSC and FCD. Overall, these findings highlight the importance of evaluating the outcome of individual variants on mTOR activity levels and support personalized medicine strategies based on patient variants and mTOR activity level for TSC, FCD, and potentially other mTORopathies.
SIGNIFICANCE STATEMENT Tuberous sclerosis complex (TSC) and focal cortical dysplasia (FCD) are epileptogenic cortical malformations caused by pathogenic variants in mechanistic target of rapamycin (mTOR) pathway genes leading to differential mTOR hyperactivation. Here, we present novel findings that neuronal mTOR hyperactivity levels correlate with the severity of epilepsy and associated neuropathology in a mouse model of TSC and FCD. Our findings suggest the need to evaluate the outcome of individual variants on mTOR activity levels in clinical assessments and support personalized medicine strategies based on patient variants and mTOR activity level. Additionally, we present useful modifications to a previously described mouse model of TSC and FCD that allows for titration of seizure frequency and generation of a mild to severe epilepsy phenotype as applicable for preclinical drug testing and mechanistic studies.
Keywords: epilepsy, focal cortical dysplasia, microglia, mTOR, personalized medicine, tuberous sclerosis complex
Introduction
Tuberous sclerosis complex (TSC) and focal cortical dysplasia (FCD) are neurodevelopmental disorders characterized by focal malformations of cortical development (FMCDs) that are highly associated with epilepsy and cognitive impairments (Leventer et al., 2008). TSC- and FCD-related epilepsies are often intractable to medical therapy and account for the majority of childhood epilepsy cases requiring surgical treatment (Harvey et al., 2008; Blümcke et al., 2017). FMCDs in TSC, known as tubers, and in FCD, particularly type II (FCDII), share a number of neuropathological features, including cortical dyslamination due to neuronal misplacement; the presence of enlarged, dysmorphic neurons; glial activation; and enhanced mechanistic target of rapamycin (mTOR) signaling (Aronica et al., 2012; Vezzani et al., 2012; Crino, 2015). The mTOR pathway is a key regulator of cell growth and development and aberrant mTOR signaling is implicated in the cellular and molecular changes leading to cortical malformations and epilepsy in TSC and FCD (Lipton and Sahin, 2014).
TSC tubers and FCDII are caused by brain somatic (with or without germline) mutations in regulators of the mTOR pathway, leading to constitutive mTOR hyperactivation (Crino, 2015; Marsan and Baulac, 2018). TSC tubers result from mutations in TSC1 or TSC2 (Orlova and Crino, 2010), whereas FCDII has more recently been linked to mutations in MTOR, DEPDC5, NPRL2, NPRL3, PIK3CA, PTEN, TSC1, and TSC2 (Marsan and Baulac, 2018). More than 2500 pathogenic variants in TSC1 and TSC2 and numerous variants in other mTOR pathway genes have been identified to date (Northrup et al., 1999; Marsan and Baulac, 2018). Different variants can lead to different clinical phenotypes, with some patients presenting with few signs and symptoms and others exhibiting severe manifestations (Curatolo et al., 2015; Iffland and Crino, 2016; Rosset et al., 2017). The broad phenotypic spectrum in TSC and FCD can be attributed to differences in the developmental time points at which the mutations occur, the proportion of cells with the mutations, or the outcome of the gene variants on mTOR activity. Indeed, data from studies in patient tissue and cell lines suggest that distinct variants may confer different degrees of mTOR pathway activation (Nellist et al., 2005; Grabiner et al., 2014; Lim et al., 2015, 2017; Mirzaa et al., 2016). However, to our knowledge, no studies have examined the relationship between cellular mTOR activity levels and the severity of TSC and FCD phenotypes. Although mTOR hyperactivation in animal models consistently recapitulates human TSC and FCD features, including neuronal hypertrophy, cortical dyslamination, and epilepsy (Wong and Roper, 2016), it is unknown whether this is an all-or-nothing occurrence in which mTOR hyperactivation induces the phenotypes after reaching a certain threshold or a graded event in which increasing mTOR activity levels leads to progressively worsening phenotypes. Given that the mTOR pathway is an emerging therapeutic target for TSC- and FCD-related epilepsy, understanding how cellular levels of mTOR hyperactivity influence disease outcome may be crucial for establishing optimal treatment strategies.
In this study, we investigated whether there is a correlation between mTOR hyperactivity levels and the severity of disease phenotypes using experimental FMCDs recapitulating TSC tubers and FCDII in mice. In this model, a constitutive active Rheb (RhebCA), the canonical activator of mTOR complex 1 (mTORC1), was expressed in developing cortical neurons via in utero electroporation to mimic persistent mTOR activation in disease states. By using different concentrations of RhebCA plasmids, we were able to induce different levels of mTOR hyperactivation in a dose-dependent manner. We found that increasing mTOR hyperactivity levels caused a corresponding increase in neuronal soma size and misplacement. Additionally, increased mTOR hyperactivity levels were associated with higher seizure frequencies. These findings highlight the importance of evaluating individual gene variants in the context of mTOR activity levels and support personalized medicine strategies based on patient variants and mTOR activity level.
Materials and Methods
Animals.
All animal procedures were performed in accordance with Yale University Institutional Animal Care and Use Committee's regulations. All experiments were performed on CD-1 mice of either sex (Charles River Laboratories).
In utero electroporation and DNA plasmids.
Timed-pregnant embryonic day 15 (E15) (±0.5) mice were anesthetized with isoflurane and a midline laparotomy was performed to expose the uterine horns. A DNA plasmid solution consisting of either pCAG-GFP (Addgene, catalog #11150; 4.8 μg/μl) or pCAG-GFP (1–3 μg/μl) + pCAG-RhebCA (also known as pCAG-RhebS16H; gift from T. Maehama and K. Hanada; Maehama et al., 2008, 1.5, 2.5, or 3.5 μg/μl) together with 0.03% Fast Green dye (for visualizing injection) was injected (1–1.5 μl) into the right lateral ventricle of each embryo using a glass pipette. Following injection, a 5 mm tweezer-type electrode was positioned on the embryo head and six 42 V, 50 ms pulses at 950 ms intervals were applied using a pulse generator (ECM830; BTX) to electroporate the plasmid constructs into neural progenitor cells. We preferentially targeted expression in layer 2/3 (L2/3) in the medial prefrontal cortex (mPFC). The uterine horns were returned to the abdominal cavity and embryos were allowed to continue with their normal development. Pups were screened at postnatal day 0 (P0) to verify successful electroporation via expression of GFP on a fluorescence-enabled stereomicroscope (SZX16; Olympus) before recruitment for video-EEG monitoring.
EEG head mount implantation.
Prefabricated EEG head mounts (Pinnacle Technology, catalog #8201-EEG) were implanted in 2- to 4-month-old mice. Mice were anesthetized with isoflurane and positioned on a stereotaxic frame using ear bars. A rostrocaudal midline incision was made in the skin to expose the skull surface. Four pilot holes (two bilateral holes 1 mm anterior to bregma and two bilateral holes 5 mm posterior to bregma, each 1.5 mm lateral to sagittal suture) were tapped through the skull to dura using a 23-gauge needle to accommodate the EEG head mount. The head mount was attached on top of the skull by threading four stainless steel screws (Pinnacle Technology, catalog #8209) into the pilot holes. Silver conductive paint (Electron Microscopy Science) was applied around the screw threads to ensure a solid connection with the head mount. The entire implant was insulated using dental acrylic. Mice were allowed to recover in their home cages for at least 5 days before video-EEG monitoring.
Video-EEG monitoring and analysis.
Mice were housed in individual recording chambers in a light-, temperature-, and humidity-controlled room during video-EEG monitoring. Mice were freely moving inside the chambers and had ad libitum access to food and water. Synchronous video-EEG recording was acquired using a three-channel EEG tethered system (Pinnacle Technology, catalog #8200-K1-iSE3) and Sirenia Acquisition software (Pinnacle Technology). Mice were continuously recorded 24 h/day for 5–7 consecutive days. The average recording duration per animal was 120.5 ± 29.6 h (±SD).
Seizure frequency and duration were analyzed using Sirenia Seizure Basic software (Pinnacle Technology). All analyses were performed blinded to experimental groups. The entire EEG traces were manually reviewed for the occurrence of seizures, defined as a sudden onset of high-amplitude activity with a characteristic pattern of progressive frequency and amplitude changes over the course of the event lasting ≥10 s. Seizure onset was defined by high-amplitude firing >2× baseline and seizure termination was defined by a return to baseline activity. Video data were inspected for behavioral correlates including myoclonic jerks, tonic-clonic activities, convulsions, and loss of postural control (rearing and falling) and were used as secondary verification of seizures. For each individual animal, the mean number of seizures per day was obtained by dividing the total number of seizures by total recording hours and multiplied by 24. For each individual animal, mean seizure duration was obtained by measuring and averaging the duration of each seizure within that animal.
The average starting ages of EEG recording for each of the experimental groups were as follows: 12.2 ± 0.93 (control), 11.3 ± 0.27 (low RhebCA), 11.2 ± 0.38 (intermediate RhebCA), and 14.8 ± 3.80 (high RhebCA) weeks (± SD). Although the average age was higher in the high RhebCA group (range 9.4–19 weeks) compared with the other RhebCA groups (p = 0.0058; one-way ANOVA with Tukey's post hoc test), there was no significant correlation between seizures and age within this group for the given age range (r = 0.3756, p = 0.2272; Spearman's rank–order correlation, data not shown).
Brain slice preparation and immunofluorescence staining.
After the end of video-EEG monitoring, mice were deeply anesthetized with sodium pentobarbital (85 mg/kg, i.p.) and transcardially perfused with ice-cold PBS, pH 7.4, followed by ice-cold 4% PFA diluted in PBS. Whole brains were dissected and postfixed in 4% PFA for 2 h and then cryoprotected in 30% sucrose diluted in PBS for 24–48 h at 4°C until brains sank.
Brains were serially cut into 50-μm-thick coronal sections using a freezing microtome and stored in PBS + 0.01% sodium azide. One series of sections was mounted on slides and coverslipped with mounting media (ProLong Diamond Antifade Mountant; Invitrogen) to evaluate neuronal soma size, placement, and count. Other series of sections were used for immunofluorescence staining to evaluate expression of phosphorylated ribosomal protein S6 (p-S6; mTORC1 marker) and ionized calcium-binding adaptor molecule 1 (IBA1; microglia marker). For staining, free-floating sections were washed in PBS + 0.1% Triton X-100 (PBS-T) for 2 × 10 min and permeabilized in PBS + 0.3% Triton X-100 for 20 min. Sections were then incubated in blocking buffer (5% goat serum + 0.3% BSA + 0.3% Triton X-100 in PBS) for 1 h at room temperature followed by incubation in primary antibodies (anti-rabbit p-S6, S240/244, Cell Signaling Technology, catalog #5364, 1:2000, or anti-rabbit IBA1, Wako, catalog #019–19741, 1:1000, diluted in blocking buffer) overnight at 4°C. The following day, sections were washed in PBS-T for 3 × 10 min and incubated in secondary antibodies (goat anti-rabbit IgG-647 plus; Invitrogen, catalog #A3273, 1:1000, diluted in blocking buffer) for 1 h at room temperature. Sections were then washed in PBS-T for 3 × 10 min, incubated in DAPI for 10 min, and rinsed in PBS before being air-dried and coverslipped with mounting media (ProLong Diamond Antifade Mountant; Invitrogen).
Microscopy and image analysis.
Fluorescence images were acquired using a confocal microscope (Fluoview FV1000; Olympus) with 10× and 20× objective lenses. All image analyses were done using ImageJ and were performed blinded to experimental groups. Cell counts were performed using the Cell Counter plugin for ImageJ. Representative images were prepared using ImageJ and Adobe Photoshop. All images meant for comparison were uniformly processed.
Cortical p-S6 staining intensity was quantified from single optical section images containing both the ipsilateral (electroporated) and contralateral cortices within the same plane of view. Two to three brain sections were analyzed per animal. For each section, the mean gray value was measured in three randomly selected, nonoverlapping 120 μm × 120 μm region of interests (ROIs) surrounding the electroporated cells on the ipsilateral cortex. Three ROIs surrounding non-electroporated cells on the contralateral cortex were also measured as internal controls. ROIs on the contralateral cortex were chosen to match the position of the ROIs on the ipsilateral cortex. All ROIs were selected within 300 μm from the pial surface. Cellular p-S6 staining intensity was quantified from single optical section images of either the ipsilateral or contralateral cortex. Two to three brain sections were analyzed per animal. For each section, the mean gray value was measured in all visible, nonoverlapping GFP+ cells on the ipsilateral cortex (by tracing around the cells) and in position-matched cells on the contralateral cortex. Sixteen to 101 neurons were analyzed per animal. Data for both cortical and cellular p-S6 staining intensity are reported as a percentage of contralateral cortex to avoid confounding effects of variabilities in the quality of sectioning or staining and for directly evaluating RhebCA-induced p-S6 changes.
Neuron size and placement were analyzed from maximum intensity projection images created from a 20-μm-thick z-stack of optical sections taken at 2 μm increments of the ipsilateral cortex. Neuron size was quantified by tracing the soma and measuring the area of GFP+ cells. A total of 21–60 neurons from two to three brain sections were analyzed per animal. The percentage of neurons in L2/3 was quantified by counting all GFP+ cells within an 850 μm × 850 μm ROI surrounding the ipsilateral cortex. Cells within 300 μm from the pial surface were considered correctly located in L2/3, whereas cells outside that boundary were considered misplaced. The distribution of neurons was quantified by dividing the cortex into 10 evenly spaced bins (bin size: 85 μm width × 675 μm height) parallel to the pial surface in coronal sections and counting the number of GFP+ cells in each bin. Only cells within the gray matter of the cortex were quantified. One brain section was analyzed per animal for cell placement analyses. Data are shown as the percentage of total GFP+ cells.
To determine the number of electroporated cells per brain section, a rostrocaudal series of 10–17 coronal sections at 300 μm intervals were scanned on a fluorescence microscope for the section with the most apparent number GFP+ cells for each animal. Single optical section images were then acquired on a confocal microscope for the selected brain sections and the number of GFP+ cells in the cortex was determined.
Microglia density and size were analyzed from sum slice and maximum intensity projection images created from a 20-μm-thick z-stack of optical sections taken at 2 μm increments, respectively. Microglia density was determined by counting all IBA1+ cells within a 500 μm × 500 μm ROI surrounding each of the ipsilateral and contralateral cortices. Two brain sections were analyzed per animal. Microglia size was quantified by tracing the soma and measuring the area of all nonoverlapping IBA1+ cells within a 200 μm × 200 μm ROI surrounding the ipsilateral cortex. A total of 25–51 cells from two to three brain sections were analyzed per animal.
Experimental design and statistical analysis.
A total of 7 control, 8 low RhebCA, 7 intermediate RhebCA, and 12 high RhebCA mice were monitored with video-EEG recording. Animals were then euthanized for post hoc histology and immunohistochemical analyses. Tissue was available for 7 control, 8 low RhebCA, 6 intermediate RhebCA mice, and 5 high RhebCA mice. Only four high RhebCA mice had complete data for seizure correlation studies. Tissue from the high RhebCA group was not available for microglia analysis and is therefore not included. Multiple brain sections were evaluated per animal. The exact number of slices and cells evaluated per animal for each experiment are outlined above in the “Microscopy and image analysis” section. All statistical analyses were performed using Prism 7 software (GraphPad Software). Differences among groups were assessed by one-way ANOVA, two-way repeated-measures ANOVA (with Tukey's or Bonferroni's post hoc test), Pearson's product–moment correlation, Spearman's rank–order correlation, or Mann–Whitney U test, as appropriate. Individual tests are specified in the results, figure legends, and Table 1. Repeat measures within an animal were averaged to yield one sample replicate for statistical analysis. For all analyses, sample size (n) refers to number of animals. The significance level was set at p < 0.05. All data are presented as mean ± SEM.
Table 1.
Summary of statistical tests
| Figure | Test | Statistical value | p-value | n (no. of animals) |
Post hoc comparisons |
|||
|---|---|---|---|---|---|---|---|---|
| Groups compared | Mean 1 | Mean 2 | Significance | |||||
| 2C | One-way ANOVA | F(3,22) = 16.68 | <0.0001 | 7 (control) | Control vs low RhebCA | 95.6 | 127.8 | * |
| 8 (low RhebCA) | Control vs int RhebCA | 95.6 | 143.7 | ** | ||||
| 6 (int RhebCA) | Control vs high RhebCA | 95.6 | 180.6 | **** | ||||
| 5 (high RhebCA) | Low vs int RhebCA | 127.8 | 143.7 | ns | ||||
| Low vs high RhebCA | 127.8 | 180.6 | ** | |||||
| Int vs high RhebCA | 143.7 | 180.6 | * | |||||
| 2E | One-way ANOVA | F(3,22) = 14.92 | <0.0001 | 7 (control) | Control vs low RhebCA | 88.9 | 156.4 | ** |
| 8 (low RhebCA) | Control vs int RhebCA | 88.9 | 143.9 | * | ||||
| 6 (int RhebCA) | Control vs high RhebCA | 88.9 | 207.9 | **** | ||||
| 5 (high RhebCA) | Low vs int RhebCA | 156.4 | 143.9 | ns | ||||
| Low vs high RhebCA | 156.4 | 207.9 | * | |||||
| Int vs high RhebCA | 143.9 | 207.9 | * | |||||
| 2F | Pearson's product–moment | r = 0.8501 | <0.0001 | 7 (control) | ||||
| correlation | 8 (low RhebCA) | |||||||
| 6 (int RhebCA) | ||||||||
| 5 (high RhebCA) | ||||||||
| 2H | One-way ANOVA | F(3,22) = 284.6 | <0.0001 | 7 (control) | Control vs low RhebCA | 82.3 | 232.9 | **** |
| 8 (low RhebCA) | Control vs int RhebCA | 82.3 | 390.9 | **** | ||||
| 6 (int RhebCA) | Control vs high RhebCA | 82.3 | 486.0 | **** | ||||
| 5 (high RhebCA) | Low vs int RhebCA | 232.9 | 390.9 | **** | ||||
| Low vs high RhebCA | 232.9 | 486.0 | **** | |||||
| Int vs high RhebCA | 390.9 | 486.0 | **** | |||||
| 2I | Pearson's product–moment | r = 0.8257 | <0.0001 | 7 (control) | ||||
| correlation | 8 (low RhebCA) | |||||||
| 6 (int RhebCA) | ||||||||
| 5 (high RhebCA) | ||||||||
| 3D | One-way ANOVA | F(3,22) = 17.77 | <0.0001 | 7 (control) | Control vs low RhebCA | 99.6 | 94.9 | ns |
| 8 (low RhebCA) | Control vs int RhebCA | 99.6 | 56.0 | **** | ||||
| 6 (int RhebCA) | Control vs high RhebCA | 99.6 | 62.3 | *** | ||||
| 5 (high RhebCA) | Low vs int RhebCA | 94.9 | 56.0 | *** | ||||
| Low vs high RhebCA | 94.9 | 62.3 | ** | |||||
| Int vs high RhebCA | 56.0 | 62.3 | ns | |||||
| 3F | Two-way repeated- | Interaction | <0.0001 | 7 (control) | See graph for relevant significant | |||
| measures ANOVA | F(27,198) = 7.371 | 8 (low RhebCA) | post hoc comparisons | |||||
| Row effect: F(9,198) = 42.96 | <0.0001 | 6 (int RhebCA) | ||||||
| Column effect: F(3,22) = 0.6691 | 0.5800 | 5 (high RhebCA) | ||||||
| Subjects (matching): F(22,198) = 6.304e-006 | >0.9999 | |||||||
| 4B | Spearman's rank–order | r = 0.8709 | <0.0001 | 7 (control) | ||||
| correlation | 8 (low RhebCA) | |||||||
| 7 (int RhebCA) | ||||||||
| 12 (high RhebCA) | ||||||||
| 4C | Spearman's rank–order | r = 0.6818 | 0.0018 | 8 (low RhebCA) | ||||
| correlation | 6 (int RhebCA) | |||||||
| 4 (high RhebCA) | ||||||||
| 4D | Spearman's rank–order | r = 0.8693 | <0.0001 | 8 (low RhebCA) | ||||
| correlation | 6 (int RhebCA) | |||||||
| 4 (high RhebCA) | ||||||||
| 4E | Mann-Whitney U | U = 15 | 0.0208 | 7 (int RhebCA) | ||||
| 12 (high RhebCA) | ||||||||
| 4F | Mann-Whitney U | U = 27 | 0.4240 | 6 (int RhebCA) | ||||
| 12 (high RhebCA) | ||||||||
| 5A | One-way ANOVA | F(3,22) = 4.783 | 0.0103 | 7 (control) | Control vs low RhebCA | 74.4 | 64.5 | ns |
| 8 (low RhebCA) | Control vs int RhebCA | 74.4 | 138.5 | * | ||||
| 6 (int RhebCA) | Control vs high RhebCA | 74.4 | 88.6 | ns | ||||
| 5 (high RhebCA) | Low vs int RhebCA | 64.5 | 138.5 | ** | ||||
| Low vs high RhebCA | 64.5 | 88.6 | ns | |||||
| Int vs high RhebCA | 138.5 | 88.6 | ns | |||||
| 5B | Spearman's rank–order | r = 0.4294 | 0.0754 | 8 (low RhebCA) | ||||
| correlation | 6 (int RhebCA) | |||||||
| 4 (high RhebCA) | ||||||||
| 6B | One-way ANOVA | F(2,18) = 6.166 | 0.0091 | 7 (control) | Control vs low RhebCA | 131.7 | 144.7 | ns |
| 8 (low RhebCA) | Control vs int RhebCA | 131.7 | 167.1 | ** | ||||
| 6 (int RhebCA) | Low vs int RhebCA | 144.7 | 167.1 | ns | ||||
| 6C | Spearman's rank–order | r = 0.5502 | 0.0441 | 8 (low RhebCA) | ||||
| correlation | 6 (int RhebCA) | |||||||
| 6D | Two-way repeated- | Interaction: | 0.0414 | 7 (control) | Ipsi-contra | |||
| measures ANOVA | F(2,16) = 3.913 | 8 (low RhebCA) | Control | 131.7 | 136.0 | ns | ||
| Row effect: | 0.0494 | 4 (int RhebCA) | Low | 144.7 | 138.2 | ns | ||
| F(1,16) = 4.522 | Intermediate | 179.3 | 163.5 | * | ||||
| Column effect: | 0.0006 | Ipsi | ||||||
| F(2,16) = 12.05 | Control vs low | 131.7 | 144.7 | ns | ||||
| Subjects (matching): | 0.0022 | Control vs int | 131.7 | 179.3 | **** | |||
| F(16,16) = 4.522 | Low vs int | 144.7 | 179.3 | *** | ||||
| Contra | ||||||||
| Control vs low | 136.0 | 138.2 | ns | |||||
| Control vs int | 136.0 | 163.5 | ** | |||||
| Low vs int | 138.2 | 163.5 | * | |||||
| 6E | One-way ANOVA | F(2,18) = 7.472 | 0.0043 | 7 (control) | Control vs low RhebCA | 49.7 | 51.6 | ns |
| 8 (low RhebCA) | Control vs int RhebCA | 49.7 | 72.0 | ** | ||||
| 6 (int RhebCA) | Low vs int RhebCA | 51.6 | 72.0 | * | ||||
*p < 0.05,
**p < 0.01,
***p < 0.001,
****p < 0.0001.
Results
We previously reported that focal expression of RhebCA in developing neurons by in utero electroporation in mice results in mTORC1 hyperactivation and typical features of TSC and FCD, including neuronal hypertrophy, ectopic neuron placement, and epilepsy (Gong et al., 2015; Hsieh et al., 2016; Lin et al., 2016). Here, we extend this model to examine whether the levels of mTORC1 hyperactivity affect the severity of these phenotypes (Fig. 1A,B). To induce differential mTORC1 activity levels in neurons, we electroporated three varying concentrations of the RhebCA plasmid, 1.5, 2.5, and 3.5 μg/μl (hereafter referred as low, intermediate, and high, respectively), into the mouse cortex. We performed electroporation at E15, which selectively introduces the plasmid into neural progenitor cells that generate pyramidal neurons destined to L2/3. Moreover, we preferentially targeted expression in the mPFC because 50–90% of the focal malformations are found in the frontal lobes of patients (Braffman et al., 1992; Houser and Gomez, 1992; Baron and Barkovich, 1999; Colombo et al., 2009; Kabat and Król, 2012). In all conditions, mice were co-electroporated with a GFP reporter plasmid to visualize targeted cells. As controls, littermates were electroporated with GFP plasmid only.
Figure 1.

Experimental design and timeline. A, Simplified diagram of RhebCA-induced mTORC1 activation. Rheb is a small GTPase upstream of mTORC1. GTP-bound Rheb directly binds to and activates mTORC1. The constitutively active mutant form of Rheb, RhebCA, selects for the GTP-bound form, thereby leading to persistent activation of mTORC1. B, Timeline for experiments. Mice were electroporated with GFP (control) or varying concentrations of GFP + RhebCA plasmids at E15, targeting L2/3 neurons in the cortex. Pups were screened for successful electroporation via GFP expression at P0. Mice were monitored with video-EEG recording for 5–7 consecutive days at 2–4 months of age and afterward euthanized for histological and immunohistochemical analysis.
RhebCA expression induces mTORC1 hyperactivation and neuronal hypertrophy in a dose-dependent manner
To verify that increasing RhebCA plasmid concentration leads to increased levels of mTORC1 hyperactivation, we performed immunofluorescence staining for p-S6, a common marker of mTORC1 activity (Ma and Blenis, 2009), in brain sections from 2- to 4-month-old control- and RhebCA-electroporated mice (Fig. 2A). Because seizure activity may lead to secondary increases in p-S6 throughout the brain (Buckmaster et al., 2009; Zeng et al., 2009), we assessed p-S6 staining intensity on the ipsilateral cortex and normalized to that of the non-electroporated contralateral side for each animal to directly evaluate RhebCA-induced p-S6 changes. In addition, we assessed p-S6 intensity both at the levels of cortical tissue (Fig. 2B,C) and electroporated cells (Fig. 2D,E) to compare the two approaches. We found significant increases in both cortical and cellular p-S6 intensity in the low, intermediate, and high RhebCA conditions compared with control. Moreover, the high RhebCA condition displayed significantly increased cortical and cellular p-S6 intensity compared with both the low and intermediate RhebCA conditions (p < 0.0001; one-way ANOVA with Tukey's post hoc test; Fig. 2B–E, Table 1). Additional correlation analyses between cortical and cellular p-S6 intensity demonstrate a significant correlation between the two variables (r = 0.8501, p < 0.0001; Pearson's product–moment correlation; Fig. 2F, Table 1), suggesting that both analysis approaches are valid. Together, these data support that increasing RhebCA plasmid concentration leads to increased mTORC1 hyperactivity levels.
Figure 2.

RhebCA expression induces S6 phosphorylation and somatic hypertrophy in a dose-dependent manner. A, Diagram of a coronal brain section illustrating the electroporated region (green) of the cortex. Ipsilateral side is the electroporated cortex; contralateral side is the non-electroporated cortex. Red square indicates the area imaged in Figure 2B. Ipsi, Ipsilateral; Contra, contralateral; ctx, cortex; cc, corpus callosum; ml, midline. B, Inverted grayscale images of p-S6 staining in ipsilateral and contralateral cortices from control and low, intermediate, and high RhebCA mice. Scale bar, 200 μm. C, Quantification of p-S6 staining intensity in ipsilateral cortices. p < 0.0001 by one-way ANOVA. Tukey's post hoc test: *p < 0.05, **p < 0.01, ****p < 0.0001. D, Images of p-S6 staining (red) and GFP+ cells (green) in ipsilateral cortices from control and low, intermediate, and high RhebCA mice. Scale bar, 100 μm. E, Quantification of p-S6 staining intensity in GFP+ (electroporated) cells. p < 0.0001 by one-way ANOVA. Tukey's post hoc test: *p < 0.05, **p < 0.01, ****p < 0.0001. F, Scatterplot of cellular versus cortical p-S6 staining intensity. r = 0.8501, p < 0.0001 by Pearson's product–moment correlation. G, Images of GFP+ cells from control and low, intermediate, and high RhebCA mice. White squares on the top panel are enlarged on the bottom panel. Scale bar, 50 μm (top and bottom). H, Quantification of GFP+ cell soma size. p < 0.0001 by one-way ANOVA. Tukey's post hoc test: ****p < 0.0001. I, Scatterplot of GFP+ cell soma size versus cortical p-S6 staining intensity. r = 0.8257, p < 0.0001 by Pearson's product–moment correlation. Error bars indicate ± SEM. Con, Control; Int, intermediate.
Because mTORC1 hyperactivation causes neuronal hypertrophy (Kwon et al., 2003; Tavazoie et al., 2005; Kassai et al., 2014), we also measured the soma size of GFP+ electroporated neurons as a proxy for mTORC1 activity. We found significantly increased soma size of electroporated neurons in the low, intermediate, and high RhebCA conditions compared with control. Moreover, the soma size of the intermediate RhebCA neurons was significantly larger than those of the low RhebCA neurons, whereas the soma size of the high RhebCA neurons was significantly larger than both the low and intermediate RhebCA neurons, indicating that increasing RhebCA concentration leads to increased neuronal soma size (p < 0.0001; one-way ANOVA with Tukey's post hoc test; Fig. 2G,H, Table 1). Further evaluation of the relationship between neuronal soma size and p-S6 level revealed a significant positive correlation between these two markers of mTORC1 activity (r = 0.8257, p < 0.0001; Pearson's product moment-correlation; Fig. 2I, Table 1). Collectively, these data demonstrate that RhebCA expression induces mTORC1 hyperactivation and neuronal hypertrophy in a dose-dependent manner in vivo.
High but not low levels of mTORC1 hyperactivity cause ectopic neuronal placement
We next evaluated the distribution of electroporated neurons in brain sections from 2- to 4-month-old control- and RhebCA-electroporated mice (Fig. 3A). As expected, all electroporated neurons in the control condition correctly integrated to L2/3. Similarly, most electroporated neurons in the low RhebCA condition were properly positioned in L2/3, whereas only ∼50% of electroporated neurons in the intermediate and high RhebCA conditions were found in L2/3. Quantification of the percentage of electroporated neurons in L2/3 revealed no statistical differences between the low RhebCA and control conditions. In contrast, the intermediate and high RhebCA conditions were significantly different from both the control and low RhebCA conditions, but not from each other (p < 0.0001; one-way ANOVA with Tukey's post hoc test; Fig. 3B–D, Table 1). The binned distribution of electroporated neurons across the cortex was similar in control and low RhebCA conditions but significantly different in the intermediate and high RhebCA conditions relative to control (p < 0.0001, interaction; two-way repeated-measures ANOVA; Fig. 3E,F, Table 1). Together, these data indicate that higher but not lower levels of mTORC1 hyperactivation lead to ectopic neuronal placement. The finding that the low RhebCA condition, which sufficiently induced neuronal hypertrophy (Fig. 2G,H), did not cause neuronal misplacement suggests that there is a different threshold level of mTORC1 hyperactivation required to induce neuronal misplacement compared with neuronal hypertrophy.
Figure 3.

High but not low levels of mTORC1 hyperactivity cause ectopic neuronal placement. A, Diagram of a coronal brain section illustrating the electroporated region (green) of the cortex. Red square indicates the area imaged in Figure 3B. Ipsi, Ipsilateral; Contra, contralateral; ctx, cortex; cc, corpus callosum; ml, midline. B, Inverted grayscale images of GFP+ neurons in ipsilateral cortices from control and low, intermediate, and high RhebCA mice. Scale bar, 100 μm. C, Diagram of the ROI used to quantify the percentage of GFP+ cells in L2/3. D, Quantification of the percentage of GFP+ cells in L2/3. p < 0.0001 by one-way ANOVA. Tukey's post hoc test: **p < 0.01, ***p < 0.001, ****p < 0.0001. E, Diagram of the binning system used to quantify GFP+ cell distribution in the cortex. F, Quantification of GFP+ cell distribution in the cortex. The number of GFP+ cells in each bin (1–10) was divided by the total number of GFP+ cells. p < 0.0001 by two-way repeated-measures ANOVA. Tukey's post hoc test (simple main effects within cortex bins compared with control): *p < 0.01, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars indicate ± SEM. Con, Control; Int, intermediate.
Increasing levels of mTORC1 hyperactivity lead to increased seizure frequency
To determine whether increasing levels of mTORC1 hyperactivity leads to a more severe seizure phenotype, we performed continuous video-EEG recording in 2- to 4-month-old control- and RhebCA-electroporated mice for 5–7 consecutive days. We detected no seizures in the control (n = 7/7) and low RhebCA (n = 8/8) mice. In contrast, 86% of the intermediate RhebCA mice (n = 6/7) and 100% of the high RhebCA mice (n = 12/12) exhibited spontaneous, recurrent seizures (Fig. 4A). Quantification of the daily seizure frequency revealed a significant positive correlation between RhebCA plasmid concentration and seizure frequency (r = 0.8709, p < 0.0001; Spearman's rank–order correlation; Fig. 4B, Table 1). Further analyses revealed significant positive correlations between seizure frequency and p-S6 intensity (r = 0.6818, p < 0.0018; Spearman's rank–order correlation; Fig. 4C, Table 1), as well as between seizure frequency and neuronal soma size (r = 0.8693, p < 0.0001; Spearman's rank–order correlation; Fig. 4D, Table 1). The number of seizures per day was significantly increased in the high RhebCA compared with the intermediate RhebCA condition (p = 0.0208; Mann–Whitney U test; Fig. 4E, Table 1). However, no statistical differences in the duration of seizures were observed between the two conditions (p = 0.4240; Mann–Whitney U test; Fig. 4F, Table 1). The daily seizure frequency within individual animals remained overall consistent over the course of the recording period in both the intermediate and high RhebCA conditions (Fig. 4G). Collectively, these findings support that increasing levels of mTORC1 hyperactivity leads to increased seizures in mice. The low RhebCA condition did not result in seizures, in contrast to the intermediate and high RhebCA conditions, suggesting that there is a threshold level of mTORC1 hyperactivation required for seizure induction. Furthermore, our data from the intermediate and high RhebCA conditions support that, past the threshold level, there is a graded event in which increased mTORC1 hyperactivity levels leads to increased seizure frequency.
Figure 4.

Increasing levels of mTORC1 hyperactivity lead to increased seizure frequency. A, Representative EEG traces from control and low, intermediate, and high RhebCA mice. Blue rectangles denote traces enlarged on the right. B, Correlation between RhebCA plasmid concentration and mean number of seizures per day. r = 0.8709, p < 0.0001 by Spearman's rank–order correlation. C, Scatterplot of mean number of seizures per day versus cortical p-S6 intensity. r = 0.6818, p = 0.0018 by Spearman's rank–order correlation. D, Scatterplot of mean number of seizures per day versus GFP+ cell soma size. r = 0.8693, p < 0.0001 by Spearman's rank–order correlation. E, Mean number of seizures per day. *p = 0.0208 by Mann–Whitney U test. F, Mean seizure duration. One of the intermediate RhebCA mice did not exhibit seizures and was therefore not included in this analysis. p = 0.4240 by Mann–Whitney U test. G, Heat map showing the number of seizures per day over the total recording period (5–7 days) for individual animals. Error bars indicate ± SEM. Int, Intermediate.
Given that the proportion of cells with aberrant mTORC1 activation (i.e., number of electroporated cells) may affect seizure severity, we compared the number of GFP+ cells in the brain section with the highest GFP+ cell count from a rostrocaudal series of sections for each animal. We found that the intermediate RhebCA condition had a higher GFP+ cell count than the control and low RhebCA conditions, whereas none of the other groups was different from each other (p = 0.0103; one-way ANOVA with Tukey's post hoc test; Fig. 5A, Table 1). Nonetheless, there was no significant correlation between the number of GFP+ cells and seizure frequency among the RhebCA conditions (r = 0.4294, p = 0.0754; Spearman's rank–order correlation; Fig. 5B, Table 1), supporting that the observed intergroup differences in seizure frequency are due to differences in cellular mTORC1 hyperactivity levels rather than differences in the proportion of electroporated cells.
Figure 5.
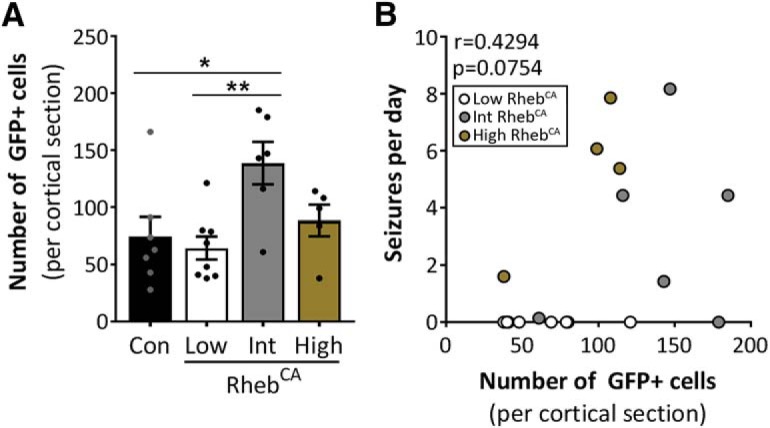
The number of electroporated cells does not affect intergroup differences in seizure frequency. A, Quantification of the number of GFP+ cells in single cortical sections containing the highest GFP+ cell count from control and low, intermediate, and high RhebCA mice. p = 0.0103 by one-way ANOVA. Tukey's post hoc test: *p < 0.05, **p < 0.01. B, Scatterplot of mean number of seizures per day versus number of GFP+ cells per single cortical section. r = 0.4294, p = 0.0754 by Spearman's rank–order correlation. Error bars indicate ± SEM. Con, Control; Int, intermediate.
RhebCA-induced seizures are associated with increased microglial activation
Microglia activation, including alterations in microglia density and morphology, have been described in human TSC tubers and FCDII brain specimens (Boer et al., 2006, 2008a,b; Choi et al., 2009; Iyer et al., 2010), but it is unclear whether these microglia changes occur in the present mouse model. We thus immunostained for the microglia-specific protein IBA1 in brain sections from 2- to 4-month-old control, low RhebCA, and intermediate RhebCA mice. Tissue was not available for the high RhebCA condition and was therefore not included. We observed prominent increases in IBA1+ microglia staining in the intermediate RhebCA condition compared with the control and low RhebCA conditions (Fig. 6A). Quantification of IBA1+ cell density in the ipsilateral cortex revealed a significant increase in the intermediate RhebCA condition compared with control (p = 0.0091; one-way ANOVA with Tukey's post hoc test; Fig. 6B, Table 1), suggesting the presence of microglia activation. While analyzing tissue from the intermediate RhebCA condition, we noticed a large intragroup difference in IBA1+ staining pattern, with some animals displaying marked increases in microglia density and others appearing similar to the control (open circles in Fig. 6B). To determine whether these changes could be associated with seizure activity, we performed correlation analyses between seizure frequency and IBA1+ cell density. Indeed, we found a significant correlation between IBA1+ cell density and seizures (r = 0.5502; p = 0.0441; Spearman's rank–order correlation; Fig. 6C, Table 1). To further investigate whether increased IBA1+ cell density reflects a global change induced by seizures or a localized effect associated with the electroporated, “disordered” neurons, we compared IBA1+ cell density in the ipsilateral and contralateral cortices. The two animals from the intermediate RhebCA conditions with ≤1 seizure in 7 days were excluded from this analysis. We found that IBA1+ cell density on both the ipsilateral and contralateral sides of the intermediate RhebCA condition was higher compared with the corresponding cortices in the control and low RhebCA conditions (p = 0.0414, interaction; two-way repeated-measures ANOVA with Bonferroni post hoc test; Fig. 6D, Table 1), suggesting that microglial activation are globally induced by seizures in this model. Interestingly, we also found a significant difference in IBA1+ cell density between the ipsilateral and contralateral cortices within the intermediate RhebCA condition, but not in the control and low RhebCA conditions, suggesting that the presence of disordered neurons leads to a larger microglia activation.
Figure 6.

RhebCA-induced seizures are associated with increased microglial activation. A, Images of IBA1 staining in ipsilateral and contralateral cortices from control, low RhebCA, and intermediate RhebCA mice. Top, IBA1 staining (red) and GFP+ cells (green). Bottom, Inverted grayscale images of the same IBA1 staining as in the top panels. Scale bar, 100 μm. Ipsi, Ipsilateral; Contra, contralateral. B, Quantification of IBA1+ cell density in ipsilateral cortices. Open circles within the intermediate RhebCA condition indicate animals with ≤1 seizure. p = 0.0091 by one-way ANOVA. Tukey's post hoc test: **p < 0.001. C, Scatterplot of mean number of seizures per day versus IBA1+ cell density. r = 0.5502, p = 0.0441 by Spearman's rank–order correlation. D, Quantification of IBA1+ cell density in ipsilateral and contralateral cortices. p = 0.0414 by two-way repeated-measures ANOVA. Bonferroni post hoc test: *p < 0.01, **p < 0.01, ***p < 0.001, ****p < 0.0001. E, Quantification of IBA1+ cell size in ipsilateral cortices. Open circles within the intermediate RhebCA condition indicate animals with ≤1 seizure. p = 0.0043 by one-way ANOVA. Tukey's post hoc test: *p < 0.05, **p < 0.01. F, Representative images of IBA1-labeled microglia morphology in tissue from control, low RhebCA, and intermediate RhebCA mice. Top, IBA1 staining (red) and DAPI-stained nuclei (blue). Bottom, Inverted grayscale images of the same IBA1 staining as in the top panels. The most apparent microglia morphology observed in control and low RhebCA tissue was ramified, whereas microglia in intermediate RhebCA tissue displayed a variety of morphologies, including ramified (indicated by white arrow), hypertrophic, bushy, and rod. Error bars indicate ± SEM. Con, Control; Int, intermediate.
Activated microglia undergo morphological changes from small cell bodies with ramified (branched) processes to enlarged cell bodies with long or short, thickened processes or ameboid-shaped cells with no processes (Kettenmann et al., 2011). Quantification of IBA1+ cell size in the ipsilateral cortex revealed significantly larger cells in the intermediate RhebCA condition compared with the control and low RhebCA conditions, whereas no differences were observed between the control and low RhebCA conditions (p = 0.0043; one-way ANOVA with Tukey's post hoc test; Fig. 6E, Table 1). Further inspection of IBA1-stained tissue from control, low RhebCA, and intermediate RhebCA mice revealed evident differences in the microglia morphological phenotypes between these groups. Although most microglia had the ramified phenotype in the control and low RhebCA conditions, we identified at least four different types of microglia morphologies, including ramified, hypertrophic, bushy, and rod, that are consistent with activated microglia states in the intermediate RhebCA condition (Fig. 6F). Overall, these findings further validate the presence of microglial activation in association with RhebCA-induced cortical malformations and seizures in this model.
Discussion
TSC and FCD result from a spectrum of pathogenic variants in the mTOR pathway that leads to differential mTOR hyperactivation (Crino, 2015). In this study, we demonstrate that the levels of mTOR hyperactivity correlate with the degree of neuronal hypertrophy, misplacement, and seizure frequency in an experimental TSC and FCD model. In addition, we describe the presence of microglia activation following mTOR-induced seizures.
The mTOR pathway regulates key processes involved in cell growth, including protein synthesis, energy metabolism, and autophagy, and aberrant mTOR activation has been implicated in epilepsy (Meng et al., 2013; Ostendorf and Wong, 2015; Saxton and Sabatini, 2017). Although the specific mechanisms underlying mTOR-induced epilepsy remain unclear, it is thought that structural changes following mTOR hyperactivation lead to circuit disorganization and altered intrinsic and network excitability that promote seizure generation (Lasarge and Danzer, 2014). Traditional methods for studying mTOR functions in epilepsy involve knocking out negative regulators of mTOR in transgenic mouse models to enhance mTOR activation (Feliciano et al., 2013; Ostendorf and Wong, 2015). Although these approaches have been instrumental in establishing the causative roles of mTOR signaling in epilepsy, investigating how the levels of mTOR hyperactivity influence epilepsy severity in these models have been limited to either the homozygosity or heterozygosity of the gene knock-out. In the present model, we expressed a constitutive activator of mTORC1, RhebCA, via in utero electroporation. This method allowed us to manipulate the levels of mTORC1 activity by varying the amount of electroporated RhebCA plasmids. Although we did not quantify the copy numbers that are transfected into cells under the different conditions, we speculate that higher copy numbers will be transfected with higher concentrations, thereby leading to higher levels of mTORC1 activation. Our data showing corresponding increases in p-S6 and neuronal soma size with increasing RhebCA plasmid concentration support this notion. By varying the degree of mTORC1 hyperactivity, we show that there is a threshold level of mTORC1 hyperactivation required to induce seizures, followed by a graded event whereby increasing mTORC1 hyperactivity levels leads to increased seizure frequency. Our findings that higher mTOR hyperactivity levels contribute to more severe epilepsy suggest that differential levels of mTOR hyperactivation caused by distinct pathogenic variants may contribute to the variable phenotype severities in TSC and FCD.
Therapeutic strategies based on mTOR pathway inhibition for TSC- and FCD-related epilepsy are emerging. Although clinical trials with the mTOR inhibitor everolimus improved seizure control in many TSC patients, 30–40% of patients did not satisfactorily respond to treatment (Krueger et al., 2013; Wiegand et al., 2013; French et al., 2016). The wide array of variants along the mTOR pathway in TSC and FCD has raised the question of whether everolimus and other mTOR inhibitors provide the same efficacy for TSC or FCD resulting from distinct mTOR pathway variants (Iffland and Crino, 2017). Our findings that the levels of mTOR hyperactivity influence epilepsy severity may illuminate why current therapeutics based on mTOR inhibitors have variable outcomes on patients and suggest the need to consider patient gene variants in the context of mTOR activation levels in clinical assessments. As advanced genetic tools become more common, new pathogenic variants in the mTOR pathway are likely to be identified. Functional evaluation of these variants to determine their impact on the mTOR pathway and disease outcomes may aid in the establishment of a personalized and potentially more effective treatment regimen.
mTOR hyperactivation in the developing cortex causes neuronal hypertrophy and abnormal neuronal migration leading to cortical dyslamination (Feliciano et al., 2011; Kassai et al., 2014; Tsai et al., 2014; Hsieh et al., 2016; Lin et al., 2016). We demonstrate that neuronal soma size strongly correlates with the levels of mTOR hyperactivity and is a reliable readout of cellular mTOR activity level. Our cell size data showed a sigmoidal increase with increasing RhebCA concentrations, suggesting that there is a plateau level for either mTOR activation or cell size. Nonetheless, determining the plateau level may be technically challenging as excess plasmid concentrations could be toxic to cells. Additionally, we show that high but not low levels of mTORC1 hyperactivation result in neuronal misplacement. We found most neurons in the low RhebCA condition to be in the correct cortical layers despite having mTORC1 hyperactivation and neuronal hypertrophy. This finding suggests that there is a threshold level of mTORC1 hyperactivity needed for neuronal misplacement that is different than for neuronal hypertrophy.
Until recently, the mechanisms by which mTOR hyperactivation induce cortical dyslamination has been unknown. A new study by Park et al. (2018) reported that brain somatic mutations in the MTOR gene lead to neuronal misplacement and cortical dyslamination by disrupting autophagy-mediated neuronal ciliogenesis. Therefore, defective ciliogenesis is a potential mechanism by which mTOR hyperactivation induced neuronal misplacement in our model. Although neuronal misplacement seems to parallel seizure occurrence (i.e., no misplacement or seizures in low RhebCA, misplacement and seizures in intermediate and high RhebCA), the role of cortical dyslamination in epileptogenesis is unclear. We previously used an inducible RhebCA plasmid to bypass neuronal misplacement and observed that misplacement is not required for seizures and neuronal hypertrophy (Hsieh et al., 2016). This finding was corroborated by Park et al. (2018). Nonetheless, the specific contribution of cortical dyslamination to seizure severity and other neurological manifestations associated with mTOR hyperactivation remain elusive and additional studies are needed to elucidate these issues.
Studies on brain somatic mutations along the mTOR pathway have reported that the percentage of cells carrying the mutations correlate with the extent of cortical malformations and phenotypic severity (Blümcke and Sarnat, 2016; D'Gama et al., 2017). Given that the proportion of cells with aberrant mTORC1 activation (i.e., number of electroporated cells) may affect epilepsy severity in our studies, we compared the number of GFP+ cells in individual animals. We found no correlation between GFP+ cell count and seizure frequency, suggesting the identified intergroup differences in seizures is due to differences in cellular mTOR hyperactivity levels rather than the proportion of cells with mTORC1 hyperactivation. Nonetheless, we do not preclude the possibility that, within groups of similar RhebCA concentrations, increasing the number of electroporated cells or brain region could lead to worsening seizures. Interestingly, studies by Pun et al. (2012) using genetic knock-out of Pten, a negative regulator of mTOR, in mice showed that Pten loss in 9–24% of hippocampal cells sufficiently induced seizures, whereas <1% Pten loss did not. Collectively, we speculate that both the degree of mTOR hyperactivity within cells, as shown here, and the number of affected cells contribute to epilepsy severity.
Activation of microglia, the main immune cells of the CNS, have been found in brain specimens from TSC and FCD patients (Boer et al., 2006, 2008a,b; Choi et al., 2009; Iyer et al., 2010). Our data in mice demonstrating changes in microglia density and morphology corroborate the findings in humans. Interestingly, we observed microglial changes only in animals that displayed recurrent seizures and in both the ipsilateral and contralateral cortices, suggesting that microglia activation occurs as a consequence of seizures. Whether this represents a neurotoxic or neuroprotective mechanism is unclear and constitutes an active field of research (Eyo et al., 2017; Hiragi et al., 2018).
In addition to the biological implications of our study, we present practical modifications to a previously described in utero electroporation-based mouse model that allows for the titration of seizure frequency and generation of a mild to severe epilepsy phenotype as applicable for preclinical drug testing and mechanistic studies (Hsieh et al., 2016). For example, electroporation using lower RhebCA plasmid concentrations can be used to generate mice for examining cellular or behavioral effects of mTOR hyperactivation in the absence of secondary changes due to seizures, whereas higher RhebCA concentrations can be used to generate mice with frequent seizures to evaluate potential antiepileptic treatments. We emphasize that the RhebCA concentrations described in this study are relative and not absolute. Investigator-dependent variability in plasmid preparation, injection volume, and electroporation techniques as well as electroporation parameters (i.e., voltage, number of pulses, etc.) could yield variable results. Therefore, investigators using this model should empirically determine the optimal dose for their experiments. Nonetheless, we demonstrate that a graded increase in RhebCA plasmid concentration results in increased mTOR hyperactivity levels and more severe phenotypes.
In summary, we show here that mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of TSC and FCD. Our study suggests that differences in mTOR hyperactivity levels may contribute to the broad range of phenotypic severity in TSC and FCD and support the need to evaluate mTOR hyperactivity levels resulting from distinct patient variants. Future studies to address whether increasing mTOR hyperactivity levels worsen other behavioral and cognitive phenotypes may provide additional insights into the role of mTOR signaling in these aspects of TSC and FCD. Finally, whereas we limited our study to TSC and FCD strictly based on pathological definitions, our findings are potentially broadly applicable to other mTORopathies.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke–National Institutes of Health (Grant NS086329 to A.B.), the American Epilepsy Society Postdoctoral Fellowship (to L.H.N.), and the Brown Coxe Postdoctoral Fellowship (to L.H.N).
The authors declare no competing financial interests.
References
- Aronica E, Becker AJ, Spreafico R (2012) Malformations of cortical development. Brain Pathol 22:380–401. 10.1111/j.1750-3639.2012.00581.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron Y, Barkovich AJ (1999) MR imaging of tuberous sclerosis in neonates and young infants. AJNR Am J Neuroradiol 20:907–916. [PMC free article] [PubMed] [Google Scholar]
- Blümcke I, Spreafico R, Haaker G, Coras R, Kobow K, Bien CG, Pfäfflin M, Elger C, Widman G, Schramm J, Becker A, Braun KP, Leijten F, Baayen JC, Aronica E, Chassoux F, Hamer H, Stefan H, Rössler K, Thom M, et al. (2017) Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med 377:1648–1656. 10.1056/NEJMoa1703784 [DOI] [PubMed] [Google Scholar]
- Blümcke I, Sarnat HB (2016) Somatic mutations rather than viral infection classify focal cortical dysplasia type II as mTORopathy. Curr Opin Neurol 29:388–395. 10.1097/WCO.0000000000000303 [DOI] [PubMed] [Google Scholar]
- Boer K, Spliet WG, van Rijen PC, Redeker S, Troost D, Aronica E (2006) Evidence of activated microglia in focal cortical dysplasia. J Neuroimmunol 173:188–195. 10.1016/j.jneuroim.2006.01.002 [DOI] [PubMed] [Google Scholar]
- Boer K, Troost D, Jansen F, Nellist M, van den Ouweland AM, Geurts JJ, Spliet WG, Crino P, Aronica E (2008a) Clinicopathological and immunohistochemical findings in an autopsy case of tuberous sclerosis complex. Neuropathology 28:577–590. 10.1111/j.1440-1789.2008.00920.x [DOI] [PubMed] [Google Scholar]
- Boer K, Jansen F, Nellist M, Redeker S, van den Ouweland AM, Spliet WG, van Nieuwenhuizen O, Troost D, Crino PB, Aronica E (2008b) Inflammatory processes in cortical tubers and subependymal giant cell tumors of tuberous sclerosis complex. Epilepsy Res 78:7–21. 10.1016/j.eplepsyres.2007.10.002 [DOI] [PubMed] [Google Scholar]
- Braffman BH, Bilaniuk LT, Naidich TP, Altman NR, Post MJ, Quencer RM, Zimmerman RA, Brody BA (1992) MR imaging of tuberous sclerosis: pathogenesis of this phakomatosis, use of gadopentetate dimeglumine, and literature review. Radiology 183:227–238. 10.1148/radiology.183.1.1549677 [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Ingram EA, Wen X (2009) Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci 29:8259–8269. 10.1523/JNEUROSCI.4179-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Nordli DR Jr, Alden TD, DiPatri A Jr, Laux L, Kelley K, Rosenow J, Schuele SU, Rajaram V, Koh S (2009) Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J Neuroinflammation 6:38. 10.1186/1742-2094-6-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo N, Salamon N, Raybaud C, Ozkara C, Barkovich AJ (2009) Imaging of malformations of cortical development. Epileptic Disord 11:194–205. 10.1684/epd.2009.0262 [DOI] [PubMed] [Google Scholar]
- Crino PB. (2015) mTOR signaling in epilepsy: insights from malformations of cortical development. Cold Spring Harb Perspect Med 5:a022442. 10.1101/cshperspect.a022442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curatolo P, Moavero R, Roberto D, Graziola F (2015) Genotype/Phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol 22:259–273. 10.1016/j.spen.2015.10.002 [DOI] [PubMed] [Google Scholar]
- D'Gama AM, Woodworth MB, Hossain AA, Bizzotto S, Hatem NE, LaCoursiere CM, Najm I, Ying Z, Yang E, Barkovich AJ, Kwiatkowski DJ, Vinters HV, Madsen JR, Mathern GW, Blümcke I, Poduri A, Walsh CA (2017) Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep 21:3754–3766. 10.1016/j.celrep.2017.11.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Murugan M, Wu LJ (2017) Microglia-neuron communication in epilepsy. Glia 65:5–18. 10.1002/glia.23006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciano DM, Su T, Lopez J, Platel JC, Bordey A (2011) Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J Clin Invest 121:1596–1607. 10.1172/JCI44909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciano DM, Lin TV, Hartman NW, Bartley CM, Kubera C, Hsieh L, Lafourcade C, O'Keefe RA, Bordey A (2013) A circuitry and biochemical basis for tuberous sclerosis symptoms: from epilepsy to neurocognitive deficits. Int J Dev Neurosci 31:667–678. 10.1016/j.ijdevneu.2013.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, Curatolo P, de Vries PJ, Dlugos DJ, Berkowitz N, Voi M, Peyrard S, Pelov D, Franz DN (2016) Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 388:2153–2163. 10.1016/S0140-6736(16)31419-2 [DOI] [PubMed] [Google Scholar]
- Gong X, Zhang L, Huang T, Lin TV, Miyares L, Wen J, Hsieh L, Bordey A (2015) Activating the translational repressor 4E-BP or reducing S6K-GSK3beta activity prevents accelerated axon growth induced by hyperactive mTOR in vivo. Hum Mol Genet 24:5746–5758. 10.1093/hmg/ddv295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH, Sabatini DM (2014) A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 4:554–563. 10.1158/2159-8290.CD-13-0929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey AS, Cross JH, Shinnar S, Mathern GW; ILAE Pediatric Epilepsy Surgery Survey Taskforce (2008) Defining the spectrum of international practice in pediatric epilepsy surgery patients. Epilepsia 49:146–155. 10.1111/j.1528-1167.2007.01421.x [DOI] [PubMed] [Google Scholar]
- Hiragi T, Ikegaya Y, Koyama R (2018) Microglia after seizures and in epilepsy. Cells 7:E26. 10.3390/cells7040026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser OW, Gomez MR (1992) CT and MR imaging of intracranial tuberous sclerosis. J Dermatol 19:904–908. 10.1111/j.1346-8138.1992.tb03803.x [DOI] [PubMed] [Google Scholar]
- Hsieh LS, Wen JH, Claycomb K, Huang Y, Harrsch FA, Naegele JR, Hyder F, Buchanan GF, Bordey A (2016) Convulsive seizures from experimental focal cortical dysplasia occur independently of cell misplacement. Nat Commun 7:11753. 10.1038/ncomms11753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iffland PH 2nd, Crino PB (2016) Sending mixed signals: the expanding role of molecular cascade mutations in malformations of cortical development and epilepsy. Epilepsy Curr 16:158–163. 10.5698/1535-7511-16.3.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iffland PH 2nd, Crino PB (2017) Focal cortical dysplasia: gene mutations, cell signaling, and therapeutic implications. Annu Rev Pathol 12:547–571. 10.1146/annurev-pathol-052016-100138 [DOI] [PubMed] [Google Scholar]
- Iyer A, Zurolo E, Spliet WG, van Rijen PC, Baayen JC, Gorter JA, Aronica E (2010) Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 51:1763–1773. 10.1111/j.1528-1167.2010.02547.x [DOI] [PubMed] [Google Scholar]
- Kabat J, Król P (2012) Focal cortical dysplasia: review. Pol J Radiol 77:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassai H, Sugaya Y, Noda S, Nakao K, Maeda T, Kano M, Aiba A (2014) Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep 7:1626–1639. 10.1016/j.celrep.2014.04.048 [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91:461–553. 10.1152/physrev.00011.2010 [DOI] [PubMed] [Google Scholar]
- Krueger DA, Wilfong AA, Holland-Bouley K, Anderson AE, Agricola K, Tudor C, Mays M, Lopez CM, Kim MO, Franz DN (2013) Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol 74:679–687. 10.1002/ana.23960 [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Baker SJ (2003) mTor is required for hypertrophy of pten-deficient neuronal soma in vivo. Proc Natl Acad Sci U S A 100:12923–12928. 10.1073/pnas.2132711100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasarge CL, Danzer SC (2014) Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front Mol Neurosci 7:18. 10.3389/fnmol.2014.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventer RJ, Guerrini R, Dobyns WB (2008) Malformations of cortical development and epilepsy. Dialogues Clin Neurosci 10:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, Cho YW, Kim S, Kim HM, Kim JA, Kim J, Rhee H, Kang SG, Kim HD, Kim D, Kim DS, Lee JH (2015) Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 21:395–400. 10.1038/nm.3824 [DOI] [PubMed] [Google Scholar]
- Lim JS, Gopalappa R, Kim SH, Ramakrishna S, Lee M, Kim WI, Kim J, Park SM, Lee J, Oh JH, Kim HD, Park CH, Lee JS, Kim S, Kim DS, Han JM, Kang HC, Kim HH, Lee JH (2017) Somatic mutations in TSC1 and TSC2 cause focal cortical dysplasia. Am J Hum Genet 100:454–472. 10.1016/j.ajhg.2017.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TV, Hsieh L, Kimura T, Malone TJ, Bordey A (2016) Normalizing translation through 4E-BP prevents mTOR-driven cortical mislamination and ameliorates aberrant neuron integration. Proc Natl Acad Sci U S A 113:11330–11335. 10.1073/pnas.1605740113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton JO, Sahin M (2014) The neurology of mTOR. Neuron 84:275–291. 10.1016/j.neuron.2014.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J (2009) Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10:307–318. 10.1038/nrm2672 [DOI] [PubMed] [Google Scholar]
- Maehama T, Tanaka M, Nishina H, Murakami M, Kanaho Y, Hanada K (2008) RalA functions as an indispensable signal mediator for the nutrient-sensing system. J Biol Chem 283:35053–35059. 10.1074/jbc.M805822200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsan E, Baulac S (2018) Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol Appl Neurobiol 44:6–17. 10.1111/nan.12463 [DOI] [PubMed] [Google Scholar]
- Meng XF, Yu JT, Song JH, Chi S, Tan L (2013) Role of the mTOR signaling pathway in epilepsy. J Neurol Sci 332:4–15. 10.1016/j.jns.2013.05.029 [DOI] [PubMed] [Google Scholar]
- Mirzaa GM, Campbell CD, Solovieff N, Goold C, Jansen LA, Menon S, Timms AE, Conti V, Biag JD, Adams C, Boyle EA, Collins S, Ishak G, Poliachik S, Girisha KM, Yeung KS, Chung BHY, Rahikkala E, Gunter SA, McDaniel SS, et al. (2016) Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol 73:836–845. 10.1001/jamaneurol.2016.0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nellist M, Sancak O, Goedbloed MA, Rohe C, van Netten D, Mayer K, Tucker-Williams A, van den Ouweland AM, Halley DJ (2005) Distinct effects of single amino-acid changes to tuberin on the function of the tuberin-hamartin complex. Eur J Hum Genet 13:59–68. 10.1038/sj.ejhg.5201276 [DOI] [PubMed] [Google Scholar]
- Northrup H, Koenig MK, Pearson DA, Au KS (1999) Tuberous sclerosis complex. Research In: GeneReviews (Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds). Seattle, WA: University of Washington. [PubMed] [Google Scholar]
- Orlova KA, Crino PB (2010) The tuberous sclerosis complex. Ann N Y Acad Sci 1184:87–105. 10.1111/j.1749-6632.2009.05117.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostendorf AP, Wong M (2015) mTOR inhibition in epilepsy: rationale and clinical perspectives. CNS Drugs 29:91–99. 10.1007/s40263-014-0223-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Lim JS, Ramakrishina S, Kim SH, Kim WK, Lee J, Kang HC, Reiter JF, Kim DS, Kim HH, Lee JH (2018) Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron 99:83–97.e7. 10.1016/j.neuron.2018.05.039 [DOI] [PubMed] [Google Scholar]
- Pun RY, Rolle IJ, Lasarge CL, Hosford BE, Rosen JM, Uhl JD, Schmeltzer SN, Faulkner C, Bronson SL, Murphy BL, Richards DA, Holland KD, Danzer SC (2012) Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 75:1022–1034. 10.1016/j.neuron.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosset C, Netto CBO, Ashton-Prolla P (2017) TSC1 and TSC2 gene mutations and their implications for treatment in tuberous sclerosis complex: a review. Genet Mol Biol 40:69–79. 10.1590/1678-4685-gmb-2015-0321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 169:361–371. 10.1016/j.cell.2017.03.035 [DOI] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL (2005) Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8:1727–1734. 10.1038/nn1566 [DOI] [PubMed] [Google Scholar]
- Tsai V, Parker WE, Orlova KA, Baybis M, Chi AW, Berg BD, Birnbaum JF, Estevez J, Okochi K, Sarnat HB, Flores-Sarnat L, Aronica E, Crino PB (2014) Fetal brain mTOR signaling activation in tuberous sclerosis complex. Cereb Cortex 24:315–327. 10.1093/cercor/bhs310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Auvin S, Ravizza T, Aronica E (2012) Glia-neuronal interactions in ictogenesis and epileptogenesis: role of inflammatory mediators. In: Jasper's basic mechanisms of the epilepsies (Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, eds). Bethesda, MD: National Center for Biotechnology Information (US). [PubMed] [Google Scholar]
- Wiegand G, May TW, Ostertag P, Boor R, Stephani U, Franz DN (2013) Everolimus in tuberous sclerosis patients with intractable epilepsy: a treatment option? Eur J Paediatr Neurol 17:631–638. 10.1016/j.ejpn.2013.06.002 [DOI] [PubMed] [Google Scholar]
- Wong M, Roper SN (2016) Genetic animal models of malformations of cortical development and epilepsy. J Neurosci Methods 260:73–82. 10.1016/j.jneumeth.2015.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, Wong M (2009) The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci 29:6964–6972. 10.1523/JNEUROSCI.0066-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]